

Functional expression of either CFTR or the calcium-activated chloride channel TMEM16A attenuates expression and secretion of the proinflammatory cytokines IL-6, IL-8, and CXCL1/2 in respiratory epithelia. Thus augmented proinflammatory cytokine secretion caused by defective anion transport may contribute to lung inflammation in cystic fibrosis.
Abstract
Cystic fibrosis (CF) is caused by the functional expression defect of the CF transmembrane conductance regulator (CFTR) chloride channel at the apical plasma membrane. Impaired bacterial clearance and hyperactive innate immune response are hallmarks of the CF lung disease, yet the existence of and mechanism accounting for the innate immune defect that occurs before infection remain controversial. Inducible expression of either CFTR or the calcium-activated chloride channel TMEM16A attenuated the proinflammatory cytokines interleukin-6 (IL-6), IL-8, and CXCL1/2 in two human respiratory epithelial models under air–liquid but not liquid–liquid interface culture. Expression of wild-type but not the inactive G551D-CFTR indicates that secretion of the chemoattractant IL-8 is inversely proportional to CFTR channel activity in cftr∆F508/∆F508 immortalized and primary human bronchial epithelia. Similarly, direct but not P2Y receptor–mediated activation of TMEM16A attenuates IL-8 secretion in respiratory epithelia. Thus augmented proinflammatory cytokine secretion caused by defective anion transport at the apical membrane may contribute to the excessive and persistent lung inflammation in CF and perhaps in other respiratory diseases associated with documented down-regulation of CFTR (e.g., chronic obstructive pulmonary disease). Direct pharmacological activation of TMEM16A offers a potential therapeutic strategy to reduce the inflammation of CF airway epithelia.
INTRODUCTION
Cystic fibrosis (CF) is caused by mutations that impair the biosynthesis, function, and/or stability of the cystic fibrosis transmembrane conductance regulator (CFTR), a cAMP-regulated chloride channel (Riordan et al., 1989; Welsh and Smith, 1993; Zielenski and Tsui, 1995). Attenuated chloride and bicarbonate secretion across the apical plasma membrane (PM) of CF airway epithelia lead to dehydration and increased viscosity of the airway surface liquid (ASL), as well as impaired mucociliary clearance, and bacterial colonization in concert with excessive inflammatory response of the CF respiratory epithelia (Matsui et al., 1998; Tarran et al., 2001; Derichs et al., 2011). The augmented release of proinflammatory cytokines promotes neutrophil chemotaxis and degranulation, further compromising mucociliary clearance and enhancing inflammation (Berger, 2002; Elizur et al., 2008; Downey et al., 2009). This positive feedback loop, which culminates in irreversible tissue damage and respiratory insufficiency, represents the major cause of mortality in CF (Ratjen and Döring, 2003; Rowe et al., 2005). A major unresolved question is whether the hyperinflammatory state of airway epithelia is initiated by the CFTR functional expression defect and precedes infection or is merely the consequence of impaired mucociliary clearance and bacterial colonization (Machen, 2006; Ratjen, 2006).
An early onset of increased proinflammatory cytokine secretion into the bronchoalveolar lavage in CF infants has been reported, but whether this occurs before infection remains controversial (Muhlebach and Noah, 2002; Armstrong et al., 2005). Impaired bacterial clearance and hyperinflammation were proposed to be consequences of lung infection in the CF mouse, pig, and ferret (van Heeckeren et al., 2006; Stoltz et al., 2010; Sun et al., 2010). It is intriguing that pancreas in newborn CF pigs shows hallmarks of increased immune and inflammatory response without apparent infection (Abu-El-Haija et al., 2011). Elevated interleukin-8 (IL-8) production has been reported in CF cell lines and primary cultures both constitutively and after activation of Toll-like or tumor necrosis factor α receptors, implying a CFTR-dependent alteration in the innate immune response (Ribeiro et al., 2005; Vandivier et al., 2009; Roussel et al., 2011). However, this could not be reproduced in some cellular models (Pizurki et al., 2000; Hybiske et al., 2007; Fulcher et al., 2009), whereas in others the CFTR appeared to have a permissive role in IL-8 secretion (John et al., 2010, 2011). Resolution of this fundamental and long-lasting controversy is hampered by the modification of the epigenome configuration after repeated infection and inflammation, which could modulate the innate immune response in freshly isolated airway epithelial cells and primary cultures (Adcock et al., 2007; Brigati et al., 2010). Furthermore, whether CFTR expression/signaling (Estell et al., 2003; Bensalem et al., 2005; Hallows et al., 2006; Mehta, 2007) or its transport function (Perez et al., 2007; Hunter et al., 2010; Vega-Carrascal et al., 2011) is necessary and sufficient to suppress proinflammatory cytokine secretions remains an unanswered question.
The recently identified TMEM16A channel (also called Ano1) confers calcium-activated chloride conductance to the PM of respiratory epithelia (Caputo et al., 2008; Schroeder et al., 2008; Yang et al., 2008). Transient mobilization of intracellular Ca2+ by G protein–coupled receptor (GPCR) signaling is widely used to promote TMEM16A activation (Caputo et al., 2008; Yang et al., 2008), and P2Y receptor (P2YR)–mediated activation of TMEM16A has been explored as a potential therapeutic approach in CF (Yerxa et al., 2002; Accurso et al., 2011).
We examined the effect of CFTR variants and TMEM16A expression on proinflammatory cytokine secretion by using human airway epithelia models lacking endogenous CFTR. Inducible expression systems were developed to tightly control the anion channel expression and minimize the effect of genetic heterogeneity, epigenetic variation, and phenotypic drift of epithelia. Here we show that functional CFTR expression attenuates IL-8 secretion from cftr∆F508/∆F508 immortalized and primary human bronchial epithelia under air–liquid culture (ALC) but not liquid–liquid culture (LLC). Similarly, direct but not P2YR-mediated activation of TMEM16A suppressed IL-8 secretion. Taken together, these findings provide a novel link between transepithelial anion transport and IL-8 secretion and suggest that constitutive activation of TMEM16A may be a useful therapeutic target for anti-inflammatory treatment in CF.
RESULTS
Cellular model with inducible CFTR expression for investigating the innate immune response of human bronchial epithelia
We selected CFBE41o– (CFBE) cells, a well-characterized CF airway cell line, to examine the consequence of CFTR expression on proinflammatory cytokine secretion. CFBE cells were originally derived by immortalizing human bronchial epithelial cells from a patient with cftrΔF508/ΔF508 genetic background and have no detectable expression of the mutant protein (Ehrhardt et al., 2006). To develop cells with adjustable CFTR expression, we sequentially transduced CFBE cells with lentiviral particles encoding a tetracycline-controlled transactivator (Gossen and Bujard, 1992) and with wild-type (wt) CFTR bearing an extracellular triple hemagglutinin (3HA) tag (Sharma et al., 2004). The inducible CFTR expression system minimized genetic and epigenetic heterogeneity caused by clonal selection (Babnigg et al., 2000) and the possibility of phenotypic drifts during the propagation of the cells.
Wild-type CFTR-3HA expression was undetectable by immunoblot (IB) in CFBE cells, when the transactivator was expressed alone (TetON+) or in combination with wt CFTR-3HA (iCFTR) without doxycycline (Dox) induction (Figure 1A). Increasing concentrations of Dox induced progressively higher accumulation of the complex-glycosylated form of CFTR, as measured by IB with anti-HA or anti-CFTR antibody (Ab) after 3 d of induction (Figure 1, A and B). At maximal Dox induction the CFTR expression level was comparable to that seen for endogenous CFTR in Calu-3 cells (see Figure 2D). CFTR expression reached a steady-state level after 3 d of Dox induction (Supplemental Figure S1A). Comparable kinetics and Dox concentration dependence were found for CFTR appearance at the PM, determined by cell surface enzyme-linked immunosorbent assay (ELISA; Figure 1C and Supplemental Figure S2A; Okiyoneda et al., 2010).
FIGURE 1:

Inducible CFTR expression in CF human bronchial epithelia (CFBE) attenuates the proinflammatory cytokines IL-8, IL-6, and CXCL1/2. (A) IB of CFBE transduced with transactivator (TetON) or in combination with inducible CFTR carrying an extracellular 3HA tag (iCFTR) with (+) or without (–) Dox (500 ng/ml)–induced expression for 3 d. (B, C) Dox concentration dependence of CFTR cellular expression determined by IB (B) or by cell surface ELISA (C). Densitometric analysis of the complex-glycosylated CFTR expression is expressed as percentage of the maximum (B, bottom). (D) Apical localization of CFTR (green) in polarized CFBE cells by confocal microscopy. ZO1 was used as tight junction marker (red), and nuclei were stained with DAPI (blue). Ap, apical; b, basal. Bar, 5 μm. (E) Maximal CFTR current (Isc) was measured through stimulation with 10 μM forskolin, followed by inhibition with 20 μM Inh172 (quantification depicted in bar chart) after basolateral permeabilization with amphotericin B (100 μM). (F) Antibody array comparing the basolateral cytokine secretion of polarized CFBE with or without induced CFTR expression. The basolateral media were conditioned for the time period 4–8 h after switch to ALC; the dotted line represents twofold background, and the prespotted positive control was used to normalize between arrays. (G–J) CFTR expression–dependent mRNA levels of IL-8 (G), IL-6 (H), CXCL1 (I), and CXCL2 (J) in CFBE cells kept under LLC or switched for 8 h to ALC, determined by quantitative PCR as described in Materials and Methods. Values show means ± SEM from three independent experiments (B, E, G–J), means ± SD of two independent experiments (F), or means ± SD of one representative experiment (C). *p < 0.05; **p < 0.01; ***p < 0.001.
FIGURE 2:

Functional CFTR expression attenuates IL-8 secretion in human bronchial epithelia under ALC. (A) CFTR expression–dependent basolateral IL-8 secretion of polarized CFBE cells subjected for 24 h to ALC or kept under LLC. (B) Time dependence of the basolateral IL-8 secretion after the switch from LLC to ALC. (C) CFTR expression level dependence of IL-8 secretion of polarized CFBE epithelia kept under ALC. (D) IB of CFBE or H441 cells transduced with inducible wild-type (iCFTR) or G551D (iG551D) CFTR with (+) or without (–) Dox in comparison to the endogenous CFTR expression in Calu3 cells. (E) The dependence of wt and G551D CFTR PM densities on Dox concentration as determined by cell-surface ELISA. (F, G) wt or G551D expression–dependent basolateral IL-8 secretion of polarized (F) CFBE or (G) H441 cells switched to ALC or kept under LLC for the indicated times. IL-8 levels were determined by ELISA. Values represent means ± SEM from three (A, D, F, G) or two (C) independent experiments or means ± SD of one representative experiment (B, E). n.s., not significant; *p < 0.05; ***p < 0.001.
Indirect immunostaining and laser confocal fluorescence microscopy confirmed that wt CFTR was predominantly confined to the apical PM in filter-grown, polarized CFBE cells (Figure 1D).
Short-circuit current measurement (Isc) verified that the CFTR-3HA channels were active. Transactivator-transduced (TetON+) CFBE cells had negligible cAMP-stimulated chloride transport activity (0.03 ± 0.4 μA/cm2). CFTR chromosomal integration (iCFTR-), however, was sufficient to result in a small forskolin-inducible Isc (4.9 ± 0.4 μA/cm2) in the absence of Dox induction. This could be attributed to induction-independent low level of transcriptional and translational activity of the transgene. Remarkably, Dox induced a ∼30-fold increase in Isc (139 ± 9 μA/cm2; Figure 1E). The cAMP-dependent protein kinase (PKA)–activated current was sensitive to CFTR inhibitor 172 (Inh172), a CFTR blocker (Ma et al., 2002).
CFTR expression attenuates the proinflammatory cytokines IL-8, IL-6, and CXCL1/2 in CFBE14o- epithelia under ALC
The CF lung inflammation is characterized by elevated secretion of proinflammatory cytokines, including the chemokine IL-8, with a pivotal role in recruiting an excessive number of neutrophils into the airway lumen (Ratjen and Döring, 2003; Elizur et al., 2008; Downey et al., 2009). To examine whether reestablishing CFTR function would lead to a decrease of the innate immune response of CF epithelial cells in the absence of infection, the secretion profile of 42 cytokines from CFBE epithelia was determined. Induction of CFTR expression significantly attenuated basolateral secretion of the proinflammatory cytokines IL-8, IL-6, and CXCL1-3 by CFBE cells grown on permeable filter supports under ALC, mimicking the native environment of the respiratory epithelia (Figure 1F; Dvorak et al., 2011). Remarkably, the CFTR-suppressive effect on proinflammatory cytokine secretion was absent in epithelia grown under LLC (Supplemental Figure S1B). Although we cannot rule out that CFTR may influence the biogenesis, trafficking, and/or PM fusion of secretory vesicles containing proinflammatory cytokines (Stanley and Lacy, 2010), these possibilities are unlikely because CFTR induction significantly decreased the mRNA level of IL-8, IL-6, CXCL1, and CXCL2 in CFBE epithelia kept under ALC (Figure 1, G–J). CFTR expression did not significantly decrease CXCL3 mRNA (Supplemental Figure S1D). To generate a second expression model, we transduced the human papillary adenocarcinoma cell line NCI-H441 (H441) of Clara cell origin (Gazdar et al., 1990) with no detectable PKA-stimulated channel activity with inducible wt CFTR (Figure 2D and Supplemental Figure S2C). Similar to CFBE epithelia, inducing CFTR expression in H441 monolayers attenuated basolateral IL-8, IL-6, and CXCL1-3 secretion under ALC but had little effect on cells grown in LLC (Supplemental Figure S1D).
In subsequent experiments we focused on the secretion of the chemokine IL-8, a potent stimulant for neutrophil recruitment. Consistent with the reduction of mRNA levels shown in Figure 1G, basolateral IL-8 secretion determined by ELISA was attenuated in CFTR-expressing CFBE cells grown under ALC. The suppressive effect was absent in epithelia under LLC and within the 2 h after transferring the monolayers from LLC to ALC (Figure 2, A and B). CFTR PM density and constitutive channel function was only slightly affected by shift from LLC to ALC. Returning the epithelia to LLC completely abrogated the suppressive effect of CFTR in IL-8 secretion (Supplemental Figure S1, E and F).
As little as ∼10% of the maximum CFTR expression (at 5 ng/ml Dox) was sufficient to suppress IL-8 secretion (Figure 2C; also see Figure 1, B and C). An important observation was that although CFTR suppression of IL-8 secretion was detected in most cell clones, the absolute amount of IL-8 secretion and its fractional attenuation by CFTR differed considerably (Supplemental Figure S1G). Thus the integration site of the virus cDNA and/or genetic and epigenetic variability of the host cell may influence the regulation of IL-8 secretion in clonally isolated cell populations. To avoid potential bias introduced by clonal selection, all subsequent studies used mixtures of at least 100 individual clones.
CFTR channel function is required to suppress IL-8 secretion
To assess whether CFTR apical PM expression is involved as a signaling platform (Li and Naren, 2005; Mehta, 2007) or as an anion translocation pathway, we established the effect of the nonfunctional G551D CFTR mutant expression on IL-8 secretion. G551D and wt CFTR have similar biosynthetic and endocytic processing, but the G551D CFTR cannot be activated by PKA stimulation (Anderson and Welsh, 1992; Barriere et al., 2009). The concentration and time dependences of G551D and wt CFTR expression in CFBE cells were similar (Figure 2E and Supplemental Figure S2A). As expected, G551D CFTR failed to confer PKA-stimulated PM iodide conductance in the absence of potentiator in both CFBE and H441 epithelia (Supplemental Figure S2, B and C). Induction of wt but not G551D CFTR significantly attenuated IL-8 secretion under ALC but not under LLC in both model systems (Figure 2, F and G), strongly suggesting that CFTR channel activity is indispensable for suppressing IL-8 secretion in these polarized respiratory epithelia.
If CFTR channel activity plays a critical regulatory role in IL-8 secretion, CFTR inhibition should enhance IL-8 secretion similar to that observed in noninduced CFBE epithelia. This was indeed the case. Both the robust forskolin-activated and the small constitutive Isc current detected in the absence of activator were inhibited by three structurally distinct CFTR blockers: Inh172, the pyrimido-pyrrolo-quinoxalinedione PPQ102 (Tradtrantip et al., 2009), and the benzopyrimido-pyrrolo-oxazinedione compound BPO27 (Snyder et al., 2011; Figure 3, A–C). PPQ102 and BPO27 abolished the suppressive effect of wt CFTR on IL-8 secretion in iCFTR+ CFBE monolayers. These inhibitors had no influence on the IL-8 production by TetON or iCFTR- cells, ruling out nonspecific effects (Figure 3D). In contrast, Inh172 produced a twofold CFTR-independent activation of IL-8 secretion, conceivably by eliciting reactive oxygen species production in mitochondria (Kelly et al., 2010; Supplemental Figure S2D).
FIGURE 3:

CFTR function is required to suppress IL-8 secretion. (A) PPQ102 (25 μM) or BPO27 (25 μM) inhibits CFTR activity as measured by Isc after stimulation with 10 μM forskolin. (B) The constitutive activity of CFTR in iCFTR+ CFBE14o- cells was unmasked by CFTR inhibitor 172 (Inh172, 20 μM), PPQ102 (25 μM), or BPO27 (25 μM). Measurements were performed in intact monolayers. (C) Quantification of the maximal inhibition as determined in B. (D) Basolateral IL-8 secretion of polarized CFBE cells after channel inhibition with PPQ102 or BPO27. Ratios between basolateral IL-8 secretion with or without induced CFTR expression are depicted, and significance was tested using the ratios derived from three independent experiments. (E) CFTR channel activation in CFBE epithelia with leaked expression of CFTR with forskolin (1 μM), IBMX (0.5 mM), or cpt-cAMP (0.5 mM) determined by Isc. (F) Basolateral IL-8 secretion of polarized CFBE cells under ALC after CFTR channel activation with 1 μM forskolin, 0.5 mM IBMX, or 0.5 mM cpt-cAMP. Numbers indicate the ratio between cells with or without induced CFTR expression. Values show means ± SEM from three (C–E) or two (F) independent experiments. *p < 0.05.
To suppress IL-8 secretion from iCFTR- CFBE cells, we tested a panel of PKA agonists. The low amount of constitutively translated CFTR was activated by forskolin (adenylate cyclase activator), 3-isobutyl-1-methyl-xanthine (IBMX; inhibitor of phosphodiesterase), or 8-(4-chlorophenylthio)-adenosine-3′,5′-cyclical monophosphate (cpt-cAMP), a cell-permeant cAMP analogue. According to Isc measurements, the residual activity represents only ∼3% of the maximally induced CFTR channel activity (Figure 3E). All three PKA agonists with distinct pharmacological targets were able to suppress IL-8 secretion in iCFTR- CFBE cells without interfering with the CFTR-independent IL-8 secretion of TetON+ cells (Figure 3F). Taken together, these results suggest that maximal activation of a small number of CFTR channels or the constitutive partial activity of a large number of CFTR channels are equally efficacious in suppressing the proinflammatory cytokine secretion.
Non–cell-autonomous mechanism of CFTR-induced suppression of IL-8 secretion in CFBE and primary human bronchial epithelia
Based on the discovery that ALC is necessary for CFTR-induced suppression of the IL-8 secretion, it was plausible to assume that CFTR may influence the ion composition and physicochemical properties of the ASL via a non–cell-autonomous mechanism. This means that CFTR expression in only a subset of cells would suppress the IL-8 secretion of the entire epithelial monolayer. To test this hypothesis, we assessed the effect of increasing proportions of CFTR-expressing cells (iCFTR+) in a mixed-cell population that contained iCFTR+ and TetON cells at ratios of 1:9 and 1:4 on IL-8 secretion (Figure 4, A–C). After 5 d on filter supports these ratios resulted in a mixed-cell population containing ∼10% or ∼20% of CFTR-expressing cells, respectively, as determined by immunostaining and cell surface ELISA (Figure 4, A and B). This was sufficient to suppress the basolateral IL-8 secretion by 34 and 77%, respectively, relative to that of monolayers consisting only of CFTR-expressing cells. This indicates a significantly higher IL-8 attenuation than would be expected for a cell-autonomous mechanism that predicts linear correlation between the number of CFTR-expressing cells and the amount of IL-8 suppression (Figure 4C).
FIGURE 4:

A limited number of wt CFTR–expressing cells suppresses the overall IL-8 secretion of CFBE epithelia and primary HBE. (A, B) CFTR expression monitored by confocal microscopy (A) or CFTR PM density (B) in polarized CFBE TetON or iCFTR cells, as well as in 9:1 or 4:1 mixtures of both, after 5 d induction with Dox. (C) Basolateral IL-8 secretion of polarized CFBE TetON, iCFTR cells, or mixtures of both. The theoretical values estimating a linear correlation between the number of CFTR-expressing cells and IL-8 secretion are depicted for comparison and were used for significance testing. (D, E) IB (D) and confocal microscopy pictures (E) of primary CF HBE transduced with lentiviral particles transferring wt, G551D CFTR (green), or empty vector cDNA at a MOI of 4 or 8. ZO1 was used as tight junction marker (red). (F) Basolateral IL-8 secretion from CF HBE (n = 8) transduced at a MOI of 4 and grown on filter supports for 15 d. Left, absolute IL-8 values; right, the percentage change in comparison to empty vector. Values indicate means ± SEM from two (B), three (C), or eight (F) independent experiments. n.s., not significant; **p < 0.01; ***p < 0.001. Bar, 50 μm.
To confirm the results obtained with inducible epithelial models, we transduced primary human bronchial epithelia (HBE) cells isolated from lung tissue of cftrΔF508/ΔF508 CF individuals (Fulcher et al., 2005) with lentiviral particles (multiplicity of infection [MOI], 4 and 8) conferring constitutive wt or G551D CFTR expression. IB and immunostaining indicated that wt or G551D CFTRs were expressed in ∼10% of the epithelial cells after 15 d culture on filter supports (Figure 4, D and E). Although the basolateral IL-8 secretion varied considerably among individuals, normalization with control cells that had been transduced with lentiviral particles containing empty vector showed that wt CFTR reduced the mean IL-8 secretion by 24% (Figure 4F). In contrast, G551D CFTR expression had no effect (Figure 4F). These results support the relevance of CFTR modulation of the innate immune response regulation in primary airway epithelia and substantiate the notion that CFTR-mediated ion transport of a subpopulation of cells is sufficient to modulate the inflammatory state of the monolayer, likely by influencing ASL properties.
Endogenous TMEM16A activation fails to suppress IL-8 secretion by CFBE monolayers
On the basis of the suppressive effect of CFTR channel activity on IL-8 secretion, we hypothesized that TMEM16A-mediated anion transport may similarly attenuate IL-8 production in respiratory epithelia. This was tested by modulating the activity of endogenous TMEM16A in CFBE epithelia.
We monitored TMEM16A activity by measuring Isc upon P2YR activation with ATP or UTP after using three different methods for channel inhibition or activation. 1) The Ca2+-dependent chloride secretion caused by the P2YR agonists (ATP or UTP) was profoundly reduced by short hairpin RNA (shRNA) TMEM16A (Figures 5A and S3A). Gene silencing decreased the TMEM16A mRNA by >70% (Supplemental Figure S3D) and unmasked K+ secretion that was blocked with TREM34, an inhibitor of the Ca2+-activated potassium channel KCa3.1 (Supplemental Figure S3A). 2) Buffering cytosolic Ca2+ concentration transients with BAPTA acetomethylester (BAPTA-AM) was equally efficient in inhibiting ATP- or UTP-induced Ca2+-activated chloride and potassium secretion (Figure 5A and Supplemental Figure S3B). 3) As a complementary approach, TMEM16A was activated by ATP or UTP alone or after long-term IL-4 treatment to stimulate TMEM16A transcription (Caputo et al., 2008; Figure 5A and Supplemental Figure S3, C and E).
FIGURE 5:

Altering the endogenous TMEM16A activity in CFBE epithelia by shRNA, inhibitor, or transcriptional activation cannot modulate IL-8 secretion. (A) ATP or UTP (each 100 μM) stimulated peak current quantification in CFBE cells in combination with TMEM16A shRNA expression, BAPTA-AM (10 μM, 30 min), or IL-4 (10 ng/ml, 24 h) pretreatment. (B, C) CFTR expression–dependent basolateral IL-8 secretion of polarized CFBE cells under ALC in combination with (B) 10 μM BAPTA-AM treatment or (C) shRNA expression targeting TMEM16A or SCNN1A. (D) IL-8 secretion in CFBE epithelia with or without induced CFTR expression or with shRNA silencing of TMEM16A in combination with IL-4 treatment to increase endogenous TMEM16A expression and channel activation with 100 μM ATP or UTP. Significance tested in comparison to untreated cells. Values indicate means ± SEM from three to five independent experiments. *p <0.05; ***p < 0.001.
Inhibition of TMEM16A by BAPTA-AM or shRNA or depleting nucleoside triphosphates with extracellular apyrase all failed to stimulate IL-8 secretion in TetON CFBE cells and did not change the differential IL-8 secretion in presence or absence of CFTR (Figure 5, B and C, and Supplemental Figure S3F). Similarly, IL-8 secretion remained unaltered upon agonist-dependent activation of TMEM16A without or with IL-4 treatment (Figure 5D and Supplemental Figure S3G). These results suggest that endogenous TMEM16A activity confers negligible chloride conductance relative to that of CFTR in CFBE epithelia and therefore is not sufficient to modulate IL-8 secretion. To enhance the PM anion transport activity, we next evaluated the effect of TMEM16A overexpression on IL-8 secretion.
TMEM16A is localized at the apical and lateral membranes of polarized epithelia
To establish whether the localization of heterologously expressed TMEM16A mimics its endogenous counterpart, a prerequisite for anion secretion, we used immunohistochemistry, domain-specific biotinylation, and Isc measurements. Inducible overexpression of TMEM16A in CFBE epithelia was accomplished by using the splice isoform (abc) containing a partial internal FLAG epitope (DYKDDDK) close to the N-terminus (Caputo et al., 2008; Figure 6A). For immunostaining, a myc epitope was attached to the C-terminus. Doxycycline-induced TMEM16A-myc was expressed with the predicted molecular mass and was confined to the apical and lateral PM membrane in stably transduced CFBE epithelia (Figure 6, A and B), as well as in transiently transduced Madin–Darby canine kidney (MDCK) cells (Figure 6C), an established model for epithelial polarity.
FIGURE 6:

TMEM16A is localized at the apical and lateral PM of polarized epithelia. (A) Schematic depiction of the TMEM16A proteins and IB of inducible expression in CFBE cells. (B, C) Apical and lateral localization of TMEM16A (red) in (B) polarized CFBE or (C) MDCK cells by confocal microscopy. Occludin or ZO1 was used as tight junction marker (green), lateral membranes were labeled with E-cadherin (green), and nuclei were stained with DAPI (blue). Ap, apical; b, basal. Bars, 10 μm. (D) Expression of TMEM16A in the apical and basolateral membranes of polarized CFBE probed by cell surface biotinylation as described in Materials and Methods. (E) ATP-stimulated change (100 μM) in apical or basolateral chloride currents after permeabilization of the basolateral or apical membranes with nystatin (100 μM) in presence of an outward-directed chloride gradient. Values represent means ± SEM from three to five independent experiments. IP, immunoprecipitation.
Polarized distribution of TMEM16A was determined by domain-specific PM biotinylation on filter-grown CFBE epithelia (Sargiacomo et al., 1989). Quantitative IB analysis revealed that ∼65% of TMEM16A was confined to apical PM, whereas 35% was localized to the basolateral PM in parental or noninduced cells (Figure 6D). Overexpression of TMEM16A at low Dox level (∼70-fold increased over endogenous level at 5 ng/ml Dox) modestly shifted the apical-to-basolateral expression ratio to 50:50. Strong TMEM16A overexpression (∼200-fold over endogenous level at 500 ng/ml Dox) reverted the apical-to-basolateral ratio to 40:60 (Figure 6D and Supplemental Figure S4B).
Inducible overexpression of TMEM16A led to a Dox-dependent increase in ATP- or UTP-stimulated Ca2+-activated chloride secretion that paralleled the TMEM16A protein expression in CFBE cells (Supplemental Figure S4, A and B). To evaluate the polarized distribution of TMEM16A functionally, we permeabilized the basolateral or the apical PM with nystatin prior to the Isc measurement. After apical permeabilization, the ATP-stimulated basolateral current was sensitive to the TMEM16A inhibitor A01 but not to Na+-K+-Cl– cotransporter, Na+/K+-ATPase, or K+ channel inhibition, indicating that TMEM16A mediated chloride transport (Supplemental Figure S4D). Based on domain-specific current densities, the apical-to-basolateral PM conductance ratio was found to be 2:1 for both endogenous and leaked TMEM16A (Figure 6E). The functional distribution of the highly overexpressed channel was decreased to a ratio of 1.5:1 (Figure 6E). Jointly, these biochemical and functional assays confirmed the immunohistochemical localization results and showed that TMEM16A is expressed at the apical and lateral PM of polarized respiratory monolayers.
TMEM16A overexpression attenuates proinflammatory cytokine secretion of CFBE epithelia under ALC
The effect of TMEM16A overexpression on proinflammatory cytokine secretion was evaluated next. TMEM16A induction attenuated basolateral secretion of the proinflammatory cytokines IL-8, IL-6, CXCL1-3, and CCL2 in CFBE cells grown on permeable filter supports under ALC but had little effect on cells in LLC (Supplemental Figure S5A). This was confirmed by measuring the relative mRNA abundance of cytokines (Supplemental Figure S5, B–H). Similar to the mRNA levels, IL-8 secretion was attenuated proportionately with TMEM16A expression and reached 50% inhibition of parental (TetON) or non-induced (iTMEM16A-) cells, resembling the suppressive effect of CFTR (Figure 7, A and B; also see Figure 2A). The TMEM16A-mediated attenuation was abrogated under LLC and in cells loaded with the high-affinity Ca2+ chelator 5,5′-dimethyl-BAPTA-AM (KD = 40 nM for Ca2+ binding; Figure 7, A and B).
FIGURE 7:

The constitutive activity of overexpressed TMEM16A suppresses IL-8 secretion in CFBE epithelia. (A) CFBE cells transduced with transactivator alone (TetON) or in combination with inducible TMEM16A with (+) or without (–) Dox (500 ng/ml)–induced expression were subjected to ALC or kept under LLC. Basolateral IL-8 secretion was determined by ELISA. (B) IL-8 secretion of polarized CFBE cells at different TMEM16A expression level (0, 5, or 500 ng/ml Dox) or after lowering of cytosolic Ca2+ with 5,5′-dimethyl BAPTA-AM (10 μM). (C) Constitutive TMEM16A activity was determined by Isc measurements in intact CFBE monolayers with (5 or 500 ng/ml Dox) or without TMEM16A expression after the addition of the A01 inhibitor (100 μM). (D) Bar graph summarizes the constitutive TMEM16A activity with or without pretreatment with 10 μM 5,5′-dimethyl BAPTA-AM. (E, F) Maximal fluorescence quenching rate after addition of iodide buffer to CFBE cells expressing the halide-sensitive YFP and TMEM16A induced with the indicated Dox concentrations. As controls, TMEM16A activity was reduced by 10 μM 5,5′-dimethyl BAPTA-AM (E) or using A01 (F). (G) Correlation between IL-8 secretion and constitutive Isc of CFBE epithelia expressing different levels of CFTR or TMEM16A in combination with channel activation or inhibition. Values show means ± SEM from three or four independent experiments. BAPTA, 5,5′-dimethyl BAPTA-AM; frk, forskolin; cAMP, cpt-cAMP. *p < 0.05; **p < 0.01.
The use of A01, a TMEM16A-specific inhibitor (Namkung et al., 2011a), or the Ca2+chelator 5,5′-dimethyl-BAPTA-AM verified that elevated chloride secretion in Dox-induced, TMEM16A-expressing CFBE epithelia was independent of ligand-induced P2YR activation (Figure 7, C and D). The Isc generated by constitutively active TMEM16A was ∼50% that of detectable in CFTR-overexpressing cells in the absence of exogenous PKA stimulation (see Figure 3, B and C). Consistent results were obtained by measuring the halide conductance with cytosolic YFP-H148Q/I152L/F46L, a halide-sensitive fluorescent protein (Ferrera et al., 2009; Figure 7, E and F, and Supplemental Figure S5I). The increased halide conductance of CFBE epithelia exposed to Dox induction was sensitive to both 5,5′-dimethyl-BAPTA-AM and A01 (Figure 7, E and F). These observations strongly suggest that constitutive activity of overexpressed TMEM16A is responsible for IL-8 suppression by compensating for the loss of CFTR transport activity in CFBE cells. Combining the data, we find that an inverse correlation is apparent between the magnitude of IL-8 secretion relative to TetON cells and the Isc carried by the TMEM16A or CFTR channels at various Dox-induction and inhibition levels (Figure 7G).
Direct but not P2YR-dependent TMEM16A activation suppresses IL-8 secretion
Given that P2YR-dependent activation of the endogenous TMEM16A was unable to attenuate IL-8 secretion of CFBE epithelia, we examined whether TMEM16A overexpression could confer P2YR agonist-mediated regulation of IL-8 secretion. To this end, we activated overexpressed TMEM16A by ATP, UTP, or the nonhydrolyzable analogues ATPγS or UTPγS, which stimulated TMEM16A with comparable efficiency (Supplemental Figure S5J). To our surprise, neither ATP nor ATPγS activation of P2YRs potentiated the TMEM16A suppressor effect on IL-8 secretion in Dox-induced CFBE epithelia (Figure 8, A and B). Apical exposure to UTP exerted a modest but significant inhibition on IL-8 secretion independent of TMEM16A expression (Figure 8A; also see Figure 5D). This was not observed with UTPγS (Figure 8B), suggesting that UTP or its hydrolytic intermediate may trigger a signaling cascade that attenuates IL-8 secretion independent of TMEM16A.
FIGURE 8:

P2YR activation cannot potentiate the suppressive effect of TMEM16A on IL-8 secretion. (A, B) TMEM16A expression level–dependent IL-8 secretion under ALC in polarized CFBE in combination with channel activation with (A) 100 μM ATP or UTP or (B) 100 μM stable nucleoside triphosphate analogue ATPγS or UTPγS. (C) Time-dependent desensitization of 100 μM ATP–stimulated peak Isc measured in polarized CFBE epithelia expressing different levels of TMEM16A. (D) Resensitization of ATP-stimulated TMEM16A activity. After initial stimulation (100 μM ATP, 5 min) ATP was removed and the cells were allowed to resensitize for the indicated period before restimulation with 100 μM ATP. Inset, representative traces of one experiment. (E, F) Activation of TMEM16A by stimulation with histamine receptor agonist (100 μM histamine, E) or muscarinic receptor agonist (100 μM carbachol [CCh], F) in the presence or absence of 100 μM ATP (15-min pretreatment), 10 μM BAPTA-AM (1-h pretreatment), or 10 μM A01 (5-min pretreatment) determined by Isc measurement. (G) Quantification of the peak Isc after 15-min pretreatment with ATP. (H, I) Cytoplasmic calcium mobilization measured by Fluo-4 fluorescence. CFBE cells with or without ATP pretreatment (100 μM, 15 min) were stimulated with 100 μM ATP (H) or 100 μM histamine (I). (J) Quantification of peak Fluo-4 fluorescence after 15-min pretreatment with ATP. Bar charts depict means ± SEM from three to five independent experiments as percentage of controls. *p < 0.05.
The Isc measurements of both endogenous and overexpressed TMEM16A activity revealed that P2YR agonist–induced chloride secretion was transient and coupled to rapid inactivation in monolayers (Figure 8C and Supplemental Figures S3, A–C, and S4A). The inactivation kinetics was independent of the TMEM16A expression levels, implying that basolateral chloride influx was not rate limiting (Figure 8C). ATP stimulation for 15 min led to near-complete homologous desensitization of P2YRs, indicated by the marginal chloride secretion elicited by repeated ATP exposure (Figure 8G and inset in D). Resensitization after the removal of extracellular ATP was slow, with 50% recovery of the ATP-stimulated Isc requiring ∼30 min (Figure 8D). Homologous desensitization likely occurs upstream of phospholipase C activation, considering the limited heterologous desensitization of the TMEM16A-mediated Isc, activated by histamine and the muscarinic receptors in the presence of ATP (Figure 8, E–G). In accord, cytosolic Ca2+ mobilization was inhibited only by 20–25% for carbachol or histamine stimulation after ATP pretreatment, whereas it was attenuated by 75–80% for a second ATP or UTP exposure (Figure 8, H–J). Thus, rapid inactivation of P2YR explains the inability of receptor agonist to suppress IL-8 secretion despite the preserved responsiveness of TMEM16A to activation by other Ca2+-mobilizing GPCRs in CFBE monolayers.
In sharp contrast to the ligand-induced transient stimulation of the P2YRs, the Ca2+-signaling–independent activator F (Namkung et al., 2011b) led to a significantly more sustained activation of TMEM16A (Figure 9, A and B). As a consequence, activator F was able to attenuate IL-8 secretion in CFBE cells with endogenous or low heterologous TMEM16A channel expression (Figure 9, C and D). Similar results were obtained for H441 cells with inducible overexpression of TMEM16A (Figure 9E and Supplemental Figure S5K). This effect is specific, since it was diminished by the shRNA-mediated ablation of endogenous TMEM16A expression in CFBE (Figure 9D), suggesting that constitutive activation of endogenous TMEM16A may reduce the innate immune response and tissue damage of the CF lung.
FIGURE 9:

Activator F results in prolonged stimulation of TMEM16A and decreases IL-8 secretion in CFBE and H441 expressing low TMEM16A cannel density. (A, B) Sample traces (A) and inactivation kinetics (B) of TMEM16A in polarized CFBE stimulated with 100 μM ATP or 20 μM activator F. Isc was determined in intact epithelia. (C–E) IL-8 secretion in polarized CFBE (C, D) or H441 (E) with different levels of TMEM16A expression (induced with 0, 5, or 500 ng/ml Dox; C, E) or endogenous TMEM16A or shRNA silencing (D) with or without channel activation by activator F (20 μM, added at 0 h ALC). Values show means ± SEM from three independent experiments. **p < 0.01; ***p < 0.001.
DISCUSSION
The question of whether hyperinflammation is a sole consequence of ASL dehydration and bacterial colonization or whether these processes can be primed by the CFTR functional expression defect has been extensively investigated by a number of laboratories but remains controversial (Pizurki et al., 2000; Ribeiro et al., 2005; Hybiske et al., 2007; Fulcher et al., 2009; Vandivier et al., 2009; Roussel et al., 2011). Several factors could account for the conflicting results.
The expression of proinflammatory cytokines might be modulated by the epigenome configuration, which is influenced by repeated infection and inflammation in CF. The hypoxic environment associated with infection and inflammation induces the production of hypoxia-inducible factor 1 (Frede et al., 2007; Brigati et al., 2010), which down-regulates the histone deacetylase 2 (HDAC2) (Charron et al., 2009), leading to epigenetic modifications of proinflammatory cytokine genes. The inflammatory response was indeed amplified by shifting the relative activity of histone acetylase and HDAC in asthma, as well as in chronic obstructive pulmonary disease (COPD; Barnes et al., 2005; Ito et al., 2005).
A number of studies have been performed on clonally isolated CF and non-CF cell lines. These cell lines were obtained by either complementation of CF cells with wt CFTR (e.g., the IB-3 and C38 cell line pair) or by CFTR inactivation in non-CF cells (e.g., 16HBEo- sense and antisense cells). Isolation of individual clones from heterologous populations, however, may favor unrepresentative phenotypes because clonal variations can exceed the underlying differences in the regulated IL-8 secretion (as illustrated in Supplemental Figure S1G) or regarding Ca2+ signaling (Babnigg et al., 2000). To bypass these potentially confounding mechanisms and evaluate the direct effect of CFTR loss on proinflammatory cytokine secretion, we inducibly expressed CFTR and TMEM16A chloride channels in the human respiratory epithelial cell lines CFBE and H441.
Our results indicate that inducible expression of CFTR suppressed the expression and release of the proinflammatory cytokines IL-8, IL-6, and CXCL1/2. Focusing on IL-8, compelling evidence supports the notion that the transport function rather than CFTR signaling is required to inhibit the tonic cytokine secretion by these cell lines. 1) Expression of CFTR as low as ∼10% of that in Calu-3 cells was sufficient to suppress IL-8 secretion by ∼50%. 2) The functionally inactive but normally processed G551D variant cannot substitute for wt CFTR in suppressing IL-8 release. 3) Inhibiting the constitutive activity of wt CFTR with PPQ102 or BPO27 (Snyder et al., 2011) curtailed the suppressive effect on IL-8 secretion. 4) Concordantly, PKA activation by forskolin, IBMX, or cpt-cAMP was sufficient to suppress IL-8 secretion by stimulating the transport function of the low-copy-number CFTR at the apical PM even without Dox induction. 5) Overexpression of TMEM16A could substitute for the suppressive role of CFTR in IL-8 secretion. 6) Finally, these results were confirmed by the suppressive effect of wt but not G551D CFTR expression on IL-8 secretion by primary HBE cells isolated form cftrΔF508/ΔF508 CF individuals.
The regulatory cascade that triggers IL-8 secretion in CF respiratory epithelia in the absence of infection remains to be deciphered. Considering that the CFTR suppressor effect was restricted to respiratory epithelia cultured under ALC but not LLC, it is conceivable that besides ionic or compositional changes, physicochemical alterations (e.g., surface tension, osmotic and oxidoreductive state) of the ASL and/or the apical PM may constitute upstream signaling for IL-8 secretion. Among other factors, signal transduction pathways involved could be modulated by ion channel and receptor protein kinase or phosphatase functions affected by changes in the membrane potential and pH (Bocharov et al., 2008; Sandoval et al., 2011), as well as by the oxidoreductive state of the ASL (Blanchetot et al., 2002). The supraproportional effect of few individual wt CFTR-expressing cells on the IL-8 secretion by CFBE cells in mixed populations, as well as in transduced primary HBE cultures, suggests that an average reduction in the anion transport capacity at the apical membrane leads to global changes in signaling of epithelial sheets. This notion is strengthened by the proportionality between IL-8 suppression and CFTR-mediated chloride conductance. These results, collectively, link the loss of CFTR-mediated ion transport to the hyperinflammation observed in CF and provide credence to the suggestion that the basic defect in CF can modulate the innate immune response of airway epithelia in the absence of infection. A similar conclusion was favored by the augmented lung inflammation of mice overexpressing the epithelial sodium channel β subunit. Increased sodium absorption was associated with increased secretion of the macrophage inflammatory protein-2, the mouse orthologue of IL-8, and keratinocyte chemoattractant by bronchial epithelia, as well as with profound lung infiltration with neutrophils in the absence of bacterial infection (Mall et al., 2004).
The critical role of apical PM anion conductance in reducing the inflammatory state of CF bronchial epithelia was underscored by the effect of TMEM16A activity on IL-8 secretion. The subcellular localization of TMEM16A is cell type dependent. In acinar cells of the submandibular and salivary glands the channel is predominantly apical (Yang et al., 2008; Huang et al., 2009), whereas in cholangiocytes TMEM16A is localized at the apical and lateral PM (Dutta et al., 2011). The latter pattern coincides with the preferentially apical and lateral distribution of TMEM16A in polarized CFBE and MDCK cells. Thus, maintained activity of apical TMEM16A could compensate, in principle, for the loss of function of CFTR channels in CF.
Our in vitro data using TMEM16A inhibition or induction of TMEM16A expression by IL-4 treatment alone, as well as in combination with P2YR agonists, indicate that endogenous TMEM16A activity is normally insufficient to modulate IL-8 secretion in human respiratory epithelia. In contrast, tonic activity of overexpressed TMEM16A significantly reduced IL-8, IL-6, and CXCL1-3 secretion under ALC. Additional nucleotide stimulation was ineffective in further attenuating IL-8 release, consistent with the conclusion that P2YR-induced cytoplasmic Ca2+ signaling is transient and the coupled TMEM16A activation is too short-lived to effect cytokine release. The transient activation of TMEM16A by apical nucleotides provokes rapid fluid secretion, but the increased ASL height returns to baseline within 1 h of stimulation (Tarran et al., 2001). These observations, taken together, may provide a plausible explanation for the limited efficacy of denufosol, a P2Y2R agonist, in a recent phase III clinical trial (Accurso et al., 2011).
Nonhydrolyzable nucleoside triphosphate analogues and repeated agonist stimulations showed that TMEM16A inactivation cannot be attributed to nucleotide breakdown but instead to rapid desensitization of the P2YR signaling. Agonist-induced phosphorylation of the P2YR promotes the recruitment of β-arrestin-2 to and subsequent internalization of the receptor (Flores et al., 2005; Hoffmann et al., 2008; Morris et al., 2011). Heterologous desensitization of other GPCRs was modest, indicating that desensitization occurred at the P2YR level. To stimulate Ca2+-activated chloride channels in respiratory epithelia, local administration of duramycin, a Ca2+ ionophore, as an alternative approach to GPCR activation was attempted (Cloutier et al., 1993; Grasemann et al., 2007). Elevated cytosolic Ca2+ concentrations, however, promote proinflammatory cytokine secretion and therefore may counteract the beneficial effect of ion transport correction (Gewirtz et al., 2000; Sakamoto et al., 2007). These limitations could be overcome by the activator F, which probably activates TMEM16A allosterically without triggering cytosolic Ca2+ signaling (Namkung et al., 2011b). Activator F was able to attenuate IL-8 secretion via endogenous TMEM16A in CFBE, substantiating the proposed functional link between apical PM ion transport capacity and the innate immunity defect of CF respiratory epithelia.
In a broader context our results suggest that the proinflammatory cytokine secretion is regulated among other factors by the anion transport capacity of the apical PM in airway epithelia. This mechanism may contribute to excessive lung inflammation not only in cystic fibrosis, but perhaps in other chronic lung diseases (e.g., cigarette smoke–induced COPD), where CFTR down-regulation has been documented (Cantin et al., 2006; Clunes et al., 2012). Direct activation of TMEM16A may provide therapeutic benefit in these conditions if the channel activation is restricted to respiratory epithelial cells.
MATERIALS AND METHODS
Reagents and antibodies
CFTR inhibitors PPQ102 and BPO27 (Tradtrantip et al., 2009; Snyder et al., 2011) and the TMEM16A inhibitor A01 and activator F have been described (Namkung et al., 2011a, b). ATPγS tetralithium salt, UTPγS trisodium salt, CFTRinh 172, and TRAM 34 were purchased from Tocris Bioscience (Ellisville, MO). BAPTA-AM and 5,5′-dimethyl BAPTA were obtained from Invitrogen (Carlsbad, CA). Human IL-4 was obtained from R&D Systems (Minneapolis, MN), and all other chemicals were purchased from Sigma-Aldrich (St. Louis, MO).
The following antibodies were used: monoclonal mouse anti-CFTR (L12B4; Chemokine, East Orange, NJ), monoclonal mouse anti-HA (MMS101R; Covance, Berkeley, CA), monoclonal mouse anti-myc (9E10; Santa Cruz Biotechnology, Santa Cruz, CA), monoclonal mouse anti-FLAG (M2; Sigma-Aldrich), monoclonal rabbit anti-TMEM16A (SP31; Abcam, Cambridge, MA), monoclonal rat anti-HSPA4 (1B5; Stressgen, San Diego, CA), monoclonal mouse anti-Na+/K+-ATPase (H3; Santa Cruz Biotechnology), polyclonal rabbit anti-occludin (Zymed; San Francisco, CA), monoclonal rat anti-ZO1 (MAB1520; Chemicon, Temecula, CA), and monoclonal mouse anti–E-cadherin (3B8; a kind gift from W. Gallin, University of Alberta, Edmonton, Canada).
Cell culture and stable cell line generation
The human CF bronchial epithelial cell line CFBE, with a cftrΔF508/ΔF508 genotype (a generous gift from D. Gruenert, University of California, San Francisco, San Francisco, CA), was maintained in MEM (Invitrogen) supplemented with 10% fetal bovine serum (FBS; Invitrogen), 2 mM l-glutamine and 10 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES). For propagation the cells were cultured in plastic flasks coated with an extracellular matrix (ECM mix) consisting of 10 μg/ml human fibronectin (EMD, San Diego, CA), 30 μg/ml PureCol collagen preparation (Advanced BioMatrix, San Diego, CA), and 100 μg/ml bovine serum albumin (Sigma-Aldrich) diluted in LHC basal medium (Invitrogen). MDCK cells were grown in DMEM medium containing 10% FBS and 10 mM HEPES, and H441 cells (American Type Culture Collection, Manassas, VA) were cultured in RPMI-1640 medium supplemented with 10% FBS and 10 mM HEPES.
CFBE and H441 cell lines containing inducible CFTR or TMEM16A were generated using the Lenti-X TetON Advanced Inducible Expression System (Clontech, Mountain View, CA). Briefly, wt or G551D CFTR harboring an extracellular 3HA tag (Sharma et al., 2004) was subcloned into pLVX-tight-puro using a NotI/blunt-end strategy. A myc-tag coding sequence was fused in-frame to the C-terminus of TMEM16A splice variant abc containing a partial internal FLAG epitope (Caputo et al., 2008) by PCR and cloned into the NotI/MluI restriction sites. PCR-amplified constructs were verified by sequencing. For the generation of stable knockdown cell lines shRNAmir constructs for TMEM16A (V2LHS_155384), and SCNN1A (V2HS_93913) were purchased from Open Biosystems (Huntsville, AL) and transferred into a modified pGIPZ vector containing a hygromycin selection cassette. Lentiviral particles were produced in the HEK293T cells with the Lenti-X Packaging System (Clontech) following the manufacturer's instructions. The cell lines were generated by consecutive transduction with viral particles containing the cDNA for the tetracycline-controlled transactivator, inducible CFTR, or TMEM16A and shRNAmir, followed by selection with G418 (200 μg/ml; InvivoGen, San Diego, CA), puromycin (3 μg/ml, InvivoGen), and hygromycin B (200 μg/ml, InvivoGen), respectively.
Isolation and culture of HBE cells
Human lung tissues were obtained from 8 cftrΔF508/ΔF508 CF individuals after lung transplantation under the protocol and consent form approved by the Institutional Review Board of the Research Ethics Office of McGill University. Isolation, culture, and differentiation of HBE were adapted from procedures previously described (Fulcher et al., 2005). Briefly, CF airway epithelial cells were isolated from bronchial tissue by enzyme digestion and were cultured in bronchial epithelial growth medium on type I collagen-coated plastic flasks (Vitrogen 100, PureCol; Advanced BioMatrix), then trypsinized, counted, and cryopreserved. Passage 1 (P1) or cryopreserved P1 cells were transduced with lentiviral particles mediating cDNA transfer of wt and G551D CFTR harboring an extracellular 3HA tag or empty TranzVector (Tranzyme, Durham, NC; Wu et al., 2000) at MOI of 4 or 8 and subsequently transferred onto collagen IV–coated, 6-mm Transwell filters (Corning, Tewksbury MA) in air–liquid interface medium at a density of 1.2 × 105 cells/filter. During the first 5 d, the air–liquid interface medium was changed every day, and then the apical media was removed and the cells were grown at an air–liquid interface for an additional 10 d before use. The isolation and growth media were complemented with antibiotics that were adapted according to recent patient antibiograms; however, only penicillin and streptomycin were added to the air-–liquid interface medium.
Screening of human cytokines and IL-8 assay
CFBE or H441 cells were plated on ECM mix–coated, 12-mm Transwell filters (Corning) at a density of 1 × 105 cells/cm2 under LLC. Protein expression was induced 24 h after plating (day 1) with 500 ng/ml Dox unless otherwise specified, and medium was changed to serum-free Opti-MEM (Invitrogen) supplemented with 2 mM l-glutamine at day 4 to avoid cross-contaminating samples with bovine serum cytokines. Epithelia were switched to ALC at day 5, and the basolateral medium was conditioned for the indicated times. The secretion of 42 cytokines into the basolateral medium was determined using a antibody array (human cytokine antibody array 3; RayBiotech, Norcross GA) following the manufacturer's protocol, except for the detection, which was performed with an IRDye 800 streptavidin conjugate (LI-COR Biosciences, Lincoln, NE), followed by imaging and quantification with an Odyssey Infrared Imaging System (LI-COR Biosciences). IL-8 levels were determined by ELISA (eBioscience, San Diego, CA) following the manufacturer's instructions, except for the final detection with a SuperSignal ELISA Femto luminescence substrate (Pierce, Rockford, IL) on a VICTOR Light plate reader (PerkinElmer, Waltham, MA).
Isc measurement
CFBE cells were plated on ECM mix–coated, 12-mm Snapwell filters (Corning) at a density of 1 × 105 cells/cm2. Polarized epithelia (≥5 d postconfluence) were mounted in Ussing chambers, bathed in Krebs-bicarbonate Ringer (KBR, ion composition 140 mM Na+, 120 mM Cl−, 5.2 mM K+, 25 mM HCO3−, 2.4 mM HPO4, 0.4 mM H2PO4, 1.2 mM Ca2+, 1.2 mM Mg2+, 5 mM glucose, pH 7.4) and continuously bubbled with 95% O2 and 5% CO2. To impose a chloride gradient, NaCl was replaced by Na+ gluconate or with K+ gluconate to establish a potassium gradient in the absence of chloride. To functionally isolate the apical or basolateral membranes, we permeabilized the opposite PM domain with 100 μM amphotericin B (for stimulated CFTR activity measurements). Measurements were performed under Isc conditions at 37°C, recorded with the Acquire and Analyze package (Physiologic Instruments, San Diego, CA), and expressed as current/cm2. Unless otherwise noted, measurements were performed after imposing an apical-to-basolateral chloride gradient in the presence of 100 μM amiloride.
Apical and basolateral PM functional channel density measurement
To determine the functional density of TMEM16A at the apical or basolateral PM by Isc measurement, we permeabilized the opposite PM (basolateral and apical, respectively) with 100 μM nystatin. Efficient apical PM permeabilization was confirmed by the BaCl2-sensitive increase of the basolateral potassium current (Rochat et al., 2004; also see Supplemental Figure S4C). ATP-stimulated changes in Isc were measured in the presence of an outward-directed chloride gradient, and peak current quantification was performed to calculate the apical-to-basolateral current ratios.
Immunostaining and confocal microscopy
Polarized epithelial monolayers were fixed with 4% paraformaldehyde and permeabilized with 0.2% Triton X-100 or 0.1% saponin, followed by blocking with 1% BSA in phosphate-buffered saline (PBS) with 0.1 mM CaCl2 and 1 mM MgCl2, pH 7.4 (PBSCM). CFTR-3HA–expressing CFBE cells were costained with anti-HA and anti-ZO1 antibodies. To detect TMEM16A-myc localization, we visualized the channel by indirect immunostaining using primary anti-myc antibody, whereas tight junctions were stained with anti-occludin or anti-ZO1 antibodies. We used Alexa Fluor 488– and Alexa Fluor 555–conjugated secondary antibodies (Invitrogen). Costaining of TMEM16A and E-cadherin was accomplished using sequential incubation, first with anti-myc antibody and Alexa Fluor 555–conjugated goat anti-mouse immunoglobulin G (Invitrogen). The free epitopes of the anti-myc antibodies were blocked with goat anti-mouse Fab fragment (Jackson ImmunoResearch, West Grove, PA). Finally anti–E-cadherin antibody incubation was followed by Alexa Fluor 488–labeled goat anti-mouse antibody (Invitrogen). Nuclei were stained with 4′,6-diamidino-2-phenylindole (DAPI). Images were obtained on an LSM710 microscope (Carl Zeiss, Jena, Germany) equipped with a Plan-Apochromat 100×/1.40 oil differential interference contrast objective. Typically 20–30 optical xy-sections were acquired and reconstituted using the Zen 2009 software package, and representative xz-sections are shown. For better visibility the gamma setting for ZO1, occludin, E-cadherin, and DAPI stainings was adjusted.
CFTR cell surface density measurement
The PM density of wt and G551D CFTR was determined by cell-surface ELISA (Okiyoneda et al., 2010). Cells were seeded in 96-well plates at a density of 2 × 104 cells/well and induced for CFTR expression with the indicated Dox concentrations for 3 d. The extracellular 3HA tag was detected by incubation with anti-HA antibody followed by incubation with horseradish peroxidase (HRP)–conjugated secondary antibody in PBSCM containing 0.5% bovine serum albumin on ice. Excess antibody was removed by extensive washing, and specific binding was determined with Amplex Red (Invitrogen) HRP substrate. The fluorescence intensity was measured at 544 nm excitation and 590 nm emission wavelengths using a POLARstar OPTIMA (BMG Labtech, Ortenberg, Germany) fluorescence plate reader, and values determined from parental cells were subtracted as background.
Cell surface biotinylation to determine TMEM16A apical-to-basolateral ratios and IB analysis
CFBE cells with or without induced TMEM16A expression with 0, 5, or 500 ng/ml Dox were grown on 24-mm polyester Transwell filters for at least 3 d postconfluence. Basolateral or apical PM proteins were biotinylated for 15 min on ice with 1 mg/ml EZ Link sulfo-SS-NHS-biotin (Thermo Fisher Scientific, Waltham, MA) in buffer H (154 mM NaCl, 3 mM KCl, 10 mM HEPES, 1 mM MgCl2, 0.1 mM CaCl2, 10 mM glucose, pH 7.8), followed by excess biotin reagent quenching with 100 mM glycine in PBSCM. Cells were lysed in RIPA buffer (150 mM NaCl, 20 mM Tris-HCl, 1% Triton X-100, 0.1% SDS, and 0.5% sodium deoxycholate, pH 8.0) containing protease inhibitors, and postnuclear lysates were collected after centrifugation. The biotinylated proteins were isolated on streptavidin-agarose beads (Invitrogen) at 4°C with end-over-end rotation for 1 h. Proteins were eluted with 2× Laemmli sample buffer, separated by SDS–PAGE, and transferred to nitrocellulose membrane. IBs were probed with rabbit anti-TMEM16A, mouse anti-Na+/K+-ATPase, or rat anti-HSPA4 antibodies, followed by IRDye 680 anti-rabbit, IRDye 800 anti-mouse, or IRDye 680 anti-rat antibodies (LI-COR Biosciences). IBs were imaged and quantified with the Odyssey Infrared Imaging System (LI-COR Biosciences).
Quantitative PCR
Total RNA was extracted from polarized CFBE grown on Transwell filters using the miRNeasy kit (Qiagen, Valencia, CA), and reverse transcription of 1 μg of total RNA was performed with the QuantiTect reverse transcription kit (Qiagen) following the manufacturer's recommendations. The abundance of transcripts was determined using SYBR Advantage qPCR premix (Clontech) with an Mx3005P real-time cycler (Stratagene, Santa Clara, CA). Primer sequences were either retrieved from the PrimerDepot (http://primerdepot.nci.nih.gov) or designed using the PearlPrimer software (Marshall, 2004) and are summarized in Table 1. PCR data were analyzed by efficiency-corrected comparative quantification (Pfaffl, 2001) and reported as relative change in mRNA abundance.
TABLE 1:
Primers used for quantitative PCR.
| Human mRNA | Forward primer (5′ to 3′) | Reverse primer (5′ to 3′) |
|---|---|---|
| GAPDH | CATGAGAAGTATGACAACAGCCT | AGTCCTTCCACGATACCAAAGT |
| IL-8 | TTTTGCCAAGGAGTGCTAAAGA | AACCCTCTGCACCCAGTTTTC |
| IL-6 | AACCTGAACCTTCCAAAGATGG | TCTGGCTTGTTCCTCACTACT |
| CXCL1 | AGGGAATTCACCCCAAGAAC | ACTATGGGGGATGCAGGATT |
| CXCL2 | TGCCAGTGCTTGCAGACC | TCTTAACCATGGGCGATGC |
| CXCL3 | TGGTCACTGAACTGCGCTG | GATGCGGGGTTGAGACAAG |
| CCL2 | CAGCCAGATGCAATCAATGCC | TGGAATCCTGAACCCACTTCT |
| CCL5 | ATCCTCATTGCTACTGCCCTC | GCCACTGGTGTAGAAATACTCC |
| SCNN1A | AGTGTGGCTGTGCCTACATCTT | GGAGAAGTCAACCTGGAGCTTA |
| TMEM16A | GCAAACATGGAGGACCACTT | GCCGTATTACCAGCCATCAT |
Cytosolic calcium measurements
CFBE cells seeded into black 96-well microplates at a density of 2 × 104 cells/well were cultured for 3 d postconfluence. Cytosolic calcium levels were determined using the Fluo-4 NW calcium assay kit (Invitrogen) following the manufacturer's protocol. The Fluo-4 fluorescence was measured at 488 ± 5 nm excitation and 516 ±6 nm emission wavelengths in an Infinite M1000 fluorescence plate reader (Tecan, Männedorf, Switzerland) equipped with syringe pumps. Fluorescence values from non–dye-loaded cells were subtracted as background, and the relative changes in cytosolic calcium levels are expressed as percentage of basal calcium signals.
Halide permeability determination of the PM
The halide sensor YFP-F46L/H148Q/I152L (Namkung et al., 2010) was amplified by PCR and cloned into the XhoI/BamHI restriction sites of the pLVX-IRES-Hyg lentiviral vector (Clontech). Lentiviral particles were produced as described, and CFBE cells containing the transactivator alone or in combination with inducible TMEM16A were transduced, followed by selection with hygromycin B (200 μg/ml). Yellow fluorescent protein (YFP)-expressing cells or controls were seeded to 96-well microplates at a density of 2 × 104 cells/well and induced for TMEM16A expression with the indicated Dox concentrations for 3 d. For the YFP quenching assay, cells were incubated in 100 μl/well PBS–chloride (140 mM NaCl, 2.7 mM KCl, 8.1 mM Na2HPO4, 1.5 mM KH2PO4, 1.1 mM MgCl2, 0.7 mM CaCl2, and 5 mM glucose, pH 7.4), followed by wellwise injection of 100 μl of PBS–iodide in which NaCl was replaced with NaI. The fluorescence was monitored over 50 s with a 5-Hz acquisition rate at 485 nm excitation and 520 nm emission wavelengths using a POLARstar OPTIMA fluorescence plate reader. For analysis, the fluorescence of non-YFP–expressing cells was subtracted, and the quenching curves were fitted to the following exponential equation:
 |
where Fq is the residual fluorescence after complete YFP quenching, t is time, R1 and R2 are quenching rates, and F1 and F2 are the relative contributions of R1 and R2 to fluorescence quenching, respectively. The overall quenching rate was determined by using
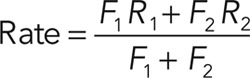 |
Statistical analysis
Results are presented as mean ± SEM for the indicated number of experiments. Statistical analysis was performed by two-tailed Student's t test with the means of at least three independent experiments, and the 95% confidence level was considered significant.
Supplementary Material
Acknowledgments
We acknowledge H. Salah's contribution to the initial phase of the study, D. Gruenert for the parental CFBE14o- cell line, and W. Gallin for the anti–E-cadherin antibody. G.V. was supported by an EMBO fellowship and in part by a Fonds de Recherche du Québec-Santé Postdoctoral Training Award. F.B. was a recipient of a Richard & Edith Strauss Fellowship. Work performed in the authors’ laboratories was supported by the National Institutes of Health–National Institute of Diabetes and Digestive and Kidney Diseases, the Canadian Institutes of Health Research, the Cystic Fibrosis Foundation Therapeutics, the Canadian Foundation for Innovation, the Cystic Fibrosis Canada Breathe Program, and National Institutes of Health Grant HL73856. G.L. is holder of a Canada Research Chair.
Abbreviations used:
- ALC
air–liquid culture
- ASL
airway surface liquid
- CF
cystic fibrosis
- CFTR
cystic fibrosis transmembrane conductance regulator
- cpt-cAMP
8-(4-chlorophenylthio)-adenosine-3′,5′-cyclical monophosphate
- Dox
doxycycline
- ECM
extracellular matrix
- GPCR
G protein–coupled receptor
- HBE
human bronchial epithelia
- IB
immunoblot
- IBMX
3-isobutyl-1-methyl-xanthine
- IL-8
interleukin-8
- Inh172
CFTR inhibitor 172
- LLC
liquid–liquid culture
- MOI
multiplicity of infection
- P2YR
P2Y receptor
- PKA
cAMP-dependent protein kinase
- PM
plasma membrane
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E12-06-0424) on September 12, 2012.
REFERENCES
- Abu-El-Haija M, Sinkora M, Meyerholz DK, Welsh MJ, McCray PB Jr, Butler J, Uc A. An activated immune and inflammatory response targets the pancreas of newborn pigs with cystic fibrosis. Pancreatology. 2011;11:506–515. doi: 10.1159/000332582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Accurso FJ, Moss RB, Wilmott RW, Anbar RD, Schaberg AE, Durham TA, Ramsey BW. Denufosol tetrasodium in patients with cystic fibrosis and normal to mildly impaired lung function. Am J Respir Crit Care Med. 2011;183:627–634. doi: 10.1164/rccm.201008-1267OC. [DOI] [PubMed] [Google Scholar]
- Adcock IM, Tsaprouni L, Bhavsar P, Ito K. Epigenetic regulation of airway inflammation. Curr Opin Immunol. 2007;19:694–700. doi: 10.1016/j.coi.2007.07.016. [DOI] [PubMed] [Google Scholar]
- Anderson MP, Welsh MJ. Regulation by ATP and ADP of CFTR chloride channels that contain mutant nucleotide-binding domains. Science. 1992;257:1701–1704. doi: 10.1126/science.1382316. [DOI] [PubMed] [Google Scholar]
- Armstrong DS, Hook SM, Jamsen KM, Nixon GM, Carzino R, Carlin JB, Robertson CF, Grimwood K. Lower airway inflammation in infants with cystic fibrosis detected by newborn screening. Pediatr Pulmonol. 2005;40:500–510. doi: 10.1002/ppul.20294. [DOI] [PubMed] [Google Scholar]
- Babnigg G, Heller B, Villereal ML. Cell-to-cell variation in store-operated calcium entry in HEK-293 cells and its impact on the interpretation of data from stable clones expressing exogenous calcium channels. Cell Calcium. 2000;27:61–73. doi: 10.1054/ceca.1999.0093. [DOI] [PubMed] [Google Scholar]
- Barnes PJ, Adcock IM, Ito K. Histone acetylation and deacetylation: importance in inflammatory lung diseases. Eur Respir J. 2005;25:552–563. doi: 10.1183/09031936.05.00117504. [DOI] [PubMed] [Google Scholar]
- Barriere H, Bagdany M, Bossard F, Okiyoneda T, Wojewodka G, Gruenert D, Radzioch D, Lukacs GL. Revisiting the role of cystic fibrosis transmembrane conductance regulator and counterion permeability in the pH regulation of endocytic organelles. Mol Biol Cell. 2009;20:3125–3141. doi: 10.1091/mbc.E09-01-0061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensalem N, et al. Down-regulation of the anti-inflammatory protein annexin A1 in cystic fibrosis knock-out mice and patients. Mol Cell Proteomics. 2005;4:1591–1601. doi: 10.1074/mcp.M500019-MCP200. [DOI] [PubMed] [Google Scholar]
- Berger M. Lung inflammation early in cystic fibrosis: bugs are indicted, but the defense is guilty. Am J Respir Crit Care Med. 2002;165:857–858. doi: 10.1164/ajrccm.165.7.2202030a. [DOI] [PubMed] [Google Scholar]
- Blanchetot C, Tertoolen LG, den Hertog J. Regulation of receptor protein-tyrosine phosphatase alpha by oxidative stress. EMBO J. 2002;21:493–503. doi: 10.1093/emboj/21.4.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bocharov EV, Mayzel ML, Volynsky PE, Goncharuk MV, Ermolyuk YS, Schulga AA, Artemenko EO, Efremov RG, Arseniev AS. Spatial structure and pH-dependent conformational diversity of dimeric transmembrane domain of the receptor tyrosine kinase EphA1. J Biol Chem. 2008;283:29385–29395. doi: 10.1074/jbc.M803089200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigati C, Banelli B, di Vinci A, Casciano I, Allemanni G, Forlani A, Borzi L, Romani M. Inflammation, HIF-1, and the epigenetics that follows. Mediators Inflamm. 2010;2010:263914. doi: 10.1155/2010/263914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantin AM, Hanrahan JW, Bilodeau G, Ellis L, Dupuis A, Liao J, Zielenski J, Durie P. Cystic fibrosis transmembrane conductance regulator function is suppressed in cigarette smokers. Am J Respir Crit Care Med. 2006;173:1139–1144. doi: 10.1164/rccm.200508-1330OC. [DOI] [PubMed] [Google Scholar]
- Caputo A, Caci E, Ferrera L, Pedemonte N, Barsanti C, Sondo E, Pfeffer U, Ravazzolo R, Zegarra-Moran O, Galietta LJ. TMEM16A, a membrane protein associated with calcium-dependent chloride channel activity. Science. 2008;322:590–594. doi: 10.1126/science.1163518. [DOI] [PubMed] [Google Scholar]
- Charron CE, et al. Hypoxia-inducible factor 1alpha induces corticosteroid-insensitive inflammation via reduction of histone deacetylase-2 transcription. J Biol Chem. 2009;284:36047–36054. doi: 10.1074/jbc.M109.025387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cloutier MM, Guernsey L, Sha'afi RI. Duramycin increases intracellular calcium in airway epithelium. Membr Biochem. 1993;10:107–118. doi: 10.3109/09687689309150258. [DOI] [PubMed] [Google Scholar]
- Clunes LA, et al. Cigarette smoke exposure induces CFTR internalization and insolubility, leading to airway surface liquid dehydration. FASEB J. 2012;26:533–545. doi: 10.1096/fj.11-192377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derichs N, Jin BJ, Song Y, Finkbeiner WE, Verkman AS. Hyperviscous airway periciliary and mucous liquid layers in cystic fibrosis measured by confocal fluorescence photobleaching. FASEB J. 2011;25:2325–2332. doi: 10.1096/fj.10-179549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downey DG, Bell SC, Elborn JS. Neutrophils in cystic fibrosis. Thorax. 2009;64:81–88. doi: 10.1136/thx.2007.082388. [DOI] [PubMed] [Google Scholar]
- Dutta AK, et al. Identification and functional characterization of TMEM16A, a Ca2+-activated Cl- channel activated by extracellular nucleotides, in biliary epithelium. J Biol Chem. 2011;286:766–776. doi: 10.1074/jbc.M110.164970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorak A, Tilley AE, Shaykhiev R, Wang R, Crystal RG. Do airway epithelium air-liquid cultures represent the in vivo airway epithelium transcriptome? Am J Respir Cell Mol Biol. 2011;44:465–473. doi: 10.1165/rcmb.2009-0453OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrhardt C, Collnot EM, Baldes C, Becker U, Laue M, Kim KJ, Lehr CM. Towards an in vitro model of cystic fibrosis small airway epithelium: characterisation of the human bronchial epithelial cell line CFBE41o. Cell Tissue Res. 2006;323:405–415. doi: 10.1007/s00441-005-0062-7. [DOI] [PubMed] [Google Scholar]
- Elizur A, Cannon CL, Ferkol TW. Airway inflammation in cystic fibrosis. Chest. 2008;133:489–95. doi: 10.1378/chest.07-1631. [DOI] [PubMed] [Google Scholar]
- Estell K, Braunstein G, Tucker T, Varga K, Collawn JF, Schwiebert LM. Plasma membrane CFTR regulates RANTES expression via its C-terminal PDZ-interacting motif. Mol Cell Biol. 2003;23:594–606. doi: 10.1128/MCB.23.2.594-606.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrera L, Caputo A, Ubby I, Bussani E, Zegarra-Moran O, Ravazzolo R, Pagani F, Galietta LJ. Regulation of TMEM16A chloride channel properties by alternative splicing. J Biol Chem. 2009;284:33360–33368. doi: 10.1074/jbc.M109.046607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flores RV, Hernandez-Perez MG, Aquino E, Garrad RC, Weisman GA, Gonzalez FA. Agonist-induced phosphorylation and desensitization of the P2Y2 nucleotide receptor. Mol Cell Biochem. 2005;280:35–45. doi: 10.1007/s11010-005-8050-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frede S, Berchner-Pfannschmidt U, Fandrey J. Regulation of hypoxia-inducible factors during inflammation. Methods Enzymol. 2007;435:405–419. doi: 10.1016/S0076-6879(07)35021-0. [DOI] [PubMed] [Google Scholar]
- Fulcher ML, Gabriel S, Burns KA, Yankaskas JR, Randell SH. Well-differentiated human airway epithelial cell cultures. Methods Mol Med. 2005;107:183–206. doi: 10.1385/1-59259-861-7:183. [DOI] [PubMed] [Google Scholar]
- Fulcher ML, Gabriel SE, Olsen JC, Tatreau JR, Gentzsch M, Livanos E, Saavedra MT, Salmon P, Randell SH. Novel human bronchial epithelial cell lines for cystic fibrosis research. Am J Physiol Lung Cell Mol Physiol. 2009;296:L82–L91. doi: 10.1152/ajplung.90314.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazdar AF, Linnoila RI, Kurita Y, Oie HK, Mulshine JL, Clark JC, Whitsett JA. Peripheral airway cell differentiation in human lung cancer cell lines. Cancer Res. 1990;50:5481–5487. [PubMed] [Google Scholar]
- Gewirtz AT, Rao AS, Simon PO Jr, Merlin D, Carnes D, Madara JL, Neish AS. Salmonella typhimurium induces epithelial IL-8 expression via Ca(2+)-mediated activation of the NF-kappaB pathway. J Clin Invest. 2000;105:79–92. doi: 10.1172/JCI8066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gossen M, Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc Natl Acad Sci USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grasemann H, Stehling F, Brunar H, Widmann R, Laliberte TW, Molina L, Doring G, Ratjen F. Inhalation of Moli1901 in patients with cystic fibrosis. Chest. 2007;131:1461–1466. doi: 10.1378/chest.06-2085. [DOI] [PubMed] [Google Scholar]
- Hallows KR, Fitch AC, Richardson CA, Reynolds PR, Clancy JP, Dagher PC, Witters LA, Kolls JK, Pilewski JM. Up-regulation of AMP-activated kinase by dysfunctional cystic fibrosis transmembrane conductance regulator in cystic fibrosis airway epithelial cells mitigates excessive inflammation. J Biol Chem. 2006;281:4231–4241. doi: 10.1074/jbc.M511029200. [DOI] [PubMed] [Google Scholar]
- Hoffmann C, Ziegler N, Reiner S, Krasel C, Lohse MJ. Agonist-selective, receptor-specific interaction of human P2Y receptors with beta-arrestin-1 and -2. J Biol Chem. 2008;283:30933–30941. doi: 10.1074/jbc.M801472200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang F, Rock JR, Harfe BD, Cheng T, Huang X, Jan YN, Jan LY. Studies on expression and function of the TMEM16A calcium-activated chloride channel. Proc Natl Acad Sci USA. 2009;106:21413–21418. doi: 10.1073/pnas.0911935106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter MJ, Treharne KJ, Winter AK, Cassidy DM, Land S, Mehta A. Expression of wild-type CFTR suppresses NF-kappaB-driven inflammatory signalling. PLoS One. 2010;5:e11598. doi: 10.1371/journal.pone.0011598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hybiske K, Fu Z, Schwarzer C, Tseng J, Do J, Huang N, Machen TE. Effects of cystic fibrosis transmembrane conductance regulator and DeltaF508CFTR on inflammatory response, ER stress, and Ca2+ of airway epithelia. Am J Physiol Lung Cell Mol Physiol. 2007;293:L1250–L1260. doi: 10.1152/ajplung.00231.2007. [DOI] [PubMed] [Google Scholar]
- Ito K, et al. Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N Engl J Med. 2005;352:1967–1976. doi: 10.1056/NEJMoa041892. [DOI] [PubMed] [Google Scholar]
- John G, Chillappagari S, Rubin BK, Gruenert DC, Henke MO. Reduced surface toll-like receptor-4 expression and absent interferon-gamma-inducible protein-10 induction in cystic fibrosis airway cells. Exp Lung Res. 2011;37:319–326. doi: 10.3109/01902148.2011.569968. [DOI] [PubMed] [Google Scholar]
- John G, Yildirim AO, Rubin BK, Gruenert DC, Henke MO. TLR-4-mediated innate immunity is reduced in cystic fibrosis airway cells. Am J Respir Cell Mol Biol. 2010;42:424–431. doi: 10.1165/rcmb.2008-0408OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelly M, Trudel S, Brouillard F, Bouillaud F, Colas J, Nguyen-Khoa T, Ollero M, Edelman A, Fritsch J. Cystic fibrosis transmembrane regulator inhibitors CFTR(inh)-172 and GlyH-101 target mitochondrial functions, independently of chloride channel inhibition. J Pharmacol Exp Ther. 2010;333:60–69. doi: 10.1124/jpet.109.162032. [DOI] [PubMed] [Google Scholar]
- Li C, Naren AP. Macromolecular complexes of cystic fibrosis transmembrane conductance regulator and its interacting partners. Pharmacol Ther. 2005;108:208–223. doi: 10.1016/j.pharmthera.2005.04.004. [DOI] [PubMed] [Google Scholar]
- Ma T, Thiagarajah JR, Yang H, Sonawane ND, Folli C, Galietta LJ, Verkman AS. Thiazolidinone CFTR inhibitor identified by high-throughput screening blocks cholera toxin-induced intestinal fluid secretion. J Clin Invest. 2002;110:1651–1658. doi: 10.1172/JCI16112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machen TE. Innate immune response in CF airway epithelia: hyperinflammatory? Am J Physiol Cell Physiol. 2006;291:C218–C230. doi: 10.1152/ajpcell.00605.2005. [DOI] [PubMed] [Google Scholar]
- Mall M, Grubb BR, Harkema JR, O'Neal WK, Boucher RC. Increased airway epithelial Na+ absorption produces cystic fibrosis-like lung disease in mice. Nat Med. 2004;10:487–493. doi: 10.1038/nm1028. [DOI] [PubMed] [Google Scholar]
- Marshall OJ. PerlPrimer: cross-platform, graphical primer design for standard, bisulphite and real-time PCR. Bioinformatics. 2004;20:2471–2472. doi: 10.1093/bioinformatics/bth254. [DOI] [PubMed] [Google Scholar]
- Matsui H, Grubb BR, Tarran R, Randell SH, Gatzy JT, Davis CW, Boucher RC. Evidence for periciliary liquid layer depletion, not abnormal ion composition, in the pathogenesis of cystic fibrosis airways disease. Cell. 1998;95:1005–1015. doi: 10.1016/s0092-8674(00)81724-9. [DOI] [PubMed] [Google Scholar]
- Mehta A. The cystic fibrosis transmembrane recruiter the alter ego of CFTR as a multi-kinase anchor. Pflugers Arch. 2007;455:215–221. doi: 10.1007/s00424-007-0290-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris GE, Nelson CP, Everitt D, Brighton PJ, Standen NB, Challiss RA, Willets JM. G protein-coupled receptor kinase 2 and arrestin2 regulate arterial smooth muscle P2Y-purinoceptor signalling. Cardiovasc Res. 2011;89:193–203. doi: 10.1093/cvr/cvq249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muhlebach MS, Noah TL. Endotoxin activity and inflammatory markers in the airways of young patients with cystic fibrosis. Am J Respir Crit Care Med. 2002;165:911–915. doi: 10.1164/ajrccm.165.7.2107114. [DOI] [PubMed] [Google Scholar]
- Namkung W, Phuan PW, Verkman AS. TMEM16A inhibitors reveal TMEM16A as a minor component of calcium-activated chloride channel conductance in airway and intestinal epithelial cells. J Biol Chem. 2011a;286:2365–2374. doi: 10.1074/jbc.M110.175109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Thiagarajah JR, Phuan PW, Verkman AS. Inhibition of Ca2+-activated Cl- channels by gallotannins as a possible molecular basis for health benefits of red wine and green tea. FASEB J. 2010;24:4178–4186. doi: 10.1096/fj.10-160648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Namkung W, Yao Z, Finkbeiner WE, Verkman AS. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011b;25:4048–4062. doi: 10.1096/fj.11-191627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okiyoneda T, Barriere H, Bagdany M, Rabeh WM, Du K, Hohfeld J, Young JC, Lukacs GL. Peripheral protein quality control removes unfolded CFTR from the plasma membrane. Science. 2010;329:805–810. doi: 10.1126/science.1191542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez A, Issler AC, Cotton CU, Kelley TJ, Verkman AS, Davis PB. CFTR inhibition mimics the cystic fibrosis inflammatory profile. Am J Physiol Lung Cell Mol Physiol. 2007;292:L383–L395. doi: 10.1152/ajplung.00403.2005. [DOI] [PubMed] [Google Scholar]
- Pfaffl MW. A new mathematical model for relative quantification in real-time RT-PCR. Nucleic Acids Res. 2001;29:e45. doi: 10.1093/nar/29.9.e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pizurki L, Morris MA, Chanson M, Solomon M, Pavirani A, Bouchardy I, Suter S. Cystic fibrosis transmembrane conductance regulator does not affect neutrophil migration across cystic fibrosis airway epithelial monolayers. Am J Pathol. 2000;156:1407–1416. doi: 10.1016/S0002-9440(10)65009-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratjen F. What's new in CF airway inflammation: an update. Paediatr Respir Rev. 2006;7(Suppl 1):S70–S72. doi: 10.1016/j.prrv.2006.04.170. [DOI] [PubMed] [Google Scholar]
- Ratjen F, Döring G. Cystic fibrosis. Lancet. 2003;361:681–689. doi: 10.1016/S0140-6736(03)12567-6. [DOI] [PubMed] [Google Scholar]
- Ribeiro CM, Paradiso AM, Schwab U, Perez-Vilar J, Jones L, O'Neal W, Boucher RC. Chronic airway infection/inflammation induces a Ca2+i-dependent hyperinflammatory response in human cystic fibrosis airway epithelia. J Biol Chem. 2005;280:17798–17806. doi: 10.1074/jbc.M410618200. [DOI] [PubMed] [Google Scholar]
- Riordan JR, et al. Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 1989;245:1066–1073. doi: 10.1126/science.2475911. [DOI] [PubMed] [Google Scholar]
- Rochat T, Lacroix JS, Jornot L. N-acetylcysteine inhibits Na+ absorption across human nasal epithelial cells. J Cell Physiol. 2004;201:106–116. doi: 10.1002/jcp.20066. [DOI] [PubMed] [Google Scholar]
- Roussel L, Martel G, Berube J, Rousseau S. P. aeruginosa drives CXCL8 synthesis via redundant toll-like receptors and NADPH oxidase in CFTRF508 airway epithelial cells. J Cyst Fibros. 2011;10:107–113. doi: 10.1016/j.jcf.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Rowe SM, Miller S, Sorscher EJ. Cystic fibrosis. N Engl J Med. 2005;352:1992–2001. doi: 10.1056/NEJMra043184. [DOI] [PubMed] [Google Scholar]
- Sakamoto N, Hayashi S, Gosselink J, Ishii H, Ishimatsu Y, Mukae H, Hogg JC, van Eeden SF. Calcium dependent and independent cytokine synthesis by air pollution particle-exposed human bronchial epithelial cells. Toxicol Appl Pharmacol. 2007;225:134–141. doi: 10.1016/j.taap.2007.07.006. [DOI] [PubMed] [Google Scholar]
- Sandoval M, Burgos J, Sepulveda FV, Cid LP. Extracellular pH in restricted domains as a gating signal for ion channels involved in transepithelial transport. Biol Pharm Bull. 2011;34:803–809. doi: 10.1248/bpb.34.803. [DOI] [PubMed] [Google Scholar]
- Sargiacomo M, Lisanti M, Graeve L, Le Bivic A, Rodriguez-Boulan E. Integral and peripheral protein composition of the apical and basolateral membrane domains in MDCK cells. J Membr Biol. 1989;107:277–286. doi: 10.1007/BF01871942. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Cheng T, Jan YN, Jan LY. Expression cloning of TMEM16A as a calcium-activated chloride channel subunit. Cell. 2008;134:1019–1029. doi: 10.1016/j.cell.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma M, et al. Misfolding diverts CFTR from recycling to degradation: quality control at early endosomes. J Cell Biol. 2004;164:923–933. doi: 10.1083/jcb.200312018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder DS, Tradtrantip L, Yao C, Kurth MJ, Verkman AS. Potent, metabolically stable benzopyrimido-pyrrolo-oxazine-dione (bpo) cftr inhibitors for polycystic kidney disease. J Med Chem. 2011;54:5468–5477. doi: 10.1021/jm200505e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley AC, Lacy P. Pathways for cytokine secretion. Physiology (Bethesda) 2010;25:218–229. doi: 10.1152/physiol.00017.2010. [DOI] [PubMed] [Google Scholar]
- Stoltz DA, et al. Cystic fibrosis pigs develop lung disease and exhibit defective bacterial eradication at birth. Sci Transl Med. 2010;2:29ra31. doi: 10.1126/scitranslmed.3000928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun X, et al. Disease phenotype of a ferret CFTR-knockout model of cystic fibrosis. J Clin Invest. 2010;120:3149–3160. doi: 10.1172/JCI43052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarran R, Grubb BR, Parsons D, Picher M, Hirsh AJ, Davis CW, Boucher RC. The CF salt controversy: in vivo observations and therapeutic approaches. Mol Cell. 2001;8:149–158. doi: 10.1016/s1097-2765(01)00286-6. [DOI] [PubMed] [Google Scholar]
- Tradtrantip L, Sonawane ND, Namkung W, Verkman AS. Nanomolar potency pyrimido-pyrrolo-quinoxalinedione CFTR inhibitor reduces cyst size in a polycystic kidney disease model. J Med Chem. 2009;52:6447–6455. doi: 10.1021/jm9009873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Heeckeren AM, Schluchter MD, Xue W, Davis PB. Response to acute lung infection with mucoid Pseudomonas aeruginosa in cystic fibrosis mice. Am J Respir Crit Care Med. 2006;173:288–296. doi: 10.1164/rccm.200506-917OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandivier RW, et al. Dysfunctional cystic fibrosis transmembrane conductance regulator inhibits phagocytosis of apoptotic cells with proinflammatory consequences. Am J Physiol Lung Cell Mol Physiol. 2009;297:L677–L686. doi: 10.1152/ajplung.00030.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega-Carrascal I, Reeves EP, Niki T, Arikawa T, McNally P, O'Neill SJ, Hirashima M, McElvaney NG. Dysregulation of TIM-3-galectin-9 pathway in the cystic fibrosis airways. J Immunol. 2011;186:2897–2909. doi: 10.4049/jimmunol.1003187. [DOI] [PubMed] [Google Scholar]
- Welsh MJ, Smith AE. Molecular mechanisms of CFTR chloride channel dysfunction in cystic fibrosis. Cell. 1993;73:1251–1254. doi: 10.1016/0092-8674(93)90353-r. [DOI] [PubMed] [Google Scholar]
- Wu X, Wakefield JK, Liu H, Xiao H, Kralovics R, Prchal JT, Kappes JC. Development of a novel trans-lentiviral vector that affords predictable safety. Mol Ther. 2000;2:47–55. doi: 10.1006/mthe.2000.0095. [DOI] [PubMed] [Google Scholar]
- Yang YD, et al. TMEM16A confers receptor-activated calcium-dependent chloride conductance. Nature. 2008;455:1210–1215. doi: 10.1038/nature07313. [DOI] [PubMed] [Google Scholar]
- Yerxa BR, et al. Pharmacology of INS37217 [P(1)-(uridine 5′)-P(4)- (2′-deoxycytidine 5′)tetraphosphate, tetrasodium salt], a next-generation P2Y(2) receptor agonist for the treatment of cystic fibrosis. J Pharmacol Exp Ther. 2002;302:871–880. doi: 10.1124/jpet.102.035485. [DOI] [PubMed] [Google Scholar]
- Zielenski J, Tsui LC. Cystic fibrosis: genotypic and phenotypic variations. Annu Rev Genet. 1995;29:777–807. doi: 10.1146/annurev.ge.29.120195.004021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.