

Abstract
Lymphangioleiomyomatosis (LAM) is a rare progressive lung disease of women. LAM is caused by mutations in the tuberous sclerosis genes, resulting in activation of the mTOR complex 1 signaling network. Over the past 11 years, there has been remarkable progress in the understanding of LAM and rapid translation of this knowledge to an effective therapy. LAM pathogenic mechanisms mirror those of many forms of human cancer, including mutation, metabolic reprogramming, inappropriate growth and survival, metastasis via blood and lymphatic circulation, infiltration/invasion, sex steroid sensitivity, and local and remote tissue destruction. However, the smooth muscle cell that metastasizes, infiltrates, and destroys the lung in LAM arises from an unknown source and has an innocent histological appearance, with little evidence of proliferation. Thus, LAM is as an elegant, monogenic model of neoplasia, defying categorization as either benign or malignant.
Introduction
LAM is a slowly progressive neoplasm that targets the lung, causing cystic destruction and respiratory failure over one to two decades (1–4). LAM occurs in about 3.4–7.8 per million women (5) (although it is likely to be substantially underdiagnosed) and in at least 30% of women with tuberous sclerosis complex (TSC) (6–8). Clinically significant LAM occurs almost exclusively in women, although radiographic evidence of cystic lung disease consistent with LAM and a few biopsy-documented cases of LAM have been reported in men with (9–11) and without (12) TSC. The average age at diagnosis is about 35 years, typically delayed by 3–5 years due to confusion with more common causes of dyspnea, including asthma or chronic obstructive lung disease (13, 14).
LAM manifestations differ in patients with and without TSC and include recurrent pneumothorax, chylous pleural effusions, and abdominal tumors, including renal angiomyolipomas and lymphangiomyomas; it can also be discovered incidentally on abdominal or chest CTs performed for unrelated purposes (Table 1). Lymphatic obstruction can lead to collection of chylous fluid in the pleural, pericardial, or peritoneal spaces or to fistulous lymphatic connections with hollow viscera including the gastrointestinal or genitourinary tracts. Lung function declines at rates that vary between 3% and 15% per year (15–18), accelerated in some patients by hormonal fluxes associated with menstruation, pregnancy, or birth control pill use (19). By 10 years from diagnosis, about 55% of LAM patients experience shortness of breath with daily activities, 20% require supplemental oxygen, and 10% have died (20).
Table 1.
LAM is a multisystem disorder that affects the lungs, pleural space, kidney, liver, lymphatics, and uterus

High-resolution CT scanning reveals diffuse, thin-walled cystic changes that may vary from a few scattered cysts to almost complete replacement of the pulmonary parenchyma with coalescent cysts (Figure 1, A–D, and ref. 21). Typical findings on lung biopsy include smooth muscle cell infiltration of lymphatics, airways, vessels, and alveolar septa (22). The invading “LAM cells” are identified by their spindle-shaped or epithelioid morphology, abundant eosinophilic cytoplasm, and low proliferative index. They stain with antibodies to smooth muscle actin and desmin; with HMB-45, an antibody that recognizes an epitope within the protein gp-100 in the melanogenesis pathway (23); and in many cases, with antibodies to estrogen or progesterone receptors (refs. 24–26 and Figure 1E). The smooth muscle cells within the kidney lesions of patients with angiomyolipomas have a nearly identical morphologic appearance and immunohistochemical profile, and primary and immortalized angiomyolipoma cells are often used as surrogates for LAM cells in laboratory studies. LAM cells also express the lymphangiogenic proteins VEGF-C and VEGF-D and abut slit-like spaces coursing through and surrounding LAM nodules (27, 28). These clefts are lined with endothelial cells that stain with antibodies against the receptor for VEGF-C and VEGF-D, VEGFR-3, as well as for LYVE-1 and podoplanin, marking them as lymphatic channels. Serum levels of VEGF-D are elevated in patients with LAM, and have utility as diagnostic and perhaps prognostic and predictive biomarkers (29–31).
Figure 1. The clinical and pathologic features of LAM.
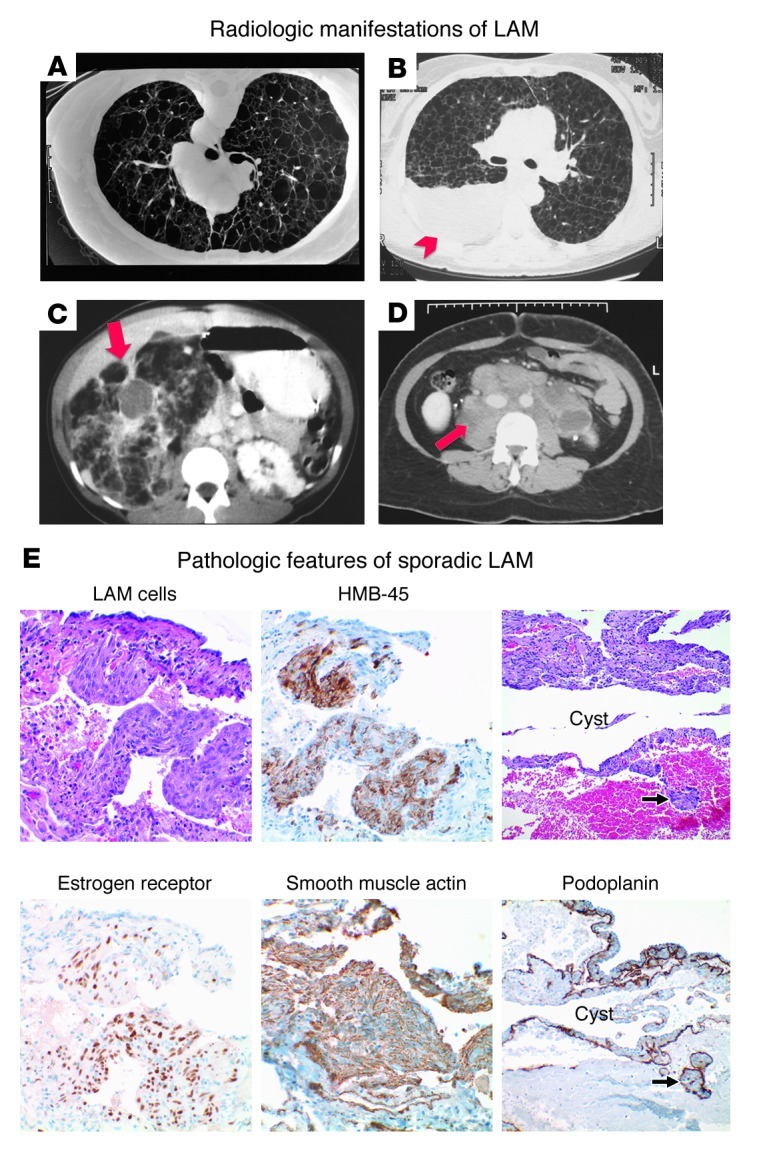
(A–D) Representative radiologic features of LAM include (A) multiple thin-walled cysts and pneumothorax, (B) pleural effusions (arrowhead), (C) renal angiomyolipomas (arrow), and (D) retroperitoneal lymphadenopathy (arrow). (E) High-power view of cystic change with surrounding LAM in the lung. LAM cells express the melanocystic antigen, HMB-45, as well as estrogen receptor and the smooth muscle cell antigen, smooth muscle actin. An immunohistochemical stain for podoplanin highlights lymphatic channels within cystic lesions and LAM cells clusters within the lymphatic lumen. Original magnification, ×400 (left and middle columns); ×200 (right column). Histology and immunohistochemistry courtesy of Kathryn Wikenheiser-Brokamp, Cincinnati Children’s Hospital Medical Center and the University of Cincinnati.
The genetic basis of LAM
Understanding the clinical and genetic relationship between TSC and LAM has been pivotal to progress in LAM pathogenesis and therapy. TSC is an autosomal dominant tumor suppressor syndrome with high penetrance associated with seizures, cognitive impairment, skin lesions, and benign “hamartomatous” tumors of the brain, heart, and kidney (32). The earliest reported case of LAM was described in a patient with TSC (TSC-LAM) who presented with bilateral spontaneous pneumothoraces and died of acute respiratory failure (33). Through most of the last century, the prevalence of LAM in TSC patients was thought to be low, affecting only about 2.5% of females with TSC. However, screening of TSC populations has demonstrated that at least one-third of women with TSC have cystic changes compatible with LAM, consistent with a prevalence of about 200,000 cases worldwide (based on an estimated TSC prevalence of 1 in 6,000) (6–8). In 1937, the first case of LAM in a patient who did not have TSC was reported (34). Over time, it has become clear that most patients with LAM who seek medical evaluation have this less prevalent “sporadic” form of LAM (S-LAM), which is estimated to affect about 10,000 patients worldwide (5). The reason for the paradoxical relationship between prevalence and frequency of clinical presentation in patients with TSC-LAM and S-LAM is not clear, but it is possible that TSC-LAM and S-LAM have different natural histories, or that other health priorities such as cognitive impairment, seizures, or renal failure affect attention to lung disease in TSC-LAM patients.
Our understanding of the genetic basis of LAM was greatly accelerated by the cloning of the tuberous sclerosis genes TSC1 (35) and TSC2 (36), in the 1990s. TSC-causing mutations are widely distributed across these large genes, composed of 23 and 41 exons, respectively. TSC-LAM occurs in women with germline mutations in either TSC1 or TSC2 (37); however, the majority have germline mutations in TSC2. TSC2 mutations are also more prevalent in the TSC population and tend to cause more severe manifestations (38). LAM cells from some women with TSC-LAM exhibit chromosome 16p13 loss of heterozygosity, indicative of inactivation of the wild-type TSC2 allele (39). Therefore, the pathogenesis of TSC-LAM is consistent with the Knudson ‘two-hit’ tumor suppressor gene mechanism (40), as are most other lesions in TSC, including angiomyolipomas, rhabdomyomas, and subependymal giant cell astrocytomas (41). Importantly, a recent genetic analysis of angiomyolipomas for regions of genomic loss and of activating and inactivating mutations revealed only TSC2 mutations and not mutations in TSC1, RHEB, or other candidate loci, consistent with a necessary and sufficient role for TSC mutations in the pathogenesis of the tumor (42).
By definition, women with S-LAM do not have TSC2 germline mutations (43), yet angiomyolipomas and para-aortic lymph nodes from patients with S-LAM have loss of heterozygosity in the TSC2 region of chromosome 16p13 (44), and inactivating somatic TSC2 mutations have been identified in microdissected LAM cells from the lung (45, 46). Consistent with these observations, FISH analyses of circulating LAM cells isolated from the peripheral blood of women with S-LAM have revealed that the majority have loss of heterozygosity in the TSC2 region of chromosome 16p13 (47). These findings strongly suggest a model whereby inactivation of both alleles of TSC2 is the cause of LAM in the majority of both the TSC-associated and sporadic cases.
TSC gene mutations in LAM cells lead to activation of the TORC1 signaling network
The TSC2 gene encodes tuberin (48), a highly evolutionarily conserved GTPase-activating protein (GAP), and TSC1 encodes hamartin, which heterodimerizes with tuberin (49) and appears to be essential for its function (Figure 2A). Rheb, a Ras homolog, is maintained in an inactive state by tuberin (50, 51). The TSC1/TSC2/Rheb triad constitutes a critical cellular signaling node, serving as a gatekeeper by sensing upstream inputs including growth factor activation, oxygen tension (52), amino acid availability, and ATP levels to regulate the downstream functions of TORC1 (reviewed in refs. 53–55). The direct targets of TORC1 continue to be defined and include P70 S6 kinase and 4EBP1, which regulates protein translation; ULK1, which is a master regulator of autophagy (56–58); and growth factor receptor–bound protein 10 (GRB10), an adaptor protein that contributes to feedback regulation of PI3K signaling (59, 60). Several other feedback loops that regulate mTOR pathway signaling have also been described, many of which have potentially important implications for the response to TORC1-targeted therapies (61). Studies from many groups have demonstrated that activation of TORC1 in LAM cells and/or other TSC2-deficient cells leads to phosphorylation of ribosomal protein S6, growth factor–independent growth, increased cell size (62), enhanced cell survival, and suppressed autophagy (56, 63). Evidence of dysregulation of this “canonical” TSC/Rheb/TORC1 signaling network has also been consistently observed in tumor cells from animal models of TSC (64–66), in angiomyolipomas from women with S-LAM (67, 68), and in LAM cells isolated from explanted lungs of LAM patients (69). Sirolimus, a highly specific inhibitor of TORC1, suppresses growth of spontaneously occurring renal tumors in the Tsc2+/– Eker rat model (65) and in Tsc1+/– and Tsc2+/– mice (70), as well as TSC2-deficient xenograft tumors in immune-deficient mice (56, 70, 71). Based on these preclinical data, trials of sirolimus therapy in humans with tuberous sclerosis or LAM began in 2003.
Figure 2. Signaling networks in LAM cells.

(A) LAM cells with biallelic mutational inactivation of the TSC1 or TSC2 gene have activation of the small GTPase and Ras homolog, Rheb. Rheb activates the “canonical” mTORC1 signaling network, leading to increased protein translation and decreased autophagy. Sirolimus (rapamycin) inhibits some of the functions of mTORC1. The impact of sirolimus on the targets of TORC1 may be cell type–specific and kinetically dynamic. New targets of the kinase domain of TORC1 continue to be identified. (B) Several TORC1-independent signaling functions of the TSC-Rheb node have been proposed, including activation of Notch and Rho and inhibition of B-Raf. Candidate TORC1-independent cellular activities of TSC-Rheb include aggresome accumulation and primary cilium formation.
In addition to the canonical TSC/Rheb/TORC1 pathway, data from a variety of experimental systems have pointed toward the existence of noncanonical functions of TSC1/2 and Rheb (reviewed in ref. 63 and Figure 2B). The molecular mechanisms and clinical significance of these are generally less well understood than the canonical mechanisms, but include aggresome formation (72), regulation of the primary cilium (73, 74), regulation of the cytoskeleton and RhoA via hamartin (75) or TOR complex 2 (TORC2) (71), and regulation of cellular differentiation and proliferation via B-Raf (76, 77) and Notch (63, 78).
Pathways affecting proliferation and survival of LAM cells: apoptotic susceptibility and links to proliferation stimuli
Resistance to cell death is a critical capability for neoplastic cells. Paradoxically, compelling data indicate that TSC2-deficient cells are more susceptible than their wild-type counterparts to apoptosis triggered by ER stress (72, 79) and glucose deprivation (80, 81). Elevated p53 transcription and translation rates may also contribute to stress-induced apoptosis (82). In contrast, under some circumstances, such as serum deprivation, TSC-deficient cells appear to be resistant to apoptosis. This may be mediated through FKBP38 (83). Regulation of the apoptotic potential of LAM cells is context-dependent and intimately related to metabolic reprogramming and the nature of mitogenic stimuli. A variety of factors are likely to contribute to the proliferation of LAM cells, including β-catenin, which is activated in LAM (84–86), associated with upregulation of cyclin D1 (84), and contributes to LAM cell invasiveness (87); HMGA2, an architectural transcription factor that is misexpressed in a number of mesenchymal neoplasms (88); Polo-like kinase–1 (PLK1) (89) and PLK2 (90), which interact directly with TSC1 in a cell cycle–dependent manner; cyclin-dependent kinase inhibitor p27, which is mislocalized to the cytoplasm in TSC-deficient cells (91, 92); and prolactin, a hormone and smooth muscle mitogen that is elevated in the serum of patients with LAM (93). A better understanding of these apoptotic and proliferative factors could lead to clearly targetable nodes.
Autophagy-dependent cell survival
Autophagy can play both pro-survival and pro-death roles during tumor initiation and tumor progression (Figure 2C). TORC1 is a key inhibitor of autophagy via ULK1 (94–99), and markers such as the adaptor protein p62/sequestrome 1 (p62/SQSTM1) indicate that autophagy levels are low in TSC-deficient LAM cells (56). Mice that are heterozygous for both TSC2 and Beclin-1 (an autophagy effector) show a decrease in the number of renal cystadenomas compared with mice heterozygous for Tsc2 alone, suggesting that inhibition of autophagy decreases the survival and/or proliferation of TSC2-deficient cells. In addition, treatment with the autophagy inhibitor chloroquine results in a decrease in the size of renal tumors in TSC2-deficient mice (56). Treatment with sirolimus, on the other hand, may promote LAM cell survival by activating autophagy. Consistent with this concept, the combination of sirolimus and chloroquine appears to be particularly effective at inhibiting the growth of TSC2-deficient cells, both in vitro and in vivo (56). Taken together, these data suggest that low levels of autophagy in TSC2-deficient LAM cells serve to limit their growth and survival, and that dependence on autophagy could represent a therapeutically targetable “Achilles heel.” Interestingly, p62/SQSTM1, which accumulates in cells with defective autophagy, may enhance the tumorigenic potential of TSC2-deficient cells (100). p62/SQSTM1 also activates Nrf2 and NF-κB and may thereby mediate the production of pro-survival cytokines that allow LAM cells to resist cell death (101–103).
Estrogen-induced effects on cell survival and proliferation
One of the leading mechanistic hypotheses for the remarkable propensity of LAM to affect females is that estrogen enhances the neoplastic potential and survival of LAM cells. Estradiol promotes the proliferation of TSC2-deficient cells in rat models of TSC-LAM, both in vitro and in vivo (100, 104, 105) and has been observed to activate non-genomic signaling networks in primary cultures of angiomyolipoma cells ex vivo (106). Recent evidence suggests that estrogen-dependent signaling networks provide one of the central mechanisms by which LAM cells resist apoptosis. In TSC2-deficient cells, estrogen activates MAPK, perhaps in part through production of reactive oxygen species (107), and promotes resistance to anoikis-induced apoptosis through (MAPK-dependent) degradation of Bim (106). Furthermore, estrogen enhances the survival of intravenously injected TSC2-deficient cells and the recovery of circulating TSC2-deficient cells in the plasma of mice bearing xenograft tumors (106).
RhoA and LAM cell survival
TSC2-deficient LAM cells may also evade apoptosis via activation of RhoA (Figure 2B). Goncharova et al. found that downregulation of RhoA in TSC2-deficient rat–derived cells increases apoptosis and upregulates the proapoptotic proteins Bim, Bok, and Puma (71). Interestingly, the RhoA activation appears to be dependent on TORC2, rather than TORC1, and inhibition of RhoA using simvastatin induces apoptosis in vitro. Most importantly, the combination of sirolimus and simvastatin inhibited xenograft tumor growth and completely blocked the recurrence of xenograft tumors after treatment withdrawal (71). This finding may have fundamental translational significance for combinatorial therapeutic strategies to induce the death of LAM cells, potentially obviating the need for continuous, life-long suppressive therapies.
Metabolic reprogramming of LAM cells mimics that seen in cancer
Otto Warburg recognized in 1930 that cancer cells utilize glycolysis rather than oxidative phosphorylation for energy production, even in aerobic conditions, despite the far lower yield of ATP per molecule of glucose (108). This “Warburg effect” has received increasing attention over the last five years as a hallmark feature of most cancer cells and other rapidly dividing cells (109). TORC1 is a master regulator of cellular metabolism through several mechanisms that may allow LAM cells to redirect energy metabolism toward biosynthetic programs. Activation of mTOR in TSC-deficient cells appears to promote the Warburg effect by increasing HIF-1α and sterol regulatory element–binding proteins (SREBP1 and SREBP2) (110) that are involved in glycolysis, the oxidative arm of the pentose phosphate pathway, and lipid biosynthesis (81). Surprisingly, angiomyolipomas (111) and LAM tissues (112) have relatively low fluorodeoxygluose (FDG) uptake on PET scanning, compared with most human neoplasms. Decreased glucose uptake is due in part to defective membrane localization of glucose transporter proteins (Glut1, -2, and -4) (111). TSC1/2-deficient cells are also hypersensitive to glucose deprivation (81). The reliance on glucose is closely linked to a dependence on glutamine metabolism via the TCA cycle, and inhibitors of glutamate dehydrogenase, such as the green tea component epigallocatechin-3-gallate (EGCG), can induce the death of TSC-deficient cells both in vitro (80, 113) and in vivo (114). Furthermore, TSC2-deficient cells have recently been found to preferentially express pyruvate kinase M2 (PKM2) (115), which is expressed by most cancer cells and enhances glycolytic flux by slowing the TCA cycle. Agents that selectively inhibit glucose or glutamate uptake or utilization, or that activate PKM2, and thereby target these “metabolic vulnerabilities” could be exploited to induce the death of LAM cells.
Novel mechanisms of invasion, metastasis, and immune evasion facilitate the spread of LAM
Mounting evidence suggests that LAM cells behave in a manner that is reminiscent of low-grade sarcoma cells, based on their smooth muscle features (spindled morphology, and smooth muscle actin and desmin staining) and their neoplastic, metastatic, and destructive potential. Genetic analyses of microdissected LAM lesions in the lungs and kidneys of a few women with sporadic LAM have revealed identical TSC2 mutations in the two locations (43). This discovery led to the paradigm-shifting “benign metastasis” model of LAM pathogenesis, in which LAM cells spread from angiomyolipoma to the lung, or from a peripheral source to both lung and kidney, despite their innocent histological appearance and low proliferative potential (116). Consistent with the benign metastasis model, two independent genetic studies of the cells that constitute recurrent LAM lesions in the donor lung of human LAM patients who had been transplanted demonstrated that they derive from the recipient rather than from the allografts (46, 117).
The source of LAM cells has remained unclear, akin to a cancer of unknown primary. LAM cells express melanocytic antigens, consistent with a potential origin in the neural crest. Other neural crest lineage tissues appear to be impacted in TSC patients, leading to pitting of the dental enamel and hypomelanotic skin lesions (32). The propensity of LAM cells to migrate and metastasize is reminiscent of the highly migratory behavior of neural crest progenitor cells during embryonic development. The uterus is a particularly attractive candidate as a source of these neural crest progenitor cells, and this explanation would make sense of their estrogen receptor expression and responsiveness (118). LAM, angiomyolipomas, and clear cell “sugar” tumors of the lung have been recently classified as perivascular epithelioid cell tumors, or PEComas, mesenchymal tumors composed of histologically and immunohistochemically distinctive cells (with no known normal anatomic counterpart) that express myoid and melanocytic markers. PEComas are most commonly found in the uterus and peritoneum (119–122). A recent study found a very high prevalence of uterine PEComa lesions in patients with S-LAM and TSC-LAM, fueling speculation that uterus may be the “primary tumor” source of LAM cells in many cases (118). Several investigators have proposed that the angiomyolipoma may be a source in some patients, but only about 30% of women with S-LAM have radiographically detectable angiomyolipomas. The thoracic duct is frequently extensively infiltrated with LAM cells at autopsy (22), suggesting that LAM may arise from a site within the lymphatic system (Figure 3). LAM cell clusters in chylous fluid (123, 124) are composed of TSC mutation–bearing smooth muscle actin–positive spindle cells enveloped by lymphatic endothelial cells. Kumasaka et al. have proposed that LAM cells, through their expression of VEGF-C and VEGF-D, drive a lymphangiogenic program that demarcates the LAM cells into endothelium-rimmed islands (27, 28). After budding into the lumen, LAM cell clusters leapfrog up the lymphatic tree through serial cycles of implantation and shedding, and are transported by lymphatic flow to the venous circulation and the pulmonary microvasculature (125). The VEGFR-3–expressing endothelial cells that envelope the LAM cell clusters may serve to shield mutation-bearing LAM cells from immune surveillance against novel or ectopic surface antigens they express, such as the glycolipid GD-3 (126). Long dwell times of these “tumor emboli” in the pulmonary capillaries may facilitate metastasis by a process similar to that proposed for the angiogenesis-driven, “invasion-independent” mechanism described for endothelial cell–lined renal cell carcinoma clusters that gain access to the interstitium via surrounding venules (127, 128).
Figure 3. Hypothesis for LAM progression.

LAM cells have smooth muscle cell features and originate from an unknown source; renal angiomyolipomas and uterine lesions have been proposed as potential primaries. These cells proliferate and drive a lymphangiogenic program that results in demarcation of tissue by chaotic lymphatic channels and the formation of LAM cell islands surrounded by lymphatic endothelium, which then bud into the lumen of the lymphatic system (i). These LAM cell clusters ascend the lymphatic tree by serial cycles of implantation and shedding (ii) and are transported by lymphatic flow to the venous circulation (iii) and ultimately impact in the pulmonary microvasculature (iv). Modified from ref. 28.
Cyst formation in the lung
LAM cell infiltration and elaboration of matrix-degrading enzymes likely drive the formation of cysts in the lung and contributes to both the obstructive and restrictive physiologic defects that are characteristic of LAM. Immunohistochemical staining of LAM lesions demonstrates overexpression of MMPs and metalloproteinase inducers, as well as a paucity of the tissue inhibitor of metalloproteinases TIMP-1 (129–131). Although MMP-2 is the most abundantly expressed MMP in LAM tissue, serum levels of MMP-9, but not MMP-2, are elevated in patients (132–134). LAM cells also exhibit strong immunoreactivity for cathepsin K, a protease that is downstream of mTOR in osteoclasts (135). Whether all of the airspace enlargement in LAM is the result of proteases secreted by LAM cells is not entirely clear. The abundance of lymphatic spaces and expression of VEGF-C, VEGF-D, VEGFR-3, Lyve-1, and podoplanin within LAM lung lesions has led to the hypothesis that disorganized lymphangiogenesis may underlie the program of metalloproteinase expression and lung remodeling in LAM (Figure 4). Elucidation of the spatial and temporal expression of proteases during lymphatic development may therefore shed light on the mechanisms of cystic remodeling in LAM and perhaps also lead to therapeutic strategies for targeting these mechanisms.
Figure 4. Mechanisms of airspace enlargement in LAM.

Two models of airspace enlargement in LAM are presented; these may not be mutually exclusive. (A) LAM cells secrete proteases including MMPs and cathepsin K, which degrade the extracellular matrix and induce apoptosis of alveolar epithelial cells. (B) LAM cells express lymphangiogenic growth factors, VEGF-C and VEGF-D, recruit lymphatic endothelial cells, drive the formation of lymphatic vascular channels and distort the lung architecture. Original magnification, ×200.
Clinical trials of sirolumus in LAM
The Cincinnati Angiomyolipoma Sirolimus Trial (CAST) was initiated in 2003 (136). Patients with angiomyolipomas due to either TSC or LAM were treated with escalating doses of sirolimus for one year, followed by one year of observation off therapy. Renal angiomyolipoma volume decreased by about 50% on the drug and then increased back to near baseline levels when sirolimus was stopped. Similar results were seen in subsequent trials (137, 138). On the basis of an unexpected lung function response in CAST, and the appreciable rate of adverse events, a pivotal trial was designed to determine the risks and benefits of sirolimus in patients with LAM. The Multicenter International LAM Efficacy of Sirolimus (MILES) trial was a double-blind, randomized, controlled trial of sirolimus in 89 adult females with LAM and abnormal lung function (139). During the treatment period, lung function stabilized with sirolimus treatment, and it declined by about 11% in the placebo group. After discontinuation of sirolimus, lung function decline resumed in the sirolimus group and paralleled that in the placebo group. Serum VEGF-D was markedly reduced by sirolimus, but tended to increase again when the drug was withdrawn. Adverse events were more common with sirolimus, but the frequency of serious adverse events was balanced between the groups. The data suggest that sirolimus therapy may attenuate tumor cell infiltration or proliferation within the lung, but does not result in durable remission. The tumor regression seen in CAST and the other renal trials is instructive in this regard, and suggests that sirolimus therapy may be cytostatic and reduce cell size, but will not lead to apoptosis and cell death in TSC-deficient cells. It is possible that mTOR inhibitors must be given continuously to maintain cellular homeostasis and avoid angiomyolipoma regrowth and lung function decline; indeed, there is early evidence of sustained benefit from long-term therapy (137, 140) The decreases in VEGF-D in patients receiving sirolimus (138, 139) are intriguing in light of the strong lymphangiogenic phenotype observed in LAM and marked improvement in chylous effusions and lymphangioleiomyoma volume in LAM patients with lymphatic involvement (140). Finally, the trial utilized a multisite international approach to clinical trial design in LAM, with more than a dozen participating sites in three countries. This investigator-initiated network facilitated the efficient testing of therapeutic strategies in LAM, and may also serve as a model for research on other rare diseases (141).
The way forward
Less than 15 years elapsed between the identification of the genetic etiology of LAM and the discovery of an effective therapy. However, additional action is necessary to identify strategies that will benefit women with LAM in the fastest possible time. Better cellular and animal models are needed, and additional biomarkers that predict disease progression and response of LAM to targeted therapies will be critical for future clinical trial design. Earlier diagnosis through screening of populations of women with TSC and women with sporadic LAM who may present with pneumothorax or nonspecific respiratory symptoms (142) is essential to facilitate treatment before irreversible lung damage occurs. Quantitative imaging techniques to measure lung destruction and LAM cell burden are needed, as current pulmonary function testing methods are insensitive, nonspecific, and effort dependent. Finding effective treatments for LAM will also require an iterative “bedside to bench and back to bedside” effort by investigative teams with broad expertise in basic, translational, and clinical science. Emerging preclinical evidence has set the stage for testing of kinase inhibitors, estrogen-targeted therapies, autophagy inhibitors, and lymphangiogenesis inhibitors (Figure 5). Combination strategies based on cancer treatment paradigms that target codependent or redundant cellular pathways to induce the death of LAM cells, with the objective of remission induction rather than disease suppression, are currently being tested in animal models. FDA-approved drugs with well-understood safety profiles are already available for many of the targets under consideration. Fortunately, mechanisms are already in place to rapidly and efficiently test promising therapeutic strategies through the coordinated efforts of an international team of investigators and an organized, motivated LAM patient community.
Figure 5. Future directions in therapy for LAM.

(A) Potential cell-autonomous therapeutic approaches in LAM include TORC1 inhibitors that may more effectively inhibit TORC1 (including kinase domain inhibitors) and/or have favorable toxicity and/or pharmacokinetic features; autophagy inhibitors; inhibitors of the putative “noncanonical” functions of TSC and Rheb, including Notch activation and Rho activation; direct inhibitors of Rheb’s activity (such as farnesyl transferase inhibitors). (B) Potential non-cell-autonomous therapeutic targets in LAM include inhibition of the lymphatic recruitment and vascular remodeling via inhibition of VEGF or VEGFR; inhibition of MMPs, cathepsin K, and other proteases that contribute to alveolar destruction; inhibition of LAM cells utilizing melanocyte or neural crest antigens as targets; and estrogen antagonism.
Acknowledgments
We thank Kathryn Wikenheiser-Brokamp and Gayle Hill of the University of Cincinnati Department of Pathology for performing the histology and immunohistochemistry experiments. Research support for E.P. Henske and F.X. McCormack was provided by The Adler Foundation (to E.P. Henske and F.X. McCormack), The LAM Foundation (to E.P. Henske and F.X. McCormack), The LAM Treatment Alliance (to E.P. Henske), The Tuberous Sclerosis Alliance (to E.P. Henske and F.X. McCormack), Department of Defense Tuberous Sclerosis Complex Research Program (to E.P. Henske and F.X. McCormack), The National Institute of Diabetes and Digestive and Kidney Diseases (to E.P. Henske), Veteran’s Administration (to F.X. McCormack), and The National Heart, Lung and Blood Institute (to E.P. Henske and F.X. McCormack).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):3807–3816. doi:10.1172/JCI58709.
References
- 1.Kitaichi M, Nishimura K, Itoh H, Izumi T. Pulmonary lymphangioleiomyomatosis: a report of 46 patients including a clinicopathologic study of prognostic factors. Am J Respir Crit Care Med. 1995;151(2 pt 1):527–533. doi: 10.1164/ajrccm.151.2.7842216. [DOI] [PubMed] [Google Scholar]
- 2.McCormack FX. Lymphangioleiomyomatosis: a clinical update. Chest. 2008;133(2):507–516. doi: 10.1378/chest.07-0898. [DOI] [PubMed] [Google Scholar]
- 3.Johnson SR. Lymphangioleiomyomatosis. Eur Respir J. 2006;27(5):1056–1065. doi: 10.1183/09031936.06.00113303. [DOI] [PubMed] [Google Scholar]
- 4.Taveira-DaSilva AM, Steagall WK, Moss J. Lymphangioleiomyomatosis. Cancer Control. 2006;13(4):276–285. doi: 10.1177/107327480601300405. [DOI] [PubMed] [Google Scholar]
- 5.Harknett EC, et al. Use of variability in national and regional data to estimate the prevalence of lymphangioleiomyomatosis. QJM. 2011;104(11):971–979. doi: 10.1093/qjmed/hcr116. [DOI] [PubMed] [Google Scholar]
- 6.Franz DN, et al. Mutational and radiographic analysis of pulmonary disease consistent with lymphangioleiomyomatosis and micronodular pneumocyte hyperplasia in women with tuberous sclerosis. Am J Respir Crit Care Med. 2001;164(4):661–668. doi: 10.1164/ajrccm.164.4.2011025. [DOI] [PubMed] [Google Scholar]
- 7.Costello LC, Hartman TE, Ryu JH. High frequency of pulmonary lymphangioleiomyomatosis in women with tuberous sclerosis complex. Mayo Clin Proc. 2000;75(6):591–594. doi: 10.4065/75.6.591. [DOI] [PubMed] [Google Scholar]
- 8.Moss J, et al. Prevalence and clinical characteristics of lymphangioleiomyomatosis (LAM) in patients with tuberous sclerosis complex. Am J Respir Crit Care Med. 2001;164(4):669–671. doi: 10.1164/ajrccm.164.4.2101154. [DOI] [PubMed] [Google Scholar]
- 9.Adriaensen ME, Schaefer-Prokop CM, Duyndam DA, Zonnenberg BA, Prokop M. Radiological evidence of lymphangioleiomyomatosis in female and male patients with tuberous sclerosis complex. Clin Radiol. 2011;66(7):625–628. doi: 10.1016/j.crad.2011.02.009. [DOI] [PubMed] [Google Scholar]
- 10.Aubry MC, et al. Pulmonary lymphangioleiomyomatosis in a man. Am J Respir Crit Care Med. 2000;162(2 pt 1):749–752. doi: 10.1164/ajrccm.162.2.9911006. [DOI] [PubMed] [Google Scholar]
- 11.Muzykewicz DA, Sharma A, Muse V, Numis AL, Rajagopal J, Thiele EA. TSC1 and TSC2 mutations in patients with lymphangioleiomyomatosis and tuberous sclerosis complex. J Med Genet. 2009;46(7):465–468. doi: 10.1136/jmg.2008.065342. [DOI] [PubMed] [Google Scholar]
- 12.Schiavina M, et al. Pulmonary lymphangioleiomyomatosis in a karyotypically normal man without tuberous sclerosis complex. Am J Respir Crit Care Med. 2007;176(1):96–98. doi: 10.1164/rccm.200610-1408CR. [DOI] [PubMed] [Google Scholar]
- 13.Johnson SR, et al. European Respiratory Society guidelines for the diagnosis and management of lymphangioleiomyomatosis. Eur Respir J. 2010;35(1):14–26. doi: 10.1183/09031936.00076209. [DOI] [PubMed] [Google Scholar]
- 14.Ryu JH, et al. The NHLBI lymphangioleiomyomatosis registry: characteristics of 230 patients at enrollment. Am J Respir Crit Care Med. 2006;173(1):105–111. doi: 10.1164/rccm.200409-1298OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Urban T, et al. Pulmonary lymphangioleiomyomatosis. A study of 69 patients. Groupe d’Etudes et de Recherche sur les Maladies “Orphelines” Pulmonaires (GERM”O”P). Medicine (Baltimore). 1999;78(5):321–337. doi: 10.1097/00005792-199909000-00004. [DOI] [PubMed] [Google Scholar]
- 16.Taveira-DaSilva AM, Stylianou MP, Hedin CJ, Hathaway O, Moss J. Decline in lung function in patients with lymphangioleiomyomatosis treated with or without progesterone. Chest. 2004;126(6):1867–1874. doi: 10.1378/chest.126.6.1867. [DOI] [PubMed] [Google Scholar]
- 17.Johnson SR, Tattersfield AE. Decline in lung function in lymphangioleiomyomatosis: relation to menopause and progesterone treatment. Am J Respir Crit Care Med. 1999;160(2):628–633. doi: 10.1164/ajrccm.160.2.9901027. [DOI] [PubMed] [Google Scholar]
- 18.Lazor R, Valeyre D, Lacronique J, Wallaert B, Urbane T, Cordier JF. Low initial KCO predicts rapid FEV1 decline in pulmonary lymphangioleiomyomatosis. Respir Med. 2004;98(6):536–541. doi: 10.1016/j.rmed.2003.11.013. [DOI] [PubMed] [Google Scholar]
- 19.Shen A, Iseman MD, Waldron JA, King TE. Exacerbation of pulmonary lymphangioleiomyomatosis by exogenous estrogens. Chest. 1987;91(5):782–785. doi: 10.1378/chest.91.5.782. [DOI] [PubMed] [Google Scholar]
- 20.Johnson SR, Whale CI, Hubbard RB, Lewis SA, Tattersfield AE. Survival and disease progression in UK patients with lymphangioleiomyomatosis. Thorax. 2004;59(9):800–803. doi: 10.1136/thx.2004.023283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Seaman DM, Meyer CA, Gilman MD, McCormack FX. Diffuse cystic lung disease at high-resolution CT. AJR Am J Roentgenol. 2011;196(6):1305–1311. doi: 10.2214/AJR.10.4420. [DOI] [PubMed] [Google Scholar]
- 22.Corrin B, Leibow AA, Friedman PJ. Pulmonary lymphangiomyomatosis: a review. Am J Pathol. 1975;79(2):348–382. [PMC free article] [PubMed] [Google Scholar]
- 23.Matsumoto Y, Horiba K, Usuki J, Chu SC, Ferrans VJ, Moss J. Markers of cell proliferation and expression of melanosomal antigen in lymphangioleiomyomatosis. Am J Respir Cell Mol Biol. 1999;21(3):327–336. doi: 10.1165/ajrcmb.21.3.3693. [DOI] [PubMed] [Google Scholar]
- 24.Logginidou H, Ao X, Russo I, Henske EP. Frequent estrogen and progesterone receptor immunoreactivity in renal angiomyolipomas from women with pulmonary lymphangioleiomyomatosis. Chest. 2000;117(1):25–30. doi: 10.1378/chest.117.1.25. [DOI] [PubMed] [Google Scholar]
- 25.Ohori NP, Yousem SA, Sonmez-Alpan E, Colby TV. Estrogen and progesterone receptors in lymphangioleiomyomatosis, epithelioid hemangioendothelioma, and sclerosing hemangioma of the lung. Am J Clin Pathol. 1991;96(4):529–535. doi: 10.1093/ajcp/96.4.529. [DOI] [PubMed] [Google Scholar]
- 26.Matsui K, et al. Downregulation of estrogen and progesterone receptors in the abnormal smooth muscle cells in pulmonary lymphangioleiomyomatosis following therapy. An immunohistochemical study. Am J Respir Crit Care Med. 2000;161(3 pt 1):1002–1009. doi: 10.1164/ajrccm.161.3.9904009. [DOI] [PubMed] [Google Scholar]
- 27.Kumasaka T, et al. Lymphangiogenesis in lymphangioleiomyomatosis: its implication in the progression of lymphangioleiomyomatosis. Am J Surg Pathol. 2004;28(8):1007–1016. doi: 10.1097/01.pas.0000126859.70814.6d. [DOI] [PubMed] [Google Scholar]
- 28.Kumasaka T, et al. Lymphangiogenesis-mediated shedding of LAM cell clusters as a mechanism for dissemination in lymphangioleiomyomatosis. Am J Surg Pathol. 2005;29(10):1356–1366. doi: 10.1097/01.pas.0000172192.25295.45. [DOI] [PubMed] [Google Scholar]
- 29.Young LR, Inoue Y, McCormack FX. Diagnostic potential of serum VEGF-D for lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):199–200. doi: 10.1056/NEJMc0707517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Young LR, et al. Serum vascular endothelial growth factor-D prospectively distinguishes lymphangioleiomyomatosis from other diseases. Chest. 2010;138(3):674–681. doi: 10.1378/chest.10-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Seyama K, et al. Vascular endothelial growth factor-D is increased in serum of patients with lymphangioleiomyomatosis. Lymphat Res Biol. 2006;4(3):143–152. doi: 10.1089/lrb.2006.4.143. [DOI] [PubMed] [Google Scholar]
- 32.Crino PB, Nathanson KL, Henske EP. The tuberous sclerosis complex. N Engl J Med. 2006;355(13):1345–1356. doi: 10.1056/NEJMra055323. [DOI] [PubMed] [Google Scholar]
- 33.Lautenbacher R. Dysembriomes metotipiques des reins, carcinose submiliere aigue puomon avec amphyseme generalise et double pneumothorax. Ann Med Interne (Paris). 1918;5:435–450. [Google Scholar]
- 34.von Stossel E. Uber muskulare Cirrhose der Lunge (Muscular cirrhosis of the lung). Beitr Klin Tuberk. 1937;90:432–442. [Google Scholar]
- 35.van Slegtenhorst M, et al. Identification of the tuberous sclerosis gene TSC1 on chromosome 9q34. Science. 1997;277(5327):805–808. doi: 10.1126/science.277.5327.805. [DOI] [PubMed] [Google Scholar]
- 36.European Chromosome 16 Tuberous Sclerosis Consortium. Identification and characterization of the tuberous sclerosis gene on chromosome 16. Cell. 1993;75(7):1305–1315. doi: 10.1016/0092-8674(93)90618-Z. [DOI] [PubMed] [Google Scholar]
- 37.Strizheva GD, Carsillo T, Kruger WD, Sullivan EJ, Ryu JH, Henske EP. The spectrum of mutations in TSC1 and TSC2 in women with tuberous sclerosis and lymphnagiomyomatosis. Am J Respir Crit Care Med. 2001;163(1):253–258. doi: 10.1164/ajrccm.163.1.2005004. [DOI] [PubMed] [Google Scholar]
- 38.Dabora SL, et al. Mutational analysis in a cohort of 224 tuberous sclerosis patients indicates increased severity of TSC2, compared with TSC1, disease in multiple organs. Am J Hum Genet. 2001;68(1):64–80. doi: 10.1086/316951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu J, Astrinidis A, Henske EP. Chromosome 16 loss of heterozygosity in tuberous sclerosis and sporadic lymphangiomyomatosis. Am J Respir Crit Care Med. 2001;164(8 pt 1):1537–1540. doi: 10.1164/ajrccm.164.8.2104095. [DOI] [PubMed] [Google Scholar]
- 40.Knudson AG. Two genetic hits (more or less) to cancer. Nat Rev Cancer. 2001;1(2):157–162. doi: 10.1038/35101031. [DOI] [PubMed] [Google Scholar]
- 41.Henske EP, et al. Allelic loss is frequent in tuberous sclerosis kidney lesions but rare in brain lesions. Am J Hum Genet. 1996;59(2):400–406. [PMC free article] [PubMed] [Google Scholar]
- 42.Qin W, Bajaj V, Malinowska I, Lu X, MacConaill L, Wu CL, Kwiatkowski DJ. Angiomyolipoma have common mutations in TSC2 but no other common genetic events. PLoS One. 2011;6(9):e24919. doi: 10.1371/journal.pone.0024919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Astrinidis A, et al. Mutational analysis of the tuberous sclerosis gene TSC2 in patients with pulmonary lymphangioleiomyomatosis. J Med Genet. 2000;37(1):55–57. doi: 10.1136/jmg.37.1.55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Smolarek TA, Wessner LL, McCormack FX, Mylet JC, Menon AG, Henske EP. Evidence that lymphangiomyomatosis is caused by TSC2 mutations: chromosome 16p13 loss of heterozygosity in angiomyolipomas and lymph nodes from women with lymphangiomyomatosis. Am J Hum Genet. 1998;62(4):810–815. doi: 10.1086/301804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Carsillo T, Astrinidis A, Henske EP. Mutations in the tuberous sclerosis complex gene TSC2 are a cause of sporadic pulmonary lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2000;97(11):6085–6090. doi: 10.1073/pnas.97.11.6085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bittmann I, Rolf B, Amann G, Lohrs U. Recurrence of lymphangioleiomyomatosis after single lung transplantation: new insights into pathogenesis. Hum Pathol. 2003;34(1):95–98. doi: 10.1053/hupa.2003.50. [DOI] [PubMed] [Google Scholar]
- 47.Crooks DM, et al. Molecular and genetic analysis of disseminated neoplastic cells in lymphangioleiomyomatosis. Proc Natl Acad Sci U S A. 2004;101(50):17462–17467. doi: 10.1073/pnas.0407971101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Manning BD, Tee AR, Logsdon MN, Blenis J, Cantley LC. Identification of the tuberous sclerosis complex-2 tumor suppressor gene product tuberin as a target of the phosphoinositide 3-kinase/akt pathway. Mol Cell. 2002;10(1):151–162. doi: 10.1016/S1097-2765(02)00568-3. [DOI] [PubMed] [Google Scholar]
- 49.Plank TL, Yeung RS, Henske EP. Hamartin, the product of the tuberous sclerosis 1 (TSC1) gene, interacts with tuberin and appears to be localized to cytoplasmic vesicles. Cancer Res. 1998;58(21):4766–4770. [PubMed] [Google Scholar]
- 50.Castro AF, Rebhun JF, Clark GJ, Quilliam LA. Rheb binds tuberous sclerosis complex 2 (TSC2) and promotes S6 kinase activation in a rapamycin- and farnesylation-dependent manner. J Biol Chem. 2003;278(35):32493–32496. doi: 10.1074/jbc.C300226200. [DOI] [PubMed] [Google Scholar]
- 51.Inoki K, Li Y, Xu T, Guan KL. Rheb GTPase is a direct target of TSC2 GAP activity and regulates mTOR signaling. Genes Dev. 2003;17(15):1829–1834. doi: 10.1101/gad.1110003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Alexander A, et al. ATM signals to TSC2 in the cytoplasm to regulate mTORC1 in response to ROS. Proc Natl Acad Sci U S A. 2010;107(9):4153–4158. doi: 10.1073/pnas.0913860107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dann SG, Selvaraj A, Thomas G. mTOR Complex1-S6K1 signaling: at the crossroads of obesity, diabetes and cancer. Trends Mol Med. 2007;13(6):252–259. doi: 10.1016/j.molmed.2007.04.002. [DOI] [PubMed] [Google Scholar]
- 54.Mieulet V, Lamb RF. Tuberous sclerosis complex: linking cancer to metabolism. Trends Mol Med. 2010;16(7):329–335. doi: 10.1016/j.molmed.2010.05.001. [DOI] [PubMed] [Google Scholar]
- 55.Yecies JL, Manning BD. Transcriptional control of cellular metabolism by mTOR signaling. Cancer Res. 2011;71(8):2815–2820. doi: 10.1158/0008-5472.CAN-10-4158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Parkhitko A, et al. Tumorigenesis in tuberous sclerosis complex is autophagy and p62/sequestosome 1 (SQSTM1)-dependent. Proc Natl Acad Sci U S A. 2011;108(30):12455–12460. doi: 10.1073/pnas.1104361108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Yu J, Parkhitko A, Henske EP. Autophagy: an “Achilles” heel of tumorigenesis in TSC and LAM. Autophagy. 2011;7(11):1400–1401. doi: 10.4161/auto.7.11.17652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yu J, Parkhitko AA, Henske EP. Mammalian target of rapamycin signaling and autophagy: roles in lymphangioleiomyomatosis therapy. Proc Am Thorac Soc. 2010;7(1):48–53. doi: 10.1513/pats.200909-104JS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hsu PP, et al. The mTOR-regulated phosphoproteome reveals a mechanism of mTORC1-mediated inhibition of growth factor signaling. Science. 2011;332(6035):1317–1322. doi: 10.1126/science.1199498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Yu Y, et al. Phosphoproteomic analysis identifies Grb10 as an mTORC1 substrate that negatively regulates insulin signaling. Science. 2011;332(6035):1322–1326. doi: 10.1126/science.1199484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Manning BD, Logsdon MN, Lipovsky AI, Abbott D, Kwiatkowski DJ, Cantley LC. Feedback inhibition of Akt signaling limits the growth of tumors lacking Tsc2. Genes Dev. 2005;19(15):1773–1778. doi: 10.1101/gad.1314605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Malhowski AJ, et al. Smooth muscle protein-22-mediated deletion of Tsc1 results in cardiac hypertrophy that is mTORC1-mediated and reversed by rapamycin. Hum Mol Genet. 2011;20(7):1290–1305. doi: 10.1093/hmg/ddq570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Neuman NA, Henske EP. Non-canonical functions of the tuberous sclerosis complex-Rheb signalling axis. EMBO Mol Med. 2011;3(4):189–200. doi: 10.1002/emmm.201100131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kenerson H, Dundon TA, Yeung RS. Effects of rapamycin in the Eker rat model of tuberous sclerosis complex. Pediatr Res. 2005;57(1):67–75. doi: 10.1203/01.PDR.0000147727.78571.07. [DOI] [PubMed] [Google Scholar]
- 65.Kenerson HL, Aicher LD, True LD, Yeung RS. Activated Mammalian target of rapamycin pathway in the pathogenesis of tuberous sclerosis complex renal tumors. Cancer Res. 2002;62(20):5645–5650. [PubMed] [Google Scholar]
- 66.Kwiatkowski DJ, et al. A mouse model of TSC1 reveals sex-dependent lethality from liver hemangiomas, and up-regulation of p70S6 kinase activity in Tsc1 null cells. Hum Mol Genet. 2002;11(5):525–534. doi: 10.1093/hmg/11.5.525. [DOI] [PubMed] [Google Scholar]
- 67.Karbowniczek M, Yu J, Henske EP. Renal angiomyolipomas from patients with sporadic lymphangiomyomatosis contain both neoplastic and non-neoplastic vascular structures. Am J Pathol. 2003;162(2):491–500. doi: 10.1016/S0002-9440(10)63843-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.El-Hashemite N, Zhang H, Henske EP, Kwiatkowski DJ. Mutation in TSC2 and activation of mammalian target of rapamycin signalling pathway in renal angiomyolipoma. Lancet. 2003;361(9366):1348–1349. doi: 10.1016/S0140-6736(03)13044-9. [DOI] [PubMed] [Google Scholar]
- 69.Goncharova EA, et al. Tuberin regulates p70 S6 kinase activation and ribosomal protein S6 phosphorylation. A role for the TSC2 tumor suppressor gene in pulmonary lymphangioleiomyomatosis (LAM). J Biol Chem. 2002;277(34):30958–30967. doi: 10.1074/jbc.M202678200. [DOI] [PubMed] [Google Scholar]
- 70.Lee L, et al. Efficacy of a rapamycin analog (CCI-779) and IFN-gamma in tuberous sclerosis mouse models. Genes Chromosomes Cancer. 2005;42(3):213–227. doi: 10.1002/gcc.20118. [DOI] [PubMed] [Google Scholar]
- 71.Goncharova EA, et al. mTORC2 is required for proliferation and survival of TSC2-null cells. Mol Cell Biol. 2011;31(12):2484–2498. doi: 10.1128/MCB.01061-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou X, Ikenoue T, Chen X, Li L, Inoki K, Guan KL. Rheb controls misfolded protein metabolism by inhibiting aggresome formation and autophagy. Proc Natl Acad Sci U S A. 2009;106(22):8923–8928. doi: 10.1073/pnas.0903621106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hartman TR, et al. The tuberous sclerosis proteins regulate formation of the primary cilium via a rapamycin-insensitive and polycystin 1-independent pathway. Hum Mol Genet. 2009;18(1):151–163. doi: 10.1093/hmg/ddn325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Boehlke C, et al. Primary cilia regulate mTORC1 activity and cell size through Lkb1. Nat Cell Biol. 2010;12(11):1115–1122. doi: 10.1038/ncb2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lamb RF, et al. The TSC1 tumour suppressor hamartin regulates cell adhesion through ERM proteins and the GTPase Rho [see comments]. Nat Cell Biol. 2000;2(5):281–287. doi: 10.1038/35010550. [DOI] [PubMed] [Google Scholar]
- 76.Karbowniczek M, Cash T, Cheung M, Robertson GP, Astrinidis A, Henske EP. Regulation of B-Raf kinase activity by tuberin and Rheb is mammalian target of rapamycin (mTOR)-independent. J Biol Chem. 2004;279(29):29930–29937. doi: 10.1074/jbc.M402591200. [DOI] [PubMed] [Google Scholar]
- 77.Karbowniczek M, Robertson GP, Henske EP. Rheb inhibits C-raf activity and B-raf/C-raf heterodimerization. J Biol Chem. 2006;281(35):25447–25456. doi: 10.1074/jbc.M605273200. [DOI] [PubMed] [Google Scholar]
- 78.Karbowniczek M, et al. The evolutionarily conserved TSC/Rheb pathway activates Notch in tuberous sclerosis complex and Drosophila external sensory organ development. J Clin Invest. 2010;120(1):93–102. doi: 10.1172/JCI40221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Ozcan U, et al. Loss of the tuberous sclerosis complex tumor suppressors triggers the unfolded protein response to regulate insulin signaling and apoptosis. Mol Cell. 2008;29(5):541–551. doi: 10.1016/j.molcel.2007.12.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Choo AY, et al. Glucose addiction of TSC null cells is caused by failed mTORC1-dependent balancing of metabolic demand with supply. Mol Cell. 2010;38(4):487–499. doi: 10.1016/j.molcel.2010.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Inoki K, Zhu T, Guan KL. TSC2 mediates cellular energy response to control cell growth and survival. Cell. 2003;115(5):577–590. doi: 10.1016/S0092-8674(03)00929-2. [DOI] [PubMed] [Google Scholar]
- 82.Lee CH, et al. Constitutive mTOR activation in TSC mutants sensitizes cells to energy starvation and genomic damage via p53. EMBO J. 2007;26(23):4812–4823. doi: 10.1038/sj.emboj.7601900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ma D, Bai X, Zou H, Lai Y, Jiang Y. Rheb GTPase controls apoptosis by regulating interaction of FKBP38 with Bcl-2 and Bcl-XL. J Biol Chem. 2010;285(12):8621–8627. doi: 10.1074/jbc.M109.092353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Mak BC, Takemaru K, Kenerson HL, Moon RT, Yeung RS. The tuberin-hamartin complex negatively regulates beta-catenin signaling activity. J Biol Chem. 2003;278(8):5947–5951. doi: 10.1074/jbc.C200473200. [DOI] [PubMed] [Google Scholar]
- 85.Mak BC, Kenerson HL, Aicher LD, Barnes EA, Yeung RS. Aberrant beta-catenin signaling in tuberous sclerosis. Am J Pathol. 2005;167(1):107–116. doi: 10.1016/S0002-9440(10)62958-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Flavin RJ, Cook J, Fiorentino M, Bailey D, Brown M, Loda MF. beta-Catenin is a useful adjunct immunohistochemical marker for the diagnosis of pulmonary lymphangioleiomyomatosis. Am J Clin Pathol. 2011;135(5):776–782. doi: 10.1309/AJCPPC9EX1ZHMRMA. [DOI] [PubMed] [Google Scholar]
- 87.Barnes EA, Kenerson HL, Mak BC, Yeung RS. The loss of tuberin promotes cell invasion through the ss-catenin pathway. Am J Respir Cell Mol Biol. 2010;43(5):617–627. doi: 10.1165/rcmb.2008-0335OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.D’Armiento J, et al. Identification of the benign mesenchymal tumor gene HMGA2 in lymphangiomyomatosis. Cancer Res. 2007;67(5):1902–1909. doi: 10.1158/0008-5472.CAN-06-1122. [DOI] [PubMed] [Google Scholar]
- 89.Astrinidis A, Senapedis W, Henske EP. Hamartin, the tuberous sclerosis complex 1 gene product, interacts with polo-like kinase 1 in a phosphorylation-dependent manner. Hum Mol Genet. 2006;15(2):287–297. doi: 10.1093/hmg/ddi444. [DOI] [PubMed] [Google Scholar]
- 90.Matthew EM, et al. The p53 target Plk2 interacts with TSC proteins impacting mTOR signaling, tumor growth and chemosensitivity under hypoxic conditions. Cell Cycle. 2009;8(24):4168–4175. doi: 10.4161/cc.8.24.10800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Rosner M, Freilinger A, Hengstschlager M. The tuberous sclerosis genes and regulation of the cyclin-dependent kinase inhibitor p27. Mutat Res. 2006;613(1):10–16. doi: 10.1016/j.mrrev.2006.03.001. [DOI] [PubMed] [Google Scholar]
- 92.Soucek T, et al. Tuberous sclerosis causing mutants of the TSC2 gene product affect proliferation and p27 expression. Oncogene. 2001;20(35):4904–4909. doi: 10.1038/sj.onc.1204627. [DOI] [PubMed] [Google Scholar]
- 93.Terasaki Y, et al. Effects of prolactin on TSC2-null Eker rat cells and in pulmonary lymphangioleiomyomatosis. Am J Respir Crit Care Med. 2010;182(4):531–539. doi: 10.1164/rccm.200911-1737OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kim J, Kundu M, Viollet B, Guan KL. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. doi: 10.1038/ncb2152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Ganley IG, Lam du H, Wang J, Ding X, Chen S, Jiang X. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009;284(18):12297–12305. doi: 10.1074/jbc.M900573200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Jung CH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. doi: 10.1091/mbc.E08-12-1249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hosokawa N, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. doi: 10.1091/mbc.E08-12-1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Jung CH, Ro SH, Cao J, Otto NM, Kim DH. mTOR regulation of autophagy. FEBS Lett. 2010;584(7):1287–1295. doi: 10.1016/j.febslet.2010.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Jung CH, Seo M, Otto NM, Kim DH. ULK1 inhibits the kinase activity of mTORC1 and cell proliferation. Autophagy. 2011;7(10):1212–1221. doi: 10.4161/auto.7.10.16660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Yu JJ, et al. Estrogen promotes the survival and pulmonary metastasis of tuberin-null cells. Proc Natl Acad Sci U S A. 2009;106(8):2635–2640. doi: 10.1073/pnas.0810790106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.White E, Karp C, Strohecker AM, Guo Y, Mathew R. Role of autophagy in suppression of inflammation and cancer. Curr Opin Cell Biol. 2010;22(2):212–217. doi: 10.1016/j.ceb.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Moscat J, Diaz-Meco MT. Feedback on fat: p62-mTORC1-autophagy connections. Cell. 2011;147(4):724–727. doi: 10.1016/j.cell.2011.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Moscat J, Diaz-Meco MT. p62 at the crossroads of autophagy, apoptosis, and cancer. Cell. 2009;137(6):1001–1004. doi: 10.1016/j.cell.2009.05.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hunter DS, et al. Influence of exogenous estrogen receptor ligands on uterine leiomyoma: evidence from an in vitro/in vivo animal model for uterine fibroids. Environ Health Perspect. 2000;108(suppl 5):829–834. doi: 10.1289/ehp.00108s5829. [DOI] [PubMed] [Google Scholar]
- 105.Hodges LC, Hunter DS, Bergerson JS, Fuchs-Young R, Walker CL. An in vivo/in vitro model to assess endocrine disrupting activity of xenoestrogens in uterine leiomyoma. Ann N Y Acad Sci. 2001;948:100–111. doi: 10.1111/j.1749-6632.2001.tb03991.x. [DOI] [PubMed] [Google Scholar]
- 106.Yu J, Astrinidis A, Howard S, Henske EP. Estradiol and tamoxifen stimulate LAM-associated angiomyolipoma cell growth and activate both genomic and nongenomic signaling pathways. Am J Physiol Lung Cell Mol Physiol. 2004;286(4):L694–L700. doi: 10.1152/ajplung.00204.2003. [DOI] [PubMed] [Google Scholar]
- 107.Finlay GA, et al. Estrogen-induced smooth muscle cell growth is regulated by tuberin and associated with altered activation of platelet-derived growth factor receptor-beta and ERK-1/2. J Biol Chem. 2004;279(22):23114–23122. doi: 10.1074/jbc.M401912200. [DOI] [PubMed] [Google Scholar]
- 108.Warburg O, Dickens F, Kaiser-Wilhelm-Institut für Biologie . The Metabolism Of Tumours; Investigations From The Kaiser Wilhelm Institute For Biology, Berlin-dahlem. London, United Kingdom: Constable & Co. Ltd.; 1930. [Google Scholar]
- 109.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144(5):646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 110.Duvel K, et al. Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol Cell. 2010;39(2):171–183. doi: 10.1016/j.molcel.2010.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Jiang X, et al. The tuberous sclerosis complex regulates trafficking of glucose transporters and glucose uptake. Am J Pathol. 2008;172(6):1748–1756. doi: 10.2353/ajpath.2008.070958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Young LR, et al. Utility of [18F]2-fluoro-2-deoxyglucose-PET in sporadic and tuberous sclerosis-associated lymphangioleiomyomatosis. Chest. 2009;136(3):926–933. doi: 10.1378/chest.09-0336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Choo AY, Blenis J. Not all substrates are treated equally: implications for mTOR, rapamycin-resistance and cancer therapy. Cell Cycle. 2009;8(4):567–572. doi: 10.4161/cc.8.4.7659. [DOI] [PubMed] [Google Scholar]
- 114.Yang P, Li H. Epigallocatechin–3–gallate ameliorates hyperglycemia–induced embryonic vasculopathy and malformation by inhibition of Foxo3a activation. Am J Obstet Gynecol. 2010;203(1):75.e1–75.e6. doi: 10.1016/j.ajog.2010.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sun Q, et al. Mammalian target of rapamycin up-regulation of pyruvate kinase isoenzyme type M2 is critical for aerobic glycolysis and tumor growth. Proc Natl Acad Sci U S A. 2011;108(10):4129–4134. doi: 10.1073/pnas.1014769108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Henske EP. Metastasis of benign tumor cells in tuberous sclerosis complex. Genes Chromosomes Cancer. 2003;38(4):376–381. doi: 10.1002/gcc.10252. [DOI] [PubMed] [Google Scholar]
- 117.Karbowniczek M, et al. Recurrent lymphangiomyomatosis after transplantation: genetic analyses reveal a metastatic mechanism. Am J Respir Crit Care Med. 2003;167(7):976–982. doi: 10.1164/rccm.200208-969OC. [DOI] [PubMed] [Google Scholar]
- 118.Hayashi T, et al. Prevalence of uterine and adnexal involvement in pulmonary lymphangioleiomyomatosis: a clinicopathologic study of 10 patients. Am J Surg Pathol. 2011;35(12):1776–1785. doi: 10.1097/PAS.0b013e318235edbd. [DOI] [PubMed] [Google Scholar]
- 119.Bonetti F, et al. Clear cell (“sugar”) tumor of the lung is a lesion strictly related to angiomyolipoma — the concept of a family of lesions characterized by the presence of the perivascular epithelioid cells (PEC). Pathology. 1994;26(3):230–236. doi: 10.1080/00313029400169561. [DOI] [PubMed] [Google Scholar]
- 120.Hornick JL, Fletcher CD. PEComa: what do we know so far? Histopathology. 2006;48(1):75–82. doi: 10.1111/j.1365-2559.2005.02316.x. [DOI] [PubMed] [Google Scholar]
- 121.Armah HB, Parwani AV. Malignant perivascular epithelioid cell tumor (PEComa) of the uterus with late renal and pulmonary metastases: a case report with review of the literature. Diagn Pathol. 2007;2:45. doi: 10.1186/1746-1596-2-45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Wagner AJ, et al. Clinical activity of mTOR inhibition with sirolimus in malignant perivascular epithelioid cell tumors: targeting the pathogenic activation of mTORC1 in tumors. J Clin Oncol. 2010;28(5):835–840. doi: 10.1200/JCO.2009.25.2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Valensi QJ. Pulmonary lymphangiomyoma, a probable forme frust of tuberous sclerosis. A case report and survey of the literature. Am Rev Respir Dis. 1973;108(6):1411–1415. doi: 10.1164/arrd.1973.108.6.1411. [DOI] [PubMed] [Google Scholar]
- 124.Itami M, Teshima S, Asakuma Y, Chino H, Aoyama K, Fukushima N. Pulmonary lymphangiomyomatosis diagnosed by effusion cytology. A case report. Acta Cytol. 1997;41(2):522–528. doi: 10.1159/000332550. [DOI] [PubMed] [Google Scholar]
- 125.Seyama K, Kumasaka T, Kurihara M, Mitani K, Sato T. Lymphangioleiomyomatosis: a disease involving the lymphatic system. Lymphat Res Biol. 2010;8(1):21–31. doi: 10.1089/lrb.2009.0018. [DOI] [PubMed] [Google Scholar]
- 126.Klarquist J, et al. Melanoma-associated antigen expression in lymphangioleiomyomatosis renders tumor cells susceptible to cytotoxic T cells. Am J Pathol. 2009;175(6):2463–2472. doi: 10.2353/ajpath.2009.090525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Sugino T, et al. An invasion-independent pathway of blood-borne metastasis: a new murine mammary tumor model. Am J Pathol. 2002;160(6):1973–1980. doi: 10.1016/S0002-9440(10)61147-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sugino T, et al. Morphological evidence for an invasion-independent metastasis pathway exists in multiple human cancers. BMC Med. 2004;2:9. doi: 10.1186/1741-7015-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Hayashi T, et al. Immunohistochemical study of matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs) in pulmonary lymphangioleiomyomatosis. Hum Pathol. 1997;28(9):1071–1078. doi: 10.1016/S0046-8177(97)90061-7. [DOI] [PubMed] [Google Scholar]
- 130.Fukuda Y, Ishizaki M, Kudoh S, Kitaichi M, Yamanaka N. Localization of matrix metalloproteinases-1, -2, and -9 and tissue inhibitor of metalloproteinase-2 in interstitial lung diseases. Lab Invest. 1998;78(6):687–698. [PubMed] [Google Scholar]
- 131.Glassberg MK, et al. Activation of the estrogen receptor contributes to the progression of pulmonary lymphangioleiomyomatosis via matrix metalloproteinase-induced cell invasiveness. J Clin Endocrinol Metab. 2008;93(5):1625–1633. doi: 10.1210/jc.2007-1283. [DOI] [PubMed] [Google Scholar]
- 132.Pimenta SP, Baldi BG, Acencio MM, Kairalla RA, Carvalho CR. Doxycycline use in patients with lymphangioleiomyomatosis: safety and efficacy in metalloproteinase blockade. J Bras Pneumol. 2011;37(4):424–430. doi: 10.1590/S1806-37132011000400003. [DOI] [PubMed] [Google Scholar]
- 133.Odajima N, Betsuyaku T, Yoshida T, Hosokawa T, Nishimura M. High levels of extracellular matrix metalloproteinase inducer are expressed in lymphangioleiomyomatosis. Hum Pathol. 2010;41(7):935–943. doi: 10.1016/j.humpath.2009.12.009. [DOI] [PubMed] [Google Scholar]
- 134.Odajima N, Betsuyaku T, Nasuhara Y, Inoue H, Seyama K, Nishimura M. Matrix metalloproteinases in blood from patients with LAM. Respir Med. 2009;103(1):124–129. doi: 10.1016/j.rmed.2008.07.017. [DOI] [PubMed] [Google Scholar]
- 135.Chilosi M, et al. Cathepsin-k expression in pulmonary lymphangioleiomyomatosis. Mod Pathol. 2009;22(2):161–166. doi: 10.1038/modpathol.2008.189. [DOI] [PubMed] [Google Scholar]
- 136.Bissler JJ, et al. Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N Engl J Med. 2008;358(2):140–151. doi: 10.1056/NEJMoa063564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Davies DM, et al. Sirolimus therapy for angiomyolipoma in tuberous sclerosis and sporadic lymphangioleiomyomatosis: a phase 2 trial. Clin Cancer Res. 2011;17(12):4071–4081. doi: 10.1158/1078-0432.CCR-11-0445. [DOI] [PubMed] [Google Scholar]
- 138.Dabora SL, et al. Multicenter phase 2 trial of sirolimus for tuberous sclerosis: kidney angiomyolipomas and other tumors regress and VEGF- D levels decrease. PLoS One. 2011;6(9):e23379. doi: 10.1371/journal.pone.0023379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McCormack FX, et al. Efficacy and safety of sirolimus in lymphangioleiomyomatosis. N Engl J Med. 2011;364(17):1595–1606. doi: 10.1056/NEJMoa1100391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Taveira–DaSilva AM, Hathaway O, Stylianou M, Moss J. Changes in lung function and chylous effusions in patients with lymphangioleiomyomatosis treated with sirolimus. Ann Intern Med. 2011;154(12):797–805. doi: 10.1059/0003-4819-154-12-201106210-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gupta S, Bayoumi AM, Faughnan ME. Rare lung disease research: strategies for improving identification and recruitment of research participants. Chest. 2011;140(5):1123–1129. doi: 10.1378/chest.11-1094. [DOI] [PubMed] [Google Scholar]
- 142.Hagaman JT, Schauer DP, McCormack FX, Kinder BW. Screening for lymphangioleiomyomatosis by high-resolution computed tomography in young, nonsmoking women presenting with spontaneous pneumothorax is cost-effective. Am J Respir Crit Care Med. 2010;181(12):1376–1382. doi: 10.1164/rccm.200910-1553OC. [DOI] [PubMed] [Google Scholar]