

Abstract
Background
Heart failure is a growing cause of morbidity and mortality. Cardiac PI3-kinase signaling promotes cardiomyocyte survival and function but is paradoxically activated in heart failure, suggesting chronic activation of this pathway may become maladaptive. Here we investigated the downstream PI3-kinase effector, SGK1 (serum- and glucocorticoid-regulated kinase-1), in heart failure and its complications.
Methods and Results
We found that cardiac SGK1 is activated in human and murine heart failure. We investigated the role of SGK1 in the heart using cardiac-specific expression of constitutively-active or dominant-negative SGK1. Cardiac-specific activation of SGK1 in mice increased mortality, cardiac dysfunction, and ventricular arrhythmias. The pro-arrhythmic effects of SGK1 were linked to biochemical and functional changes in the cardiac sodium channel and could be reversed by treatment with ranolazine, a blocker of the late sodium current. Conversely, cardiac-specific inhibition of SGK1 protected mice after hemodynamic stress from fibrosis, heart failure, and sodium channel alterations.
Conclusions
SGK1 appears both necessary and sufficient for key features of adverse ventricular remodeling and may provide a novel therapeutic target in cardiac disease.
Keywords: arrhythmia, heart failure, ion channels or ion channel, signal transduction
Introduction
Cardiovascular disease remains the dominant cause of morbidity and mortality in industrialized nations. Heart failure is associated with altered electrical properties of cardiomyocytes1, 2 including prolongation of action potential duration (APD). These changes, in combination with cardiac structural changes, are termed ‘electrical remodeling’, and comprise the triggers and substrates of ventricular arrhythmia, an important cause of sudden cardiac death. Electrical remodeling frequently occurs in association with adverse mechanical remodeling and cardiac dysfunction. In heart failure, an increase in persistent sodium current (late INa or INaL)1 can contribute to APD prolongation, although the pathways leading to INaL are incompletely understood. Thus, our understanding of electrical and mechanical remodeling in heart disease remains incomplete.
Acute activation of PI3-kinase signaling promotes cardiomyocyte survival and function3-5. Surprisingly, proximal PI3-kinase signaling is enhanced in patients with cardiac dysfunction and heart failure6, 7, raising the possibility that initially compensatory activation of this pathway becomes maladaptive and contributes to adverse remodeling. In this context, SGK1 is a particularly intriguing candidate. SGK1 is a PI3-kinase-dependent, serine-threonine kinase that is structurally similar to Akt8. SGK1 expression is transcriptionally regulated by mineralocorticoid signaling9, an important contributor to heart failure and arrhythmia. SGK1 regulates sodium ion transport in the kidney10 and in heterologous expression systems11, and thus could mechanistically link PI3-kinase signaling, heart failure, and arrhythmia.
We previously found that cardiac SGK1 is activated early after pressure overload induced by transverse aortic constriction (TAC) and that acute activation promotes cardiomyocyte survival5. Here we demonstrate that SGK1 is also persistently activated in TAC induced heart failure (TAC-HF) in mice, as well as in human heart disease. To examine the functional role of chronic SGK1 activation, we generated cardiac-specific gain- and loss-offunction models through expression of constitutively active (CA) and dominant negative (DN) SGK1 mutants8. SGK1 activation comparable to that seen in failing hearts is sufficient to induce hallmarks of mechanical and electrical remodeling. Interestingly, aspects of adverse remodeling could be reversed by ranolazine, suggesting a key role for the sodium current in the phenotype of the SGK1-CA transgenic mice. Conversely, genetic SGK1 inhibition mitigated the development of heart failure and fibrosis after TAC, and abrogated heart failure-associated biochemical changes in the sodium channel. Together these data suggest an important role for SGK1 in both the adverse electrical and mechanical remodeling seen in heart failure.
Methods
Generation of SGK1-CA and SGK1-KD mice
All studies were approved by IACUC. HA-tagged CA (S422D) and KD (K127M) mutants of SGK1 were subcloned downstream of the MHC promoter in pbS2SK+ (a generous gift from Dr. Jeff Robbins). Linearized plasmids were microinjected into C57/BL6 oocytes and transferred to pseudopregnant mice as previously described12. Three independent lines were identified for each construct.
Transverse Aortic Constriction
12 week old male mice were subjected to TAC, as previously described using a 25G needle13. Perioperative mortality was not different between the wild-type and any of the transgenic lines studied.
Ischemia/Reperfusion Experiments
12 week old mice were subjected to 30 minutes LAD ligation and 24 hour reperfusion as previously described14. The animals were monitored with continuous telemetry (Scisense data recording) during the time of ischemia and the first 45 minutes of reperfusion or until the animals had recovered from anesthesia.
Ranolazine pellet implantation
Two ranolazine pellets (14-day release, 140 mg/pellet, Innovative Research of America) were implanted subcutaneously in the intra-scapular region. This dosage and formulation allows for stable plasma therapeutic levels of ranolazine (3-4 μM) from 4 days onward after implantation.
Cardiac function
Echocardiograms were performed on unanesthetized mice using a GE Vivid5 with a 15L8 linear array transducer (13.0 MHz, imaging depth 10 mm) at a frame-rate of 166/sec.
Hemodynamic studies were conducted on anesthetized mice (ketamine 100mg/kg; xylazine 5mg/kg) using a 1.5Fr SciScience PV loop catheter (see Supplemental Methods).
In vivo Electrophysiology Studies
Scisense octapolar EP catheters were advanced via the internal jugular vein to the right heart of mice anesthetized as above. Using an OctalBioamp stimulator (Medtronic) and AD data acquisition system, EP studies were conducted as described15. Briefly, unipolar and bipolar electrograms were obtained from the RA, RV, and LV, amplified and filtered (EVR recorder). Provocative testing was performed with double and triple extra-stimulation as well as rapid pacing.
Tissue Harvesting
Mice were anesthetized and exsanguinated while the heart was perfusion-fixed in situ with 4% paraformaldehyde at 100cm H2O (20 min) or removed and cryopreserved in liquid nitrogen for RNA and protein analysis.
Human Studies
Discarded pathological tissues were obtained and studied under an IRB-approved protocol.
Protein and RNA analysis
Cardiac protein lysates were prepared, electrophoresed, and transferred to membranes for immunoblotting as described16 and used with the following antibodies: total SGK1, p(thr256)SGK1, GSK3β, p(ser9)GSK3βp(ser318)Foxo3A, p(ser253)Foxo3A (all from Cell Signaling); Ryanodine receptor, Nav1.5, Cav1.2a (all from Alomone); NCX (Gene Tex); SERCA (gift from Dr. del Monte); SP-19, monoclonal antibody against SCN5a (Sigma); Nedd4-2, phospholamban, p(S16)Phospholamban, caveolin-3, GAPDH, HAtag (ABCAM). Immunoblots were scanned and bands quantified by densitometry. Only samples run on the same gel were compared for quantification.
Immunoprecipitation
Ventricular proteins were prepared in Cell Signaling lysis buffer (1% Triton) supplemented with protease (Thermo Scientific) and phosphatase inhibitors (Roche). 500mg protein was pre-cleared with Protein A/G slurry (Calbiochem), immunoprecipitated with SP-19 (2mg) or control IgG (3 hours, 4°C). Protein A/G slurry (40mL) was then added to each tube and nutated for 90 minutes at 4°C. After several washes with PBS buffer/0.1% TritonX-100 followed by PBS/protease inhibitors (Thermo scientific), protein was eluted with Laemli sample buffer (37°C, 30min), separated using 4-20% gradient SDS-PAGE (BioRad), and immunoblotted as above.
SGK1 Kinase Assay
Ventricular proteins (500 mg) were immunoprecipitated with 2mg anti-SGK1 antibody (Upstate) using Upstate Catch-and-Release columns. Immunoprecipitates were eluted using non-denaturing buffer and incubated with 1mg SGK1 substrate (GSK3α/β fusion protein, Cell Signaling) and 200mM ATP (30 minutes, 30°C). Reaction products were separated by SDSPAGE and detected by immunoblotting using an antibody against phospho-ser 9/21 GSK3α/β (Cell Signaling).
Sucrose Gradient Analysis
Ventricular lysates were subjected to discontinuous sucrose gradient, and heavy membrane pellets as well as supernatant fractions collected and subjected to immunoblotting using a modified version of a published protocol17 (details in Supplemental Methods).
Histochemistry
Mid-ventricular short axis sections were fixed (4% paraformaldehyde) and 5mm sections stained with Masson’s trichrome to visualize fibrosis, WGA staining to outline cardiomyocytes, and hematoxylin-eosin for cytoarchitecture. A pre-specified, genotype-blinded image-selection method using NIH SCION software was used to quantify fibrosis (see Supplemental Methods).
Whole-cell patch clamp recording
Isolated ventricular myocytes were current and voltage clamped using the whole-cell patchclamp configuration as previously described18, 19. Voltage and current clamp control and data acquisition are performed using custom-written software (see Supplemental Methods).
Biotin Surface Labeling
Neonatal rat ventricular myocytes (NRVMs) were prepared as previously described5 and plated at 1×106 cells/60 mm dish. Cells were grown in serum-containing media (DMEM, 10% Horse serum, 5% FBS, 1% Glutamate) for 24 hours, then switched to serum-free media and infected with the indicated adenoviral vectors (MOI 50). After 48 hours, Pierce Cell Surface Protein Isolation Kit (Thermo Scientific) was used. Briefly, cells were washed and labeled with Sulfo-NHS-SS-Biotin (30 min, 4°C). Cells were then washed, lysed, and biotin-labeled proteins collected on a Neutravidin-agarose column. After elution, biotin-labeled proteins were subjected to SDS-PAGE and immunoblotting as above.
Statistics
Unless otherwise specified, data are expressed as mean±SEM. Distributions for continuous variables within each group were tested for normality using D’Agostino and Pearson omnibus normality test or Shapiro-Wilkes normality test if sample size was too small for the former. Equality of variance between groups was tested using Bartlett’s test for equal variance. Mean comparisons between two groups were compared using the Student’s t-test (normal distribution) or Mann-Whitney (non-normal distribution (Prism 5.0d for MAC). For multiple comparisons, one-way ANOVA was performed, followed by the appropriate post hoc test as specified in figure legends (Stata/IC 11.2 for windows or Prism 5.0d for MAC). For those subgroups with too few numbers to assess normality statistically, we validated parametric tests with appropriate nonparametric tests (Kruskal-Wallis test with post-hoc Dunnett’s multiple comparison test or Mann-Whitney test for comparing two groups). For comparisons with two crossed factors we performed two-way ANOVA with post hoc Bonferroni test for multiple comparison. In experiments where two-way ANOVA test could not be performed due to low sample size, we performed multiple t-tests with adjustment for multiple comparisons using a Bonferroni correction. For myocyte experiments, analysis was performed using repeated measures ANOVA with random effect. Mortality data was quantified using a Z-test of proportions. For experiments with Ranolazine in vivo, two-factor repeated measures ANOVA was performed; in addition, we also performed paired-T-tests between the ‘pre-‘and ‘post-’ groups for ranolazine and placebo groups.
Results
Cardiac SGK1 is activated in heart failure and diet-induced insulin resistance
We previously found that both activated or phosphorylated (thr-256)-SGK1 (pSGK1) and total SGK1 increase early after TAC-induced pressure-overload (TAC)5. SGK1 activity also remained elevated in chronic models of heart failure following TAC (TAC-HF) (Fig. 1A). In contrast, neither phosphonor total SGK1 were altered in a model of exercise-induced physiological hypertrophy (Supplemental Fig. 1).
Figure 1.
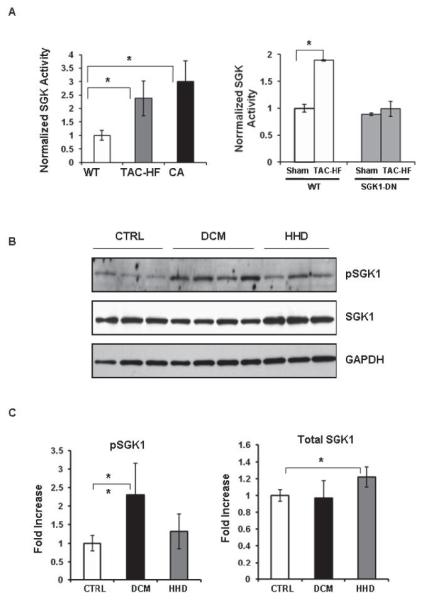
SGK1 activity is increased in murine and human heart disease. (A) SGK1 kinase assays were performed on SGK1 immunoprecipitated from heart lysates. Data shown are mean±SEM normalized to WT sham-treated values in each group from 3 independent experiments (*p<0.05, Welch’s t-test with Bonferroni adjustment). (B) Immunoblots of ventricular lysates from humans with healthy hearts who died of other causes (CTRL), patients with dilated cardiomyopathy (DCM) at the time of transplant, or patients with hypertension and increased LV mass (HHD), using an antibody against total- or phospho-SGK1. (C) Cumulative quantitated data (mean±SD for n=6-10 samples/group) normalized to GAPDH are represented as fold-change over control (*p<0.05, **p<0.001; one-way ANOVA; Fischer-Hayter post hoc test).
To see if SGK1 is altered in human heart disease, we examined ventricular tissue from patients with hypertensive heart disease who died from non-cardiac causes (HHD, n=7) as well as explants from patients with heart failure and dilated cardiomyopathy (DCM, n=10) at the time of orthotopic transplantation. Healthy unused donor hearts served as controls (n=7). The cohorts were age and gender matched. Coronary angiograms had confirmed lack of significant epicardial coronary artery disease in the patients with DCM, and gravimetry confirmed a trend towards increased LV mass in patients with HHD (CTRL 363±133 gm, HHD 518±112 gm, p =0.07). In HHD hearts, there was an increase in total SGK1 (1.22±0.12, p<0.05) without a change in pSGK1 compared with controls (Fig. 1B, C). DCM hearts had a 2.3-fold increase in pSGK1 (2.3±0.85-fold, p<0.005 vs controls) without alterations in total SGK1 (Fig. 1B, C).
These data suggest that SGK1 is abnormally regulated on multiple levels in human heart disease and murine models of hemodynamic stress but not in physiological hypertrophy.
Cardiac-specific SGK1 Transgenic Mice
To investigate the functional role of SGK1 in the heart, we generated transgenic mice (TG) with cardiac-specific expression of either a constitutively-active (S433D) or a dominant-negative (K127M) SGK1 mutant8 driven by the α-myosin heavy chain promoter20. Two of three independent SGK1-CA lines had increased early mortality and could not be maintained. A third line manifested normal viability and stable Mendelian inheritance, and was investigated further. Two independent SGK1-DN lines had similar transgene expression and baseline phenotypes, and thus only one of these lines was characterized in detail. Immunoblotting for an incorporated hemagglutinin (HA) epitope tag confirmed cardiac-specific expression of both transgenes (Supplemental Fig. 2A). SGK1 kinase activity was increased in SGK1-CA hearts at baseline to a level comparable to that seen in hearts from mice with TAC-induced HF (2.3 fold for TAC-HF, 2.9 fold for SGK1-CA TG mice vs WT mice, p<0.05 for both, Fig. 1A). SGK1 activity in SGK1-DN hearts was no different from the low level seen in WT at baseline, but the increase in SGK1 kinase activity seen in TAC-HF WT mice was completely blocked in SGK1-DN mice (Fig. 1A). These activity measurements were further supported by the observation that SGK1-CA increased phosphorylation of an SGK1-specific substrate (Foxo3A Ser-315/31821), while SGK1-DN blocked phosphorylation of this site after TAC (Supplemental Fig. 2B,C). Phosphorylation of he Foxo3A-Ser-253 site, which is favored by Akt, was not altered (Supplemental Fig. 2B). Thus the SGK1-CA mice provide constitutive activation of SGK1 in a pathophysiologically relevant range at baseline, while the SGK1-DN mice act as an effective dominant negative blocking the increase in SGK1 activity otherwise seen after TAC.
Gross and microscopic cardiac structure appeared normal in young adult (3-6 month old) SGK1-CA and –DN mice, including normal HW/BW, HW/TL, and cardiomyocyte size. There was also no increase in fibrosis by Mason-Trichrome staining at these ages (Supplemental Table 1 and Supplemental Fig. 3). Echocardiography revealed normal left ventricular wall and diastolic chamber dimensions in both SGK1-CA and SGK1-DN TG at these ages. However, fractional shortening (FS) was reduced in SGK1-CA mice compared with the age-matched WT littermates (WT: 64±5%; SGK1-CA: 56±8%, p≤0.001). SGK1-DN animals had normal function by echocardiography (Supplemental Table 1). To exclude potential confounding effects of fibrosis and gender, we focused subsequent experiments on 3-6 month old male animals of each genotype.
Invasive hemodynamic studies confirmed that measures of systolic (dP/dtmax and PRSW) and diastolic (dP/dtmin, taug) function were depressed in SGK1-CA but not SGK1-DN mice (Supplemental Table 2). Thus at baseline, young adult SGK1-CA mice have mild but significant systolic and diastolic dysfunction without overt hypertrophy or fibrosis, while the SGK1-DN mice have normal cardiac structure and function.
SGK1 activation modulates the response to TAC
To assess the role of SGK1 activation in the development of heart failure, we subjected SGK1-CA and –DN mice to TAC. Animals were euthanized either 4 (for SGK1-CA and WT littermates) or 7 weeks (for SGK1-DN and WT littermates) after TAC. The time-points were dictated by our IACUC policies which require animals be euthanized when they develop severe heart failure. At 4 weeks, TAC induced an increase in HW/BW and wall thickness in both SGK1-CA mice and WT littermates (Supplemental Fig. 4A, B). However, SGK1-CA hearts were dilated with thinner walls and markedly reduced function (Supplemental Fig. 4C, D). There was a non-significant trend toward increased fibrosis in the SGK1-CA compared with WT littermates after TAC (Supplemental Fig. 4E). These data suggest that the baseline cardiac dysfunction seen with chronic SGK1 activation is markedly exacerbated by the additional stress imposed by TAC and accelerates progression to HF.
Both WT and SGK1-DN mice tolerated TAC better than SGK1-CA mice, and thus could be followed longer after the intervention. Seven weeks after TAC, HW/BW ratios and LV wall thickness increased in both SGK1-DN and WT littermates compared to sham-operated controls (Fig. 2A, B), although the change in HW/BW was less marked in SGK1-DN mice (Fig. 2A). Echocardiography seven weeks after TAC revealed reduced fractional shortening (FS) and increased LV dilatation (LVDD) in WT but not SGK1-DN mice (Fig. 2C, D), likely accounting for the differences noted in HW/BW. SGK1-DN mice also had less fibrosis than WT mice after TAC (Fig. 2E). Thus, SGK1 inhibition protects against the development of heart failure, cardiac dilation, and fibrosis following pressure overload.
Figure 2.
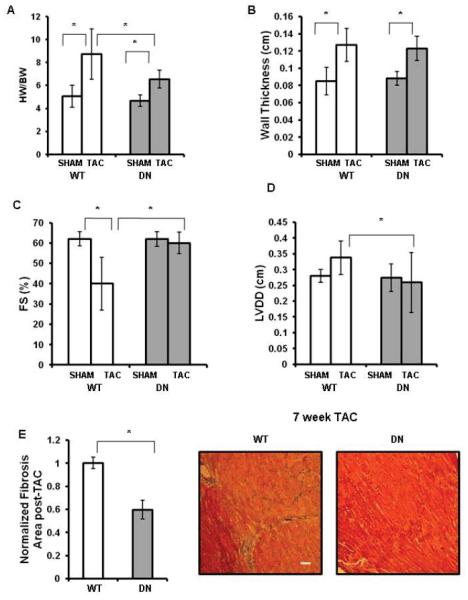
SGK1-DN mice are protected from heart failure after aortic constriction. (A-E): Gravimetric, echocardiographic, and histological data from SGK1-DN and WT littermates 7 weeks after TAC or SHAM operation. (A): HW/BW ratios showed significantly more LV mass in WT mice compared with SGK1-DN mice after TAC; (B-D): Wall thickness, FS, and LVDD as measured by echocardiography 7 weeks after TAC showed SGK1-DN mice are protected from LV dilatation and cardiac dysfunction compared with WT mice; (E): Fibrosis is reduced in SGK1-DN compared to WT mice after TAC (inset shows representative Masson-Trichrome stain). *p<0.05 (two-way ANOVA with post hoc Bonferroni) n=10 in TAC groups, n=5 in sham groups.
Altered cardiac and cellular electrophysiology in SGK1-CA mice
Because two of the three SGK1-CA transgenic lines had increased mortality and SGK1 is known to interact with ion channels, we investigated the electrophysiological effects of chronic SGK1 activation. Electrocardiograms (EKGs) in SGK1-CA mice revealed increases in R wave amplitude, QRS duration, and QTc interval (QTc; 137±32 in SGK1-CA vs. 100±12 ms in WT; p<0.05) without a change in RR or PR intervals (Supplemental Fig. 5A). In addition, spontaneous ventricular tachycardia (VT) was noted in a 12-month old SGK1-CA mouse. SGK1-DN EKGs were not different from WT littermates (Supplemental Fig. 5B) and did not manifest ventricular arrhythmias. Holter monitoring for 2 weeks showed a trend towards increased premature ventricular activity, but due to the sporadic nature of sudden cardiac death, we were not able to correlate mortality with ventricular arrhythmias in the SGK1-CA mice at baseline (Supplemental Fig. 6).
To provoke ventricular arrhythmia, we performed intracardiac electrophysiological studies (EPS) using rapid ventricular pacing and programmed ventricular extra-stimuli15 in SGK1-CA, SGK1-DN, and WT littermates. Half of the six SGK1-CA mice studied had inducible polymorphic ventricular tachycardia (PMVT) with pacing to 90msec cycle lengths (CLs). In contrast, more aggressive protocols with pacing to 60msec CLs did not induce ventricular tachycardia (VT) in any of nine WT and SGK1-DN mice studied (p<0.05 by Z-test for proportions) (Fig. 3A, B).
Figure 3.
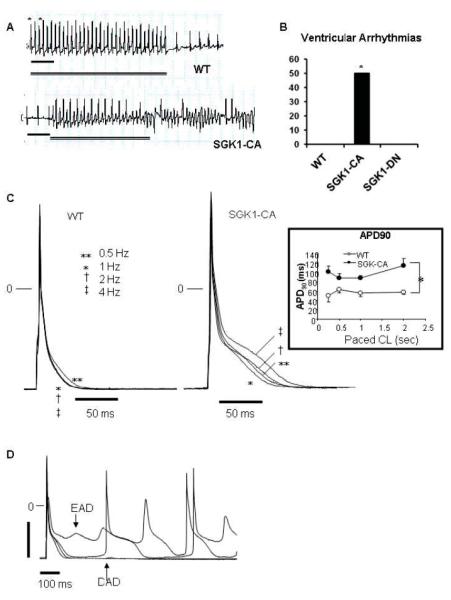
Propensity to ventricular arrhythmias, action potential prolongation, and afterdepolarizations are increased in SGK1-CA mice. (A) Representative tracings for WT and SGK1-CA mice from the distal (RV) pole during rapid pacing protocol (with pacing cycle lengths [CL] down to 60 msec in WT mice and 90 msec in the SGK1-CA mice). Asterisks denote pacing, double line indicates duration of pacing and scale bar denotes 200 msec. (B) Cumulative results for VT/VF inducibility from age-matched WT (0/5), SGK1-CA (3/6) and SGK1-DN (0/4) mice are shown (*p<0.05 by Z-test of proportions), in the absence of fibrosis or structural abnormalities (Supplemental Fig. S3). (C) Superimposed action potentials (APs) at CL 0.5-4 Hz in WT and SGK1-CA ventricular cardiomyocytes. Inset: APD at 90% repolarization (APD90) was longer in SGK1-CA compared with WT ventricular CMs, *p<0.05 by repeated measures ANOVA. (D) Representative EADs or DADs in SGK1-CA myocytes paced at 0.5 Hz. For quantification of EADs/DADs in comparison to WT see Fig. 5D.
The induction of PMVT with pacing rather than programmed extra-stimuli in the absence of fibrosis and structural heart disease was suggestive of triggered electrical activity. Hence we next examined the electrophysiological characteristics of cardiomyocytes isolated from SGK1-CA mice. Whole cell patch clamp of cardiomyocytes isolated from 3-5 month SGK1-CA mice demonstrated action potential prolongation over a range of pacing frequencies (0.5 to 4 Hz) compared with cardiomyocytes from WT littermates (Fig. 3C). Early (EADs) and delayed (DADs) after depolarizations, a hallmark of triggered electrical activity, were also more frequent in SGK1-CA than WT cardiomyocytes (Fig. 3D). Thus the increased ventricular arrhythmia seen in the SGK1-CA mice likely reflects an increase in triggered activity secondary to action potential prolongation.
Regulation of INa by SGK1
An increase in APD could be secondary to altered potassium, calcium, or sodium currents. Whole cell patch-clamp analyses of cardiomyocytes isolated from SGK1-CA transgenics or WT littermates showed no differences in the peak potassium or calcium current densities (Ito, IK1, or ICa, Supplemental Fig. 7). We also observed no difference in major calcium handling proteins in cardiac sarcoplasmic reticulum or membrane preparations in SGK1-CA mice except for an increase in the sodium-calcium exchanger (NCX1) (Supplemental Fig. 7). Overall these data suggest that altered potassium or calcium currents are unlikely to account for APD prolongation and arrhythmias in SGK1-CA mice.
In contrast, whole cell patch clamp studies revealed substantial changes in sodium currents in SGK1-CA cardiomyocytes. Peak INa current density manifested a −10 mV hyperpolarizing shift (Fig. 4A, B), in addition to increased peak current density when compared to WT cardiomyocytes (Fig. 4C). No difference was found in the reversal potential of INa between the SGK1-CA and WT mice cardiomyocytes.
Figure 4.
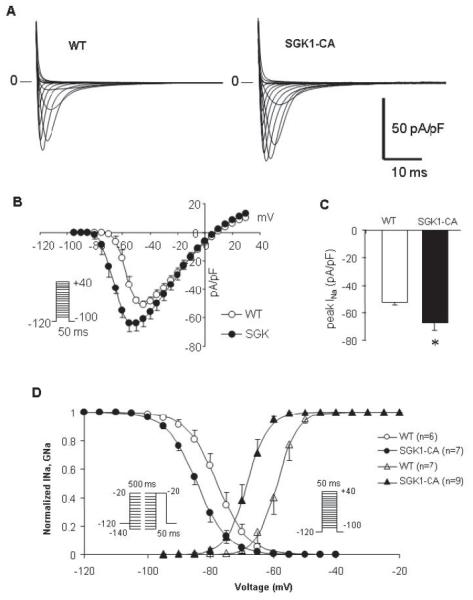
Sodium current density, activation and inactivation are altered in SGK1-CA cardiomyocytes. (A) Representative INa currents in WT and SGK1-CA cardiomyocytes (10mM [Na+]o). (B) Current-Voltage (I-V) relationship for WT (n=7) and SGK1-CA (n=10) cardiomyocytes. (C) Cumulative quantitation reveals peak INa density is increased in SGK1-CA n=10) compared with WT (n=7) cardiomyocytes (*p<0.05, T-test). (D) Superimposed normalized conductance-voltage (GNa-V) and steady state inactivation curves from WT (open circles; n=6 and open triangles; n=7 respectively) and SGK1-CA (closed circles; n=7 and closed triangles; n=9 respectively) cardiomyocytes. There is a significant hyperpolarizing shift noted both in the voltage dependence of activation and steady-state inactivation of INa in SGK1-CA compared with WT.
To assess the effect of SGK1 activation on channel gating, we measured voltagedependent activation and steady-state inactivation of INa. Cardiomyocytes from SGK1-CA mice showed a hyperpolarizing shift of −10 mV in the voltage-dependence of Na+ activation as illustrated in the conductance (GNa) curves (V1/2: −68.1±4.0 vs. −58.8±4.4mV, p<0.001), without altering the slope factor k (Fig. 4D and Supplemental Table 3). There was also a smaller negative voltage shift of −5 mV shift in INa steady state inactivation in the SGK1-CA compared with WT cardiomyocytes (V1/2; −83.9±3.6 vs. −77.8±4.3mV, p<0.05) (Fig. 4D, Supplemental Table 3). The combination of a large hyperpolarizing shift in the conductance-voltage relationship with a smaller hyperpolarizing shift of the steady-state inactivation leads to an increase in the ‘window current’, which would increase Na+ conductance over a wider range of membrane potentials22, 23.
SGK1 activation did not affect recovery from, or entry into fast or intermediate inactivation (IIM) (Supplemental Fig. 8A,B, Supplemental Table 3). Similarly, neither the fast nor slow time constants of INa decay were altered by chronic SGK1 activation (Supplemental Fig. 8C, Supplemental Table 3).
An increase in the persistent Na current, INaL and increases in window currents have been described in genetic arrhythmia and cardiomyopathy syndromes caused by mutations in the Nav1.5 sodium channel encoded by SCN5a24, 25 as well as in acquired heart failure26. SGK1-CA cardiomyocytes displayed a 3.6-fold increase in INaL compared to WT (0.87±0.29% vs. 0.24±0.13%, p<0.01, Fig. 5A). This increase is comparable to that seen with arrhythmogenic SCN5a mutations or acquired heart failure26.
Figure 5.
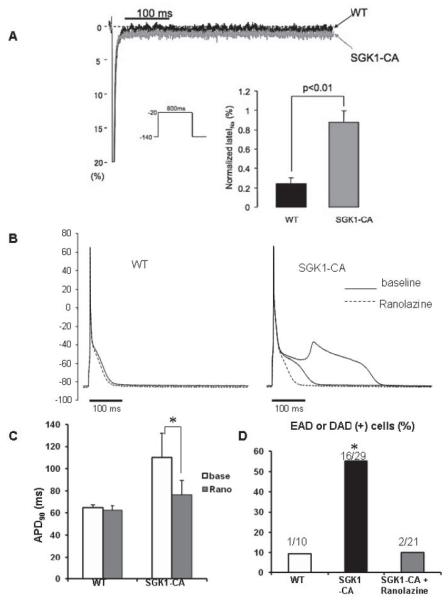
SGK1 activation increases INaL while ranolazine normalizes APD and suppresses afterdepolarizations in SGK1-CA cardiomyocytes. (A) Representative normalized INaL (% of peak) superimposed for WT and SGK1-CA cardiomyocytes. Inset shows that normalized INaL was larger in SGK1-CA (n=6) than in WT cardiomyocytes (n=5). (B) Superimposed APs in cardiomyocytes from WT and SGK1-CA mice before (baseline: solid line) and after ranolazine (dotted line) at 0.5Hz pacing. Ranolazine (1μmol/L) normalized APD and suppressed EAD in SGK but did not affect APs in WT myocytes. (C) Ranolazine (1μmol/L) normalized APD90 in SGK1-CA cardiomyocytes (*p<0.05, repeated measures ANOVA), but did not affect APD90 in WT cardiomyocytes. (D) EADs and DADs were more frequent in SGK1-CA compared with WT cardiomyocytes. Ranolazine (1 μmol/L) reduced after-depolarizations in SGK1-CA cardiomyocytes (*p<0.05 by χ2 test) to levels seen in WT cardiomyocytes.
Alteration of INa plays a key role in the phenotype of SGK1-CA TG mice
To test the functional importance of increased INaL in SGK1-CA cardiomyocytes, we used ranolazine at low concentrations to selectively block INaL, without affecting peak INa27. Ranolazine treatment normalized APD in SGK1-CA cardiomyocytes without affecting APD in WT cardiomyocytes (Fig. 5B, C). Moreover, ranolazine treatment reduced the number of SGK1-CA cardiomyocytes with after-depolarizations (EADs and DADs) to the background level seen in WT cardiomyocytes (Fig. 5D). These data strongly suggest that SGK1 activation leads to APD prolongation and increased after-depolarizations primarily through increasing INaL.
To investigate the role of increased INaL in this model in vivo, we examined the effects of ranolazine on SGK1-CA TG mice at baseline and in a model of ischemia-reperfusion, where INaL has been implicated in ventricular arrhythmias28, 29. SGK1-CA TG mice had substantially higher mortality (80%) compared to either WT (24%, p=0.002) or SGK1-DN mice (12.5%, p=0.0006) (Fig. 6A). Because the majority of SGK1-CA mice were dying in the first hour after reperfusion, we subjected the mice to continuous telemetry during the 30 minutes of ischemia and the first 45 minutes of reperfusion or until the mice had recovered from anesthesia. Interestingly, SGK1-CA mice had a higher incidence of lethal ventricular arrhythmias during ischemia-reperfusion compared to WT mice (4/5 in SGK1-CA versus 0/4 in WT, p=0.02) (Fig. 6A, B).
Figure 6.
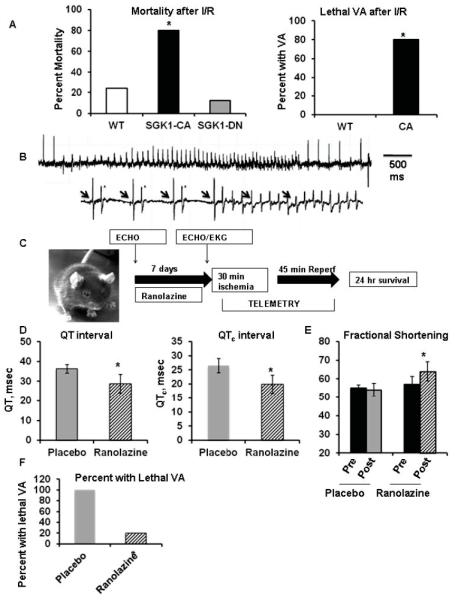
Increased susceptibility to ventricular arrhythmias and cardiac dysfunction in SGK1-CA mice can be reversed by ranolazine. (A) Mortality is increased in SGK1-CA mice after ischemia-reperfusion (I/R: 30 minute LAD ligation/24 hour reperfusion) Left panel: mortality in SGK1-CA (8/10), WT (7/29) and SGK1-DN (2/16) mice, *p<0.005 by Z-test of proportions. Right panel: Telemetry demonstrated that SGK1-CA mice develop lethal ventricular arrhythmia (VA) (4/5) after I/R, that were not seen in WT mice (0/4) *p=0.02 by Z-test. (B) Representative tracings of VA in SGK1-CA mice during reperfusion. Lower tracing shows ventricular bigeminy (asterisks) prior to onset of ventricular tachycardia with AV dissociation (arrows denote p-waves). (C) Schema for in vivo ranolazine experiments. (D) Ranolazine decreases QT and QTc intervals in SGK1-CA mice in comparison to placebo (*p<0.05 by unequal variance Ttest or Mann-Whitney test). (E) Fractional shortening was better in SGK1-CA mice after 7 days of ranolazine treatment than in mice treated with placebo (*p<0.05 by two-way repeated measures ANOVA). In paired T-tests, mice treated with ranolazine showed a non-significant trend towards improved fractional shortening (p=0.055), while there was no change in placebotreated mice (p=0.59). (F) Incidence of lethal VA after I/R was decreased in ranolazine-treated SGK1-CA mice in comparison to placebo (*p<0.05 by Z-test of proportions).
To investigate the role of INaL in vivo, we implanted ranolazine slow-release pellets subcutaneously in SGK1-CA mice in comparison to placebo pellets (Fig. 6C). Seven days after implantation, both the QT and QTc intervals were decreased in the ranolazine-compared with placebo-treated animals (Fig. 6D). Remarkably, echocardiography also revealed that fractional shortening was better in ranolazine-compared to placebo-treated mice (Fig. 6E). Evaluation of change from baseline fractional shortening by paired T-tests demonstrated a non-significant trend toward improvement in ranolazine-treated mice (p=0.055) but no difference in placebotreated animals (p=0.59). The small number of animals in this complex protocol obviously limits the statistical power for such inferences. Nevertheless, these findings are provocative and suggest the role of SGK1-mediated changes in sodium currents in adverse mechanical remodeling should be addressed more fully in future studies. Finally, ranolazine treatment markedly reduced incidence of lethal ventricular arrhythmia (1/5) compared to either untreated (4/5, Fig. 6A) or placebo-treated SGK1-CA mice (3/3) (Fig. 6F). Taken together, these data strongly support the hypothesis that increased INaL is responsible for the ventricular arrhythmia seen in SGK1-CA mice, paralleling our in vitro studies, and suggest that INaL may also contribute to the cardiac dysfunction seen with SGK1 activation in vivo.
SGK1 is necessary and sufficient for heart failure-associated alterations in Nav1.5
To understand the biochemical basis for the functional changes seen in INa, we examined Nav1.5, the primary pore-forming subunit of the cardiac voltage-gated sodium channel complex. While there was no change in overall cardiac Nav1.5 protein expression in SGK1-CA or –DN mice compared to WT, sucrose gradient fractionation30 demonstrated that the subcellular distribution of Nav1.5 was altered by both TAC-induced heart failure and SGK1 activation in a similar way. In sham-operated WT mice, a large proportion of Nav1.5 was localized to lipid rafts (LR) with a modest amount of Nav1.5 in the heavy membrane (HM) fraction, likely comprising both intercalated disc and sarcoplasmic reticulum proteins (Supplemental Fig. 9A, top panel). TACinduced heart failure and SGK1-CA hearts had similar increases in Nav1.5 in the HM fraction (Supplemental Fig. 9A, middle and bottom panels). In contrast, while the baseline Nav1.5 distribution in SGK1-DN hearts was similar to that in sham-operated WT, the increase in HM Nav1.5 seen in WT mice following TAC was markedly decreased in SGK1-DN hearts (Supplemental Fig. 9B). Thus chronic SGK1 activation and TAC-induced HF increase trafficking of Nav1.5 to the HM fraction, while SGK1 inhibition blocks this response to TAC, suggesting SGK1 is both necessary and sufficient for these changes.
The increase in HM Nav1.5 would be expected to increase Na+-channel surface availability. To test this directly, we used biotin to label surface-exposed proteins in neonatal rat ventricular myocytes (NRVMs) expressing either SGK1-CA or GFP, and selectively collected labeled surface proteins with an avidin-coupled column. As controls, the transmembrane protein, Glut 4, was labeled and recovered robustly from the surface of both GFP- and SGK1-CAexpressing NRVMs, while GSK3β, a cytoplasmic protein, was not (Fig. 7A). SGK1-CA expression increased the amount of biotin-labeled Nav1.5 in NRVMs in comparison to GFPtransduced cells (Fig. 7A). These data are consistent with the hypothesis that SGK1 activation increases the amount of Nav1.5 available at the cell surface.
Figure 7.
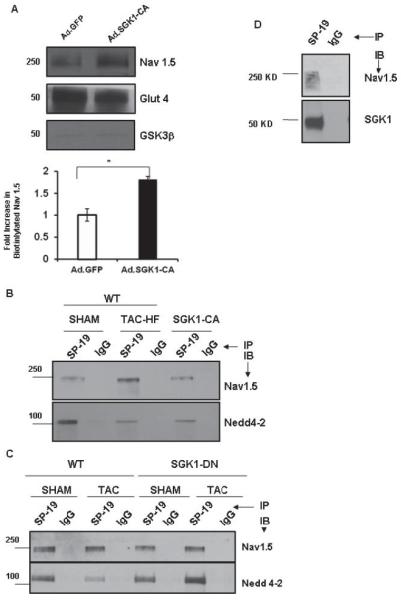
SGK1 activation is necessary and sufficient for alterations in cardiac sodium channel seen in failing hearts. (A) Cardiomyocytes were infected with either Ad.SGK1-CA or Ad.GFP, and biotin labeled to tag surface (e.g. Nav1.5, Glut4) but not cytoplasmic proteins (e.g. GSK3β). Biotin-labeled proteins were captured using an avidin column and subjected to immunoblotting. SGK1-CA expression increased surface expression of Nav1.5, *p<0.05, n=3 independent experiments. (B) Total heart lysates from WT (sham-operated), TAC-HF, or unoperated SGK1-CA mice were immunoprecipitated with a pan-Na+-channel antibody (SP-19). Immunoblotting with antibodies to Nav1.5 or Nedd4-2 showed a decrease in Nedd4-2 binding in TAC-HF and SGK1-CA hearts. (C) Cardiac lysates from SGK1-DN transgenic or WT mice subjected to TAC or sham-operation were immunoprecipitated with SP-19 and immunoblotted with the antibody specific Nav1.5 or Nedd4-2 (bottom panel). The decrease in Nedd4-2 binding to Nav1.5 seen in failing WT hearts is completely prevented in SGK1-DN hearts after TAC. (D) SGK1 directly associates with Nav1.5: Sodium channel was immunoprecipitated from heart lysates using SP-19 or control IgG antibody-coupled columns and subjected to immunoblotting as labeled. Figures are representative of 3 independent experiments.
Activation of SGK1 in renal tubular cells leads to phosphorylation of the ubiquitin ligase, Nedd4-231, 32 and decreased binding of the phospho-Nedd4-2 to the PY motif of ENaC, which increases surface expression and channel activity of ENaC33. Nav1.5 has also been shown to bind to Nedd4-2 via a conserved PY motif in its C-terminal tail34, and in a heterologous expression system, decreased binding of Nedd4-2 to Nav1.5 leads to increased Nav1.5 membrane expression35. We therefore examined if altered Nedd4-2 binding plays a role in Nav1.5 trafficking in response to TAC or SGK1 activation in vivo. Nedd4-2 co-immunoprecipitated with the Na channel in sham-operated WT hearts and there was a marked decrease in Nedd4-2 binding to Na channels in TAC-HF and SGK1-CA hearts (Fig. 7B). In contrast, SGK1-DN preserved Nedd 4-2 binding to Na channels after TAC (Fig. 7C). Thus SGK1 activation appears both necessary and sufficient for the alterations in Nedd4-2 binding to Nav1.5 seen after TACinduced HF.
It seems likely that the observed alterations in Nedd4-2 binding and surface-available Na channel contribute to overall increases in INa but not the observed changes in channel gating and kinetics. To investigate other possible contributing mechanisms, we next examined whether SGK1 physically interacts with Nav1.5 protein. Immunoprecipitation experiments demonstrated that endogenous SGK1 protein binds the Na channel (Fig. 7D). Since isoform-specific Na channel antibodies for immunoprecipitation are not available, it is possible that SGK1 binds not only Nav1.5 but also less abundant channel isoforms present in the heart. This physical association raised the possibility that SGK1 could phosphorylate Nav1.5. To begin to explore this possibility, we determined the SGK1 consensus target sequence using a peptide library36. The preferred phosphorylation target sequence for SGK1 is similar to that for Akt (RKRRnS/T) but differs in the secondary amino acids (Supplemental Fig. 9C). Using the newly defined targe motif in ScanSite, we identified two consensus sites in SCN5a (S483 and S664, both in the DI-II linker, which have been previously noted11) and an additional novel but weaker consensus site (S1590) in the D4/S3 region (Supplemental Fig. 9D)
Overall, our data support the model that SGK1 is both necessary and sufficient for changes seen in Na-channel-Nedd4-2 binding and trafficking in TAC-induced HF. In addition, SGK1 directly associates with the sodium channel, which harbors multiple consensus target sequences for SGK1 phosphorylation. Cumulatively these biochemical changes are likely to account for the electrophysiological alterations observed in SGK1-CA cardiomyocytes and mice. Determining the specific functional contributions of these protein-protein interactions as well as the putative SGK1 phosphorylation sites will be of interest for future studies.
Discussion
Heart failure and the ventricular arrhythmias that can accompany it are growing causes of morbidity and mortality around the world37. Acute activation of proximal PI-3 Kinase signaling has beneficial effects on cardiomyocyte survival and function4, 5, 38 but is paradoxically increased in heart failure and diabetes7, raising the possibility that chronic activation of these initially compensatory pathways may become maladaptive. In this context, we investigated the role of SGK1, a serine-threonine kinase that we had previously shown is activated early in pressureoverload and acutely promotes cardiomyocyte survival5. We found that SGK1 phosphorylation is also increased in animal models of chronic heart failure and human dilated cardiomyopathy. In contrast, SGK1 was not altered in exercise-induced physiological hypertrophy.
Transgenic mice with constitutive cardiac activation of SGK1 spontaneously developed everal hallmarks of adverse ventricular remodeling including systolic and diastolic dysfunction as well as susceptibility to ventricular arrhythmias. The pro-arrhythmic effects of SGK1 activation were linked to biochemical and functional changes in the cardiac sodium channel that culminated in action potential prolongation and after-depolarizations, as well as an increase in INaL. APD prolongation and after-depolarizations were completely normalized by treatment with ranolazine at doses that block late but not peak INa. Finally, SGK1-CA mice treated with ranolazine in vivo not only showed decreased EKG abnormalities and susceptibility to ventricular arrhythmias, but also had better cardiac function, suggesting that altered sodium flux, specifically increased INaL is an important contributor to both the electrical and mechanical phenotypes seen with chronic SGK1 activation in the heart. Consistent with this hypothesis, human SCN5a mutations can cause not only arrhythmia but also cardiomyopathy23, 39. However, it seems likely that other SGK1 targets also contribute to cardiac dysfunction and/or fibrosis. The genetic models described in this work should provide useful platforms for identification of such pathways and dissecting their functional contributions to the phenotypes observed.
We also found that SGK1 activation led to biochemical changes in the sodium channel that recapitulated changes seen in TAC-induced heart failure and included reduced association with the ubiquitin ligase, Nedd4-2, and altered channel trafficking resulting in an increase of surface available Nav1.5. Conversely, inhibition of SGK1 in the SGK1-DN mice blocked the biochemical changes in Nav1.5 after TAC-induced HF, supporting the specificity of these findings and suggesting SGK1 is also necessary for at least some aspects of electrical remodeling. SGK1 bound Nav1.5, and peptide mapping identified several candidate SGK1 targets in the sodium channel. Intriguingly, one of these is in a region important in channel inactivation gating23 and adjacent to a site mutated (D1595H in the Domain IV/S3 region) in a cardiomyopathy syndrome associated with arrhythmias23,39. Testing the functional role of these putative phosphorylation sites will be of interest for future studies.
Taken together, the current study underscores the importance of SGK1 in both cardiac dysfunction and arrhythmia, Other kinases are also important modulators of Nav1.5, including PKA40, PKC41, and CamKII42, 43, and thus inhibiting SGK1 alone may not fully protect against these phenotypes. Nevertheless, our genetic data suggest that inhibition of SGK1 warrants further investigation as a therapeutic target in these settings particularly given the success of therapeutically targeting kinases on other settings.
Supplementary Material
Acknowledgements
We thank Dr. Ling Li for expert technical support and Dr. Serafima Zaltsman for managing the mouse colonies. SD is a trainee of the Clinical Investigators Training Program: BIDMC–Harvard/MIT HST, in collaboration with Pfizer, Inc and Merck&Co.
Funding Sources: This research was supported by a Leducq Foundation Network of Research Excellence (AR) and the NIH [AR(R01HL094677, R21HL104370), SD (KO8HL089319), PE, GT(R01HL050411)]. AR also gratefully acknowledges support from Judith and David Ganz. AR is a principal faculty member of the Harvard Stem Cell Institute and an Associate Member of the Broad Institute.
Footnotes
Conflict of Interest Disclosures: None.
Journal Subject Codes: [132] Arrhythmias-basic studies; [138] Cell signalling/signal transduction; [145] Genetically altered mice; [148] Heart failure - basic studies; [152] Ion channels/membrane transport
References
- 1.Undrovinas AI, Maltsev VA, Kyle JW, Silverman N, Sabbah HN. Gating of the late na+ channel in normal and failing human myocardium. J Mol Cell Cardiol. 2002;34:1477–1489. doi: 10.1006/jmcc.2002.2100. [DOI] [PubMed] [Google Scholar]
- 2.Cesario DA, Brar R, Shivkumar K. Alterations in ion channel physiology in diabetic cardiomyopathy. Endocrinol Metab Clin North Am. 2006;35:601–610. ix–x. doi: 10.1016/j.ecl.2006.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Matsui T, Li L, MonteF d, Fukui Y, Franke TF, Hajjar RJ, Rosenzweig A. Adenoviral gene ransfer of activated phosphatidylinositol 3′-kinase and akt inhibits apoptosis of hypoxic cardiomyocytes in vitro. Circulation. 1999;100:2373–2379. doi: 10.1161/01.cir.100.23.2373. [DOI] [PubMed] [Google Scholar]
- 4.Matsui T, Tao J, del Monte F, Lee K-H, Li L, Picard M, Force TL, Franke TF, Hajjar RJ, Rosenzweig A. Akt activation preserves cardiac function and prevents injury after transient cardiac ischemia in vivo. Circulation. 2001;104:330–335. doi: 10.1161/01.cir.104.3.330. [DOI] [PubMed] [Google Scholar]
- 5.Aoyama T, Matsui T, Novikov M, Park J, Hemmings B, Rosenzweig A. Serum and glucocorticoid-responsive kinase-1 regulates cardiomyocyte survival and hypertrophic response. Circulation. 2005;111:1652–1659. doi: 10.1161/01.CIR.0000160352.58142.06. [DOI] [PubMed] [Google Scholar]
- 6.Haq S, Choukroun G, Lim H, Tymitz KM, del Monte FF, Gwathmey J, Grazette L, Michael A, Hajjar R, Force T, Molkentin JD. Differential activation of signal transduction pathways in human hearts with hypertrophy versus advanced heart failure. Circulation. 2001;103:670–677. doi: 10.1161/01.cir.103.5.670. [DOI] [PubMed] [Google Scholar]
- 7.Cook SA, Varela-Carver A, Mongillo M, Kleinert C, Khan MT, Leccisotti L, Strickland N, Matsui T, Das S, Rosenzweig A, Punjabi P, Camici PG. Abnormal myocardial insulin signalling in type 2 diabetes and left-ventricular dysfunction. Eur Heart J. 2010;31:100–111. doi: 10.1093/eurheartj/ehp396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Park J, Leong ML, Buse P, Maiyar AC, Firestone GL, Hemmings BA. Serum and glucocorticoid-inducible kinase (sgk) is a target of the pi 3-kinase-stimulated signaling pathway. Embo J. 1999;18:3024–3033. doi: 10.1093/emboj/18.11.3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Webster MK, Goya L, Ge Y, Maiyar AC, Firestone GL. Characterization of sgk, a novel member of the serine/threonine protein kinase gene family which is transcriptionally induced by glucocorticoids and serum. Mol Cell Biol. 1993;13:2031–2040. doi: 10.1128/mcb.13.4.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wulff P, Vallon V, Huang DY, Volkl H, Yu F, Richter K, Jansen M, Schlunz M, Klingel K, Loffing J, Kauselmann G, Bosl MR, Lang F, Kuhl D. Impaired renal na(+) retention in the sgk1-knockout mouse. J Clin Invest. 2002;110:1263–1268. doi: 10.1172/JCI15696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Boehmer C, Wilhelm V, Palmada M, Wallisch S, Henke G, Brinkmeier H, Cohen P, Pieske B, Lang F. Serum and glucocorticoid inducible kinases in the regulation of the cardiac sodium channel scn5a. Cardiovasc Res. 2003;57:1079–1084. doi: 10.1016/s0008-6363(02)00837-4. [DOI] [PubMed] [Google Scholar]
- 12.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard MH, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 13.Arany Z, Novikov M, Chin S, Ma Y, Rosenzweig A, Spiegelman BM. Transverse aortic constriction leads to accelerated heart failure in mice lacking ppar-gamma coactivator 1alpha. Proc Natl Acad Sci U S A. 2006;103:10086–10091. doi: 10.1073/pnas.0603615103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Matsui T, Li L, Wu JC, Cook SA, Nagoshi T, Picard M, Liao R, Rosenzweig A. Phenotypic spectrum caused by transgenic overexpression of activated akt in the heart. J Biol Chem. 2002;277:22896–22901. doi: 10.1074/jbc.M200347200. [DOI] [PubMed] [Google Scholar]
- 15.Berul CI, Aronovitz MJ, Wang PJ, Mendelsohn ME. In vivo cardiac electrophysiology studies in the mouse. Circulation. 1996;94:2641–2648. doi: 10.1161/01.cir.94.10.2641. [DOI] [PubMed] [Google Scholar]
- 16.Morissette MR, Cook SA, Foo S, McKoy G, Ashida N, Novikov M, Scherrer-Crosbie M, Li L, Matsui T, Brooks G, Rosenzweig A. Myostatin regulates cardiomyocyte growth through modulation of akt signaling. Circ Res. 2006;99:15–24. doi: 10.1161/01.RES.0000231290.45676.d4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Song KS, Li S, Okamoto T, Quilliam LA, Sargiacomo M, Lisanti MP. Co-purification and direct interaction of ras with caveolin, an integral membrane protein of caveolae microdomains. Detergent-free purification of caveolae microdomains. J Biol Chem. 1996;271:9690–9697. doi: 10.1074/jbc.271.16.9690. [DOI] [PubMed] [Google Scholar]
- 18.Kaab S, Nuss HB, Chiamvimonvat N, O’Rourke B, Pak PH, Kass DA, Marban E, Tomaselli GF. Ionic mechanism of action potential prolongation in ventricular myocytes from dogs with pacing-induced heart failure. Circ Res. 1996;78:262–273. doi: 10.1161/01.res.78.2.262. [DOI] [PubMed] [Google Scholar]
- 19.Aiba T, Hesketh GG, Barth AS, Liu T, Daya S, Chakir K, Dimaano VL, Abraham TP, O’Rourke B, Akar FG, Kass DA, Tomaselli GF. Electrophysiological consequences of dyssynchronous heart failure and its restoration by resynchronization therapy. Circulation. 2009;119:1220–1230. doi: 10.1161/CIRCULATIONAHA.108.794834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Subramaniam A, Jones WK, Gulick J, Wert S, Neumann J, Robbins J. Tissue-specific regulation of the alpha-myosin heavy chain gene promoter in transgenic mice. J Biol Chem. 1991;266:24613–24620. [PubMed] [Google Scholar]
- 21.Brunet A, Park J, Tran H, Hu LS, Hemmings BA, Greenberg ME. Protein kinase sgk mediates survival signals by phosphorylating the forkhead transcription factor fkhrl1 (foxo3a) Mol Cell Biol. 2001;21:952–965. doi: 10.1128/MCB.21.3.952-965.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abriel H, Cabo C, Wehrens XH, Rivolta I, Motoike HK, Memmi M, Napolitano C, Priori SG, Kass RS. Novel arrhythmogenic mechanism revealed by a long-qt syndrome mutation in the cardiac na(+) channel. Circ Res. 2001;88:740–745. doi: 10.1161/hh0701.089668. [DOI] [PubMed] [Google Scholar]
- 23.Nguyen TP, Wang DW, Rhodes TH, George AL., Jr Divergent biophysical defects caused by mutant sodium channels in dilated cardiomyopathy with arrhythmia. Circ Res. 2008;102:364–371. doi: 10.1161/CIRCRESAHA.107.164673. [DOI] [PubMed] [Google Scholar]
- 24.Chandra R, Starmer CF, Grant AO. Multiple effects of kpq deletion mutation on gating of human cardiac na+ channels expressed in mammalian cells. Am J Physiol. 1998;274:H1643–1654. doi: 10.1152/ajpheart.1998.274.5.H1643. [DOI] [PubMed] [Google Scholar]
- 25.Dumaine R, Wang Q, Keating MT, Hartmann HA, Schwartz PJ, Brown AM, Kirsch GE. Multiple mechanisms of na+ channel--linked long-qt syndrome. Circ Res. 1996;78:916–924. doi: 10.1161/01.res.78.5.916. [DOI] [PubMed] [Google Scholar]
- 26.Valdivia CR, Chu WW, Pu J, Foell JD, Haworth RA, Wolff MR, Kamp TJ, Makielski JC. Increased late sodium current in myocytes from a canine heart failure model and from failing human heart. J Mol Cell Cardiol. 2005;38:475–483. doi: 10.1016/j.yjmcc.2004.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Fredj S, Sampson KJ, Liu H, Kass RS. Molecular basis of ranolazine block of lqt-3 mutant sodium channels: Evidence for site of action. Br J Pharmacol. 2006;148:16–24. doi: 10.1038/sj.bjp.0706709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kloner RA, Dow JS, Bhandari A. First direct comparison of the late sodium current blocker ranolazine to established antiarrhythmic agents in an ischemia/reperfusion model. J Cardiovasc Pharmacol Ther. 2011;16:192–196. doi: 10.1177/1074248410386485. [DOI] [PubMed] [Google Scholar]
- 29.Belardinelli L, Shryock JC, Fraser H. Inhibition of the late sodium current as a potential cardioprotective principle: Effects of the late sodium current inhibitor ranolazine. Heart. 2006;92(Suppl 4):iv6–iv14. doi: 10.1136/hrt.2005.078790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yarbrough TL, Lu T, Lee HC, Shibata EF. Localization of cardiac sodium channels in caveolin-rich membrane domains: Regulation of sodium current amplitude. Circ Res. 2002;90:443–449. doi: 10.1161/hh0402.105177. [DOI] [PubMed] [Google Scholar]
- 31.Staub O, Verrey F. Impact of nedd4 proteins and serum and glucocorticoid-induced kinases on epithelial na+ transport in the distal nephron. J Am Soc Nephrol. 2005;16:3167–3174. doi: 10.1681/ASN.2005050454. [DOI] [PubMed] [Google Scholar]
- 32.Snyder PM, Olson DR, Thomas BC. Serum and glucocorticoid-regulated kinase modulates nedd4-2-mediated inhibition of the epithelial na+ channel. J Biol Chem. 2002;277:5–8. doi: 10.1074/jbc.C100623200. [DOI] [PubMed] [Google Scholar]
- 33.Diakov A, Korbmacher C. A novel pathway of epithelial sodium channel activation involves a serum- and glucocorticoid-inducible kinase consensus motif in the c terminus of the channel’s alpha-subunit. J Biol Chem. 2004;279:38134–38142. doi: 10.1074/jbc.M403260200. [DOI] [PubMed] [Google Scholar]
- 34.Abriel H, Kamynina E, Horisberger JD, Staub O. Regulation of the cardiac voltage-gated na+ channel (h1) by the ubiquitin-protein ligase nedd4. FEBS Lett. 2000;466:377–380. doi: 10.1016/s0014-5793(00)01098-x. [DOI] [PubMed] [Google Scholar]
- 35.van Bemmelen MX, Rougier JS, Gavillet B, Apotheloz F, Daidie D, Tateyama M, Rivolta I, Thomas MA, Kass RS, Staub O, Abriel H. Cardiac voltage-gated sodium channel nav1.5 is regulated by nedd4-2 mediated ubiquitination. Circ Res. 2004;95:284–291. doi: 10.1161/01.RES.0000136816.05109.89. [DOI] [PubMed] [Google Scholar]
- 36.Hutti JE, Jarrell ET, Chang JD, Abbott DW, Storz P, Toker A, Cantley LC, Turk BE. A rapid method for determining protein kinase phosphorylation specificity. Nat Methods. 2004;1:27–29. doi: 10.1038/nmeth708. [DOI] [PubMed] [Google Scholar]
- 37.Redfield MM. Heart failure--an epidemic of uncertain proportions. N Engl J Med. 2002;347:1442–1444. doi: 10.1056/NEJMe020115. [DOI] [PubMed] [Google Scholar]
- 38.Nagoshi T, Matsui T, Aoyama T, Leri A, Anversa P, Li L, Ogawa W, del Monte F, Gwathmey JK, Grazette L, Hemmings BA, Kass DA, Champion HC, Rosenzweig A. Pi3k rescues the detrimental effects of chronic akt activation in the heart during ischemia/reperfusion injury. J Clin Invest. 2005;115:2128–2138. doi: 10.1172/JCI23073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Olson TM, Michels VV, Ballew JD, Reyna SP, Karst ML, Herron KJ, Horton SC, Rodeheffer RJ, Anderson JL. Sodium channel mutations and susceptibility to heart failure and atrial fibrillation. JAMA. 2005;293:447–454. doi: 10.1001/jama.293.4.447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou J, Yi J, Hu N, George AL, Jr., Murray KT. Activation of protein kinase a modulates trafficking of the human cardiac sodium channel in xenopus oocytes. Circ Res. 2000;87:33–38. doi: 10.1161/01.res.87.1.33. [DOI] [PubMed] [Google Scholar]
- 41.West JW, Numann R, Murphy BJ, Scheuer T, Catterall WA. A phosphorylation site in the na+ channel required for modulation by protein kinase c. Science. 1991;254:866–868. doi: 10.1126/science.1658937. [DOI] [PubMed] [Google Scholar]
- 42.Wagner S, Dybkova N, Rasenack EC, Jacobshagen C, Fabritz L, Kirchhof P, Maier SK, Zhang T, Hasenfuss G, Brown JH, Bers DM, Maier LS. Ca2+/calmodulin-dependent protein kinase ii regulates cardiac na+ channels. J Clin Invest. 2006;116:3127–3138. doi: 10.1172/JCI26620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maltsev VA, Reznikov V, Undrovinas NA, Sabbah HN, Undrovinas A. Modulation of late sodium current by ca2+, calmodulin, and camkii in normal and failing dog cardiomyocytes: Similarities and differences. Am J Physiol Heart Circ Physiol. 2008;294:H1597–1608. doi: 10.1152/ajpheart.00484.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.