

Abstract
DC-mediated NKT cell activation is critical in initiating the immune response following kidney ischemia/reperfusion injury (IRI), which mimics human acute kidney injury (AKI). Adenosine is an important antiinflammatory molecule in tissue inflammation, and adenosine 2A receptor (A2AR) agonists protect kidneys from IRI through their actions on leukocytes. In this study, we showed that mice with A2AR-deficient DCs are more susceptible to kidney IRI and are not protected from injury by A2AR agonists. In addition, administration of DCs treated ex vivo with an A2AR agonist protected the kidneys of WT mice from IRI by suppressing NKT production of IFN-γ and by regulating DC costimulatory molecules that are important for NKT cell activation. A2AR agonists had no effect on DC antigen presentation or on Tregs. We conclude that ex vivo A2AR–induced tolerized DCs suppress NKT cell activation in vivo and provide a unique and potent cell-based strategy to attenuate organ IRI.
Introduction
Kidney DCs, residing in the interstitial extracellular compartment, are professional APCs and play a critical role in initiating an early immune response against pathogens as well as maintaining immunological tolerance to self antigens. DCs are activated by danger-associated molecular patterns (DAMPS) or pathogen-associated molecular patterns (PAMPS) (1). In the presence of PAMPS or DAMPS, DCs are key initiators, potentiators, and effectors of the innate immune system, which comprises neutrophils, monocytes/macrophages, DCs, NK cells, and NKT cells, in kidney ischemia/reperfusion injury (IRI) and induce injury either directly or through inflammatory signals (2).
NKT cells represent a subset of innate-like lymphocytes that share receptor structures and functions with both conventional T cells and NK cells. NKT cells typically recognize endogenous or exogenous glycolipid antigens bound by the MHC class I–like protein CD1d on APCs. Type I NKT cells express an invariant TCR-α chain encoded by a Vα14-Jα18 rearrangement in mice, whereas the type II NKT cells possess a diverse TCR. CD1d-restricted type I NKT cells, but not type II NKT cells, recognize the strong agonist/glycolipid α-galactosylceramide (αGC), originally isolated from a marine sponge (3). A unique property of NKT cells is their capacity to rapidly produce both Th1-type (IL-2 and IFN-γ) and Th2-type (IL-4 and IL-10) cytokines upon TCR engagement.
NKT cells initiate inflammation following IRI in liver, lung, and/or kidney (4, 5). By studying NKT cell–deficient mice and by blocking DC-NKT cell interaction, we demonstrated that CD1d-restricted NKT cells are necessary for kidney IRI. NKT cells are activated rapidly after IRI, and the initial NKT cell response is followed by a secondary activation of other immune cells and by cytokine production, which crucially influences the subsequent inflammatory cascade in kidney IRI (6). Modulating NKT cell function is a strategy used in the treatment of autoimmune diseases and cancer (7, 8) and in the generation of various vaccines (9). Therefore, specifically modulating NKT cell function has broad clinical significance.
Although DCs contribute to immune activation, they also induce tolerance. Tolerogenic DCs have been used as potential therapeutic tools and represent a new and promising immunotherapeutic approach for ameliorating or preventing graft rejection or treating autoimmune disorders, cancers, and other serious conditions (10). Immature myeloid DCs that express low surface levels of MHC class II and costimulatory molecules induce T cell tolerance, whereas mature myeloid DCs, which express much higher levels of these molecules, induce T cell immunity. BM-derived DCs (BMDCs) have been rendered tolerogenic by exposure to cytokines, growth factors, or pharmacological mediators, or by genetic engineering (11, 12). Tolerogenic DCs can produce T cell death, T cell anergy, or Treg expansion and can regulate autoreactive or alloreactive T cell responses and promote or restore antigen-specific tolerance in experimental animal models (13). Tolerogenic DCs of either donor or host origin can promote transplant tolerance induction (14).
Kidney-resident DCs reside in the interstitium throughout the kidney (15, 16) in close apposition to tubular epithelial cells, endothelial cells, macrophages, and fibroblasts (17), where they can respond to changes in the local microenvironment. DC phenotype is likely determined by direct cell contact or through soluble mediators. For example, fibroblasts express an abundance of CD73 (17), the enzyme that catalyzes the final step in the breakdown of ATP/ADP to adenosine. Adenosine concentrations increase dramatically in inflamed and remodeling tissues (18, 19). Extracellular adenosine binds to adenosine receptors (ARs), the A1AR, A2AR, A2BR, and A3R (20). Activation of adenosine 2A receptor (A2AR), a Gs-coupled receptor, increases intracellular cAMP, which is a potent inhibitor of the NF-κB pathway downstream of immunoreceptors (21) and therefore may contribute to the antiinflammatory effects of A2AR agonists (4, 22). Ohta et al. showed that A2AR agonists attenuate tissue-specific and systemic inflammation (23); this antiinflammatory effect is mediated through inhibition of TLR-induced transcription of proinflammatory cytokines (24). We also showed that A2AR agonists ameliorate kidney IRI through effects on BM-derived cells (25, 26) and that NKT cell activation is necessary for the innate immune response in kidney IRI (4, 6). Our data showed that activation of A2AR expressed either on DCs or T cells suppresses the immune response in allograft rejection (27). In addition, activation of A2ARs mediates inhibition of T cell proliferation and expansion and NK cell cytotoxicity (28, 29). Even short-term exposure to adenosine is sufficient to inhibit TCR-triggered effector CD4+ and CD8+ T cell functions (30), which by demonstrating a memory effect of activation of A2AR on immune cells, provides support for the concept of prolonged effect of adoptive transfer of A2AR agonist–pretreated DCs in the current study.
In the current study, we hypothesized that A2AR agonists, e.g., ATL313, act on DCs loaded with NKT cell antigen αGC (DCs-αGC) to induce tolerance, and these tolerogenic DCs may be administered to mice to block NKT cell activation and prevent acute kidney injury (AKI). Conceptually this cell-based therapeutic approach used in the treatment of cancer and autoimmune disease as well as AKI may be useful in minimizing side effects of systemically administered drugs and in targeting specific immune cells in kidney IRI. Our data showed that these ATL313-treated antigen-pulsed DCs (DCs-αGC-ATL313) have regulatory properties and protected kidney from IRI in vivo by suppressing IFN-γ production. ATL313 had no effect on DC CD1d/glycolipid complex formation and therefore didn’t interfere with antigen presentation and NKT cell recognition. However, ATL313 downregulated positive and upregulated negative costimulatory molecule expression on DCs-αGC. IL-10 produced from systemic sources contributed to kidney protection. We found that IL-10 can be produced from spleen B220+ B cells and CD11c+ cells. Our study indicates that (a) renal interstitial DC phenotype is determined in part by the expression of A2ARs and activation by selective agonists reduces IRI and (b) ex vivo A2AR agonist–generated tolerogenic DCs provide what we believe to be a new therapeutic strategy in the prevention of AKI by suppressing NKT cell–mediated innate immune responses.
Results
Activation of A2AR expressed on CD11c+ DCs is important for kidney IRI.
We have shown that activation of A2AR on BM-derived cells protects kidneys from IRI (26). Using CD11c-DTR transgenic mice and administration of diphtheria toxin to selectively deplete DCs, we have also shown that DCs are necessary for the development of injury in the kidney following IR (31). We now show that sort-purified CD11c+ kidney DCs express all 4 subtypes of ARs (A1R, A2AR, A2BR, and A3R), but mRNA expression of these receptors was not changed after ischemia and 24 hours of reperfusion (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI63170DS1). To further examine the role of A2AR on DCs, we generated mice lacking 1 or both A2AR alleles only on CD11c+ DCs and performed subthreshold kidney IRI; conditional deletion of A2ARs using the Adora2afl/fl mice has been described (32), and genotyping of the DC A2AR KO has been confirmed (Y.J. Day and J. Linden, unpublished results). The small increase in injury (“mild injury” or subthreshold IRI, produced by a modest period of ischemia), indicated by elevated plasma creatinine in CD11c-Cre (WT) mice, was markedly increased in CD11c-CreAdora2afl/WT and CD11c-CreAdora2afl/fl mice (Figure 1), which is similar to our prior results in global Adora2a–/– mice, and suggested that absence of the A2AR-dependent protective effect of endogenous adenosine renders A2AR-deficient mice more susceptible to injury (26). In addition, the A2AR agonist ATL313 was not protective in either CD11c-CreAdora2afl/WT or CD11c-CreAdora2afl/fl mice, which is similar to the ATL313 effect on global Adora2a–/– mice (26). Thus the data indicate that DC A2ARs activated by endogenously released adenosine or selective A2AR agonists mediate tissue protection.
Figure 1. Activation of A2AR expressed on CD11c+ DCs is important for kidney IRI.

A2AR agonist ATL313 (10 ng/kg/min, osmotic mini-pump, s.c.) was administered to mice 24 hours prior to kidney IRI surgery. Following 26 minutes of kidney ischemia and 24 hours of kidney reperfusion, plasma creatinine levels were measured from vehicle- and ATL313-pretreated CD11c-Cre, CD11c-CreAdora2afl/WT, and CD11c-CreAdora2afl/fl mice. Kidney pedicles of sham-operated animals were exposed but not clamped. Values are mean ± SEM. n = 3–7. **P < 0.01.
Activation of A2AR signaling by ATL313 blocks NKT cell activation in kidney IRI.
Our previous findings showed the proinflammatory role of IFN-γ production from NKT cells in IRI (6, 33) and that targeting NKT cell suppression is an important strategy for inhibiting IRI. The tissue-protective effects of A2AR activation could involve modulation of NKT cell activation pathways. ATL146e (10 ng/kg/min, s.c.), another selective A2AR agonist with properties similar to those of ATL313, significantly decreased IFN-γ–producing NKT cell migration to the inflamed kidney following 24-hour reperfusion compared with vehicle-treated control IRI mice (Figure 2A).
Figure 2. Suppression of DC-NKT interaction in vivo by A2AR activation or by treatment of mice with A2AR-activated αGC-loaded BMDCs protects kidneys from IRI.

(A) WT mice were pretreated with A2AR agonist ATL146e (10 ng/kg/min, osmotic mini-pump, s.c.) 24 hours prior to kidney IRI surgery. IFN-γ–producing live CD45+7-AAD–CD1d-tetramer+TCRβ+ NKT cell number was measured by FACS 24 hours after sham surgery or after 28 minutes of kidney ischemia. Values are mean ± SEM. n = 3–5. (B) Pretreatment of WT mice with vehicle (saline) or the A2AR agonist ATL313 (1 ng/kg/min, osmotic mini-pump, s.c.) was initiated at the onset of adoptive transfer of BMDCs loaded with αGC (DC-αGC) or vehicle (DC); mini-pumps were removed 2 days later, and then mice were subjected to sham surgery or subthreshold (26 minutes ischemia) mild IRI. Plasma creatinine was measured 24 hours after sham or IRI surgery. Values are mean ± SEM. n = 2–4. (C and D) WT or Adora2a–/– DCs were primed ex vivo with vehicle (DC) or αGC in the presence (DC-αGC-ATL313) or absence (DC-αGC) of ATL313 (1 nM), incubated for 2.5 days, washed, and adoptively transferred to WT mice 2 days prior to kidney surgery. WT mouse kidneys were subjected to 26 minutes of subthreshold ischemia followed by 24 hours of reperfusion. (C) Plasma creatinine was measured 24 hours after sham operation or IRI. Values are mean ± SEM. n = 5–15. **P < 0.001. (D) Representative morphology (by H&E staining) of kidney outer medulla 24 hours after sham operation or IRI. Scale bar: 100 μm. Original magnification, ×2 (insets).
To investigate the effect of A2AR stimulation on DC-NKT cell interaction, BMDCs primed with the glycolipid antigen αGC (DCs-αGC) were administered to mice to activate NKT cells before subthreshold kidney IRI, a paradigm known to contribute to kidney IRI (34). ATL313 (1 ng/kg/min, s.c.) was delivered by osmotic mini-pump to WT mice at the onset of DC-αGC adoptive transfer. To restrict the effect of A2AR agonist stimulation to DC-αGC-NKT interaction prior to surgery, ATL313-loaded mini-pumps were removed after 2 days, and mice were then subjected to subthreshold (mild) injury. The increase in plasma creatinine induced in the DC-αGC control group following subthreshold injury was markedly decreased in the ATL313-treated DC-αGC group (Figure 2B). These results indicate that activation of the A2AR suppressed DC-αGC–mediated NKT cell activation in vivo, and blocking this activation protected kidneys from IRI. However, the contribution of A2AR signaling on other cells cannot be excluded, and the next set of experiments explored the direct effect of ATL313 on DC-NKT cell interactions.
DCs-αGC-ATL313 protect kidneys from IRI.
Based on the above results, we hypothesized that A2AR agonists may tolerize DCs and that protection from kidney injury may result from reduced DC-mediated NKT cell activation in the presence of tolerized DCs loaded with NKT cell agonist αGC. To isolate the A2AR effect on DCs, we treated DCs with ATL313 during priming with αGC ex vivo. WT DCs-αGC were pretreated with ATL313 (1 nM; WT DCs-αGC-ATL313) and washed extensively (to remove any residual ATL313); an optimal dose of 0.5 × 106 cells was adoptively transferred to naive WT mice 2 days prior to kidney surgery to allow sufficient time for an NKT cell response. Compared with mice that received WT DCs-αGC, administration of WT DC-αGC-ATLs significantly protected mouse kidneys from subthreshold IRI, with lower plasma creatinine levels (Figure 2C) and reduced proximal tubular cell necrosis, as revealed by H&E staining (Figure 2D). Similarly, DCs-αGC from Adora2a–/– mice (Adora2a–/– DCs-αGC) also exacerbated mild kidney IRI compared with control, but ATL313-treated Adora2a–/– DCs-αGC (Adora2a–/– DCs-αGC-ATL313) had no protective effect (Figure 2C), thus demonstrating the selective effect of ATL313. Reduced neutrophil infiltration, as indicated by FACS analysis, was also observed in mice that were protected from injury (Table 1 and Supplemental Figure 2).
Table 1.
Neutrophil numbers in kidneys from mice that received WT or Adora2a–/– DCs ± αGC ± ATL313
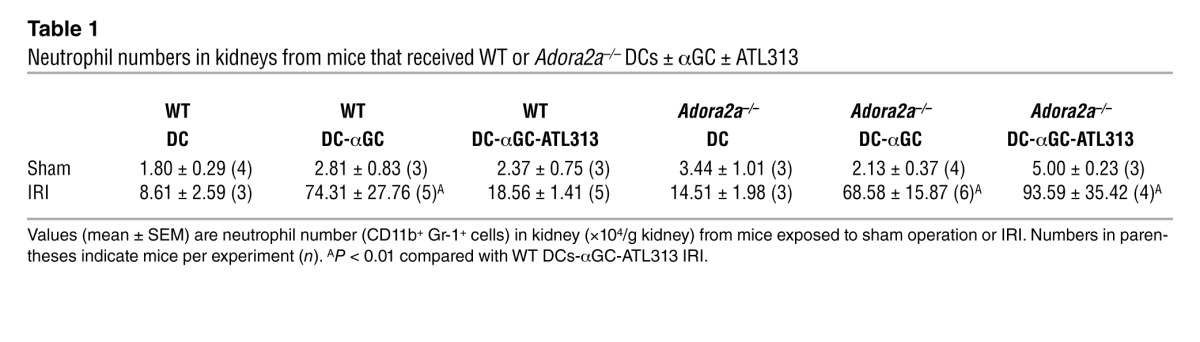
To examine the duration of efficacy of ATL313-treated DCs, particularly with regard to future clinical application, we adoptively transferred either WT or Adora2a–/– DCs-αGC-ATL313 to naive mice 7 days prior to kidney surgery. Remarkably, the protective effect of adoptively transferred DCs-αGC-ATL313 lasted for 1 week. Plasma creatinine levels were approximately 80% lower in mice that received DCs-αGC-ATL313 1 week prior to kidney IR compared with DC-αGC control (0.35 ± 0.07 vs. 2.01 ± 0.04 mg/dl, n = 3–4, P < 0.001). This protective effect was not found in Adora2a–/– DC-αGC-ATL313–treated mice (P > 0.05). These results suggest that ex vivo treatment of DCs with ATL313 results in a long-lasting change in vivo either in DC function through activation of A2ARs, which could be similar to the memory effect of A2AR signaling described in T cells (30), or in DC target cells.
The subthreshold injury experiments, in which DC-mediated NKT cell activation is revealed by exacerbation of mild ischemic injury, were designed to specifically target the protective effect of DCs-αGC-ATL313 to injury driven by mechanisms involving the DC-NKT cell-activation pathway. In the next experiments, WT DCs-αGC-ATL313 were adoptively transferred to WT mice prior to moderate kidney IR to demonstrate that DCs-αGC-ATL313 can provide protection from a longer ischemic period (28 minute ischemia/24 hour reperfusion) that alone was sufficient to produce substantial injury. In contrast to the subthreshold paradigm, the longer period of ischemia produced maximal injury; IRI alone produced a large rise in plasma creatinine that could not be further increased by administration of DCs or DCs-αGC. Kidneys of mice that received WT DCs-αGC-ATL313 (but not Adora2a–/– DCs-αGC-ATL313; NS) were significantly protected from moderate IRI compared with control mice that received either DCs-αGC or untreated DCs (Figure 3A). Therefore, we conclude that WT DCs-αGC-ATL313 are indeed ATL313-induced tolerogenic DCs.
Figure 3. DCs-αGC-ATL313 administered before or after ischemia protect kidneys from moderate IRI.

(A) The experimental process was similar to that in Figure 2, except that WT mice were subjected to 28 minutes of moderate ischemia and 24 of hours reperfusion. Plasma creatinine was measured 24 hours after sham operation or IRI. n = 2–4. (B) The experiment was similar to that in A, except that mice were subjected to renal artery clamping (instead of renal pedicle clamping) for 35 minutes; plasma creatinine was measured after 24 hours of reperfusion. n = 4 and 5 for DCs-αGC and DCs-αGC-ATL313, respectively. (C) DCs-αGC-ATL313 were administered 1 or 6 hours after moderate (28 minutes) kidney ischemia, and plasma creatinine was measured after 24 or 48 hours of reperfusion (from the end of the ischemic period). n = 3–5. *P < 0.05; **P < 0.01; ***P < 0.001. Values are mean ± SEM.
Our studies on immune mechanisms in AKI have employed a bilateral kidney IRI model involving clamping and release of both kidney pedicles (6, 16, 26). In recent years, an AKI model was established that more closely mimics human renal artery hypoperfusion during cardiac surgery (35–38). To demonstrate that our results can be recapitulated in the artery occlusion model, we successfully established a mouse renal artery IRI model. DCs-αGC or DCs-αGC-ATL313 were adoptively transferred to naive WT mice, and 2 days later, the mice were subjected to renal artery IRI surgery followed by 24-hour reperfusion. We found that DCs-αGC-ATL313 were also protective in this model; the increase in plasma creatinine (Figure 3B) and kidney tubule cell necrosis (as shown by morphology; Supplemental Figure 3A) following renal artery occlusion was reduced in mice pretreated with DCs-αGC-ATL313.
Administration of DCs-αGC-ATL313 1 or 6 hours after moderate kidney IRI protected kidneys from injury.
Our results showing a lasting effect of DCs-αGC-ATL313 could be applicable in a clinical setting for pretreatment of patients to prevent AKI (e.g., in cardiac surgery); however, just as important is the need for treatment of established AKI. We found that kidneys were still protected if mice received WT DCs-αGC-ATL313 1 or 6 hours after moderate ischemia (Figure 3C). Protection was not sustained if adoptive transfer of DCs-αGC-ATL313 was delayed to 24 hours after the IRI surgery (Supplemental Figure 3B).
Together, these data provide strong support for the concept that cell-based therapy with DCs-αGC whose function has been modulated with ATL313 ex vivo can regulate NKT cell activation in vivo for a period of at least 1 week to protect kidneys from IRI, which also may be relevant to the control of prolonged graft rejection in organ transplant, and can be delayed for up to 6 hours following kidney IR. These findings provide a substantial prospect for clinically relevant AKI or kidney transplant treatment.
DCs-αGC-ATL313 reduce NKT cell responses in vivo and in vitro.
Our previous findings that activation of IL-12/IFN-γ and IL-23/IL-17 pathways promotes neutrophil infiltration in kidney IRI and NKT cell–mediated inflammation and that IL-17A production is upstream of and stimulates IFN-γ production by NKT cells and neutrophils (33) prompted us to determine whether altered plasma IFN-γ and IL-17A/F levels contribute to the protective effects of ATL313-treated DCs. DC-αGC-ATL313 administration prior to surgery prevented the increase in IFN-γ and IL-17A/F production by DC-αGC–activated NKT cells in WT mice, while Adora2a–/– DCs-αGC-ATL313 failed to suppress IFN-γ or IL-17A/F production (Figure 4, A and B). IL-4 and IL-10 could not be detected in plasma of DC-αGC-ATL313–treated mice (data not shown), indicating that DC-αGC-ATL313–mediated kidney protection from IRI does not occur through skewing of Th1- to Th2-type cytokines.
Figure 4. Adoptive transfer of DCs-αGC-ATL313 reduces NKT cell activation.

(A and B) WT or Adora2a–/– DCs, DCs-αGC, or DCs-αGC-ATL313 were adoptively transferred to WT mice, and plasma was collected 18 hours later. Plasma IFN-γ (A) and IL-17A/F (B) levels were measured by ELISA. n = 3–6. (C) WT or Adora2a–/– DCs, DCs-αGC, or DCs-αGC-ATL313 (primed and incubated for 2.5 days with vehicle or αGC ± ATL313) were cocultured with WT liver NKT cells for 5 days; then supernatant was collected and IFN-γ level was measured by ELISA. n = 3–4. *P < 0.05; **P < 0.01. Values are mean ± SEM.
We further examined the mechanisms involved in the suppressive effect of DCs-αGC-ATL313 in cocultures of DCs and NKT cells. Coculture with WT DCs-αGC produced a dramatic increase in WT NKT cell IFN-γ production; however, DCs-αGC-ATL313 failed to induce NKT cell IFN-γ production in the cocultures (Figure 4C). Similar results on cytokine production were found when αGC-loaded WT DCs or αGC-loaded B cells (A20) were cocultured with NKT hybridoma cells (DN3A4-1.2). Cocultures of NKT hybridoma cells with WT DCs-αGC-ATL313 (56.3% ± 25.7% reduction, n = 3, P < 0.05), but not Adora2a–/– DCs-αGC-ATL313 (4.6% ± 7.1% reduction, n = 3, NS), showed decreased IL-2 production compared with cocultures with WT DCs-αGC or Adora2a–/– DCs-αGC, respectively.
DCs-αGC-ATL313 do not induce NKT cell anergy.
Protection of kidneys from IRI by ATL313-treated DCs, including the observed reduction in cytokine production, could be due to NKT cell hyporesponsiveness to antigen stimulation. To test whether DCs-αGC-ATL313 mediated NKT cell anergy, we restimulated isolated splenocytes from WT mice treated with WT DCs-αGC or DCs-αGC-ATL313. A comparable increase in IFN-γ production in response to αGC restimulation was observed in splenocytes isolated from mice treated either with WT DCs-αGC or DCs-αGC-ATL313 (3.69 ± 3.97 vs. 5.32 ± 2.72, fold of control unstimulated, NS). These data indicate that DC-αGC-ATL313–mediated kidney protection does not occur through NKT cell anergy in kidney IRI.
ATL313 affects DC positive and negative costimulatory molecule expression but not CD1d/glycolipid presentation.
Reduced DC-mediated stimulation of NKT cells could reside with altered ATL313-mediated DC function. We investigated differences in cell phenotype between DCs-αGC and DCs-αGC-ATL313 by measuring the percentage of DCs expressing positive (CD80, CD86, CD40, OX40L) and negative (ICOS, B7-H1, and B7-DC) costimulatory molecules and antigen-presenting molecules (CD1d, class II [IA]) by flow cytometry. Specifically, the upregulation of CD40 and OX40L expression that was found in WT DCs-αGC, compared with WT DCs, was significantly reduced in WT DCs-αGC-ATL313; ATL313 was ineffective in suppressing CD40 expression in Adora2a–/– DCs-αGC (Figure 5A). Interestingly, we found that ATL313 upregulated negative costimulatory molecule B7-DC expressed on DCs-αGC (Figure 5A). WT DCs-αGC-ATL313 had no effect on CD80, CD86 (not shown), CD1d, IA, ICOS, and B7-H1 expression (Supplemental Figure 4).
Figure 5. Changes in costimulatory molecule (CD40, OX40L, and B7-DC) but not CD1d/glycolipid complex expression on DCs-αGC-ATL313.

(A) WT or Adora2a–/– DCs were primed with vehicle (DC) or αGC in the presence (αGC-ATL313) or absence (αGC) of ATL313 (1 nM), incubated for 2.5 days, and analyzed by FACS. Representative flow cytometry histogram of CD40, OX40L, and B7-DC from gated CD11c+ BMDCs. (B) Alexa Fluor 647–labeled L363 was used to detect surface CD1d/αGC complex by flow cytometry. Representative flow cytometry histogram of L363 from gated CD11c+ BMDCs. Histograms are representative of three to four experiments.
To examine whether the suppressive effect of ATL313 on DC-mediated NKT cell activation might be due to alterations in cellular processing and antigen presentation of the CD1d/glycolipid complex, we used an antibody (L363) that recognizes CD1d/glycolipid complex intracellularly and on the cell surface, indicative of antigen presentation (39, 40). Labeling with antibody L363, as revealed by FACS, increased significantly both intracellularly (data not shown) and on the surface of WT or Adora2a–/– DCs after priming with αGC but was not altered by ATL313 treatment of WT DCs-αGC (Figure 5B) or Adora2a–/– DCs-αGC (not shown). Furthermore, we used a B cell line transfected with the murine CD1d molecule (A20.mCD1d) to investigate αGC trafficking in antigen-presenting cells. Similar to the BMDCs, αGC loading increased L363 expression in A20.mCD1d cells and ATL313 had no suppressive effect on surface or intracellular expression (data not shown). The data suggest that ATL313 does not affect CD1d/glycolipid complex trafficking inside the cell or presentation on the cell surface.
DC-αGC-ATL313–mediated kidney protection is due to IL-10.
Most recent studies in other models have shown that Tregs play an important role in DC-mediated tolerance by secreting IL-10. To explore the contribution of Tregs to the protective effect of ATL313-treated DCs, we first measured CD4+CD25+FOXP3+ Tregs by using FACS and found that there was no difference in numbers of Tregs in kidney and spleen from WT or Adora2a–/– mice pretreated with WT DCs, DCs-αGC, or DCs-αGC-ATL313 alone or prior to kidney ischemia and 24 hours of reperfusion (data not shown). No change in kidney Foxp3 mRNA level was detected in mice that received DCs-αGC-ATL313 compared with DCs-αGC (data not shown). However, kidney Il10 mRNA expression increased 20 hours after pretreatment with DCs-αGC-ATL313 compared with the DC-αGC group (Figure 6A). To determine a causal relation between IL-10 and kidney protection induced by DCs-αGC-ATL313, we injected a blocking antibody to IL-10 (i.v.) simultaneously with DC adoptive transfer. Blocking endogenous IL-10 with anti–IL-10 mAbs significantly reversed the protective effect of DCs-αGC-ATL313 in kidney IRI (Figure 6B). In a variety of models, increased IL-10 release from tolerogenic DCs promotes tolerogenic responses and protective function, e.g., suppressing autoimmune processes or prolonging graft function (41). To determine whether DC IL-10 production contributes to kidney protection, Il10–/– DCs-αGC and Il10–/– DCs-αGC-ATL313 were adoptively transferred to WT mice. Like WT DC-αGC, Il10–/– DCs-αGC exacerbated mild kidney injury and Il10–/– DCs-αGC-ATL313 completely reversed mild kidney injury compared with Il10–/– DCs-αGC (Figure 6C). Administration of WT DCs-αGC-ATL313 to Il10–/– mice did not prevent the increase in creatinine after IRI (Figure 6D), further confirming that IL-10 is necessary for the protective effect of DCs-αGC-ATL313 and indicating that the source is systemic rather than locally produced by the injected DCs.
Figure 6. IL-10 plays a role in DC-αGC-ATL313–mediated protection from kidney IRI.

(A) Kidney RNA was isolated from naive mice 20 hours after adoptive transfer of WT DCs, DCs-αGC, or DCs-αGC-ATL313, and Il10 mRNA levels were measured by real-time PCR. Values are fold change relative to DCs. n = 3. (B) Two days before mild IRI, neutralization IL-10 mAb or IgG2a isotype control (200 μg, i.p) was administered to mice during pretreatment with DCs-αGC or DCs-αGC-ATL313. n = 4–5. (C) αGC-loaded Il10–/– DCs treated either with vehicle or ATL313 (1 nM) or WT DCs-αGC were adoptively transferred to WT mice 2 days prior to mild (26 minutes) kidney ischemia. n = 3–9. (D) PBS, WT DCs-αGC, or WT DCs-αGC-ATL313 were administered to Il10–/– mice 2 days prior to moderate IRI. n = 3 for DCs-αGC or DCs-αGC-ATL313. Plasma creatinine (B–D) was measured following 24 hours of kidney reperfusion. *P < 0.05; ***P < 0.001. Values are mean ± SEM.
Spleen cells may be a source of IL-10 in DC-αGC-ATL313–mediated attenuation of kidney IRI.
To look for a systemic source of IL-10 in DC-αGC-ATL313–mediated kidney protection, we used IL-10 GFP (X-Vert) reporter mice. Following ischemia and 24 hours of kidney reperfusion in DC-αGC-ATL313–treated mice, we found many IL-10–producing splenocytes by flow cytometry, and the majority were CD11c+ DCs and B220+ B cells (Supplemental Figure 5A), but CD4+ and CD8+ T cells were not IL-10–GFP+ (data not shown). Interestingly, CD11c+ DCs and B220+ B cells from DC-αGC-ATL313–treated mice produced more IL-10 based on MFI of IL-10 (Supplemental Figure 5B). The phenotype of kidney IL-10–producing cells and the signal mediating IL-10 production from spleen from CD11c+ DCs and B220+ B cells in DC-αGC-ATL313–induced protection from IRI will require further study. We also noted that some IL-10–GFP+ cells, although few, were detected by immunostaining in kidney outer medulla from mice that received either PBS or DCs-αGC-ATL313 (data not shown). The significance of these cells is not known and will require further study.
DCs-αGC-ATL1223 do not alter FOXP3 or surface PD-1 expression on Tregs.
Although Treg numbers did not increase in DC-αGC-ATL313–treated mice (data not shown), it is possible that altered Treg function could contribute to kidney protection. To determine whether A2AR agonist–treated DCs-αGC reduced kidney injury by enhancing Treg function, we first determined whether coculture with ATL-treated DCs-αGC enhanced FOXP3 expression in freshly isolated Tregs; another A2AR agonist, ATL1223, which has properties similar to ATL313 and antiinflammatory effects in lung transplantation (22), was used for these experiments. No difference in the mean expression level of FOXP3, the proportion of Tregs that were FOXP3 positive at the end of coculture, or expression of surface programmed death 1 (PD-1) (a member of a family of T cell regulators that suppress immune system response) was observed between Tregs incubated with untreated DCs, ATL1223-treated DCs, DCs-αGC, or DCs-αGC-ATL1223 (data not shown). We also determined the effect of DC coculture on FoxP3 induction in naive CD4+ T cells (CD4+CD25–) and found no difference among untreated DCs, ATL1223-treated DCs, DCs-αGC, or DCs-αGC-ATL1223 in their ability to induce FOXP3 in non-Tregs (data not shown).
Discussion
Prompted by our findings that A2AR agonist ATL313 reduced kidney IRI through direct effects on DCs in vivo, we demonstrated in this study that transient treatment of DCs ex vivo with ATL313 during αGC loading resulted in a tolerogenic DC phenotype that modulated DC-mediated NKT cell activation in vivo and markedly protected kidneys from IRI produced by renal pedicle or renal artery clamping. Our data showed that ATL313 induced a change in the BMDC phenotype during DC priming with αGC, a glycolipid presented by DCs to activate NKT cells. ATL313 downregulated positive costimulatory molecule expression (CD40 and OX40L) and upregulated negative costimulatory molecule B7-DC expression on DCs, but had no effect on DC antigen presentation (surface and intracellular CD1d/αGC complexes). Adoptive transfer of DCs-αGC-ATL313 substantially inhibited kidney inflammation following IRI with less NKT cell activation, IFN-γ, and IL-17A/F production and neutrophil infiltration. DCs-αGC-ATL313 did not switch NKT cell Th1-type cytokine (IFN-γ) production to Th2-type (IL-4) or cause NKT cell anergy, but DCs-αGC-ATL313 showed reduced secretion of proinflammatory cytokines IL-6 and IL-12p40 (data not shown). DC-αGC-ATL313–treated mice had enhanced kidney IL-10 mRNA levels, and systemic IL-10 was important for protection from IRI injury, although Treg numbers did not change in vivo. We found IL-10–producing cells in spleen (CD11c+ DC and B220+ B cells) and kidney. To our knowledge, this is the first study to show the significance of using an A2AR agonist in tolerizing DCs to block the innate immunity of AKI. Importantly, this protective effect persisted for 1 week after administration of DCs -αGC-ATL313 and could be seen if treatment was delayed until 6 hours after kidney IRI, thus demonstrating clinical relevance for the use of this approach for established AKI. This study provides a proof of concept of a potentially new immunotherapeutic strategy for the prevention of tissue injury and graft rejection.
AKI is caused by a variety of conditions and has serious consequences. There is a marked increase in the incidence of AKI and unacceptably high morbidity and mortality in hospitalized patients from the past 15 years, and there is an urgent need for effective therapy (42). Numerous factors contribute to the development of AKI. IRI involves a complex cascade of events, including oxidative stress, inflammation, and interactions among many cell types (43, 44). The proinflammatory responses of renal endothelial cells and infiltrated leukocytes reduce renal blood flow through vascular congestion and promote kidney proximal tubule cell injury.
DCs are a heterogeneous group of cells important in immunity or tolerance, and the idea of using tolerized DCs in cell-based therapy of cancer, autoimmune disease, and transplantation has been under investigation for the past 2 decades (45). However, most studies have focused on the induction of T cell–tolerogenic responses. Immune regulation of innate immune response via tolerogenic DCs is critically important in bridging innate and adaptive immunity and provides the foundation for use in transplant tolerance of allograft injury (46). Extending this concept, we believe that designing protocols to tolerize DCs may be useful in the prevention and even early treatment of AKI (47).
One strategy to block immune response to IRI is to adoptively transfer tolerized DCs to modulate NKT activation and thereby limit resident DC function. We and others have shown that NKT cells contribute to organ IRI (5, 6, 48, 49). Interrupting the DC-NKT cell interaction, depleting CD1d-restricted NKT cells, or using NKT cell–deficient mice (CD1d KO) protected kidney from IRI (6). We also found IFN-γ produced from NKT cells initially activates the immune response and mediates neutrophil migration into the inflamed kidney following kidney IRI. Neutrophils are the major IL-17–producing cells in kidney IRI; IL-17 upregulates cytokine and chemokine production in kidney IRI (33). Targeting of NKT cells should be an efficient pathway to blocking the initial immune response to protect the kidney from IRI.
Adenosine is a nucleoside, locally released in response to cellular stress, such as IRI and inflammation, which allows tissues to adapt to hypoxia. Hypoxic conditions can lead to inflammation and vice versa in a variety of disorders (50). Large amounts of adenosine are released by injured cells in a hypoxic environment, and HIFs are activated by hypoxia. Hypoxia, and specifically HIF, increases extracellular concentrations of adenosine by stimulating enzymatic conversion from ATP and AMP and by reducing its cellular uptake. Acting through multiple receptors, adenosine has long been known to have antiinflammatory and tissue-protective effects (21). In a series of studies, we showed that A2AR agonists protect kidney from IRI (26, 27, 51). These findings were extended in the current study, confirming that DCs are necessary for kidney injury after IR and showing that endogenous adenosine acts at DC A2ARs to protect kidneys from injury and that the protective effect of systemic administration of the A2AR agonist ATL313 is dependent on DC A2ARs. It is possible that the protective effects of adenosine acting on kidney DCs could be modulated by changes in receptor expression; however, we found by RT-PCR that A2AR, A2BR, A1R, and A3R mRNA expression on kidney CD11c+ DCs did not change after IRI. Induction of AR expression may have occurred at an earlier time point in initiation of inflammation following kidney reperfusion that was no longer evident at 24 hours. Taken together, these observations led us to explore a new therapeutic strategy to generate tolerized DCs ex vivo and adoptively transfer them to mice to block the innate immune system and prevent kidney injury following IRI. More specifically, our strategy was to suppress NKT cell activation by using NKT cell–specific antigen–loaded (αGC) DCs treated with an A2AR agonist ex vivo.
Adenosine may act on other ARs to mediate protection. Recently, Grenz et al. demonstrated an important homeostatic role of equilibrative nucleoside transporter 1 (ENT1); they showed that ENT1 regulates uptake of adenosine and that crosstalk between renal ENTs and vascular endothelial A2BR protected kidneys from ischemic injury by improving reperfusion (52). In the current study, we extended our previous findings, that A2AR expressed on BM-derived cells contributes to suppression of kidney IRI, by demonstrating a critical role for A2AR expressed on CD11c+ DCs both in kidney IRI and as a target for ex vivo modulation of DC function to block DC-mediated NKT cell activation in kidney IRI. By reducing leukocyte infiltration and preventing increases in adhesion molecule expression (53), A2AR stimulation also reduces vascular congestion, thus improving renal blood flow by mechanisms that may complement the effects described by Grenz et al. (52). Thus, by broadly acting at multiple cellular sites to restore function and dampen inflammation, adenosine’s action at different AR subtypes on distinct hematopoietic and nonhematopoietic cells could provide powerful nonredundant (additive or synergistic) homeostatic tissue protection.
Compared with mature DCs, immature DCs interact actively with T cells and direct them into a regulatory response. There are several strategies used to maintain the immature state of DCs and generate tolerogenic DCs (1). These include culturing DCs with different cytokines (e.g., IL-10, TGF-β), growth factors (GM-CSF), or immunosuppressive agents (e.g., corticosteroids, vitamin D3, cyclosporine, tacrolimus, rapamycin), modulation of the expression of costimulatory molecules, and genetic interference. Tolerized DCs may suppress immune responses through various mechanisms. Tolerance is influenced by suppressing positive costimulatory molecules (CD80, CD86, and CD40), upregulating negative costimulatory molecules (PD-1, B7-H1, and ICOS) (54), and inhibiting cytokines, such as IL-12p70 and IL-12p40 (55). The IL-12 family member IL-27 (comprising EBI3 and IL-27–p28) is critical for tolerized DCs to produce the antiinflammatory cytokine IL-10 (56). Epstein-Barr virus–induced gene 3 (EBI3) pairs with IL-12p35 to form IL-35, which is a mediator for Treg differentiation and is important in maintaining an immunosuppressive state (57). Finally, TGF-β, retinoic acid, and vitamin D3 appear to be important in peripheral tolerance through Treg differentiation and expansion (58).
Although we did not find that DCs-αGC-ATL313 promoted kidney or spleen CD4+CD25+FOXP3+ Treg numbers, we found that systemic IL-10 played an important role in suppression of kidney IRI (58). IL-10 and heme oxygenase 1 are important in prolonging skin allograft survival (59). Precise determinations of the phenotype and function of cells that contribute to systemic IL-10 will require additional study; however, IL-10–producing cells (CD11c+ DCs and B220+ B cells) were detected in spleen. Kidney IRI has been recognized as a systemic inflammatory process, and splenectomy ameliorates acute multiple organ damage in IR models (60). The role of IL-10 production from CD11c+ DCs and B220+ B cells in tissue protection mediated by ATL313-induced tolerogenic DCs will require further studies. A subset of IL-10–producing human DCs, termed DC-10, are present in vivo and are potent inducers of antigen-specific IL-10–producing Tregs (61). IL-10–differentiated DCs (DC-10) induce tolerance at least in part by inducing T effector cells to differentiate into CD4+CD25hiFOXP3+ Tregs (62). The defective generation of IL-10–induced tolerogenic DCs and iTregs may contribute to inflammatory changes in hyper-IgE syndrome (63). Exogenously administered tolerogenic DCs may change the phenotype of the kidney resident DCs and/or further promote tolerance by inducing resident DCs to become IL-10–producing tolerogenic DCs, as we observed increased IL-10 mRNA expression in DC-αGC-ATL313–treated naive WT kidneys. To examine the possibility that non–BM-derived cells can produce IL-10 to mediate tolerogenic DC-induced protection from kidney IRI, future studies using BM chimera will be needed. DCs and marginal zone B cells may coordinate together to initiate T/NKT cell responses. IL-10 produced from B220+ cells contributed to early T cell regulation (64, 65). Although increased systemic levels of Th2 cytokines IL-4 and IL-10 could not be detected in DC-αGC-ATL313–treated mice, local tissue levels of these antiinflammatory cytokines may mediate protection from kidney IRI following infusion of ATL313-induced tolerogenic DCs.
Recent studies have demonstrated that Tregs have the ability to protect the kidney from ischemic injury and inflammation (66, 67), and our findings on IL-10 would suggest that Tregs may be involved in the protective effect of tolerogenic DCs. PD-1 is a member of a family of T cell regulators that suppress immune system responses. Treg-mediated protection can be overcome by blocking PD-1 on the surface of Tregs, suggesting that Treg surface PD-1 expression is vital for their action in this model. Furthermore, PD-1–KO Tregs have reduced ability to suppress both CD4+ and CD8+ T cell activation (68). We hypothesized that DCs-αGC-ATL313 could be protecting the kidney by enhancing the functional properties of Tregs. However, DCs-αGC-ATL1223 had no effect on FOXP3 expression in Tregs or non-Treg CD4+ T cells, and A2AR agonist–treated DCs (in the presence or absence of αGC) had no effect on the expression of PD-1 on the surface of Tregs.
There are many challenges in using the approach of tolerized DCs in blocking the innate immune system, e.g., standardizing treatment strategies, as DCs represent a heterogeneous population that express different surface markers in different tissues. Our study showed that at least a partial protective effect of ATL313-induced tolerized DCs was sustained for 1 week after adoptive transfer, which could be relevant for some clinical indications, such as prophylaxis of AKI in high-risk patients. In addition, the route of delivery of tolerized DCs may affect tolerized DC homing efficiency and their immune regulatory function. We found most of the adoptively transferred DCs migrated to the lymphoid nodes, spleen, and other organs, including kidney (our unpublished observations). Other DC subtypes, such as plasmacytoid DCs, also should be considered for therapeutic use.
In summary, through step-wise ex vivo DC manipulation and administration in an in vivo kidney IRI model, our study has identified a cell-based strategy to prevent AKI and potentially allograft rejection. We have demonstrated that A2AR agonist ATL313-induced DC tolerance shows strong biological activity to prevent NKT cell–mediated innate immune activation in vitro and in vivo and further regulate immunosuppression in kidney IRI.
Methods
Mice and surgical protocol.
WT C57BL/6 mice (6 to 8 weeks old; NCI) were used, and C57BL/6 background Il10–/–, Ifng–/–, CD11c Cre, and IL-10–IRES-eGFP reporter mice (Vert-X) were purchased from The Jackson Laboratory. Adora2a–/– mice (C57BL/6 background, 6 to 7 weeks of age), generated as previously described (69), were a gift from J.-F. Chen (Boston University, Boston, Massachusetts, USA). KO mice were backcrossed with C57BL/6 background mice for at least 8 generations.
Mice were anesthetized with a mixture (i.p.) of ketamine (120 mg/kg), xylazine (12 mg/kg), and atropine (0.324 mg/kg) and were subjected to bilateral flank incisions. Both kidney pedicles were exposed and cross-clamped for 26 minutes (subthreshold) to produce mild injury (in order to evaluate exacerbation of injury) or in some experiments for 28 minutes to produce moderate injury; then clamps were released (reperfusion) for 24 hours as described before (25). Kidney pedicles were exposed but not clamped in sham-operated mice. During the surgery, mouse core temperature was maintained at 34–36°C with a heating pad; during the recovery and reperfusion period, mice were housed in a warming incubator with ambient temperature at 30–32°C.
For renal artery IRI, mice were subjected to bilateral flank incisions. On the right side, surgical thread was tied around the renal vessels, a cut was made above the suture to extract the kidney, and the incision was closed. The ureter and kidney blood vessels were secured with minimal blood loss. Kidney artery and vein were dissected under a Zeiss Stemi 2000-C Stereo Microscope (Carl Zeiss Microscopy LLC). The kidney artery was clamped for a period of 35 minutes by using a clip with a pressure of 70 g (Roboz Surgical Instrument); then the clip was released to allow the kidney to reperfuse. Control of temperature during surgery and reperfusion was the same as in the bilateral clamping procedure.
Following 24 hours of reperfusion, animals were reanesthetized, blood was obtained from the right retroorbital sinus, and kidneys were removed for various analyses. Plasma creatinine as a measure of kidney function was determined using a colorimetric assay according to the manufacturer’s protocol (Sigma-Aldrich).
The optimal dose of neutralization antibodies (200 μg/mouse for anti–IL-10; BioXCell) or their respective isotype control (IgG2a) was administrated i.p. at the onset of BMDC adoptive transfer 2 days prior to kidney surgery. Osmotic mini-pumps (Alzet Osmotic Pumps) preloaded with sterile saline vehicle, ATL146e (at a concentration calculated to deliver 10 ng/kg/min; Adenosine Therapeutics LLC), or ATL313 (1 ng/kg/min, Adenosine Therapeutics LLC) were introduced s.c. 24 hours prior to kidney surgery or at the onset of BMDC adoptive transfer, depending on the experimental design.
Generation of CD11c-Cre Adora2afl/WT and Adora2afl/fl mice.
The Cre-loxP strategy was used to generate a CD11c+ DC–specific deletion of A2AR in mice. Briefly, homozygous Adora2afl/fl mice (C57BL/6 background; Adora2a gene modified by floxed sequences flanking exon 2) (32) were bred with CD11c-Cre transgenic mice (The Jackson Laboratory), expressing Cre recombinase under the control of the CD11c promoter, to generate CD11c-CreAdora2afl/WT and CD11c-CreAdora2afl/fl mice that lack A2ARs in DCs. CD11c-CreAdora2afl/WT, CD11c-CreAdora2afl/fl, and control mice (CD11c-Cre, Adora2afl/fl, and Adora2afl/WT) were genotyped using DNA from tail biopsies with the following primer sets: (a) for CD11c-Cre: Cre forward, 5′-AGGTGTAGAGAAGGCACTTAG, and Cre reverse, 5′-CTAATCGCCATCTTCCAGCAGG, which generated a 411-bp product; and (b) for Adora2a WT (Adora2aWT) and floxed Adora2a alleles: forward 5′-GGGCAAGATGGGAGTCATT-3′ and reverse 1-5′-ATTCTGCATCTCCCGAAACC-3′ and reverse 2-5′-AACAGTTATTCTGATCTTTCC -3′, which generated a 216-bp product for Adora2afl/fl and an 182-bp product for the WT DNA sequence.
Kidney and spleen tissue digestion and FACS analysis of leukocytes.
Kidney or spleen cell suspensions were prepared from mice subjected to IRI or sham operation, and kidney leukocyte subset cell number was calculated as described before (6). The following antibodies (1 μg/ml; eBioscience) were used to identify kidney neutrophils: anti-mouse CD45-APC-Alexa Fluor 750 (30-F11), CD11b-PE (M1/70), and GR-1-APC (Ly6G RB6-8C5). 7-AAD was used to exclude dead cells. CD45-labeled samples were further labeled with CD11c antibodies to identify DCs expressing costimulatory markers (with anti-mouse CD80, CD86, CD40, OX40L, ICOS, B7-H1, and B7-DC) and antigen-presenting molecules (with anti-mouse CD1d and MHC class II [IA]) (eBioscience), as described previously (6). CD1d tetramer loaded with PBS-57 (1:100), an analog of α-GC (NIH Tetramer Core Facility, Emory University, Atlanta, Georgia, USA) (6), and anti-mouse TCR-β (eBioscience) were used to identify NKT cells. Intracellular staining for IFN-γ was performed using the BD Biosciences Fix/Perm buffer set according to the manufacturer’s protocol and as described previously (6). Tregs were labeled with anti-mouse antibodies (clones) from eBioscience (CD45 [Ly-5] APC-eFluor 780, PD-1 [J43] PE, CD25 [PC61.5] PE, FOXP3 [FJK-16s] Alexa Fluor 647) and from BD Biosciences (CD4 [RM4-5] V500) (66). IL-10–producing GFP+ cells from kidney and spleen of Vert-X mice were identified by gating on the CD45+7-AAD– live leukocyte population; additional antibodies included anti-mouse CD11c, CD4, CD8, and B220 (eBioscience). Flow cytometry data was acquired on BD FACSCalibur (BD Bioscience) with Cytek 8 Color Flow Cytometry Upgrade (Cytek Development Inc.) and analyzed by FlowJo software 9.0 (Tree Star Inc.).
Quantitative real-time RT-PCR.
Total RNA was extracted from kidneys with TriReagent according to the manufacturer’s protocol (Molecular Research Center Inc.) and reverse transcribed with cDNA transcript kit (Invitrogen). Primers were designed using IDT PrimerQuest (Integrated DNA Technologies; www.idtdna.com). Primer sequences for Il10 and FoxP3 mRNA are listed in Supplemental Table 1. RT-PCR was performed using the iScript 1-step RT-PCR kit with SYBR Green (Bio-Rad), and samples were normalized to RPS29.
Kidney CD11c+ DC isolation and real-time PCR detection of adenosine A1R, A2AR, A2BR, and A3R receptors.
Kidneys from mice exposed to sham operation or IRI were digested as described above for FACS analysis. Kidney cell suspensions were washed, cells were resuspended in 30% Percoll (GE Healthcare Life Science), and the samples were gently laid over 70% Percoll and centrifuged at 960 g for 20 minutes at room temperature. Mononuclear cells were collected, washed, and stained with anti-mouse CD45-PECy7, CD11c-APC–Alexa Fluor 780 (eBioscience), and 7-AAD. CD45+CD11chi7AAD– cells were sorted with an i-Cyt Reflection Cell Sorter (UVA Flow Cytometry Core). RNA isolation from sorted kidney DCs was performed by using QIAGEN RNeasy Mini Kit (QIAGEN). The primers for adenosine A1R, A2AR, A2BR, and A3R receptors are shown in Supplemental Table 1.
Detection of CD1d/αGC complex with L363 mAbs.
BMDCs or A20 and CD1d-transfected (A20.CD1d) B cell lines (provided by Mitchell Kronenberg, La Jolla Institute for Allergy and Immunology) were cultured and treated with vehicle, αGC (0.1 μM), or αGC plus ATL313 (1 nM) for 2.5 days. L363 was labeled with Alexa Fluor 647 by using the APEX Alexa Fluor 647 Antibody Labeling Kit (Invitrogen). Cells were collected and Alexa Fluor 647–L363 (10 μg/ml) surface and intracellular staining were performed by using BD Cytofix/Cytoperm Kit (BD Biosciences). 7-AAD was used to exclude dead cells. Cells were analyzed by flow cytometry as described above.
Priming BMDCs with αGC.
As described before (33), BM cells were isolated from WT, Adora2a–/–, or Il10–/– mice and >90% pure DCs were obtained following 2 weeks in culture. DCs were primed with vehicle (0.1% DMSO in culture medium; DC) or 0.1 μg/ml αGC (AXXORA, LLC; DC-αGC) for 2.5 days with or without 1 nM ATL313. Cells were then washed and 0.5 × 106 cells/mouse (optimal dose) were adoptively transferred (i.v.) to naive mice 2 days prior to kidney IR surgery (subthreshold, mild [26 minutes] or moderate [28 minutes] ischemic injury).
Coculture of DCs with NKT cells in vitro.
Naive WT mouse liver tissue was digested with type I collagenase (10 μg/ml) for 20 minutes and mashed. Mononuclear cells were isolated over 35% Percoll. 10 μg/ml PE-labeled anti-CD1d tetramer (NIH Tetramer Core Facility, Emory University) was added to the liver mononuclear cells to stain NKT cells; then anti-PE microbeads were used to isolate NKT cells through MACS columns as described by the manufacturer (Miltenyi Biotec). 1 × 104 WT DCs, DCs-αGC, or DCs-αGC-ATL313 were cocultured with 5 × 104 NKT cells in 96-well round-bottom plates for 5 days, and supernatants were collected for analysis of IFN-γ by ELISA (eBioscience).
Similarly, 5 × 105/ml DCs, DCs-αGC, or DCs-αGC-ATL313 were cocultured with Vα14Vβ8.2 NKT cell DN3A4-1.2 hybridomas (1.2) in 96-well round-bottom plates, and supernatant IL-2 was measured by ELISA (eBioscience) according to published protocols (70). αGC- or αGC-ATL313–loaded B cells (A20) were also cocultured with NKT cells to test NKT cell response by measuring IL-2 levels.
NKT cell anergy test.
Splenocytes were isolated from WT mice pretreated for 2 days with either WT or Adora2a–/– DCs, DCs-αGC, or DCs-αGC-ATL313. 1 × 106 splenocytes were restimulated with vehicle or αGC (0.1 μg/ml) in 96-well round-bottom plates for 24 hours, and supernatants were collected for IFN-γ assay by ELISA (eBioscience).
Histochemistry.
Kidneys were fixed and processed for H&E staining as previously described (6) and viewed by light microscopy (Zeiss AxioSkop) under ×200 magnification. Photographs were taken and brightness/contrast adjustment was made with a SPOT RT camera (software version 3.3; Diagnostic Instruments).
Plasma cytokine detection by ELISA.
Plasma was collected from mice 20 hours after administration of DCs, DCs-αGC, or DCs-αGC-ATL313. Plasma IFN-γ, IL-17A/F, IL-4, and IL-10 levels were measured by using mouse ELISA kits (eBioscience) following the manufacturer’s protocol.
DC and Treg coculture.
DCs were isolated and cultured as described above and exposed to 10 nM ATL1223 (Dogwood Pharmaceuticals Inc., a wholly owned subsidiary of Forest Laboratories Inc. ) and/or 100 ng/ml αGC for 2 days, then washed prior to initiation of cocultures with freshly isolated Tregs. Tregs were isolated from spleens of naive WT CD45.1 mice using the Dynal CD4+ T cell–negative isolation kit followed by the CD25–positive isolation kit from Miltenyi Biotec according to the manufacturers’ protocols and as described previously (71). Cocultures consisted of 5 × 104 DCs plus 1 × 105 Tregs in a total of 250 μl of medium in round-bottom 96-well plates for 48 hours. No activation stimuli were added (e.g., anti-CD3 or anti-CD28). Subsequently, FOXP3 staining for flow cytometry was conducted using the eBioscience FOXP3 Staining Buffer Set according to the manufacturer’s protocol (66) The anti–PD-1 (29F.1A12) antibody (Biolegend) was used only prior to permeabilization of the cells.
Statistics.
GraphPad Instat 3 (GraphPad Inc.), SigmaPlot 11.0 (Systat Software Inc.), and Canvas X (ACD Systems of America Inc.) were used to analyze and present the data. Data were analyzed, after transformation if needed to generate a normal distribution, by t test or 2-way ANOVA with post hoc analysis as appropriate. P < 0.05 was used to indicate significance.
Study approval.
All experiments were performed in accordance with NIH and Institutional Animal Care and Use Guidelines and were approved by the Animal Care and Use Committee at the University of Virginia.
Supplementary Material
Acknowledgments
This work was supported in part by funds from NIH RO1DK56223, RO1DK62324, P01HL073361, Genzyme (Genzyme Renal Innovations Program), and American Heart Association National Scientist Development Grant 0835258N. We thank Steven A. Porcelli (Albert Einstein College of Medicine, New York, New York, USA) for generously providing L363 mAbs and Mitchell Kronenberg (La Jolla Institute for Allergy and Immunology) for providing the Vα14Vβ8.2 DN3A4-1.2 (1.2) NKT cell hybridomas, A20 B cell line, and transfected murine CD1d A20.CD1d cell line. We also thank the University of Virginia Research Histology Core and Flow Cytometry Core Facilities.
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):3931–3942. doi:10.1172/JCI63170.
See the related Commentary beginning on page 3852.
References
- 1.Adema GJ. Dendritic cells from bench to bedside and back. Immunol Lett. 2009;122(2):128–130. doi: 10.1016/j.imlet.2008.11.017. [DOI] [PubMed] [Google Scholar]
- 2.Rosin DL, Okusa MD. Dangers within: DAMP responses to damage and cell death in kidney disease. J Am Soc Nephrol. 2011;22(3):416–425. doi: 10.1681/ASN.2010040430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Diao H, et al. Osteopontin as a mediator of NKT cell function in T cell-mediated liver diseases. Immunity. 2004;21(4):539–550. doi: 10.1016/j.immuni.2004.08.012. [DOI] [PubMed] [Google Scholar]
- 4.Lappas CM, Day YJ, Marshall MA, Engelhard VH, Linden J. Adenosine A2A receptor activation reduces hepatic ischemia reperfusion injury by inhibiting CD1d-dependent NKT cell activation. J Exp Med. 2006;203(12):2639–2648. doi: 10.1084/jem.20061097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sharma AK, et al. Natural killer T cell-derived IL-17 mediates lung ischemia-reperfusion injury. Am J Respir Crit Care Med. 2011;183(11):1539–1549. doi: 10.1164/rccm.201007-1173OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li L, et al. NKT cell activation mediates neutrophil IFN-gamma production and renal ischemia-reperfusion injury. J Immunol. 2007;178(9):5899–5911. doi: 10.4049/jimmunol.178.9.5899. [DOI] [PubMed] [Google Scholar]
- 7.Exley MA, Nakayama T. NKT-cell-based immunotherapies in clinical trials. Clin Immunol. 2011;140(2):117–118. doi: 10.1016/j.clim.2011.04.015. [DOI] [PubMed] [Google Scholar]
- 8.Van Kaer L, Parekh VV, Wu L. Invariant NK T cells: potential for immunotherapeutic targeting with glycolipid antigens. Immunotherapy. 2011;3(1):59–75. doi: 10.2217/imt.10.85. [DOI] [PubMed] [Google Scholar]
- 9.Fletcher MT, Baxter AG. Clinical application of NKT cell biology in type I (autoimmune) diabetes mellitus. Immunol Cell Biol. 2009;87(4):315–323. doi: 10.1038/icb.2009.5. [DOI] [PubMed] [Google Scholar]
- 10.Morelli AE, Thomson AW. Tolerogenic dendritic cells and the quest for transplant tolerance. Nat Rev Immunol. 2007;7(8):610–621. doi: 10.1038/nri2132. [DOI] [PubMed] [Google Scholar]
- 11.Steinman RM, Hawiger D, Nussenzweig MC. Tolerogenic dendritic cells. Annu Rev Immunol. 2003;21:685–711. doi: 10.1146/annurev.immunol.21.120601.141040. [DOI] [PubMed] [Google Scholar]
- 12.Pulendran B, Tang H, Manicassamy S. Programming dendritic cells to induce T(H)2 and tolerogenic responses. Nat Immunol. 2010;11(8):647–655. doi: 10.1038/ni.1894. [DOI] [PubMed] [Google Scholar]
- 13.Mahnke K, Schmitt E, Bonifaz L, Enk AH, Jonuleit H. Immature, but not inactive: the tolerogenic function of immature dendritic cells. Immunol Cell Biol. 2002;80(5):477–483. doi: 10.1046/j.1440-1711.2002.01115.x. [DOI] [PubMed] [Google Scholar]
- 14.Ezzelarab M, Thomson AW. Tolerogenic dendritic cells and their role in transplantation. Semin Immunol. 2011;23(4):252–263. doi: 10.1016/j.smim.2011.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soos TJ, et al. CX3CR1+ interstitial dendritic cells form a contiguous network throughout the entire kidney. Kidney Int. 2006;70(3):591–596. doi: 10.1038/sj.ki.5001567. [DOI] [PubMed] [Google Scholar]
- 16.Li L, et al. The chemokine receptors CCR2 and CX3CR1 mediate monocyte/macrophage trafficking in kidney ischemia-reperfusion injury. Kidney Int. 2008;74(12):1526–1537. doi: 10.1038/ki.2008.500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaissling B, Le Hir M. The renal cortical interstitium: morphological and functional aspects. Histochem Cell Biol. 2008;130(2):247–262. doi: 10.1007/s00418-008-0452-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Driver AG, Kukoly CA, Ali S, Mustafa SJ. Adenosine in bronchoalveolar lavage fluid in asthma. Am Rev Respir Dis. 1993;148(1):91–97. doi: 10.1164/ajrccm/148.1.91. [DOI] [PubMed] [Google Scholar]
- 19.Blackburn MR, et al. Adenosine mediates IL-13-induced inflammation and remodeling in the lung and interacts in an IL-13-adenosine amplification pathway. J Clin Invest. 2003;112(3):332–344. doi: 10.1172/JCI16815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fredholm BB. Adenosine, an endogenous distress signal, modulates tissue damage and repair. Cell Death Differ. 2007;14(7):1315–1323. doi: 10.1038/sj.cdd.4402132. [DOI] [PubMed] [Google Scholar]
- 21.Morello S, et al. IL-1 beta and TNF-alpha regulation of the adenosine receptor (A2A) expression: differential requirement for NF-kappa B binding to the proximal promoter. J Immunol. 2006;177(10):7173–7183. doi: 10.4049/jimmunol.177.10.7173. [DOI] [PubMed] [Google Scholar]
- 22.LaPar DJ, et al. Pretreatment strategy with adenosine A2A receptor agonist attenuates reperfusion injury in a preclinical porcine lung transplantation model. J Thorac Cardiovasc Surg. 2011;142(4):887–894. doi: 10.1016/j.jtcvs.2011.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ohta A, Sitkovsky M. Role of G-protein-coupled adenosine receptors in downregulation of inflammation and protection from tissue damage. Nature. 2001;414(6866):916–920. doi: 10.1038/414916a. [DOI] [PubMed] [Google Scholar]
- 24.Lukashev D, Ohta A, Apasov S, Chen JF, Sitkovsky M. Cutting edge: Physiologic attenuation of proinflammatory transcription by the Gs protein-coupled A2A adenosine receptor in vivo. J Immunol. 2004;173(1):21–24. doi: 10.4049/jimmunol.173.1.21. [DOI] [PubMed] [Google Scholar]
- 25.Day YJ, Li Y, Rieger JM, Ramos SI, Okusa MD, Linden J. A2A adenosine receptors on bone marrow-derived cells protect liver from ischemia-reperfusion injury. J Immunol. 2005;174(8):5040–5046. doi: 10.4049/jimmunol.174.8.5040. [DOI] [PubMed] [Google Scholar]
- 26.Day YJ, et al. Renal protection from ischemia mediated by A2A adenosine receptors on bone marrow-derived cells. J Clin Invest. 2003;112(6):883–891. doi: 10.1172/JCI15483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sevigny CP, et al. Activation of adenosine 2A receptors attenuates allograft rejection and alloantigen recognition. J Immunol. 2007;178(7):4240–4249. doi: 10.4049/jimmunol.178.7.4240. [DOI] [PubMed] [Google Scholar]
- 28.Huang S, Apasov S, Koshiba M, Sitkovsky M. Role of A2a extracellular adenosine receptor-mediated signaling in adenosine-mediated inhibition of T-cell activation and expansion. Blood. 1997;90(4):1600–1610. [PubMed] [Google Scholar]
- 29.Raskovalova T, Huang X, Sitkovsky M, Zacharia LC, Jackson EK, Gorelik E. Gs protein-coupled adenosine receptor signaling and lytic function of activated NK cells. J Immunol. 2005;175(7):4383–4391. doi: 10.4049/jimmunol.175.7.4383. [DOI] [PubMed] [Google Scholar]
- 30.Koshiba M, Kojima H, Huang S, Apasov S, Sitkovsky MV. Memory of extracellular adenosine A2A purinergic receptor-mediated signaling in murine T cells. J Biol Chem. 1997;272(41):25881–25889. doi: 10.1074/jbc.272.41.25881. [DOI] [PubMed] [Google Scholar]
- 31.Li L, Okusa MD. Macrophages, dendritic cells, and kidney ischemia-reperfusion injury. Semin Nephrol. 2010;30(3):268–277. doi: 10.1016/j.semnephrol.2010.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bastia E, et al. A crucial role for forebrain adenosine A(2A) receptors in amphetamine sensitization. Neuropsychopharmacology. 2005;30(5):891–900. doi: 10.1038/sj.npp.1300630. [DOI] [PubMed] [Google Scholar]
- 33.Li L, et al. IL-17 produced by neutrophils regulates IFN-gamma-mediated neutrophil migration in mouse kidney ischemia-reperfusion injury. J Clin Invest. 2010;120(1):331–342. doi: 10.1172/JCI38702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Agudelo J, et al. Analysis of efficacy and failure in proximal humerus fractures treated with locking plates. J Orthop Trauma. 2007;21(10):676–681. doi: 10.1097/BOT.0b013e31815bb09d. [DOI] [PubMed] [Google Scholar]
- 35.Mitty HA, Shapiro RS, Parsons RB, Silberzweig JE. Renovascular hypertension. Radiol Clin North Am. 1996;34(5):1017–1036. [PubMed] [Google Scholar]
- 36.Postma CT, van Aalen J, de Boo T, Rosenbusch G, Thien T. Doppler ultrasound scanning in the detection of renal artery stenosis in hypertensive patients. Br J Radiol. 1992;65(778):857–860. doi: 10.1259/0007-1285-65-778-857. [DOI] [PubMed] [Google Scholar]
- 37.Li X, et al. Acute renal venous obstruction is more detrimental to the kidney than arterial occlusion: implication for murine models of acute kidney injury. Am J Physiol Renal Physiol. 2012;302(5):F519–F525. doi: 10.1152/ajprenal.00011.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Grenz A, et al. Use of a hanging-weight system for isolated renal artery occlusion. J Vis Exp. 2011;(53):2549. doi: 10.3791/2549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu KO, et al. Production and characterization of monoclonal antibodies against complexes of the NKT cell ligand alpha-galactosylceramide bound to mouse CD1d. J Immunol Methods. 2007;323(1):11–23. doi: 10.1016/j.jim.2007.03.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bai L, et al. Lysosomal recycling terminates CD1d-mediated presentation of short and polyunsaturated variants of the NKT cell lipid antigen alphaGalCer. Proc Natl Acad Sci U S A. 2009;106(25):10254–10259. doi: 10.1073/pnas.0901228106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lefrancois L, Obar JJ. Once a killer, always a killer: from cytotoxic T cell to memory cell. Immunol Rev. 2010;235(1):206–218. doi: 10.1111/j.0105-2896.2010.00895.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Uchino S, et al. Acute renal failure in critically ill patients: a multinational, multicenter study. JAMA. 2005;294(7):813–818. doi: 10.1001/jama.294.7.813. [DOI] [PubMed] [Google Scholar]
- 43.Eltzschig HK, Eckle T. Ischemia and reperfusion--from mechanism to translation. Nat Med. 2011;17(11):1391–1401. doi: 10.1038/nm.2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kinsey GR, Okusa MD. Role of leukocytes in the pathogenesis of acute kidney injury. Crit Care. 2012;16(2):214. doi: 10.1186/cc11228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoop JN, Robinson JH, Hilkens CM. Developing tolerogenic dendritic cell therapy for rheumatoid arthritis: what can we learn from mouse models? Ann Rheum Dis. 2011;70(9):1526–1533. doi: 10.1136/ard.2011.151654. [DOI] [PubMed] [Google Scholar]
- 46.Land WG. Emerging role of innate immunity in organ transplantation Part I: evolution of innate immunity and oxidative allograft injury. Transplant Rev (Orlando). 2012;26(2):60–72. doi: 10.1016/j.trre.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 47.Jo SK, Rosner MH, Okusa MD. Pharmacologic treatment of acute kidney injury: why drugs haven’t worked and what is on the horizon. Clin J Am Soc Nephrol. 2007;2(2):356–365. doi: 10.2215/CJN.03280906. [DOI] [PubMed] [Google Scholar]
- 48.Wallace KL, et al. NKT cells mediate pulmonary inflammation and dysfunction in murine sickle cell disease through production of IFN-gamma and CXCR3 chemokines. Blood. 2009;114(3):667–676. doi: 10.1182/blood-2009-02-205492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao Z, et al. Preactivation of NKT cells with alpha-GalCer protects against hepatic ischemia-reperfusion injury in mouse by a mechanism involving IL-13 and adenosine A2A receptor. Am J Physiol Gastrointest Liver Physiol. 2009;297(2):G249–G258. doi: 10.1152/ajpgi.00041.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Eltzschig HK, Carmeliet P. Hypoxia and inflammation. N Engl J Med. 2011;364(7):656–665. doi: 10.1056/NEJMra0910283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Okusa MD, Linden J, Macdonald T, Huang L. Selective A2A adenosine receptor activation reduces ischemia-reperfusion injury in rat kidney. Am J Physiol. 1999;277(3 pt 2):F404–F412. doi: 10.1152/ajprenal.1999.277.3.F404. [DOI] [PubMed] [Google Scholar]
- 52.Grenz A, et al. Equilibrative nucleoside transporter 1 (ENT1) regulates postischemic blood flow during acute kidney injury in mice. J Clin Invest. 2012;122(2):693–710. doi: 10.1172/JCI60214. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 53.Awad AS, et al. Selective sphingosine 1-phosphate 1 receptor activation reduces ischemia-reperfusion injury in mouse kidney. Am J Physiol Renal Physiol. 2006;290(6):F1516–F1524. doi: 10.1152/ajprenal.00311.2005. [DOI] [PubMed] [Google Scholar]
- 54.Tuettenberg A, Fondel S, Steinbrink K, Enk AH, Jonuleit H. CD40 signalling induces IL-10-producing, tolerogenic dendritic cells. Exp Dermatol. 2010;19(1):44–53. doi: 10.1111/j.1600-0625.2009.00975.x. [DOI] [PubMed] [Google Scholar]
- 55.Brandl C, Ortler S, Herrmann T, Cardell S, Lutz MB, Wiendl H. B7-H1-deficiency enhances the potential of tolerogenic dendritic cells by activating CD1d-restricted type II NKT cells. PLoS One. 2010;5(5):e10800. doi: 10.1371/journal.pone.0010800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Murugaiyan G, Mittal A, Lopez-Diego R, Maier LM, Anderson DE, Weiner HL. IL-27 is a key regulator of IL-10 and IL-17 production by human CD4+ T cells. J Immunol. 2009;183(4):2435–2443. doi: 10.4049/jimmunol.0900568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Collison LW, et al. The inhibitory cytokine IL-35 contributes to regulatory T-cell function. Nature. 2007;450(7169):566–569. doi: 10.1038/nature06306. [DOI] [PubMed] [Google Scholar]
- 58.Unger WW, Laban S, Kleijwegt FS, van der Slik AR, Roep BO. Induction of Treg by monocyte-derived DC modulated by vitamin D3 or dexamethasone: differential role for PD-L1. Eur J Immunol. 2009;39(11):3147–3159. doi: 10.1002/eji.200839103. [DOI] [PubMed] [Google Scholar]
- 59.Chauveau C, et al. Heme oxygenase-1 expression inhibits dendritic cell maturation and proinflammatory function but conserves IL-10 expression. Blood. 2005;106(5):1694–1702. doi: 10.1182/blood-2005-02-0494. [DOI] [PubMed] [Google Scholar]
- 60.White LE, Hassoun HT. Inflammatory mechanisms of organ crosstalk during ischemic acute kidney injury. Int J Nephrol. 2012;2012:505197. doi: 10.4061/2012/505197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gregori S, et al. Differentiation of type 1 T regulatory cells (Tr1) by tolerogenic DC-10 requires the IL-10-dependent ILT4/HLA-G pathway. Blood. 2010;116(6):935–944. doi: 10.1182/blood-2009-07-234872. [DOI] [PubMed] [Google Scholar]
- 62.Huang H, Dawicki W, Zhang X, Town J, Gordon JR. Tolerogenic dendritic cells induce CD4+CD25hiFoxp3+ regulatory T cell differentiation from CD4+CD25-/loFoxp3- effector T cells. J Immunol. 2010;185(9):5003–5010. doi: 10.4049/jimmunol.0903446. [DOI] [PubMed] [Google Scholar]
- 63.Saito M, et al. Defective IL-10 signaling in hyper-IgE syndrome results in impaired generation of tolerogenic dendritic cells and induced regulatory T cells. J Exp Med. 2011;208(2):235–249. doi: 10.1084/jem.20100799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Burke F, Stagg AJ, Bedford PA, English N, Knight SC. IL-10-producing B220+CD11c- APC in mouse spleen. J Immunol. 2004;173(4):2362–2372. doi: 10.4049/jimmunol.173.4.2362. [DOI] [PubMed] [Google Scholar]
- 65.Velupillai P, Harn DA. Oligosaccharide-specific induction of interleukin 10 production by B220+ cells from schistosome-infected mice: a mechanism for regulation of CD4+ T-cell subsets. Proc Natl Acad Sci U S A. 1994;91(1):18–22. doi: 10.1073/pnas.91.1.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kinsey GR, Huang L, Vergis AL, Li L, Okusa MD. Regulatory T cells contribute to the protective effect of ischemic preconditioning in the kidney. Kidney Int. 2010;77(9):771–780. doi: 10.1038/ki.2010.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kinsey GR, Li L, Okusa MD. Inflammation in acute kidney injury. Nephron Exp Nephrol. 2008;109(4):e102–107. doi: 10.1159/000142934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou Q, et al. Program death-1 signaling and regulatory T cells collaborate to resist the function of adoptively transferred cytotoxic T lymphocytes in advanced acute myeloid leukemia. Blood. 2010;116(14):2484–2493. doi: 10.1182/blood-2010-03-275446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Chen JF, et al. A(2A) adenosine receptor deficiency attenuates brain injury induced by transient focal ischemia in mice. J Neurosci. 1999;19(21):9192–9200. doi: 10.1523/JNEUROSCI.19-21-09192.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Wingender G, et al. Invariant NKT cells are required for airway inflammation induced by environmental antigens. J Exp Med. 2011;208(6):1151–1162. doi: 10.1084/jem.20102229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Kinsey GR, et al. Regulatory T cells suppress innate immunity in kidney ischemia-reperfusion injury. J Am Soc Nephrol. 2009;20(8):1744–1753. doi: 10.1681/ASN.2008111160. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.