

Abstract
CD8+ cytotoxic T cells are critical for viral clearance from the lungs upon influenza virus infection. The contribution of antigen cross-presentation by DCs to the induction of anti-viral cytotoxic T cells remains controversial. Here, we used a recombinant influenza virus expressing a nonstructural 1–GFP (NS1-GFP) reporter gene to visualize the route of antigen presentation by lung DCs upon viral infection in mice. We found that lung CD103+ DCs were the only subset of cells that carried intact GFP protein to the draining LNs. Strikingly, lung migratory CD103+ DCs were not productively infected by influenza virus and thus were able to induce virus-specific CD8+ T cells through the cross-presentation of antigens from virally infected cells. We also observed that CD103+ DC resistance to infection correlates with an increased anti-viral state in these cells that is dependent on the expression of type I IFN receptor. These results show that efficient cross-priming by migratory lung DCs is coupled to the acquisition of an anti-viral status, which is dependent on the type I IFN signaling pathway.
Introduction
The identification of the mechanisms that control the initiation of anti-influenza virus CD8+ T cell responses that clear viral infections requires knowledge of the identity of the APCs and the location and time of antigen presentation by APCs to T lymphocytes. In viral infections, DCs could potentially acquire viral antigens through direct infection (direct MHC-I presentation pathway) or through the acquisition of exogenous antigens by phagocytosis of virally infected cells or viral particles (cross-presentation pathway). Efficient cross-priming is easily demonstrated in mouse models with an impaired direct antigen presentation pathway (1–3). In addition, genetic deletion of the CD103+ lung DC subset that excels in cross-priming revealed that these cells control the priming of naive CD8+ T cells during influenza virus infection (4) or Sendai virus infection (5). However similar to lymphoid tissue CD8+ DCs, CD103+ DCs are also very potent at direct priming of CD8+ T cells (6) (J. Helft and M. Merad, unpublished observations), suggesting the possibility that the reduced CD8+ T cell responses (4, 5) resulted from the loss of direct antigen presentation normally provided by infected CD103+ DCs. Thus the physiological contribution of cross-presentation to the induction of anti-influenza virus CD8+ T cell immunity in vivo is still a matter of deb–ate.
Attempts to generate recombinant fluorescent influenza viruses have been hampered because most of the viruses expressing reporter genes have reduced levels of replication and do not show significant pathogenesis in mice (7). In this study, we visualized the route of viral antigen uptake by lung and LN DCs and examined the antigenic presentation pathway used by DCs to induce efficient CD8+ T cell immunity upon intranasal influenza virus infection. We used a new recombinant virus expressing GFP in the nonstructural 1 (NS1) segment of the A/Puerto Rico/8/34 PR8 (H1N1) virus to follow influenza virus interactions with the host APCs (8). Despite some attenuation, the NS1-GFP virus replicates efficiently in murine lungs, and the pathogenicity of NS1-GFP virus infection in mice resembles that of the parental virus (8).
Here, we found that lung CD103+ DCs that transport viral antigens to the draining LNs are protected from viral infection in vivo and acquire viral antigens through phagocytosis of infected cells. Importantly, we found that lung migratory CD103+ DCs are the only DCs to preserve viral antigens in their endocytic compartment and to control the induction of virus-specific CD8+ T cells through the cross-presentation of antigens from virally infected cells.
Results
Tracking virus antigen uptake by lung cells during influenza virus infection in vivo.
Lung phagocytes consist of alveolar macrophages and classical CD103+ and CD11b+ DC populations (refs. 9, 10, and Figure 1A). To visualize influenza virus interactions with lung phagocytes, we used an influenza virus expressing GFP in the PR8 strain (8). The GFP is expressed from segment 8 (NS) of influenza virus as a fusion protein with NS1 (NS1-GFP). NS1 is a nonstructural protein, and therefore, viral particles are not fluorescent and expression of GFP by phagocytes is indicative of direct viral infection or uptake of virally infected cells.
Figure 1. Tracking viral antigens during influenza infection in vivo.

C57BL/6 mice were infected intranasally with 106 PFUs of NS1-GFP virus. (A) Six hours post infection (p.i.), GFP levels were assessed by flow cytometry in CD45+ lung cells: MHCIIhiCD11c+CD103+ DC (gate 2), MHCIIhiCD11c+CD11b+ DC (gate 2), SSChiMHCIIloCD11c+CD103–macrophages (gate 1), and epithelial CD45– cells. Lower panels show a representative percentage of GFP+ cells gated among each lung population. Graph represents the percentage of GFP+ cells among live lung cells. (B) Images show GFP expression in lung sections 6 hours after infection stained with anti-langerin and anti-CD169 Abs and analyzed by confocal microscopy. White arrows show CD169+ macrophage and langerin+ DCs. Original magnification, ×40, zoom 6. (C) Percentage of total DCs (left panel) or GFP+ DCs (right panel) among live lung cells at the indicated time points after infection (n = 3). (D) Representative dot plots showing the percentage of each DC population in the lung 48 hours after infection (left panel) and the percentage of GFP+ cells in each lung DC subset (middle and right panels). (E) Dot plots show the percentage of GFP expression in each MLN DC subset 48 hours after infection. Gating strategy is described in Supplemental Figure 1F. (F) Absolute numbers of total DC subsets (lower panel) or GFP+ DC subsets (upper panel) in the MLNs at the indicated time points after NS1-GFP infection. (G) Images show GFP expression in MLN sections isolated 48 hours after infection, stained with anti-langerin and anti-CD169 mAbs, and analyzed by confocal microscopy. White arrows show CD169+ subcapsular macrophage and langerin+ DCs. Original magnification ×40, zoom 6.
Mice were infected intranasally with a lethal dose of NS1-GFP virus (106 PFUs). GFP+ cells in the lung and the draining mediastinal LNs (MLNs) were traced at different times after infection using flow cytometry and confocal microscopy. Six hours after infection, GFP accumulated mostly in epithelial cells and alveolar macrophages and was present at much lower levels in lung DCs (Figure 1, A and B). During the first 2 days of infection, CD103+ DCs disappeared from the lung (Figure 1, C and D), whereas the CD11b+ DC population expressing GFP increased dramatically. Unlike CD11b+ DCs found in the steady state, CD11b+ DCs that accumulated in influenza-infected lungs expressed high levels of Ly6C and likely arose from blood monocytes (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI60659DS1).
Lung migratory CD103+ DCs carrying GFP appeared in the draining MLNs at around 12 hours after infection, peaked at 48 hours after infection (Figure 1, E–G, and Supplemental Figure 1B), and remained the main DC population carrying GFP to the MLNs during the first 4 days after infection (Figure 1F and Supplemental Figure 1B). In contrast, GFP was barely detectable in CD11b+ DCs that reached the MLNs at all time points after infection (Figure 1, E and F) despite a large influx of CD11b+ DCs in the MLNs during infection (Figure 1F). The lack of GFP expression in CD11b+ DCs infiltrating the MLNs also contrasted with the high GFP expression found in the CD11b+ DCs infiltrating the lungs (Figure 1D).
To explore further the mechanisms of viral antigen transport to the MLNs upon intranasal infection, we examined the possibility that free-flowing virus can reach the LNs independently of DCs. Free-flowing virus, once it reaches the MLNs, should first encounter and infect subcapsular CD169+ macrophages, as previously described (11), and then infect LN-resident DCs. However, we failed to detect GFP expression by LN-resident subcapsular CD169+ macrophages at all time points after intranasal NS1-GFP virus infection using confocal microscopy or flow cytometry analysis (Figure 1G and Supplemental Figure 1, C and D). In addition, LN-resident CD8+ and CD4+ DCs (gating strategy shown in Supplemental Figure 1F) lacked GFP expression at all time points after infection (Figure 1, E and F, and Supplemental Figure 1B). We also failed to detect GFP signal in the draining LNs of Ccr7–/– mice, establishing that DC migration, which is abrogated in Ccr7–/– mice (12), is required to transport GFP to the MLNs (Supplemental Figure 1G). Thus, virus-derived GFP is actively transported to the draining LNs by lung migratory CD103+ DCs.
Together, these results show that the migration of DCs is the main road for viral antigen transport to the LNs and exclude a significant contribution of free virus transport through afferent lymph. Influenza virus infection is thus restricted to cells residing in the respiratory tract.
Lung migratory CD103+ DCs control anti-viral CD8+ T cell immunity.
The results above show that upon pulmonary infection with NS1-GFP virus, 7%–10% of lung migratory CD103+ DCs transport GFP to the MLNs in a CCR7-dependent manner. To evaluate the role of migratory lung DCs in the priming of anti-viral CD8+ T cells, we infected WT or Ccr7–/– mice with a lethal dose of a recombinant influenza virus expressing the MHC-I OVA peptide SIINFEKL (PR8-OTI). Forty-eight hours later, MLNs of WT or Ccr7–/– infected mice were collected and cocultured with CFSE-labeled OVA-specific TCR transgenic CD8+ T cells (OTI) (Figure 2A). OTI T cells proliferated only in the presence of LN cells isolated from WT but not Ccr7–/– mice, establishing that CCR7-dependent lung DC migration controlled the priming of anti-viral CD8+ T cells.
Figure 2. Lung tissue migratory CD103+ DCs control anti-viral CD8+ T cell immunity.

(A) WT and Ccr7–/– mice were infected with 107 PFUs of PR8-OTI virus. Forty-eight hours later, MLN cells were cocultured with CFSE-labeled CD8+ OT-I T cells for 3 days. Proliferation was measured by flow cytometry assessment of CFSE dilution (representative histograms). Graph represents the number of divided T cells per well. Each dot represents 1 mouse. (B) WT mice were infected with 107 PFUs of PR8-OTI virus. Lung migratory CD103+ DCs and CD11b+ DCs and LN-resident CD8+ and CD4+ DCs purified from the MLN 24 hours after infection were cocultured with CFSE-labeled CD8+ OT-I T cells for 3 days. Proliferation was measured by flow cytometry assessment of CFSE dilution (representative histograms). Graph represents the number of divided T cells per well after coculture with different numbers of DCs per well (DC:T ratio). Data are representative of 3 separate experiments. (C) CD45+CD11c+MHCII+ lung phagocyte populations (CD103+ DCs, CD11b+ DCs, and CD103–CD11b– alveolar macrophages) were analyzed by flow cytometry in naive WT and Batf3–/– mice. Representative percentages of each population in the CD45+CD11c+MHCII+ gate are noted. (D and E) WT and Batf3–/– mice were infected with 3 × 103 PFUs of NS1-GFP virus. Seven days later, the percentages and absolute numbers of NP-specific endogenous CD8+ T cells in the MLNs (D) and the lungs (E) were measured by flow cytometry using H2Db/ASNENMETM-specific dextramer staining. Graphs represent the percentage (%) and the absolute numbers (abs no.) of dextramer-positive CD8+ T cells among total CD3+CD8+B220– T cells. Data are representative of 3 separate experiments.
To examine the specific contribution of lung migratory DCs and LN-resident DCs to the priming of anti-viral CD8+ T cells, we purified lung migratory CD103+ and CD11b+ DCs and LN-resident CD8+ and CD4+ DCs isolated from the MLNs 24 hours (Figure 2B) or 48 hours (not shown) after intranasal infection with 107 PFUs of PR8-OTI virus. Each DC population was then cocultured for 3 days with CFSE-labeled OTI T cells. We found that lung migratory CD103+ DCs (Figure 2B) drove very potent CD8+ T cell proliferation at 24 and 48 hours after infection, whereas migratory CD11b+ DCs and LN-resident CD8+ and CD4+ DCs were much less efficient at presenting viral antigens to CD8+ T cells (Figure 2B).
To test whether depletion of lung migratory CD103+ DCs in vivo compromises the induction of anti-viral CD8+ T cell immunity, we used 2 different mouse models. Conditional depletion of lung migratory CD103+ DCs in diphtheria toxin–treated (DT-treated) transgenic mice expressing the receptor for DT under the langerin promoter (langerin-DTR), abrogated the induction of anti–viral-specific CD8+ T cells (Supplemental Figure 2, A–C), which is consistent with prior studies (4). We further confirmed the key role of migratory CD103+ DCs in the priming of viral antigens to CD8+ T cells using mice deficient for the basic leucin zipper transcription factor 3 (Batf3–/–). Batf3–/– mice specifically lack CD103+ DCs in the lung, but not CD11b+ DCs (Figure 2C and ref. 5). Similar to langerin-DTR mice treated with DT, Batf3–/– mice were unable to mount an efficient anti-viral CD8+ T cell response in the lung-draining LNs and the lungs 7 days after infection (Figure 2, D and E). Although LN-resident CD8+ DCs were also reduced in DT-treated langerin-DTR and Batf3–/– mice, the ex vivo antigenic presentation assays (Figure 2, A and B) showing that lung migratory CD103+ DCs are the main inducers of anti-viral CD8+ T cell responses suggest that the compromised ability to mount anti-viral CD8+ T cell response in langerin-DTR and Batf3–/– mice is solely due to the absence of lung migratory CD103+ DCs.
Lung migratory CD103+ DCs are protected from infection by influenza virus.
To assess whether the unique ability of CD103+ DCs to induce anti-viral CD8+ T cell immune response was due to their higher susceptibility to influenza virus infection in vivo, we measured productive viral infection of migratory and LN-resident DC populations in the draining MLNs. The influenza HA protein is essential for the budding of newly synthesized viral particles and is among the first viral proteins to be expressed on the cell surface of host infected cells. Therefore, detection of HA expression on DC cell surface marks their productive infection. Strikingly and in contrast to epithelial cells and macrophages (Figure 3, A and B), we failed to detect cell-surface expression of HA in CD103+GFP+ DCs isolated from the MLNs (Figure 3C) of mice 48 hours after infection with 106 PFUs of NS1-GFP virus. This suggests that most lung CD103+ DCs that transport GFP to the draining LNs were not productively infected by the virus. We further confirmed that CD103+GFP+ DCs were not virally infected using embryonated chicken eggs, which are permissive for viral growth ex vivo (Table 1). We failed to detect viral growth in eggs injected with purified migratory CD103+ DCs isolated from the MLNs 48 hours after infection, whereas viral growth was detected in eggs injected with alveolar macrophages purified from the same mouse (Table 1). We also failed to detect virally infected CD11b+ GFP+HA+ DCs in the draining MLNs of virally infected mice (Figure 3D and Table 1). To determine whether productive virus could reach the LNs independently of DCs, WT or Ccr7–/– mice were infected intranasally with NS1-GFP virus (106 PFUs) and different dilutions of LN single-cell suspension collected 2 days after infection were injected into embryonated chicken eggs (Supplemental Figure 3A). We were unable to detect viral growth in embryonated eggs at any dilution (Supplemental Figure 3A)
Figure 3. CD103+ migratory DCs are not infected by influenza virus and uniquely preserve viral proteins in endosomal compartments.

(A–I) Mice were infected with 106 PFUs of NS1-GFP virus. HA expression was measured by flow cytometry in CD45– lung cells (A) and alveolar macrophages (B) 15 hours after infection or in MLN CD103+ (C) and CD11b+ DCs (D) 48 hours after infection. Percentage of GFP+ cells in each population is noted in C and D. (E) Cell-surface HA expression was measured by confocal microscopy in MLN CD103+GFP+ DCs and CD11b+ DCs purified 48 hours after infection and lung alveolar macrophages purified 15 hours after infection. Original magnification, ×63, zoom 3. (F) Intracellular LAMP2 expression was analyzed by confocal microscopy in migratory CD103+GFP+ or CD11b+ DCs, isolated from the MLNs 48 hours after infection. Original magnification, ×63, zoom 3. (G) Intracellular NP expression was measured by flow cytometry in MLN cells isolated 48 hours after infection. Dot plots show percentage of intracellular NP expression among migratory CD103+ and CD11b+ DCs. (H and I) Affymetrix gene chip arrays of CD11b+ and CD103+ migratory DCs sorted from the MLNs of naive mice. Graphs represent mRNA transcript ratio between CD103+ DCs and CD11b+ DCs. Each dot represents 1 experiment. Bars represent the mRNA transcript absolute values for each gene. Data are representative of 3 separate experiments. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Table 1.
Viral growth in eggs following leukocyte injection
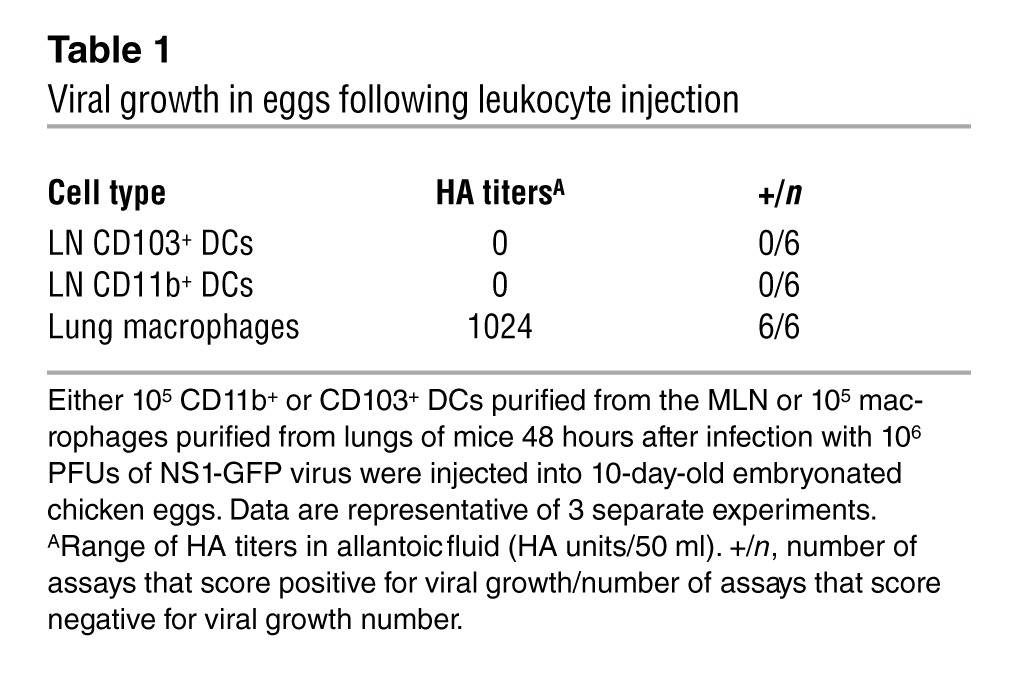
Together, these results show that lung CD103+ and CD11b+ DCs migrating to the MLNs upon intranasal influenza infection do not carry infectious viral particles and are not infected by influenza virus in vivo.
CD103+ DCs uniquely preserve viral protein in endosomal compartments.
The results above revealed that the influenza virus did not efficiently infect CD103+ DCs transporting GFP to the draining LNs. In infected cells, viral NS1 proteins should accumulate in the cytoplasm and the nucleus. To further confirm that lung migratory CD103+GFP+HA– DCs were not virally infected, we analyzed the intracellular localization of the NS1-GFP protein in CD103+GFP+ DCs purified from MLNs of mice infected with 106 PFUs of NS1-GFP virus using confocal microscopy. We found that NS1-GFP was not diffusely expressed in the nucleus and cytoplasm of CD103+GFP+ DCs (Figure 3E and Supplemental Figure 3B) as is the case for infected lung macrophages (Figure 3E and Supplemental Figure 3B). Importantly, we also found that GFP accumulated in early endocytic compartments positive for LAMP2 and EEA1 (Figure 3F and Supplemental Figure 3C). These results further establish that CD103+GFP+ DCs acquire GFP through phagocytosis of virally infected cells and not through direct viral infection. Lung CD103+ DCs seemed to be remarkably potent at preserving intact intracellular GFP during their migration to the draining LNs when compared with CD11b+ migratory DCs. Consistently, using intracellular staining, we found that in addition to GFP, CD103+GFP+ DCs were also able to preserve other viral proteins such as viral nucleoprotein (NP) (Figure 3G).
To assess whether CD103+ DCs’ ability to preserve GFP reflected their poor degradative potential compared with CD11b+ DCs, we used expression array analysis of migratory CD103+ DCs purified from the MLNs of naive mice. Interestingly, we found that lung CD103+ DCs expressed a lower number of transcripts for proteases (Figure 3H and Supplemental Figure 3D) and lysosomal markers (Figure 3I and Supplemental Figure 3D) that control lysosomal antigen degradation compared with lung CD11b+ DCs, whereas both DC subsets expressed abundant and similar expression of housekeeping genes (not shown). Together, these results suggest that CD103+ DCs are endowed with a specific ability to retain phagocytosed antigens in low degradative compartments, thus supporting efficient cross-presentation to antigen-specific CD8+ T cells (13, 14).
To directly establish the role of CD103+ DCs in the cross-presentation of lung antigens in vivo, we used antigen-coated latex beads, an experimental system that has been widely used to directly assess phagocytic cross-presentation potential of DCs (15, 16). We found that both lung DC subsets captured 1 μm fluorescent latex beads injected intratracheally, with the same efficiency in the lung (Supplemental Figure 4A), and transported them to the draining MLNs (Supplemental Figure 4B). Both DC subsets transported the latex beads to the LNs in a CCR7-dependent manner (Supplemental Figure 4, C and D), and we failed to detect any bead uptake by LN-resident CD4+ and CD8+ DCs (Supplemental Figure 4B). To directly assess the contribution of CD103+ DCs to the cross-presentation of lung particulate antigens, langerin-DTR mice were immunized with 1 μm latex beads covalently coated with the influenza viral protein NP and mixed with poly(I:C) as adjuvant 24 hours prior to PBS or DT injection. The depletion of CD103+ DCs in DT-treated mice completely abrogated the cross-priming of anti-NP CD8+ T cells 7 days after immunization (Supplemental Figure 4E). These results establish the superior ability of lung migratory CD103+ DCs to cross-present lung particulate antigens to CD8+ T cells.
Type I IFN signaling protects migratory CD103+ DCs from influenza virus infection in vivo.
The results above revealed that, in contrast to epithelial cells and alveolar macrophages of the lung, influenza virus does not efficiently infect lung CD103+ and CD11b+ DCs that migrate to the LNs. In contrast, in vitro coculture of CD103+ and CD11b+ DCs isolated from the lung of naive mice in the presence of NS1-GFP virus led to their productive infection, as both DC subsets harbored HA expression at the cell surface and diffuse NS1 expression in the nucleus and cytoplasm (Figure 4, A and B, and Supplemental Figure 5). These results suggest that active mechanisms induced upon viral infection help protect lung migratory DCs from productive viral infection.
Figure 4. Type I IFNs protect migratory DCs from influenza virus infection in vivo.

(A and B) Lung CD103+ DCs were purified from naive mice and infected in vitro with NS1-GFP virus (10 MOI). Twelve hours after infection, cell-surface expression of HA was measured in CD103+ DCs by flow cytometry (A) and confocal microscopy analysis (B). Original magnification, ×63, zoom 3. A representative percentage of GFP+ cells among CD103+ DCs is shown in A. (C–E) WT and Ifnar1–/– mice were infected with 106 PFUs of NS1-GFP virus. Dot plots and graph show the percentages of GFP+HA+ cells among CD103+ (C) and CD11b+ (D) DCs isolated from MLNs 48 hours after infection. In the graph, each dot represents 1 mouse. Data are representative of 3 separate experiments (n = 3). (E) Percentages of GFP+HA+ cells among alveolar macrophages and CD45– epithelial cells isolated from the lungs 12 hours after infection. Each dot represents 1 mouse. (F) Lung CD103+ DCs isolated from WT CD45.1+C57BL/6 or Ifnar1–/– CD45.2+C57BL/6 mice were mixed at 1:1 ratio, plated in vitro, and infected with the indicated NS1-GFP virus MOI. Plots represent the percentages of GFP+HA+ cells among WT or Ifnar1–/– DCs 12 hours after infection. Each dot represents 1 well. Data are representative of 2 separate experiments. **P ≤ 0.01, ***P ≤ 0.001. (G) WT and Stat2–/– mice were infected with 106 PFUs of NS1-GFP virus. Graph shows the percentages of GFP+HA+ cells among CD103+ DCs isolated from MLNs 48 hours after infection (n = 3 to 4). Each dot represents 1 mouse.
Type I IFNs induce cellular resistance to virus infection (17). All type I IFNs bind to the same heterodimeric receptor, IFN-α/β receptor (IFNAR), and loss of IFNAR increases lung pathology after influenza virus infection, but does not increase susceptibility to influenza virus infection in vivo (18). To examine whether type I IFNs contributed to lung DC protection from viral infection in vivo, we infected Ifnar1–/– mice with 106 PFUs of NS1-GFP virus and measured lung DC infection rate. Two days after infection, the number of GFP+HA+ infected lung CD103+ DCs present in the MLNs was substantially increased in Ifnar1–/– mice as compared with WT mice (Figure 4, C and D), suggesting that type I IFN signaling contributes to CD103+ DC resistance to influenza virus infection. Increased viral infection was specific to lung migratory DCs, since there was no significant increase of infected GFP+HA+ alveolar macrophages and lung epithelial cells in Ifnar1–/– mice at this early time point after infection (Figure 4E).
To directly compare WT and Ifnar1–/– DC susceptibility to infection in the presence of similar amounts of virus, we cocultured Ifnar1–/– and Ifnar1+/+ lung DCs with increasing doses of NS1-GFP virus in vitro. Using HA+GFP+ staining to measure infection, we found that even in the presence of similar virus titers in vitro, Ifnar1–/– CD103+ DCs were more susceptible to virus infection compared with Ifnar1+/+ CD103+ DCs (Figure 4F). To further confirm the role of IFN in CD103+ DC protection from viral infection, we determined whether compromised IFN signaling in Stat2–/– CD103+ DCs will affect their susceptibility to viral infection in vivo. We infected Stat2–/– mice with 106 PFUs of NS1-GFP and found that, similar to the phenotype found in Ifnar1–/– mice, the number of GFP+HA+ infected lung CD103+ DCs present in the MLNs was substantially increased in Stat2–/– mice as compared with WT mice (Figure 4G). Taken together, these results establish that CD103+ DCs’ increased susceptibility to viral infection in Ifnar1–/– mice does not result from higher viral load, but is a consequence of an active protective mechanism that is partly controlled by type I IFNs.
CD103+ DCs acquire an anti-viral state in vivo.
To explore the mechanisms that control CD103+ DC resistance to viral infection, we quantified the viral sensors and IFN signaling machinery of alveolar macrophages and CD103+ DCs that populate the normal lungs (Figure 5A). We found that alveolar macrophages expressed higher levels of transcripts involved in viral sensing (Rigi, Ips1, Myd88, Trif) compared with lung CD103+ DCs, whereas CD103+ DCs expressed higher levels of transcripts involved in IFN signaling (Ifnar1, Stat1, Stat2) compared with alveolar macrophages, suggesting that macrophages and CD103+ DCs are differentially equipped to respond to viral infection and IFN stimuli in vivo (Figure 5A).
Figure 5. CD103+ DCs present an anti-viral state that protects them from viral infection.

(A) Heat map of mRNA transcript relative value comparing the microarray analysis of CD103+ DCs and alveolar macrophages (Mϕ) purified from the lung naive mice (n = 3). Red represents high relative expression, while blue represents low relative expression. (B and C) Quantitative PCR (qPCR) analysis of lung alveolar macrophages and CD103+ DCs purified from WT mice 12 hours after infection with 106 PFUs of NS1-GFP virus. mRNA levels are normalized for each population to the level of housekeeping gene 18S (n = 3). (D and E) Deep sequencing mRNA analysis of lung alveolar macrophages and CD103+ DCs purified from lungs of naive WT mice (steady state) (D) or mice infected 12 hours earlier with 106 PFUs of NS1-GFP virus. (E). Heat maps present the relative mRNA expression levels between CD103+ DCs and alveolar macrophages in the indicated mice. Red represents high relative expression, while blue represents low relative expression. (F and G) WT mice (F) and Ifnar1–/– mice (G) were injected intranasally with 106 PFUs of NS1-GFP or PBS. Twelve hours later, alveolar macrophages (F) and CD103+ DCs (F and G) were purified from the lungs of naive or infected mice and cultured in 96-well plates for 12 hours with 3 or 10 MOI of NS1-GFP virus in vitro. Graphs represent the percentage of GFP-positive cells measured by flow cytometry 12 hours after in vitro infection. The baseline GFP expression level is subtracted from the GFP expression after in vitro infection. Data are representative of 3 separate experiments.
To assess the response of lung phagocytes to influenza virus infection, WT mice were infected with 106 PFUs of NS1-GFP virus and lung alveolar macrophages, and CD103+ DCs were purified 12 hours after infection. Alveolar macrophages isolated from infected lungs expressed higher levels of type I IFN-α and -β compared with CD103+ DCs (Figure 5B), which is consistent with the higher expression of viral-sensing transcripts by these cells in the steady state (Figure 5A). In contrast to alveolar macrophages and in line with the higher expression of IFNAR and IFN signaling transcripts, CD103+ DCs expressed higher levels of IFN-inducible genes as compared with alveolar macrophages (ISG15, OAS1a, Mx1) at 12 hours after infection (Figure 5C).
To characterize the anti-viral status of lung tissue phagocytes, we used RNA sequencing to perform a broader analysis of the transcriptome of purified lung CD103+ DCs and alveolar macrophages isolated from naive (Figure 5D and Supplemental Figure 6A) and infected lungs (Figure 5E and Supplemental Figure 6A). Consistent with the expression array analysis, RNA sequencing analysis established that alveolar macrophages expressed higher levels of mRNAs involved in viral sensing, whereas steady-state CD103+ DCs and CD103+ DCs isolated 12 hours after NS1-GFP virus infection (106 PFUs) expressed higher levels of IFN-induced anti-viral genes described as being directly involved in cell protection from influenza virus infection (ISG15, Mx1, ifitm1, ifitm3, PKR) (Figure 5, D and E). Based on these results, we hypothesized that IFN-induced genes expressed by lung CD103+ DCs upon lung exposure to influenza virus contributed to lung CD103+ DC protection from influenza virus infection.
To directly test this hypothesis, we developed an ex vivo infection system in which alveolar macrophages and CD103+ DCs were purified 12 hours after exposure to influenza virus in vivo and cultured in 96-well plates with NS1-GFP virus for an additional 12 hours. We monitored DC protection from virus infection by measuring NS1-GFP levels using flow cytometry analysis. Consistent with the profiling results, we found that CD103+ DCs isolated from infected lungs were protected from reinfection with NS1-GFP (Figure 5F). Alveolar macrophages were also partially protected, but to a lesser extent than CD103+ DCs. In contrast, Ifnar1–/– CD103+ DCs were much less protected from influenza virus infection compared with Ifnar1+/+ CD103+ DCs (Figure 5G).
These results confirm that CD103+ DCs specifically display an enhanced anti-viral state that is further strengthened upon viral infection and is partially controlled by type I IFN.
Discussion
We used a recombinant influenza virus expressing GFP as a fusion protein with the nonstructural protein NS1 to establish the kinetics of infection and transport to the draining LNs. Using this system, we provide evidence that lung DCs that transport viral antigens to the draining LNs are protected from influenza virus infection in vivo and that induction of viral-specific CD8+ T cell immunity is mainly dependent on cross-presentation of virally infected cells by lung migratory noninfected CD103+ DCs (Supplemental Figure 6B). We also show that lung migratory CD103+ DCs express a natural anti-viral state that is further strengthened by type I IFN released during the first few hours following influenza virus infection.
In our model, influenza virus did not migrate freely to the draining LNs and reached the LNs carried by lung migratory DCs. Lung CD103+ DCs appeared in the draining LNs as early as 12 hours after infection and started to activate viral-specific CD8+ T cells between 18 and 24 hours after infection. The early migration potential of lung CD103+ DCs is consistent with prior studies (19, 20) and is not specific to viral infection settings, as similar results were obtained when mice were immunized with particulate antigens (21).
CD103+ DCs were the only lung DC population able to transport intact NS1-GFP protein to the draining LNs. CD103+ DCs could have potentially acquired GFP through direct influenza virus infection or through phagocytosis of dying infected cells. Although in vitro studies established DC susceptibility to influenza virus infection (22, 23), strong evidence of DC infection by influenza virus in vivo is still lacking, and confusion remains on the primary route of antigen acquisition by DCs in vivo. Most studies have used intracellular staining and PCR of viral products (4, 24, 25) to measure the presence of viral antigens in DCs. However, these studies cannot formally discriminate between the different routes of antigen uptake because phagocytic compartments containing dying material may also contain intact viral protein and mRNA. Using cell-surface HA staining and nuclear localization of NS1-GFP protein expression as a measure of cellular viral infection, we found that lung CD103+ DCs that transport GFP to the LNs showed no signs of viral infection. Instead CD103+ DCs accumulated NS1-GFP protein in endocytic LAMP2- and EEA1-positive compartments and not in the nucleus, suggesting that they acquired GFP mainly through phagocytosis of virally infected cells in the lungs. CD103+ DCs’ ability to preserve intact GFP material captured in the lungs while migrating to the LNs was consistent with the low expression levels of lysosomal degradative enzymes and the localization of NS1-GFP in early endosomal compartments positive for EEA1. Together, these results suggest that CD103+ DCs’ specific ability to retain phagocytosed antigens in low-degradative compartments likely contributed to their superior ability to promote the cross-presentation of virally infected cells to LN CD8+ T cells (14).
The inability of lung CD11b+ DCs to present viral antigens to CD8+ T cells in our study contrasts with recent published findings (20). However, in the study of Ballesteros-Tato et al., CD11b+ DCs purified from infected mice were cultured with effector endogenous CD8+ T cells isolated from the MLNs of mice infected 7 days earlier, and the T cell proliferation observed in this assay may also result from the reactivation of effector CD8+ T cells primed by lung CD103+ DCs.
Importantly, our study revealed that virally infected CD103+ DCs accumulate in the MLNs of Ifnar1–/– mice but are absent from the MLNs of Ifnar1+/+ mice infected with NS1-GFP, suggesting that type I IFN not only protects DCs from viral infection, but also protect mice from systemic viral dissemination. In addition to its key anti-viral effect, type I IFN was shown to promote DC maturation (26, 27) and enhance the cross-presentation potential of DCs (28). Together, these results suggest that type I IFN signaling may simultaneously activate the acquisition of an anti-viral state while promoting DC capacity to cross-present antigens and cross-prime adaptive T cell responses.
Protecting lung DCs from viral infection may have several advantages in vivo. First, it protects DCs from NS1-mediated inhibition of maturation, which may compromise their ability to initiate adaptive immunity (29). This likely also applies to other respiratory viruses known to encode inhibitors of DC function, such as respiratory syncytial viruses (30). Second and more importantly, it may help contain the virus locally and prevent its systemic dissemination by infected DCs migrating to LNs. During influenza virus infection, induction of adaptive immunity is required to clear the infection, but systemic dissemination of the virus could severely compromise host integrity. Preventing DC infection while promoting cross-presentation of virally infected cells may therefore provide a means to achieve optimal immunity while limiting virus spread in vivo. This is further supported by a recent publication showing that viral infection of human DCs directly impairs their ability to cross-present viral antigen (31).
Cross-presentation has been thought to represent an evolutionary feature of DCs to avoid viral interference with antigen presentation (32, 33). Our findings showing that induction of viral-specific CD8+ T cell immunity is strongly dependent on the cross-presentation of virally infected cells by lung-noninfected CD103+ DCs provide a strong argument in favor of this hypothesis. Importantly, our study identifies a broad transcriptional program, partly elicited by type I IFN signaling that controls the acquisition of an anti-viral state by cross-presenting DC (34). Dissecting the molecular program that controls DC protection from viral infection should help the development of novel strategies to promote anti-viral therapeutic immunity in vivo.
Methods
Mice.
C57BL/6 mice were purchased from the Jackson Laboratory. Langerin-DTR mice were provided by B. Malissen (Centre d’Immunologie de Marseille-Luminy, Marseille, France) (35). Ccr7–/– mice were provided by M. Lowe (Cleveland Clinic, Cleveland, Ohio, USA). Rag–/– OT-I mice were purchased from Taconic. Ifnar1–/– mice (36) were housed in the Mount Sinai School of Medicine animal facility. 129SV Batf3–/– mice were provided by K.M. Murphy (37). Stat2–/– mice were provided by C. Schindler (Columbia University, New York, New York, USA). All mice were analyzed at between 8 and 12 weeks of age and housed in a specific pathogen–free environment.
Isolation of DCs.
Lungs, spleen, or LN tissues were digested with 0.4 mg/ml of type IV collagenase (Sigma-Aldrich) at 37°C for 45 minutes (lungs) or 30 minutes (spleen and LNs).
Viruses.
Influenza strain A/Puerto Rico/8/34 carrying GFP reporter in the NS segment (PR8:NS1-GFP) has been described (8). PR8:OTI (38) virus carries the chicken OVA257–264 SIINFEKL peptide in the neuraminidase (NA) segment. All viruses were grown either in 9-day-old embryonated eggs or in MDCK cells (ATCC) and titered using standard plaque assay on MDCK cells (ATCC).
Infection in vivo.
Mice were anesthetized with ketamine-xylazine and intranasally infected with the indicated virus dose diluted in 25 μl PBS. For GFP expression analysis, mice were infected with 106 PFUs of NS1-GFP virus. For in vivo T cell priming experiments, mice were infected with 3 × 103 PFUs of NS1-GFP virus. For ex vivo T cell assay, mice were infected with 107 PFUs of PR8:OTI virus.
Infection in vitro.
10,000 DCs or 200,000 total CD11c+ cells purified from lungs of naive mice were infected for 1 hour at 37°C in a 96-well plate with NS1-GFP virus at an MOI of 3 or 10 in Opti-MEM medium (Gibco Life Technologies; Invitrogen). After 1 hour, RPMI medium with 10% FBS was added to the cells.
Cell lines.
MDCK cells were maintained in MEM with 10% FBS and penicillin/streptomycin (Gibco Life Technologies; Invitrogen).
Detection of infectious virus.
Indicated cell types were purified and FACS sorted based on cell-surface markers. 100,000 cells were injected into pathogen-free 10-day-old embryonated eggs (Charles River Laboratories) and incubated for 2 days at 37°C. The presence of virus in the allantoic fluid was measured by standard HA assay using 0.5% chicken red-blood cells.
Flow cytometry.
Multiparameter analysis were performed on the LSR II (BD) and analyzed with FlowJo software (Tree Star). Monoclonal Abs specific to mouse B220, CD8α, MHC class II (I-A/I-E), CD103, CD11b, CD11c, CD45, Vα2, SiglecF, and the corresponding isotype controls were purchased from BD Biosciences or eBioscience. Anti-HA (PY-102) and anti-NP Ab (HT-103) were provided by Shared Hybridoma Facility (MSSM). Dextramers H-2 Db ASNENMETM and H-2 Kb SIINFEKL were purchased from Immudex. Prior to acquisition, cells were resuspended in PBS/BSA 0.5%/EDTA (2 mM) solution with 1 μg/ml of DAPI to exclude dead cells.
Cell sorting.
Single-cell suspensions were stained for the surface markers, and individual DC subsets were sorted with ARIA Sorter (BD) to achieve 98% purity. For sorting experiments, CD11c+ cells were positively enriched by AutoMACs with CD11c microbeads (Miltenyi Biotec).
T cell proliferation assay in vitro.
Live CD103+ and CD11b+ lung DCs were sorted as described above. Different numbers of DCs were cocultured with 1 × 105 OT-I T cells labeled with 10 μM CFSE (Molecular Probes). Three days later, the proliferation (CFSE dilution) of T cells was measured using flow cytometry.
Latex bead injection.
For DC migration tracking, mice were injected intratracheally with 50 μl of 1 μm fluorescent latex beads (Fluoresbrite Polychromatic Red Microsphere; Polysciences) mixed with 10 μg of poly(I:C) (Invivogen). For the T cell priming experiment, mice were immunized with 50 μl of 1 μm latex beads (Amino beads, Polysciences) covalently coated with NP. Prior to injection, beads were diluted 1:4 with PBS and mixed with 10 μg of poly(I:C). The protein coating of the amino beads was done following the manufacturer’s instructions. See Supplemental Methods for details.
Histology on lung section.
Mice were perfused intravenously with 4% formaldehyde/30% sucrose in PBS. Lungs were incubated in 20% sucrose/OCT (1/1 ratio) for 30 minutes and frozen in OCT. Eight μm-thick sections of lungs were permeabilized with 0.5% Triton X-100 for 10 minutes and then incubated with 2% donkey serum for 1 hour. Sections were stained for 2 hours with goat anti-langerin (Santa Cruz Biotechnology Inc.) and biotinylated anti-CD169 (Abcam) Abs. Sections were stained for 90 minutes with donkey anti-goat Cy3 and streptavidin-Cy5 (Jackson Immunoresearch Laboratories). Images were acquired with a Zeiss Axioplan2IE epi-fluorescence microscope controlled by Zeiss Axiovision software or with a Leica SP5 DM microscope. Images were deconvolved with the AutoQuant X2 AutoDeblur software.
Microscopy on cell suspension.
LN DCs and lung macrophages were purified by flow cytometry. 104 cells were incubated at 37°C on fibronectin-coated (5 μg/ml; Sigma-Aldrich) glass coverslips (Fisherbrand) for 30 minutes. Cells were stained with anti-HA Ab without permeabilization and fixed in 2% PFA for 5 minutes at room temperature. For intracellular staining of LAMP2, cells were fixed with 2% PFA for 5 minutes, permeabilized with PBS–Triton X-100 (0.2%), and stained with anti–LAMP2–Alexa Fluor 647 (eBioscience) in PBS–Triton X-100 (0.2%) for 30 minutes at room temperature. Multichannel 3D confocal images were acquired with a Leica SP5 DM and deconvolved with AutoQuant X2 AutoDeblur software (Media Cybernetics). For intracellular staining of EEA1, cells were fixed and permeabilized with BD Cytofix/Cytoperm according to the manufacturer’s protocol. Cells were stained with anti-EEA1 (Abcam) or isotype control Abs covalently coupled with Alexa Fluor 647 (Molecular Probes) in BD Cytoperm for 1 hour on ice. After extensive washing, cells were spun onto glasse slides using Cytospin 3 centrifuge (Thermo Shandon) for 3 minutes at 34 g. The images were captured using an Axioplan 2IE microscope equipped with an AxioCam camera (Carl Zeiss). Coverslips were mounted with Prolong Gold Antifade/API mounting medium (Invitrogen). The images were analyzed using ImageJ software (NIH) and Photoshop CS3 (Adobe).
Microarray analysis, normalization, and data set analysis.
LN CD103+ and CD11b+ migratory DCs and lung alveolar macrophages were purified by flow cytometry (ARIA; BD). To achieve 99% purity, cells were sorted twice. RNA was prepared from sorted cell populations using Trizol reagent as described (39). RNA was amplified and hybridized on the Affymetrix Mouse Gene 1.0 ST array according to the manufacturer’s instructions. Raw data for all populations were preprocessed and normalized using the RMA algorithm (40) implemented in the “Expression File Creator” module in the GenePattern suite (41). All data sets have been deposited at the National Center for Biotechnology Information/GEO (GEO GSE15907). Thresholds for value expression were defined using 2 methods. See Supplemental Methods for detail.
mRNA deep sequencing.
For RNA-seq, total RNA was extracted from FACS-sorted cells using Trizol Isolation Reagent (Roche Molecular Biochemicals) according to the manufacturers’ instructions. PolyA+ RNA was selected from 300–600 ng of total RNA per sample using oligo-dT Magnetic Beads (Illumina). The polyA+ RNA was fragmented by Covaris and retrotranscribed; libraries were prepared for mRNA-seq using the Illumina mRNA Sample Preparation Kit (Illumina) following the manufacturer’s instructions. The resulting libraries were subjected to end-repaired cDNA by PCR with Phusion High-Fidelity Taq Polymerase (Finnzymes). The samples were sequenced on an Illumina Hi-Seq 2000. Between 19,000,000 to 20,000,000 mouse genome mapping reads were obtained. All data sets have been deposited at the National Center for Biotechnology Information/GEO (GEO GSE38838).
Visualization tools.
Microarray and deep-sequencing transcripts were visualized with the “Heat Map Viewer” or the “Hierarchical Clustering” tool using Gene Pattern (41).
Statistics.
All data shown are presented as mean ± SEM. Mean, SEM, and Student’s t test (unpaired, 2-tailed) were calculated with Prism (GraphPad software). In scatter-plot graphs, symbols represent individual mice or assay and horizontal bars represent means. P < 0.05 is considered significant.
Study approval.
All animal procedures performed in this study were conducted in accordance with Institutional Animal Care and Use Committee (IACUC) guidelines and were approved by the IACUC of Mount Sinai School of Medicine.
Supplementary Material
Acknowledgments
M. Merad is supported by NIH grants U01 AI10008 and R01 HL086899. A. García-Sastre is supported by NIH grants R01AI046954, P01AI058113, and U19AI083025, and by Center for Research on Influenza Pathogenesis (CRIP), an National Institute of Allergy and Infectious Diseases Centers of Excellence for Influenza Research and Surveillance network supported CRIP (contract number HHSN266200700010C). K.M. Murphy is an investigator of the Howard Hughes Medical Institute. B. Manicassamy is supported by NIH K99 Pathway to Independence award (1K99AI095320-01). B.D. Brown is supported by JDRF-17-2010-770 and DP2DK083052-01. J. Agudo is supported by a Fulbright-Generalitat de Catalunya postdoctoral fellowship and a Beatriu de Pinos postdoctoral fellowship. We thank C. Schindler for kindly providing the Stat2–/– mice. We thank C. Muñoz-Fontela for providing the PR8-OTI influenza virus.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):4037–4047. doi:10.1172/JCI60659.
References
- 1.Sigal LJ, Crotty S, Andino R, Rock KL. Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature. 1999;398(6722):77–80. doi: 10.1038/18038. [DOI] [PubMed] [Google Scholar]
- 2.Yewdell JW. Designing CD8+ T cell vaccines: it’s not rocket science (yet). Curr Opin Immunol. 2010;22(3):402–410. doi: 10.1016/j.coi.2010.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Heath WR, Carbone FR. Dendritic cell subsets in primary and secondary T cell responses at body surfaces. Nat Immunol. 2009;10(12):1237–1244. doi: 10.1038/ni.1822. [DOI] [PubMed] [Google Scholar]
- 4.GeurtsvanKessel CH, et al. Clearance of influenza virus from the lung depends on migratory langerin+CD11b- but not plasmacytoid dendritic cells. J Exp Med. 2008;205(7):1621–1634. doi: 10.1084/jem.20071365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Edelson BT, et al. Peripheral CD103+ dendritic cells form a unified subset developmentally related to CD8alpha+ conventional dendritic cells. J Exp Med. 2010;207(4):823–836. doi: 10.1084/jem.20091627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dorner BG, et al. Selective expression of the chemokine receptor XCR1 on cross-presenting dendritic cells determines cooperation with CD8+ T cells. Immunity. 2009;31(5):823–833. doi: 10.1016/j.immuni.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 7.Kittel C, et al. Rescue of influenza virus expressing GFP from the NS1 reading frame. Virology. 2004;324(1):67–73. doi: 10.1016/j.virol.2004.03.035. [DOI] [PubMed] [Google Scholar]
- 8.Manicassamy B, Manicassamy S, Belicha-Villanueva A, Pisanelli G, Pulendran B, Garcia-Sastre A. Analysis of in vivo dynamics of influenza virus infection in mice using a GFP reporter virus. Proc Natl Acad Sci U S A. 2010;107(25):11531–11536. doi: 10.1073/pnas.0914994107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sung SS, Fu SM, Rose CE, Jr, Gaskin F, Ju ST, Beaty SR. A major lung CD103 (alphaE)-beta7 integrin-positive epithelial dendritic cell population expressing Langerin and tight junction proteins. J Immunol. 2006;176(4):2161–2172. doi: 10.4049/jimmunol.176.4.2161. [DOI] [PubMed] [Google Scholar]
- 10.Ginhoux F, et al. The origin and development of nonlymphoid tissue CD103+ DCs. J Exp Med. 2009;206(13):3115–3130. doi: 10.1084/jem.20091756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Norbury CC, Malide D, Gibbs JS, Bennink JR, Yewdell JW. Visualizing priming of virus-specific CD8+ T cells by infected dendritic cells in vivo. Nat Immunol. 2002;3(3):265–271. doi: 10.1038/ni762. [DOI] [PubMed] [Google Scholar]
- 12.Hintzen G, et al. Induction of tolerance to innocuous inhaled antigen relies on a CCR7-dependent dendritic cell-mediated antigen transport to the bronchial lymph node. J Immunol. 2006;177(10):7346–7354. doi: 10.4049/jimmunol.177.10.7346. [DOI] [PubMed] [Google Scholar]
- 13.Dudziak D, et al. Differential antigen processing by dendritic cell subsets in vivo. Science. 2007;315(5808):107–111. doi: 10.1126/science.1136080. [DOI] [PubMed] [Google Scholar]
- 14.Delamarre L, Pack M, Chang H, Mellman I, Trombetta ES. Differential lysosomal proteolysis in antigen-presenting cells determines antigen fate. Science. 2005;307(5715):1630–1634. doi: 10.1126/science.1108003. [DOI] [PubMed] [Google Scholar]
- 15.Kovacsovics-Bankowski M, Rock KL. A phagosome-to-cytosol pathway for exogenous antigens presented on MHC class I molecules. Science. 1995;267(5195):243–246. doi: 10.1126/science.7809629. [DOI] [PubMed] [Google Scholar]
- 16.Schnorrer P, et al. The dominant role of CD8+ dendritic cells in cross-presentation is not dictated by antigen capture. Proc Natl Acad Sci U S A. 2006;103(28):10729–10734. doi: 10.1073/pnas.0601956103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Isaacs A, Lindenmann J, Valentine RC. Virus interference. II. Some properties of interferon. Proc R Soc Lond B Biol Sci. 1957;147(927):268–273. doi: 10.1098/rspb.1957.0049. [DOI] [PubMed] [Google Scholar]
- 18.Durbin JE, et al. Type I IFN modulates innate and specific anti-viral immunity. J Immunol. 2000;164(8):4220–4228. doi: 10.4049/jimmunol.164.8.4220. [DOI] [PubMed] [Google Scholar]
- 19.Kim TS, Braciale TJ. Respiratory dendritic cell subsets differ in their capacity to support the induction of virus-specific cytotoxic CD8+ T cell responses. PLoS One. 2009;4(1):e4204. doi: 10.1371/journal.pone.0004204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ballesteros-Tato A, Leon B, Lund FE, Randall TD. Temporal changes in dendritic cell subsets, cross-priming and costimulation via CD70 control CD8(+) T cell responses to influenza. Nat Immunol. 2010;11(3):216–224. doi: 10.1038/ni.1838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jakubzick C, Helft J, Kaplan TJ, Randolph GJ. Optimization of methods to study pulmonary dendritic cell migration reveals distinct capacities of DC subsets to acquire soluble versus particulate antigen. J Immunol Methods. 2008;337(2):121–131. doi: 10.1016/j.jim.2008.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bhardwaj N, Bender A, Gonzalez N, Bui LK, Garrett MC, Steinman RM. Influenza virus-infected dendritic cells stimulate strong proliferative and cytolytic responses from human CD8+ T cells. J Clin Invest. 1994;94(2):797–807. doi: 10.1172/JCI117399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392(6671):86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 24.Hao X, Kim TS, Braciale TJ. Differential response of respiratory dendritic cell subsets to influenza virus infection. J Virol. 2008;82(10):4908–4919. doi: 10.1128/JVI.02367-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moltedo B, Li W, Yount JS, Moran TM. Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog. 2011;7(11):e1002345. doi: 10.1371/journal.ppat.1002345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Honda K, et al. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc Natl Acad Sci U S A. 2003;100(19):10872–10877. doi: 10.1073/pnas.1934678100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Montoya M, et al. Type I interferons produced by dendritic cells promote their phenotypic and functional activation. Blood. 2002;99(9):3263–3271. doi: 10.1182/blood.V99.9.3263. [DOI] [PubMed] [Google Scholar]
- 28.Bougneres L, et al. A role for lipid bodies in the cross-presentation of phagocytosed antigens by MHC class I in dendritic cells. Immunity. 2009;31(2):232–244. doi: 10.1016/j.immuni.2009.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fernandez-Sesma A, et al. Influenza virus evades innate and adaptive immunity via the NS1 protein. J Virol. 2006;80(13):6295–6304. doi: 10.1128/JVI.02381-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartz H, Turkel O, Hoffjan S, Rothoeft T, Gonschorek A, Schauer U. Respiratory syncytial virus decreases the capacity of myeloid dendritic cells to induce interferon-gamma in naive T cells. Immunology. 2003;109(1):49–57. doi: 10.1046/j.1365-2567.2003.01629.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Smed-Sorensen A, et al. Influenza a virus infection of human primary dendritic cells impairs their ability to cross-present antigen to CD8 T cells. PLoS Pathog. 2012;8(3):e1002572. doi: 10.1371/journal.ppat.1002572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heath WR, Carbone FR. Cross-presentation in viral immunity and self-tolerance. Nat Rev Immunol. 2001;1(2):126–134. doi: 10.1038/35100512. [DOI] [PubMed] [Google Scholar]
- 33.Yewdell JW, Hill AB. Viral interference with antigen presentation. Nat Immunol. 2002;3(11):1019–1025. doi: 10.1038/ni1102-1019. [DOI] [PubMed] [Google Scholar]
- 34.Zurney J, Howard KE, Sherry B. Basal expression levels of IFNAR and Jak-STAT components are determinants of cell-type-specific differences in cardiac anti-viral responses. J Virol. 2007;81(24):13668–13680. doi: 10.1128/JVI.01172-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kissenpfennig A, et al. Dynamics and function of Langerhans cells in vivo: dermal dendritic cells colonize lymph node areas distinct from slower migrating Langerhans cells. Immunity. 2005;22(5):643–654. doi: 10.1016/j.immuni.2005.04.004. [DOI] [PubMed] [Google Scholar]
- 36.Muller U, et al. Functional role of type I and type II interferons in anti-viral defense. Science. 1994;264(5167):1918–1921. doi: 10.1126/science.8009221. [DOI] [PubMed] [Google Scholar]
- 37.Hildner K, et al. Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science. 2008;322(5904):1097–1100. doi: 10.1126/science.1164206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jenkins MR, Webby R, Doherty PC, Turner SJ. Addition of a prominent epitope affects influenza A virus-specific CD8+ T cell immunodominance hierarchies when antigen is limiting. J Immunol. 2006;177(5):2917–2925. doi: 10.4049/jimmunol.177.5.2917. [DOI] [PubMed] [Google Scholar]
- 39.Yamagata T, Mathis D, Benoist C. Self-reactivity in thymic double-positive cells commits cells to a CD8 alpha alpha lineage with characteristics of innate immune cells. Nat Immunol. 2004;5(6):597–605. doi: 10.1038/ni1070. [DOI] [PubMed] [Google Scholar]
- 40.Irizarry RA, Bolstad BM, Collin F, Cope LM, Hobbs B, Speed TP. Summaries of Affymetrix GeneChip probe level data. Nucleic Acids Res. 2003;31(4):e15. doi: 10.1093/nar/gng015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Reich M, Liefeld T, Gould J, Lerner J, Tamayo P, Mesirov JP. GenePattern 2.0. Nat Genet. 2006;38(5):500–501. doi: 10.1038/ng0506-500. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.