

Abstract
In the human brain, microRNAs (miRNAs) from the microRNA-376 (miR-376) cluster undergo programmed “seed” sequence modifications by adenosine-to-inosine (A-to-I) editing. Emerging evidence suggests a link between impaired A-to-I editing and cancer, particularly in high-grade gliomas. We hypothesized that disruption of A-to-I editing alters expression of genes regulating glioma tumor phenotypes. By sequencing the miR-376 cluster, we show that the overall miRNA editing frequencies were reduced in human gliomas. Specifically in high-grade gliomas, miR-376a* accumulated entirely in an unedited form. Clinically, a significant correlation was found between accumulation of unedited miR-376a* and the extent of invasive tumor spread as measured by magnetic resonance imaging of patient brains. Using both in vitro and orthotopic xenograft mouse models, we demonstrated that the unedited miR-376a* promoted glioma cell migration and invasion, while the edited miR-376a* suppressed these features. The effects of the unedited miR-376a* were mediated by its sequence-dependent ability to target RAP2A and concomitant inability to target AMFR. Thus, the tumor-dependent introduction of a single base difference in the miR-376a* sequence dramatically alters the selection of its target genes and redirects its function from inhibiting to promoting glioma cell invasion. These findings uncover a new mechanism of miRNA deregulation and identify unedited miR-376a* as a potential therapeutic target in glioblastoma cells.
Introduction
MicroRNAs (miRNAs) are short, noncoding RNAs that mediate post-transcriptional silencing of a set of target genes. The target gene specificity of each miRNA is dictated by sequence-dependent interaction between approximately 22-nt-long mature miRNAs, especially their 6- or 7-nucleotide “seed” sequences at the 5′ end, and the 3′ untranslated regions of mRNAs (1). It has been shown that the epigenetic process of adenosine-to-inosine (A-to-I) editing of certain miRNAs can lead to a single base substitution in their seed sequence and generate variant “edited” mature miRNA species with target gene specificity drastically different from that of the unedited, genomically encoded miRNA (2). This was demonstrated for miR-376 cluster transcripts collected from normal human and mouse tissues. The miR-376 cluster encodes 4 primary miRNAs (pri-miRs), including pri-miR-376a1, -376a2, -376b, and -376c, that are processed to 5 distinct mature miRNAs, miR-376a, -376a*, -376a2-5p, -376b, and -376c. In the human brain, 9 adenosines within this miRNA cluster are subject to specific and high-level A-to-I RNA editing (2).
In gliomas, the most frequent primary brain tumors, Alu repeats and several protein-coding substrates of adenosine deaminases acting on RNA (ADARs; a family of enzymes that mediate A-to-I editing of RNAs) have been found to be edited to lower than normal frequencies (3–5). Particularly in high-grade gliomas, glioblastoma multiforme (GBMs), emerging lines of evidence suggest a link between A-to-I editing and cancer due to anomalous ADAR activity or levels (6). For example, the frequency of A-to-I editing of the Q/R site of glutamate receptor subunit B (GluR-B) is lower in GBMs (3) and astrocytomas (5) compared with normal brain tissue. It has been reported that in GBMs, glutamate receptors assembled from the unedited (Q-containing) GluR-B subunit are highly Ca2+ permeable and expression of this form of the receptor can promote glioma cell invasion (7). Apart from this, the functional consequences of under-editing or non-editing of ADAR substrates in gliomas are not clear.
We considered the possibility that miR-376 cluster transcripts may also be targets of ADAR dysfunction in gliomas. We hypothesized that disruption to the A-to-I editing of the transcripts would lead to changes in the relative abundance of edited and unedited forms of the miRNAs, which would in turn lead to altered gene expression profiles in gliomas that regulate tumor phenotypes and clinical behaviors. Herein we report that the A-to-I editing of the miR-376 cluster miRNAs is significantly reduced in high-grade human gliomas, with miR-376a* accumulating entirely in the “unedited” form in GBMs. We also explicitly demonstrate that the overexpression of unedited miR-376a* in glioma cells promotes their migration and invasion, which in an orthotopic xenograft mouse glioma model paralleled features of human GBMs such as disseminated tumor foci, extensive vascularization, and necrosis. Strikingly, the edited miR-376a* suppressed glioma cell migration and invasion in in vitro assays. As dictated by the single base difference between unedited and edited miR-376a*, we identified genes targeted in an exclusive manner by either form of miR-376a* that potentially accounted for their dramatic functional divergence. The ability of unedited miR-376a* to target RAP2A and its concomitant inability to target AMFR constitute a subset of the redirection of miRNA function toward an unfavorable target gene profile and implicate this miRNA as a therapeutic candidate.
Results
A-to-I RNA editing of miR-376 cluster transcripts is reduced in high-grade gliomas.
To investigate possible involvement of the miR-376 cluster in gliomas, we first performed data mining on the Cancer Genome Atlas dataset (8) and used the Anduril program (9) to analyze glioma miRNA expression data. We noted that expression levels of miR-376 cluster members in GBMs are in general unaffected, ranging from 0.8- to 0.94-fold of normal brain levels, except miR-376a, which was expressed at 0.5-fold (Supplemental Figure 1A; supplemental material available online with this article; doi: 10.1172/JCI62925DS1).
We then investigated whether A-to-I editing of miR-376 cluster transcripts is disrupted in GBMs. Since transcription of the miR-376 cluster produces a long single primary transcript with pri-miR-376a1, -376a2, -376c, and -376b and A-to-I editing (hereafter referred to as “editing”) occurs on the primary transcript (2), we performed direct sequencing of RT-PCR products to determine editing frequency at 9 editing sites within the 4 pri-miRNAs (Figure 1A). Sequencing of 5 normal human brain tissue samples confirmed that editing at the sites was high and occurred to the same extent as described previously (2). Compared with normal brain, in 24 high-grade human glioma samples (WHO grades III and IV), we observed that editing was altered to variable extents, appearing as reduced or absent “G” peaks at the editing sites in sequence chromatograms (Figure 1B). Quantification of editing frequencies for all sites revealed a general reduction, with frequent baseline editing of 0%–4% in the tumors (Table 1). Reduction in editing was most significant for the +9 site of pri-miR-376a1 and the +15 site of pri-miR-376a2 (Table 1). When the tumors were grouped by histopathology, reduction in editing was most consistently observed in GBMs and anaplastic astrocytomas (AAs) at the +9 site of pri-miR-376a1, averaging 3.3% ± 3.8% in GBMs (n = 12, excluding known gliosarcomas) and 10% ± 15% in AAs (n = 4), down from 47% ± 2% in the normal brain (n = 5). Gliomas classified as anaplastic oligoastrocytomas (AOAs), anaplastic oligodendrogliomas (AOGs), displayed substantial editing (48% ± 38%, n = 4) for the +9 site of pri-miR-376a1 (Figure 1C).
Figure 1. A-to-I editing of the miR-376 cluster is reduced in gliomas.

(A) Primary miRNAs encoded by the human miR-376 cluster. Mature miRNAs processed from the pri-miRNAs are indicated in green. Positions of editing sites are indicated by red arrows. (B) Chromatograms from direct sequencing of RT-PCR products of pri-miR-376a1 from normal human brain (NB), GBM (GBM 1), and AA (AA 20). The genomically encoded sequence is shown. Underlined regions correspond to two mature miRNAs, miR-376a* and miR-376a, processed from pri-miR-376a1. Editing sites are indicated by red arrows. (C) Editing frequency at the +9 site of pri-miR-376a1 (miR-376a*) in samples of NB (n = 5), GBM (n = 12), AA (n = 4), and AOAs and AOGs (n = 4). Means are designated by horizontal lines. (D) Editing frequency of mature miRNAs from the miR-376 cluster in glioma cell lines. cDNA clones (n = 50) corresponding to mature miRNA were sequenced, and frequency of editing was calculated as the percentage of clones with A-to-G change at the editing site. (E) Direct sequencing of RT-PCR products of pri-miR-376a1 from U87 cells co-transfected with a plasmid expressing ADARB1 and the human miR-376 cluster. Red arrows indicate editing sites. Site corresponding to miR-376a* is boxed. A plasmid expressing EGFP was used as a control. (F) Relative expression of ADARB1 mRNA, by qRT-PCR in NB and tumor samples. Data represent mean ± SD (n = 3).
Table 1.
Quantification of A-to-I RNA editing of primary miRNAs from miR-376 cluster in normal human brain and primary brain tumors

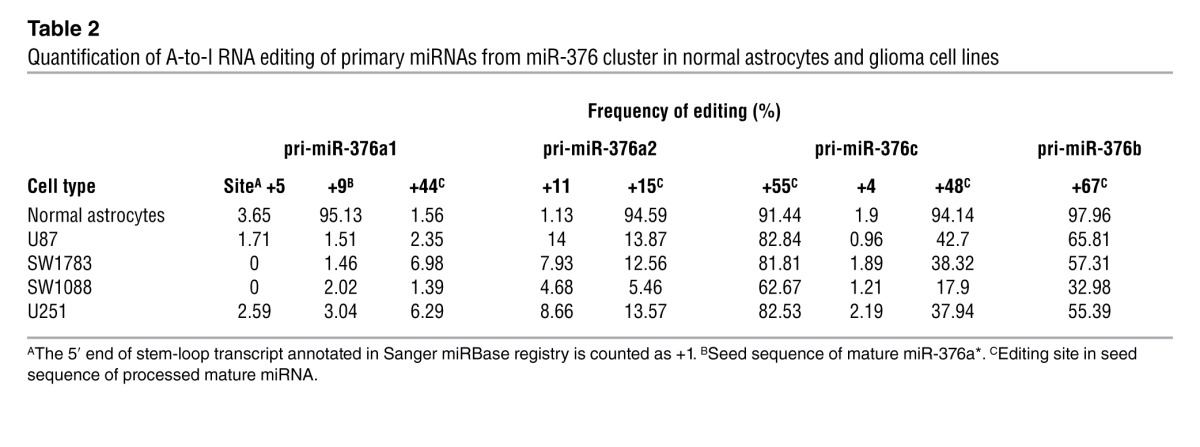
To evaluate the editing of miR-376 cluster transcripts at a cellular level, rather than in tumor tissue with mixed cell populations, we examined 4 glioma cell lines, U87, SW1783, SW1088, and U251, and observed consistent hypoediting of miR-376 cluster transcripts (Table 2). Using the cell lines also allowed us to evaluate the extent of editing of mature miRNAs with a targeted cloning strategy developed previously (2). We confirmed that the small extent of editing of primary miRNAs in the miR-376 cluster produced mature miRNAs at similarly low editing frequencies in the glioma cell lines (Figure 1D and Table 2). In particular, the mature miR-376a* was negligibly edited to 0%–5% in these tumor cells, corresponding well with the editing frequency of the +9 site of pri-miR-376a1, a site in the seed sequence of miR-376a*. In normal human astrocytes, remarkably high editing for all sites within mature miRNA sequences was observed, with 95% of miR-376a* being in the edited form (Table 2).
Table 2.
Quantification of A-to-I RNA editing of primary miRNAs from miR-376 cluster in normal astrocytes and glioma cell lines
In the human brain, editing of pri-miR-376a1 is a result of the activity of ADARB1 (10), an enzyme known to be deregulated in brain tumors (3, 4). To establish a causal link between ADARB1 function and loss of editing of miR-376a*, we overexpressed ADARB1 in U87 cells that were co-transfected with the miR-376 cluster gene. Editing at the +9 site of pri-miR-376a1 was restored from 1.5% to 95% in the glioma cells in the presence of ectopic ADARB1 (Figure 1E). Most GBMs in the tumor panel had markedly diminished levels of ADARB1 mRNA (<20% of normal levels), while most WHO grade III gliomas retained nearly 50% of normal levels of ADARB1 (Figure 1F). The other major editing enzyme, ADAR, was found to be generally underexpressed in all tumors, without any particular tumor grade–dependent expression pattern (Supplemental Figure 1B). Furthermore, we confirmed that increased ADARB1 expression, hence activity, did not impact the endogenous abundance of mature miRNAs from the miR-376 cluster, indicating that editing of pri-miRs from the miR-376 cluster does not impact their processing (Supplemental Figure 1C). Thus, our cumulative data suggest that unedited miR-376a* is a tumor-specific miRNA sequence variant, aberrantly produced due to the loss of regulated A-to-I editing in GBMs.
Reduced miR-376a* editing frequency in high-grade gliomas correlates with increased signal on T2-weighted MRI scans.
High-grade gliomas, especially GBMs, are characterized by extensive invasiveness in the brain (11). With the aim of placing the finding on miR-376a* editing frequency in a clinical context, we surveyed the above panel of glioma samples for correlation between the editing frequency at site +9 of pri-miR-376a1 (corresponding to miR-376a*) and peritumoral T2-weighted MRI signal. The regions with enhanced signal intensity on T2-weighted MRI images histologically correspond to the hypervascular tissue of viable tumor, likely due to tumor cell invasion away from the main tumor mass, vasogenic edema resulting from cytokines elaborated by invasive tumor cells, and/or foci of tumor-induced neovascularization. The samples GBM 1–4 and AA 17, 19, and 20 were excluded from this analysis due to the lack of MRI data. When tumor volumes were measured based on peritumoral T2 signals and the tumor samples were grouped based on high (>12%) and low level of editing frequency, a striking difference in distribution of tumor volumes was observed. The tumors with low percent editing of miR-376a* (n = 8) had an average volume of 537.7 ± 130.1 mm3, whereas those with high percent editing (n = 7) had an average volume of only 159.3 ± 34.05 mm3 (Figure 2, A and B). After calculating the expression levels of the unedited miR-376a* based on qRT-PCR quantification of total miR-376a* (Supplemental Figure 2A) and the editing frequency at site +9 of pri-miR-376a1 and then correlating the expression levels with the tumor volumes, a significant correlation was further identified, demonstrating that gliomas with larger tumor volumes combinatorially showed low percent editing and relatively higher expression of the unedited miR-376a* (Figure 2C). The relatively small sample size of this study cohort, however, limits the generalizability of the conclusion, and studies with a larger group of tumors will be necessary. Nonetheless, since a lower miR-376a* editing frequency in glioma is associated with more pronounced T2 signal abnormality, the editing frequency of the miRNA may potentially serve as a quantifiable biomarker for invasive high-grade gliomas.
Figure 2. Frequency of miR-376a* editing and expression of its unedited form correlate with invasive glioma spread.

(A) Distribution of tumor volume/spread of gliomas grouped by high (>12%, n = 8) and low percent editing (n = 7) of miR-376a* at site +9 of pri-miR-376a1. Individual data points indicate tumor volumes (mm3) assessed by T2-weighted MRI imaging. Horizontal lines indicate mean tumor volumes. Student’s t test P value is indicated. (B) Representative MRI scans of patients with gliomas with low or high editing frequency of miR-376a*. White X indicates the main tumor mass, and white arrow indicates region of invasive tumor spread. (C) Correlation of expression of unedited miR-376a* with tumor volume indicating invasive tumor spread. Each data point represents an individual sample, and correlation coefficient determined by Pearson’s correlation is indicated. Expression levels of unedited miR-376a* were estimated by the frequency of editing of site +9 of pri-miR-376a1 and relative expression level of total miR-376a*.
Unedited miR-376a* specifically accumulates in invasive glioma cells.
To investigate whether the unedited miR-376a* is specifically involved in the invasive process of glioma, we performed an in vivo experimental lung metastasis (ELM) assay in mice to enrich highly invasive cells from human U87 cell line (Figure 3A). This assay employs tail vein injection of tumor cells, selection of metastatic cell populations from the lung, and in vitro expansion of the selected cells to enrich highly invasive tumor cells from established tumor cell lines, the invasive feature of which is often diminished over long-term in vitro culture (12–14). Applying this strategy to the U87 cell line, we were able to establish 3 independent ELM-selected cell lines, 1M1, 1M2, and 2M1. We compared in vivo tumor growth between these selected cells and parental U87 cells after intracranial injection into nude mice. As previously reported (15), parental U87 cells formed oval-shaped tumors with well defined edges (Figure 3B). However, mice receiving ELM-selected U87 cell injection developed multiple and large aggressive tumors with invasive cell foci and infiltrative tumor boundaries (Figure 3B). Quantified in an in vitro invasion assay, the ELM-selected cells displayed a 1.5- to 2-fold greater invasive capacity than parental U87 cells and were highly migratory as confirmed by a wound healing assay (Figure 3, C and D). In agreement with the inverse correlation between glioma cell proliferation and motility (16), in vitro proliferation rates of ELM-selected U87 cells in serum-containing media were several-fold lower than those of parental U87 cells (Supplemental Figure 3A). Together, these data suggest that ELM selection is effective in enriching glioblastoma cells that are more migratory and invasive.
Figure 3. Invasive U87 glioma cells are selected using ELM assay.

(A) Strategy for selection of highly invasive glioma cells using ELM assay, resulting in establishment of 3 ELM cell lines, named 1M1, 1M2, and 2M1. (B) H&E staining of tumors formed at day 21 after brain inoculation of U87 and 2 ELM cell lines. Top row shows whole brain sections; scale bars: 2 mm. N, necrosis. Bottom panel shows disseminated tumors (black arrows) after 1M1 inoculation and pseudopalisading glioma cells around a necrotic region for 2M1. Scale bar: 500 μm. (C) Representative photomicrographs and quantification of indicated fluorescently labeled ELM cells invading Matrigel in Transwell invasion assays. Values are relative to U87 cells (n = 4). Original magnification, ×100. (D) Wound healing assay showing increased migration ability of ELM cells compared with parental U87 cells. Wound gap closure was calculated as percentage and is shown in the accompanying graph (n = 3). Original magnification, ×100. Error bars in C and D indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 by t test.
We then used the ELM-selected U87 cells and parental U87 cells to investigate whether the extent of A-to-I editing of miRNAs from the miR-376 cluster, especially miR-376a*, correlated with tumor cell invasiveness using direct sequencing of pri-miRs. We found that overall editing levels for all sites within the miR-376 cluster in the ELM-selected cells were similar to the levels in parental U87 cells (Figure 4A and Supplemental Figure 3B). A negligible “G” trace at the +9 site of pri-miR-376a1 confirmed that miR-376a* remained unedited in the ELM-selected cells, as in parental U87 cells. Levels of editing enzymes, ADAR and ADARB1, were not significantly changed in the ELM-selected cells (Figure 4B).
Figure 4. Unedited miR-376a* specifically accumulates in ELM-selected U87 cells.

(A) Editing analysis of miR-376 cluster pri-miRNAs from parental U87 and ELM cells by direct sequencing of RT-PCR products. Editing sites are highlighted by red arrows. The +9 site of pri-miR-376a1 corresponding to miR-376a* is boxed. (B) Relative expression of editing enzymes ADAR and ADARB1 in parental U87 and ELM cells measured by qRT-PCR (n = 3). Values are normalized to 18S rRNA. (C) RT-PCR for transcripts corresponding to indicated pri-miRNAs from miR-376 cluster in U87 and ELM cells. (D) Relative abundance of various mature miRNAs in U87 and ELM cells by qRT-PCR. Expression was normalized to 5S rRNA (n = 3). Error bars in B and D indicate SD. **P < 0.01 by t test.
We next measured the expression levels of pri-miRNAs and mature miRNAs from the miR-376 cluster to investigate changes in transcript abundance. Primary transcripts were moderately but not consistently increased in the ELM-selected cells (Figure 4C). Significantly, mature miR-376a* was enriched 7- to 75-fold in the ELM-selected U87 cells in comparison with parental U87 cells, but none of the other mature miRNAs from the miR-376 cluster were significantly enriched in ELM-selected cells (Figure 4D). The fold increase in miR-376a* correlated with the increase in invasion and migration capacity of ELM-selected U87 cells (Figure 3, C and D). Also, we determined the relative expression of selected miRNAs from the chr.14q32.31 region (where miR-376 cluster lies), including those ubiquitously expressed (miR-16) and those with known functions in gliomas (miR-21, miR-221, miR-10b, miR-206), and found no significant change in their expression in the ELM-selected U87 cells (Figure 4D). Collectively, this study suggests a mechanism for highly specific enrichment of unedited miR-376a* in invasive glioma cells.
Unedited miR-376a* promotes glioma cell invasion and migration, while edited miR-376a* suppresses this ability in vitro.
The specific accumulation of unedited miR-376a* in GBMs and selected invasive glioma cells prompted us to investigate whether this miRNA functionally modulates the invasive behavior of glioma cells. Cellularly, editing of miR-376a* substitutes a single adenosine (A) for inosine (I), and functional equivalence has been suggested for inosine and guanosine (G) for miRNA target selection via base pairing to cytosine for both (2). Thus, miRNA mimics for the miR-376a* in unedited form (hereafter, miR-376a*A) and for the edited form (hereafter, miR-376a*G) were designed (Figure 5A). For stable expression, a stem-loop construct was engineered encoding miR-376a*, with A or G substituted at the appropriate position, to generate miR-376a*A or miR-376a*G (Figure 5A). Expression of miR-376a* in glioma cells by transfection of miRNA mimics and in cells stably expressing miR-376a* was verified by qRT-PCR (Supplemental Figure 4, A and B).
Figure 5. Unedited miR-376a* promotes migration and invasion of glioma cells in vitro.

(A) miRNA sequences for transfection. Top: Sequences of miRNA mimics used for transient expression of miR-376a*A and miR-376a*G. Bottom: Design of stem-loop constructs for stable expression of 376a*A, miR-376a*G, and miR-control. The position of mature miRNA in the engineered precursor is shown in blue. EGFP is cocistronically expressed. pmiR, pre-miR. (B) Matrigel invasion assay of glioma cells transfected with miR-376a* or control miRNA either transiently or stably (n = 4). Values are normalized to control for each cell line. (C) miR-376a*A promotes glioma cell migration in wound healing assay, while miR-376a*G suppresses migration. Graph shows relative percent wound gap closure (n = 3). Original magnification, ×100. (D) Matrigel invasion assay of 2M1 and U87 cells after knockdown of miR-376a*A using LNA inhibitors (n = 4). Values are normalized to control for each cell line. (E) Wound healing migration assay of U87 cells and ELM-selected 2M1 cells after LNA-mediated knockdown of miR-376a*A. scr, scrambled control (n = 3). Original magnification, ×100. (F) Expression verification by qRT-PCR of genes involved in glioma migration, invasion, and angiogenesis in U87 cells transfected with miR-376a*A or -376a*G (n = 3). Error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 by t test.
Using a Matrigel invasion assay, we found that transient or stable ectopic expression of miR-376a*A enhanced invasiveness of glioma cell lines (Figure 5B). Under similar conditions, miR-376a*G suppressed the invasiveness to below basal levels. Wound healing assays revealed that migration (a prerequisite for invasion) of glioma cells was promoted by miR-376a*A and suppressed by miR-376a*G (Figure 5C). In line with the involvement of cell shape and cytoskeleton in cellular motility, distinct morphological changes in U87 and SW1783 cells were also observed upon transfection of variant forms of miR-376a* (Supplemental Figure 4C). Additionally, specific knockdown of miR-376a*A by LNA-modified oligonucleotides in 2M1 cells, which had the highest expression of miR-376a* among ELM-selected lines, resulted in a reversal of their highly invasive and migratory phenotype, while it did not affect invasion and motility of parental U87 cells, which had negligible expression of miR-376a* (Figure 5, D and E). Knockdown of miR-376a* using anti–miR-376a* inhibitors (of chemistry distinct from LNA inhibitors) also resulted in decreased migration ability of 2M1 cells specifically (Supplemental Figure 4D). Cell proliferation and cell cycle analyses showed that miR-376a*A suppressed proliferation rates of glioma cells, while miR-376a*G exerted the opposite effect (Supplemental Figure 4, E and F), limiting the possibility that the increase or decrease in invasion was due to a change in cell proliferation. Thus, miR-376a*A and 376a*G have distinct and opposing cellular functions in regulating the invasive behavior of glioma cells.
We next investigated the genome-wide transcriptional profiles in U87 cells transfected with miR-376a*A or 376a*G using Affymetrix microrarrays for a broader insight into molecular changes mediated by the two miRNA variants. DAVID analysis of the differentially expressed genes revealed that genes regulating cell adhesion and cell motion functions were significantly enriched among those upregulated by miR-376a*A, in contrast to the enrichment of cell cycle function for genes upregulated by miR-376a*G (Supplemental Table 1). A less significant but inverse enrichment was found for the same processes among the downregulated genes. We chose a set of genes that are known to pleiotropically regulate cell invasion, migration, and angiogenesis (17, 18) to validate the microarray results by qRT-PCR. miR-376a*A increased, while miR-376a*G decreased, the expression of selected genes (Figure 5F). Expression changes mediated by two forms of edited miRNA — miR-376a*G and miR-376a*I — were similar (Supplemental Figure 5). The data suggest that miR-376a*A has widespread effects on the transcriptome related to glioma cell motility, further confirming the cellular effects of the miRNA in enhancing in vitro migration and invasion.
Overexpression of unedited miR-376a* promotes aggressive growth of orthotopic gliomas in the mouse brain.
Taking advantage of the tumorigenic but noninvasive property of U87 cells in the brain, we generated U87 cells stably expressing miR-376a*A (hereafter, U87/376a*A) or miR-376a*G (hereafter, U87/376a*G) and injected them into the brains of nude mice. At day 21 after tumor inoculation, brains were collected and sectioned for histological analysis to evaluate whether ectopic expression of unedited miR-376a* promotes in vivo tumor aggressiveness. As shown in H&E-stained brain sections, U87/376a*A formed large, aggressive tumors with irregular shape and extensive intratumoral necrosis and especially displaying dissemination away from the primary inoculation site into the surrounding brain (Figure 6A and Supplemental Figure 6A). In contrast, tumors formed by U87/376a*G cells were highly uniform and well circumscribed, characteristic of expansile growth of noninvasive tumors such as U87 tumors (15). Boundaries of U87/376a*A tumors were marked by infiltrative projections associated with vascular structures and satellite tumors, while U87/376a*G tumors were well delineated from the normal brain.
Figure 6. Unedited miR-376a* promotes aggressive glioma growth in vivo.

(A) Orthotopic gliomas formed by stably transfected U87 cells in mouse brain. Representative images from two cell clones each for U87/376a*A and U87/376a*G are shown. Top: Arrows indicate tumors; N, necrosis. Scale bars: 2 mm. Bottom: Tumor boundaries at higher magnification. Black arrowheads: peritumoral vascular structures for clone D5; disseminated tumor satellites for clone D3. Scale bars: 200 μm. (B) Immunohistochemical staining for proliferation marker Ki-67. Left: Higher Ki-67 signals at tumor edge demarcated by white dotted line for U87/376a*A tumors. BV, blood vessel. Scale bars: 100 μm. Right: Ki-67 signal quantification using ImageJ. Relative levels at tumor edge and core are indicated (n = 8–12). (C) Expression of factors involved in glioma invasion and angiogenesis in orthotopic tumors measured by qRT-PCR (n = 3). Left: Upstream growth factor signaling factors. Right: Factors promoting glioma cell invasion and angiogenesis. (D) Kaplan-Meier survival analysis after intracranial implantation of glioma cells. U87/376a*A clone D5 and U87/376a*G clone 2A2 stably express miR-376a*. 1M1 and 2M1 cells were enriched from U87 cells by ELM assay as in Figure 3. (E) Median survival of animals in D. Statistical analysis was done using log-rank test (n = 10). Error bars indicate SD. *P < 0.05, ***P < 0.001 by t test. In E, error bars represent SEM; **P < 0.01 and ***P < 0.001 vs. U87/control; ††P < 0.01 and †††P < 0.001 vs. U87/376a*G; ‡P < 0.001 vs. 1M1.
U87/376a*A tumors stained for the Ki-67 proliferation marker showed high but non-uniform distribution of Ki-67+ cells that especially colonized regions around blood vessels (Figure 6B), indicating perivascular invasion (19). A roughly 2-fold enrichment of Ki-67 signal at the invasion fronts of U87/376a*A tumors in relation to the core was noted, while U87 and U87/376a*G tumors showed uniform distribution of Ki-67+ cells, consistent with expansile growth (Figure 6B). We confirmed that aggressive in vivo tumor growth associated with U87/376a*A cells was not due to a higher intrinsic proliferation rates measured in vitro (Supplemental Figure 6B). qRT-PCR analysis of tumor samples collected from the mouse brain revealed that mRNAs of several glioma invasion-promoting factors, including integrins, ECM proteins, matrix metalloproteinases, and angiogenic factors, and components of upstream growth factor signaling pathways were enriched (5- to 45-fold) in U87/376a*A tumors (Figure 6C).
Conclusive evidence regarding the ability of miR-376a*A to promote aggressive tumor growth came from the survival rates of mice bearing tumors formed by U87 clones with higher expression levels of miR-376a*A. Indeed, both U87/376a*A and ELM-selected U87 cells with enrichment of miR-376a*A caused a significantly shortened survival time of animals compared with control U87 cells (Figure 6D). Importantly, as invasiveness was the only property selected by the ELM assay, the observed differences in survival are chiefly attributable to increased tumor burden through invasive dissemination of the selected tumor cells. Furthermore, there was an inverse correlation between the fold enrichment of miR-376a*A in ELM-selected cells and survival rates of tumor-bearing mice (Figure 6E). Although, miR-376a*G suppresses cell invasion in vitro, tumors formed in vivo by U87/376a*G and the associated survival rates were similar to control tumors (Figure 6, D and E), probably due to the inherently noninvasive nature of the U87 glioma model. Collectively, these results show that expression of miR-376a*A in otherwise noninvasive U87 cells promotes aggressive tumor growth characterized by invasion, angiogenesis, and necrosis, hallmarks of human GBMs. Coincident with the presence of unedited miR-376a* in GBMs, the data support a major pro-invasive role for this miRNA.
RAP2A is a direct target of unedited miR-376a*, whereas AMFR is a direct target of edited miR-376a*.
To understand the mechanism by which unedited miR-376a* promotes glioma cell migration and invasion, we performed microarray analysis to identify genes downregulated by the miRNA. miR-376a*A and 376a*G were separately transfected in U87 and SW1783 cells, and transcripts downregulated by each of them at an early time point (P < 0.05; fold-change >1.5 at 24 hours) were identified. After removing commonly downregulated transcripts, we isolated 116 potential candidate target genes for miR-376a*A and 140 genes for miR-376a*G (Supplemental Figure 7A and Supplemental Tables 2 and 3). We then used RNA22, a program that allows flexibility of inputting custom miRNA sequences necessary to distinguish miR-376a*A and miR-376a*G, to identify genes with predicted binding sites for the two miRNAs in their 3′ UTRs. To prioritize from among several potential target genes, we narrowed down candidates with known roles in cell migration and invasion, in light of the observed cellular effects of miR-376a* on migration and invasion (Supplemental Tables 2 and 3). RAP2A (member of the RAS oncogene family) was identified as a potential unique target of miR-376a*A, since 6 binding sites for miR-376a*A were present in RAP2A 3′ UTR that failed to be predicted as miR-376a*G–binding sites, although 4 independent sites for miR-376a*G were also predicted (Figure 7A). Conservation of 4 of 6 miR-376a*A target sites was found in mammals (Supplemental Figure 7B). Next, we narrowed down autocrine motility factor receptor (AMFR) as a potential unique target of miR-376a*G but “untargetable” to miR-376a*A (Supplemental Table 3). The 3′ UTR of AMFR harbors two predicted conserved binding sites for miR-376a*G, while none were predicted for miR-376a*A (Figure 7E and Supplemental Figure 7C).
Figure 7. Unedited miR-376a* targets RAP2A, whereas edited miR-376a* targets AMFR.

(A) RAP2A 3′ UTR with predicted miR-376a*A–binding sites (red bars) and miR-376a*G–binding sites (green bars). Sequence of binding site 3 is shown aligned to miR-376a*A, and seed region is underlined. RAP2A 3′ UTR includes all predicted miR-376a*–binding sites, RAP2A 3′ UTR-1,154 includes all miR-376a*A–binding sites; excluding two miR-376a*G sites downstream; while RAP2A 3′ UTR-426 only includes the two downstream miR-376a*G sites. (B) Luciferase reporter assays in HeLa cells transfected with miR-376a* or control miRNA and indicated RAP2A 3′ UTR constructs as described in A (n = 4). Luciferase readings were normalized to β-galactosidase activity for each construct. (C) Quantification of RAP2A expression by qRT-PCR in transfected glioma cells (n = 3). 18S rRNA was used for normalization. (D) Immunoblotting of RAP2A protein in transfected glioma cells. β-Actin was used as loading control. Densitometric analysis was done by ImageJ software. (E) AMFR 3′UTR and reporter constructs with predicted miR-376a*G–binding sites (green bars). AMFR 3′ UTR-417 represents the partial AMFR 3′ UTR. (F) Luciferase reporter assay in HeLa cells transfected with miR-376a* or control miRNA and indicated AMFR 3′ UTR constructs described in A (n = 4). (G) qRT-PCR of AMFR mRNA in transfected glioma cells (n = 3). 18S rRNA was used for normalization. (H) Immunoblotting for AMFR protein levels in transfected glioma cells. β-Actin was used as loading control. Values above blots indicate fold-variation of protein expression, relative to cells treated with control, calculated by densitometric analysis using Image J and normalization of values with β-actin. Error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 by t test.
To validate the specificity of miR-376a*A targeting for RAP2A, we constructed 3 luciferase reporter vectors, one containing a 2,475-bp RAP2A 3′ UTR including all predicted miR-376a* sites; another containing a 1,154-bp 3′ UTR encompassing 6 miR-376a*A sites but excluding two downstream miR-376a*G sites; and a third 426-bp region that contained only two downstream predicted miR-376a*G sites (Figure 7A). As tested in HeLa cells, miR-376a*A was able to reduce luciferase activity by 30% and 70% for the reporter constructs containing predicted miR-376a*A sites (Figure 7B), suggesting that the predicted miR-376a*A binding sites in the RAP2A 3′ UTR are functional. Meanwhile, miR-376a*G/I failed to suppress luciferase activities of the 3′ UTR constructs containing predicted miR-376a*G sites, including the 426-bp 3′ UTR region, which exclusively encompassed predicted miR-376a*G sites (Figure 7B). This suggests that although predicted by bioinformatics, the miR-376a*G sites within the RAP2A 3′ UTR are nonfunctional. We speculate that the 2,475-bp RAP2A 3′ UTR, which is full-length and a native structure, possesses features of secondary structure or other determinants of miRNA targeting that allow miR-376a*A to target the 3′ UTR more efficiently than the 1,145-bp RAP2A 3′ UTR does, leading to enhanced suppression of luciferase activity (70% compared with 30%). We further observed that miR-376a*A, but not miR-376a*G and miR-376a*I, downregulated the expression of the RAP2A mRNA and RAP2A protein in U87 and SW1783 cells (Figure 7, C and D).
We then validated specific targeting of AMFR by miR-376a*G using luciferase reporter vectors fused to the full-length or partial AMFR 3′ UTR (Figure 7E) and observed luciferase activity suppressed by 50% in HeLa cells (Figure 7F). Accordingly, miR-376a*G and miR-376a*I, but not miR-376a*A, downregulated AMFR mRNA and protein levels in U87 and SW1783 cells (Figure 7, G and H).
Downregulation of RAP2A expression and upregulation of AMFR expression promote in vitro migration and invasion of glioma cells.
To investigate the functions of RAP2A and AMFR in glioma development in the context of miR-376a*, we performed a set of in vitro experiments in U87 and SW1783 glioma cells. We first inhibited RAP2A expression by two specific RAP2A siRNAs. Western blot analysis confirmed RAP2A knockdown by both siRNAs (Figure 8A). Importantly, the biological effects of RAP2A knockdown phenocopied the effects of unedited miR-376a* and resulted in a significant increase in invasion and migration of the glioma cells (Figure 8, B and C), suggesting that RAP2A functions as a suppressor of glioma cell invasion.
Figure 8. Effects of manipulating RAP2A expression on migration and invasion of glioma cells.

(A–C) Knockdown of RAP2A by two siRNAs (2.5 nM) validated by Western blotting in A promotes glioma cell invasion (B) and migration (C). In the invasion assays, seeded cells that invaded Matrigel were quantified after 24 hours (n = 4), and values are normalized to control for each cell line. Graph shows results of wound healing migration assay as wound gap closure relative to control (n = 3). Original magnification, ×100. (D) Immunoblotting of RAP2A in U87 and SW1783 cells co-transfected with expression vectors containing a full-length RAP2A cDNA (RAP2A 3′ UTR) or RAP2A without 3′ UTR (RAP2A ORF) and miR-376a* or control miRNA. β-Actin was used as loading control. Densitometric analysis was done by Image J software, and values were normalized to control for each construct. (E) Matrigel invasion assay of U87 and SW1783 glioma cells co-transfected with RAP2A expression vectors and miR-376a* or control miRNA. Number of invading cells was quantified after 24 hours (n = 4). Values are normalized to control for each expression construct. Error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 by t test.
In order to demonstrate that enhanced invasion of glioma cells by miR-376a*A observed in our previous experiments was due to RAP2A targeting by the miRNA through its direct interaction with the RAP2A 3′ UTR, and not indirectly caused by secondary modulation of RAP2A expression, we performed an experiment to investigate whether RAP2A expression could rescue the effects of miR-376a*A on cell invasion. Two overexpression vectors for RAP2A were used: one with the full-length RAP2A cDNA containing both the RAP2A ORF and 3′ UTR (named RAP2A 3′ UTR) and one containing the RAP2A ORF only, without the 3′ UTR (named RAP2A ORF). The constructs were co-transfected with miR-376a*A, miR-376a*G, or control miRNA into U87 and SW1783 glioma cells. Western blot analysis confirmed that the RAP2A protein was subject to downregulation by miR-376a*A only when RAP2A mRNA containing its 3′ UTR was expressed, whereas negligible downregulation of RAP2A occurred when RAP2A mRNA did not include its 3′ UTR (Figure 8D). When invasion assays were performed in U87 and SW1783 glioma cells, the invasion-enhancing effects of miR-376a*A were not compromised in the presence of the RAP2A 3′ UTR, while concomitant overexpression of the RAP2A ORF robustly abrogated the enhanced cell invasiveness induced by miR-376a*A (Figure 8E). Our findings indicate that RAP2A is an immediate downstream effector specific to miR-376a*A and likely the major target gene through which the invasion-enhancing effects are mediated, as overexpression of the RAP2A ORF, which is not subject to regulation by miR-376a*A due to the lack of 3′ UTR, was sufficient to restore the invasive capacity of glioma cells to baseline levels, even in the presence of miR-376a*A (Figure 8E). Thus, RAP2A expression is specifically repressed by unedited miR-376a*, and the repression is essential for miR-376a*A–induced invasiveness.
We also used two specific siRNAs to knock down AMFR expression (Figure 9A) and observed that the knockdown suppressed the invasion and migration of U87 and SW1783 cells to a level nearly half that of control siRNA-transfected cells (Figure 9, B and C). Thus, effects of AMFR knockdown on glioma cells phenocopied the effect of 376a*G, the edited form of miR-376a*, suggesting that blocking the migration and invasion-promoting effect of AMFR by miR-376a*G accounts for its anti-migratory property.
Figure 9. Effects of manipulating AMFR expression on migration and invasion of glioma cells.

(A–C) Silencing of AMFR by two siRNAs (2.5 nM, A) decreased glioma cell invasion in a Matrigel invasion assay (B) and cell migration in a wound healing assay (C). In the invasion assays, the number of seeded cells that invaded Matrigel was quantified after 24 hours (n = 4) and values are normalized to control for each cell line. For the wound healing migration assay, graph shows wound gap closure relative to control (n = 3). Original magnification, ×100. (D) Immunoblotting of AMFR in U87 and SW1783 cells co-transfected with expression vectors containing a full-length AMFR cDNA (AMFR 3′ UTR) or AMFR without 3′ UTR (AMFR ORF) and miR-376a* or control miRNA. β-Actin was used as loading control. Densitometric analysis was done by ImageJ software, and values are normalized to control for each construct. (E) Matrigel invasion assay of U87 and SW1783 glioma cells co-transfected with AMFR expression vectors and miR-376a* or control miRNA. Number of invading cells was quantified after 24 hours (n = 4). Values are normalized to control for each expression construct. Error bars indicate SD. *P < 0.05, **P < 0.01, ***P < 0.001 by t test.
Overexpression of AMFR concomitantly with miR-376a*G was then performed in glioma cells to investigate whether the invasion-suppressing effects of miR-376a*G could be rescued. Two AMFR expression vectors, one with the full-length AMFR cDNA (named AMFR 3′ UTR) and another containing the AMFR ORF only, without 3′ UTR (named AMFR ORF), were co-transfected with miR-376a*A, miR-376a*G, or control miRNA into U87 and SW1783 glioma cells. As assessed by Western blot analysis, AMFR as a direct target of edited miR-376a* was confirmed, since the major downregulation of AMFR was indeed mediated through its 3′ UTR (Figure 9D). In the presence of AMFR 3′ UTR, miR-376a*G continued to robustly suppress the invasiveness in both U87 and SW1783 glioma cells (Figure 9E). Concomitant overexpression of the AMFR ORF partially but significantly abrogated the reduction in cell invasiveness induced by miR-376a*G (Figure 9E). As overexpression of AMFR was not very pronounced in U87 and especially SW1783 cells (Figure 9D), the partial restoration of the phenotype could be due to continued suppression of endogenous AMFR by miR-376a*G.
Downregulation of RAP2A and upregulation of AMFR in clinical specimens and animal tumor models indicate their roles in GBM development.
To further investigate the functions of the RAP2A and AMFR genes in GBM development related to attenuated A-to-I editing of miR-376a*, we quantified AMFR and RAP2A mRNA levels in the panel of human glioma samples and the tumor samples collected from the in vivo animal models used in the above experiments. In primary tumor samples, there was an inverse correlation between the expression of miR-376a*A (derived as a function of editing frequency and expression of total miR-376a*) and RAP2A mRNA levels (Figure 10A). Furthermore, there was a positive correlation between AMFR mRNA and expression of unedited miR-376a* (Figure 10B), suggesting that as miR-376a*A accumulates in tumors (at the expense of miR-376a*G, a regulator of AMFR), it allows the accumulation of AMFR mRNA.
Figure 10. Expression of RAP2A and AMFR mRNAs in clinical specimens and animal tumor models.

(A) Correlation of RAP2A mRNA and unedited miR-376a* levels in primary tumors (n = 22). (B) Correlation of AMFR mRNA and unedited miR-376a* levels in primary tumors (n = 21). For A and B, expression of miR-376a*A was determined by factoring the editing frequency with expression of total miR-376a* measured by qRT-PCR. Pearson’s correlation coefficient is indicated. (C) Expression of AMFR and RAP2A mRNA in xenograft tumors formed by U87 cells stably expressing miR-376a* (see Figure 6A). Values are relative to gene expression in U87/376a*G. (D) Relative expression of AMFR and RAP2A mRNA in U87 and ELM-selected 2M1 cells (n = 3). For C and D, error bars indicate SD. *P < 0.05, **P < 0.01 by t test. (E) Immunoblotting of RAP2A and AMFR in U87 and ELM 2M1 cells. β-Actin was used as loading control. (F) RAP2A expression in GBMs is significantly lower compared with normal brain tissue (NB), astrocytomas (AA), oligodendrogliomas (OAG), and mixed tumor types (MIX) in the REMBRANDT database. Data represent median expression. ***P < 0.001 compared with NB by t test. (G) Kaplan-Meier survival curves for RAP2A expression from REMBRANDT (n = 142 for high and n = 148 for low) and Gravendeel (n = 134 for high and n = 138 for low) glioma patient data. Two Affymetrix probesets corresponding to RAP2A were analyzed, and P value was determined by log-rank test.
In xenograft tumors formed by U87 cells stably expressing miR-376a*A or -376a*G, AMFR was relatively lower in U87/376a*G tumors, and RAP2A, as the target of miR-376a*A, was lower in U87/376a*A tumors, confirming the specificity of targeting of the two miRNAs in vivo (Figure 10C). In ELM-derived 2M1 cells with the increased expression of miR-376a*A (Figure 3), expression levels of RAP2A mRNA and protein were lower, while the levels of AMFR mRNA and protein were higher (Figure 10, D and E).
When the expression analysis of RAP2A was extended to publicly available cancer databases, RAP2A mRNA was found downregulated, particularly in GBMs, from the analysis of a cohort of human gliomas catalogued in the REMBRANDT database (ref. 20 and Figure 10F). Significantly, in line with the invasion-promoting effect of RAP2A depletion, survival of patients with low expression of RAP2A mRNA was found to be significantly reduced from analysis of both the REMBRANDT (577 gliomas) and Gravendeel (276 gliomas) databases (20, 21), where information on patient clinical outcome is also available (Figure 10G). As accumulation of unedited miR-376a* is correlated with increased tumor invasiveness, the cumulative data suggest that the reduction of RAP2A mRNA due to targeting by unedited miR-376a* leads to shortened patient survival as a result of increased tumor cell invasiveness and aggressive growth.
The data gathered from RAP2A and AMFR studies suggest that attenuated A-to-I editing in glioma may exert a dual effect: increased inhibition to the migration-restraining effect of RAP2A by the accumulation of unedited miR-376a* (miR-376a*A), coupled with failure in blocking AMFR function of promoting cell migration due to the decrease of edited miR-376a* (miR-376a*I/G) level, as summarized in Figure 11. These effects may collectively contribute to the migratory and invasive properties of GBMs.
Figure 11. Roles of attenuated miR-376a*editing in regulation of glioma cell invasion and migration.

Our findings indicate opposite roles of AMFR and RAP2A, which are subject to epigenetic regulation by miR-376a*, in regulating cell invasion. When the A-to-I editing of miR-376a* is attenuated in glioblastoma, the accumulation of unedited miR-376a* (miR-376a*A) downregulates RAP2A, while a decreased level of edited miR-376a* (miR-376a*I/G) leads to upregulation of AMFR, collectively resulting in increased migration and invasiveness of glioma cells.
Discussion
In this study we sought to establish a link between epigenetic regulation of miRNAs via A-to-I editing and glioma invasiveness. Under normal circumstances, A-to-I editing of primary miRNAs usually negatively modulates Drosha or Dicer processing (10, 22, 23), suppressing mature miRNA abundance, but in exceptional cases it can lead to the generation of edited mature miRNAs, as is the case for miR-376 cluster miRNAs (2). Mature miRNAs from this cluster are found to be edited from 50% to 100% in studies of the whole brain and to nearly 100% in astrocytes. Another such rare substrate identified in the brain with extensive A-to-I modification (>99.9%) is the precursor messenger RNA (pre-mRNA) of the glutamate receptor B subunit ion channel (GluR-B), in which the glutamine (Q) codon CAG is changed to a CIG to specify arginine (R) at position 586 of GluR-B (24). The GluR-B pre-mRNA editing controls the Ca2+ permeability of the ion channel and is essential for brain function (25, 26). The high-level editing of miR-376 cluster miRNAs is reminiscent of the close to 100% recoding of GluR-B at the Q/R site, implying an important role for editing-mediated recoding of these miRNAs in normal brain cells and, consequently, a higher likelihood of a disruptive impact of its aberrant editing in the brain. In agreement with previous findings, we uncovered a widespread reduction in ADAR and ADARB1 mRNA levels in gliomas, especially GBMs (3, 4). As the substrates of ADARs are numerous, ADAR dysfunction will undoubtedly have pleiotropic effects. Notably, however, even GluR-B, the most well-studied target of A-to-I editing, showed a modest loss of editing, from 100% to 69%–88% in GBMs (3), in contrast to the nearly complete abolishment of editing for miR-376a* in GBMs, as we have reported here. Because a common editing machinery, comprising ADAR and ADARB1, is responsible for all A-to-I RNA editing events, the disparities in editing loss are likely due to the differential sensitivity of each site to attenuated enzyme function, stemming from variable editing efficiencies of ADARs for different sites depending on RNA sequence and structure (27) or due to the involvement of other factors that regulate ADAR activity for each site (28). Our observations of disparate changes in editing levels of substrates and the near-normal editing levels observed at sites corresponding to miR-376a, -376b, and -376c are in agreement with a recent study in which deep sequencing of miRNAs from a panel of 6 GBM samples was conducted and no change was found in the average abundance of the edited and unedited transcripts compared with normal brain tissue for miR-376a, -376b, and -376c (29).
Changes in transcript editing levels due to ADAR dysfunction most likely play a role in progression rather than initiation of malignant growth, given that mouse models of ADAR dysfunction do not have increased cancer incidence (30). The aggregation of unedited miR-376a* initiated by loss of A-to-I editing in GBMs may constitute of the lesions conferring considerable malignant advantage during cancer progression, accounting for its exceptionally low editing levels in GBMs compared with other editing substrates, including miRNAs from the miR-376 cluster. A role for unedited miR-376a* in malignant progression was further supported by its specific enrichment in ELM-selected invasive U87 cells. Mechanistically, this specific accumulation is likely due to a post-transcriptional selective bias for miR-376a*, in the form of miRNA duplex strand selection (31), independent of a change in the abundance of mature miR-376a encoded in the same precursor as miR-376a*. The observation that even in various normal tissues the ratios of miR to miR* strands are widely different is in line with differential miRNA strand selection due to the action of specific regulatory factors (31). Although, a role for miR-376a* in invasion promotion in GBMs or any other cancer has previously not been demonstrated, a recent in vitro study showed that the expression of precursor miR-376a (encoding both miR-376a and miR-376a*) or the whole miR-376 cluster promoted the migration and invasion of otherwise non-migratory tumor cells derived from in vitro transformation of non-human primate kidney cells (32). It would be interesting to delineate whether unedited miR-376a* from the pre-miR-376a is responsible for the enhanced migratory phenotype. In GBMs, glutamate receptors assembled from the unedited (Q-containing) GluR-B subunit are highly Ca2+ permeable and can promote glioma cell invasion (7), suggesting a general role of unedited substrates in promoting invasiveness. Notably, general low expression of ADAR enzymes in GBMs will affect editing of several RNAs, including miRNAs, and other as-yet-unidentified targets of ADAR dysfunction may potentially and perhaps cooperatively regulate glioma invasiveness and growth.
The presence of unedited and edited variants of miR-376a* in disease and normal states, respectively, suggests that each form establishes a distinct cell type–specific transcriptome. Our findings attribute the pro-invasive nature of unedited miR-376a* to its aberrantly gained ability to target RAP2A and its concomitant inability to target AMFR in glioma cells due to single-base alteration in its seed sequence. We identified RAP2A as a specific target of unedited miR-376a* and found that suppression of RAP2A function has a pro-invasive effect. RAP2A belongs to the Ras family of GTP-binding proteins that regulate proliferation, differentiation, and cytoskeletal rearrangements (33). RAP2A is involved in the regulation of dendrite and axonal branches and restrains dendritic outgrowth of neurons (34, 35). It has been shown that RAP2 regulates the actin cytoskeleton via two effectors, PARG1 and TNIK (36, 37). In HEK293 cells, TNIK induces actin fiber disassembly and perturbs cell spreading, a function that is promoted by RAP2A (37). A similar regulatory role for RAP2A in cancer cell migration, dependent on actin cytoskeleton remodeling, is likely and is suggested by the current work, which demonstrates in vitro that RAP2A knockdown promotes both migration and invasion of glioma cells. Our finding that unedited miR-376a* directly suppresses the RAP2A 3′ UTR reveals a link between altered editing and accumulation of unedited miRNA and identifies a key regulator of glioma cell invasion. This gene is likely the primary target of unedited miR-376a*, as relieving its regulation by miR-376a* nearly completely restored invasion of glioma cells to baseline levels. Significantly, the strong association of reduced RAP2A expression with decreased patient survival uncovers RAP2A as a novel regulator of glioma cell invasion and associated aggressiveness of gliomas, especially GBMs. Other target genes, including those identified by microarray analysis and harboring target sites, may simultaneously be under regulation of unedited miR-376a* and may contribute to aggressiveness of GBMs. Possible candidate genes with roles in GBM metabolism, invasion, or cytoskeleton include MDH1, STAT3, and VEZT and will be the subject of future studies.
Due to the change in miRNA sequence by altered editing, target genes that would normally be accessible to the edited miR-376a* become “untargetable” in GBMs, further contributing to disruption of miRNA function. One such target we identified is AMFR, a target specific to edited miR-376a* in gliomas. A mechanistic role for AMFR in promotion of glioma cell migration and invasion has not yet been demonstrated. Nonetheless, in the normal brain, there is no expression of AMFR or AMF in glial cells, while abundant mRNA levels of both are found in majority of grade III astrocytomas and GBMs (38). The observation of AMFR expression in pseudopalisading cells in necrotic regions suggests that actively migrating glioma cells express AMFR (38, 39). Significantly, we demonstrated that knockdown of AMFR efficiently inhibited migration and invasion of glioma cells in vitro. Increased expression of AMFR due to the abolishment of edited miR-376a* function caused by loss of editing could additionally be responsible for promotion of glioma migration. As miRNAs have multi-gene targeting ability, the partial rescue of decreased invasion by edited miR-376a* by overexpression of AMFR can potentially be accounted for by the continued suppression of other target genes in the presence of edited miR-376a*. Nonetheless, it was still confirmed in this study that AMFR is a direct target of unedited miR-376a*, as the downregulation of AMFR by this miRNA is indeed mediated primarily through its 3′ UTR. Cumulative effects of unedited miR-376a* on glioma invasion may, however, be the result of repression of more than one target gene.
In summary, we have uncovered a novel function of miRNA dysregulation in GBMs and demonstrated, among other genetic and post-transcriptional sequence variations of human miRNAs, a cancer-specific mature miRNA variation due to attenuated A-to-I RNA editing as a molecular mechanism underlying the migration and invasion of glioma cells. Thus, our findings have extended the spectrum of aberrations characterizing miRNAs in cancer. The unedited miR-376a* represents a promising therapeutic target for GBMs, as does the manipulation of the editing machinery to restore normal editing frequencies of multiple editing substrates. The identification of additional edited RNA molecules, including miRNAs, that can regulate features of GBM growth in a sequence-dependent manner will further the understanding of the role of A-to-I editing under normal conditions and the consequent impact of its disruption in cancer.
Methods
Tumor tissues and cells.
Twenty-four human brain tumor samples were used in this study. Seventeen sampled were obtained from the National Neuroscience Institute, Singapore. The remaining samples (GBM 1–4 and AA 17, 19, 20) were purchased (Asterand and Capital Biosciences). Five human brain total RNA samples were purchased from Ambion, Clontech, and Stratagene to serve as normal brain tissue controls. Human glioma cell lines U87, U251, SW1088, and SW1783 and human cervical cancer cell line HeLa were from ATCC. Frozen normal human astrocyte pellet was purchased from Cell Systems.
miRNA editing analysis.
Editing analysis of pri-miRs was performed using a previously described method (2). From the sequencing chromatogram, frequency of A-to-I editing was determined as the percent G peak over the sum of A and G peaks using Vector NTI software package (Invitrogen) for sequence analysis. For editing analysis of mature miRNAs, small RNA fraction was enriched from total RNA using miRacULS miRNA isolation kit (Kreatech). Small RNA cDNA library preparation by poly(A) tailing, adapter ligation, cDNA synthesis, PCR, and TOPO TA cloning of mature miRNAs was performed as previously described (2). For each mature miRNA from the miR-376 cluster, >50 cDNA clones were sequenced, and the frequency of editing was determined as the percentage of clones with G at the editing site.
Intracranial brain tumor xenografts.
Adult male and female athymic BALB/c nude mice (aged 6–8 weeks) were used to generate the human xenograft tumor model. Glioma cells (0.1 × 106) were injected into the right side of the striatum of anesthetized mice, and animal survival was monitored. For histological analysis, animals were sacrificed on day 21 after tumor inoculation by cardiac perfusion with PBS, followed by 4% paraformaldehyde in PBS. Harvested mouse brains were cryosectioned for H&E staining or immunostaining.
Selection of invasive U87 cells by ELM assay.
The ELM assay was performed on U87 glioma cells using a previously reported method (13). Briefly, 1 × 106 U87 cells in 200 μl PBS were injected intravenously through tail vein of nude mice. After 3–4 months, when signs of moribundity appeared, animals were euthanized and lung lesions were collected. Propagation of lesions was done subcutaneously in vivo for 4 weeks. Establishment of primary ELM-selected cell lines from subcutaneous tumors was done as previously described (13). Three ELM lines were established and cultured in complete DMEM medium. Early passages (5–7 passages after first plating in vitro) were used for RNA isolation, intracranial injection, and transfection. Microarray data described herein have been deposited in the NCBI Gene Expression Omnibus ( http://www.ncbi.nlm.nih.gov/geo/), with accession number GSE34482, in a MIAME-compliant format.
Statistics.
Data are presented as mean ± SD unless otherwise indicated. Statistical significance was determined by paired or unpaired 2-tailed Student’s t test, and P less than 0.05 was considered to be statistically significant. Correlations were performed using Pearson’s correlation analysis. Survival analysis of tumor-bearing mice was carried out using the log-rank test in SigmaStat 3.5.
Study approval.
All animal studies were approved by the Institutional Animal Care and Use Committee, Biological Resource Centre, Singapore, and conducted under protocols 080312 and 110612. Patient tumors were obtained following receipt of written informed consent in accordance with study protocols CIRB 2008/802/A and CIRB 2008/803/A approved by the Institutional Review Board of National Neuroscience Institute and Singapore General Hospital, Singapore.
Supplemental methods.
Details of methods for RNA isolation, transfection, migration and invasion assays, cell viability, proliferation, and cell cycle assays, luciferase reporter assay, immunohistochemistry, Western blotting, microarray, bioinformatics analyses, survival analysis in public glioma datasets, RT-PCR, and oligonucleotide sequences are provided in the Supplemental material.
Supplementary Material
Acknowledgments
This work was supported by the Institute of Bioengineering and Nanotechnology (Biomedical Research Council, Agency for Science, Technology and Research, Singapore), grants from the National Medical Research Council in Singapore (NMRC/1203/2009 and NMRC/IRG10Nov122) to S. Wang, and institutional core funding provided to B.-T. Ang from the Biomedical Research Council, Agency for Science, Technology and Research, Singapore.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):4059–4076. doi:10.1172/JCI62925.
See the related Commentary beginning on page 3842.
References
- 1.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136(2):215–233. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kawahara Y, Zinshteyn B, Sethupathy P, Iizasa H, Hatzigeorgiou AG, Nishikura K. Redirection of silencing targets by adenosine-to-inosine editing of miRNAs. Science. 2007;315(5815):1137–1140. doi: 10.1126/science.1138050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Maas S, Patt S, Schrey M, Rich A. Underediting of glutamate receptor GluR-B mRNA in malignant gliomas. Proc Natl Acad Sci U S A. 2001;98(25):14687–14692. doi: 10.1073/pnas.251531398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paz N, et al. Altered adenosine-to-inosine RNA editing in human cancer. Genome Res. 2007;17(11):1586–1595. doi: 10.1101/gr.6493107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cenci C, et al. Down-regulation of RNA editing in pediatric astrocytomas: ADAR2 editing activity inhibits cell migration and proliferation. J Biol Chem. 2008;283(11):7251–7260. doi: 10.1074/jbc.M708316200. [DOI] [PubMed] [Google Scholar]
- 6.Gallo A, Galardi S. A-to-I RNA editing and cancer: from pathology to basic science. RNA Biol. 2008;5(3):135–139. doi: 10.4161/rna.5.3.6739. [DOI] [PubMed] [Google Scholar]
- 7.Ishiuchi S, et al. Blockage of Ca(2+)-permeable AMPA receptors suppresses migration and induces apoptosis in human glioblastoma cells. Nat Med. 2002;8(9):971–978. doi: 10.1038/nm746. [DOI] [PubMed] [Google Scholar]
- 8. Cancer Genome Atlas Research Network. Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature. 2008. 455 7216 1061 1068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ovaska K, et al. Large-scale data integration framework provides a comprehensive view on glioblastoma multiforme. Genome Med. 2010;2(9):65. doi: 10.1186/gm186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kawahara Y, et al. Frequency and fate of microRNA editing in human brain. Nucleic Acids Res. 2008;36(16):5270–5280. doi: 10.1093/nar/gkn479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Furnari FB, et al. Malignant astrocytic glioma: genetics, biology, and paths to treatment. Genes Dev. 2007;21(21):2683–2710. doi: 10.1101/gad.1596707. [DOI] [PubMed] [Google Scholar]
- 12.Clark EA, Golub TR, Lander ES, Hynes RO. Genomic analysis of metastasis reveals an essential role for RhoC. Nature. 2000;406(6795):532–535. doi: 10.1038/35020106. [DOI] [PubMed] [Google Scholar]
- 13.Xie Q, et al. A highly invasive human glioblastoma pre-clinical model for testing therapeutics. J Transl Med. 2008;6:77. doi: 10.1186/1479-5876-6-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Gladson CL, Prayson RA, Liu WM. The pathobiology of glioma tumors. Annu Rev Pathol. 2010;5:33–50. doi: 10.1146/annurev-pathol-121808-102109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Radaelli E, et al. Immunohistopathological and neuroimaging characterization of murine orthotopic xenograft models of glioblastoma multiforme recapitulating the most salient features of human disease. Histol Histopathol. 2009;24(7):879–891. doi: 10.14670/HH-24.879. [DOI] [PubMed] [Google Scholar]
- 16.Giese A, Bjerkvig R, Berens ME, Westphal M. Cost of migration: invasion of malignant gliomas and implications for treatment. J Clin Oncol. 2003;21(8):1624–1636. doi: 10.1200/JCO.2003.05.063. [DOI] [PubMed] [Google Scholar]
- 17.Hoelzinger DB, Demuth T, Berens ME. Autocrine factors that sustain glioma invasion and paracrine biology in the brain microenvironment. J Natl Cancer Inst. 2007;99(21):1583–1593. doi: 10.1093/jnci/djm187. [DOI] [PubMed] [Google Scholar]
- 18.Louis DN. Molecular pathology of malignant gliomas. Annu Rev Pathol. 2006;1:97–117. doi: 10.1146/annurev.pathol.1.110304.100043. [DOI] [PubMed] [Google Scholar]
- 19.Winkler F, et al. Imaging glioma cell invasion in vivo reveals mechanisms of dissemination and peritumoral angiogenesis. Glia. 2009;57(12):1306–1315. doi: 10.1002/glia.20850. [DOI] [PubMed] [Google Scholar]
- 20. REMBRANDT. National Cancer Institute web site. http://rembrandt.nci.nih.gov . Accessed September 12, 2012.
- 21.Gravendeel LA, et al. Intrinsic gene expression profiles of gliomas are a better predictor of survival than histology. Cancer Res. 2009;69(23):9065–9072. doi: 10.1158/0008-5472.CAN-09-2307. [DOI] [PubMed] [Google Scholar]
- 22.Kawahara Y, Zinshteyn B, Chendrimada TP, Shiekhattar R, Nishikura K. RNA editing of the microRNA-151 precursor blocks cleavage by the Dicer-TRBP complex. EMBO Rep. 2007;8(8):763–769. doi: 10.1038/sj.embor.7401011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang W, et al. Modulation of microRNA processing and expression through RNA editing by ADAR deaminases. Nat Struct Mol Biol. 2006;13(1):13–21. doi: 10.1038/nsmb1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Higuchi M, Single FN, Kohler M, Sommer B, Sprengel R, Seeburg PH. RNA editing of AMPA receptor subunit GluR-B: a base-paired intron-exon structure determines position and efficiency. Cell. 1993;75(7):1361–1370. doi: 10.1016/0092-8674(93)90622-W. [DOI] [PubMed] [Google Scholar]
- 25.Brusa R, et al. Early-onset epilepsy and postnatal lethality associated with an editing-deficient GluR-B allele in mice. Science. 1995;270(5242):1677–1680. doi: 10.1126/science.270.5242.1677. [DOI] [PubMed] [Google Scholar]
- 26.Higuchi M, et al. Point mutation in an AMPA receptor gene rescues lethality in mice deficient in the RNA-editing enzyme ADAR2. Nature. 2000;406(6791):78–81. doi: 10.1038/35017558. [DOI] [PubMed] [Google Scholar]
- 27.Valente L, Nishikura K. ADAR gene family and A-to-I RNA editing: diverse roles in posttranscriptional gene regulation. Prog Nucleic Acid Res Mol Biol. 2005;79:299–338. doi: 10.1016/S0079-6603(04)79006-6. [DOI] [PubMed] [Google Scholar]
- 28.Wahlstedt H, Daniel C, Enstero M, Ohman M. Large-scale mRNA sequencing determines global regulation of RNA editing during brain development. Genome Res. 2009;19(6):978–986. doi: 10.1101/gr.089409.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Skalsky RL, Cullen BR. Reduced expression of brain-enriched microRNAs in glioblastomas permits targeted regulation of a cell death gene. PLoS One. 2011;6(9):e24248. doi: 10.1371/journal.pone.0024248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Farajollahi S, Maas S. Molecular diversity through RNA editing: a balancing act. Trends Genet. 2010;26(5):221–230. doi: 10.1016/j.tig.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Siomi H, Siomi MC. Posttranscriptional regulation of microRNA biogenesis in animals. Mol Cell. 2010;38(3):323–332. doi: 10.1016/j.molcel.2010.03.013. [DOI] [PubMed] [Google Scholar]
- 32.Teferedegne B, Murata H, Quinones M, Peden K, Lewis AM. Patterns of microRNA expression in non-human primate cells correlate with neoplastic development in vitro. PLoS One. 2010;5(12):e14416. doi: 10.1371/journal.pone.0014416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bos JL. Ras-like GTPases. Biochim Biophys Acta. 1997;1333(2):M19–M31. doi: 10.1016/s0304-419x(97)00015-2. [DOI] [PubMed] [Google Scholar]
- 34.Fu Z, Lee SH, Simonetta A, Hansen J, Sheng M, Pak DT. Differential roles of Rap1 and Rap2 small GTPases in neurite retraction and synapse elimination in hippocampal spiny neurons. J Neurochem. 2007;100(1):118–131. doi: 10.1111/j.1471-4159.2006.04195.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kawabe H, et al. Regulation of Rap2A by the ubiquitin ligase Nedd4-1 controls neurite development. Neuron. 2010;65(3):358–372. doi: 10.1016/j.neuron.2010.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Myagmar BE, et al. PARG1, a protein-tyrosine phosphatase-associated RhoGAP, as a putative Rap2 effector. Biochem Biophys Res Commun. 2005;329(3):1046–1052. doi: 10.1016/j.bbrc.2005.02.069. [DOI] [PubMed] [Google Scholar]
- 37.Taira K, et al. The Traf2- and Nck-interacting kinase as a putative effector of Rap2 to regulate actin cytoskeleton. J Biol Chem. 2004;279(47):49488–49496. doi: 10.1074/jbc.M406370200. [DOI] [PubMed] [Google Scholar]
- 38.Tanizaki Y, et al. Expression of autocrine motility factor mRNA is a poor prognostic factor in high-grade astrocytoma. Pathol Int. 2006;56(9):510–515. doi: 10.1111/j.1440-1827.2006.01999.x. [DOI] [PubMed] [Google Scholar]
- 39.Brat DJ, et al. Pseudopalisades in glioblastoma are hypoxic, express extracellular matrix proteases, and are formed by an actively migrating cell population. Cancer Res. 2004;64(3):920–927. doi: 10.1158/0008-5472.CAN-03-2073. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.