

Abstract
Diabetic cardiomyopathy (DbCM), which consists of cardiac hypertrophy and failure in the absence of traditional risk factors, is a major contributor to increased heart failure risk in type 2 diabetes patients. In rodent models of DbCM, cardiac hypertrophy and dysfunction have been shown to depend upon saturated fatty acid (SFA) oversupply and de novo sphingolipid synthesis. However, it is not known whether these effects are mediated by bulk SFAs and sphingolipids or by individual lipid species. In this report, we demonstrate that a diet high in SFA induced cardiac hypertrophy, left ventricular systolic and diastolic dysfunction, and autophagy in mice. Furthermore, treatment with the SFA myristate, but not palmitate, induced hypertrophy and autophagy in adult primary cardiomyocytes. De novo sphingolipid synthesis was required for induction of all pathological features observed both in vitro and in vivo, and autophagy was required for induction of hypertrophy in vitro. Finally, we implicated a specific ceramide N-acyl chain length in this process and demonstrated a requirement for (dihydro)ceramide synthase 5 in cardiomyocyte autophagy and myristate-mediated hypertrophy. Thus, this report reveals a requirement for a specific sphingolipid metabolic route and dietary SFAs in the molecular pathogenesis of lipotoxic cardiomyopathy and hypertrophy.
Introduction
Obesity and diabetes present two of the most important health challenges facing the Western world at this time. Patients suffering from type 2 diabetes (T2D) are subject to a number of major health risks, including a greatly increased risk of heart failure (1). This is due in part to the development of diabetic cardiomyopathy (DbCM), which occurs independently of other traditional risk factors (2). DbCM promotes cardiac remodeling and impairs cardiac function (1). Importantly, individuals with T2D and the metabolic syndrome present with dyslipidemia, and recent studies have suggested that DbCM may occur as a result of lipid overload and subsequent lipotoxic events (ref. 2, reviewed in ref. 3). In particular, oversupply of saturated fatty acids (SFAs) has been implicated in this process.
Previous studies of lipotoxic DbCM in rodents have relied on several important transgenic models. The B6.Cg-Lepob/J (ob/ob) and B6.BKS(D)-Leprdb/J (db/db) mouse models, which lack the genes encoding leptin and the leptin receptor, respectively, are both very popular models that develop obesity and a DbCM-like cardiac phenotype (reviewed in ref. 4). Other less common models, including the Atgl-knockout mouse and the LpLGPI transgenic mouse, also induce lipotoxic cardiomyopathy by perturbing cardiac lipid uptake, handling, or metabolism (reviewed in ref. 4). While these transgenic models robustly induce lipid overload in cardiomyocytes, the very disruptions that produce lipotoxic DbCM also drastically alter patterns of lipid uptake and handling in a nonphysiologic way. In contrast, wild-type mice fed standard lard-based high-fat diets (LBD) failed to develop a DbCM-like phenotype until very late time points, if at all, and insulin resistance and other aspects of the diabetic phenotype were less pronounced in this model system than in transgenic animals (4–9). This may result from the high levels of protective unsaturated fatty acids (UFAs) contained in these diets (10–12). Thus, while these transgenic model systems produce a robust DbCM phenotype, they are unable to reveal clinically relevant roles of specific lipid species in the molecular pathogenesis of DbCM.
Because of these weaknesses in present model systems, our laboratory recently implemented a new obesogenic high-fat diet that was designed to replicate the shift toward shorter SFAs observed in the plasma of obese and diabetic patients (13). Milk was considered an ideal fat source for this diet, as it contains low levels of UFAs and high levels of saturated fat. Furthermore, epidemiologic studies have demonstrated a relationship between consumption of high-fat dairy (e.g., butter, cheese, ice cream) and insulin resistance in humans (reviewed in ref. 14). Similarly, multiple studies have also linked the SFA myristate (C14:0), which is a major constituent of milk fat, to insulin resistance and the metabolic syndrome in humans (14–17). Strikingly, in wild-type C57BL/6J mice, this milk fat–based diet (MFBD) promoted profound insulin resistance and hyperglycemia by 8 weeks. This stands in contrast with mice fed a traditional LBD, which do not develop metabolic defects until very late time points (4). These findings suggested that the MFBD might promote a more severe DbCM phenotype than the LBD in wild-type mice.
Additional evidence suggests that milk fat and its constituent fatty acid, myristate, may promote cardiac dysfunction and hypertrophy. In particular, dietary patterns that feature high-fat dairy were associated with increased LV mass and reduced systolic function in humans, and plasma myristate content was positively associated with incidence of heart failure and total cardiovascular disease mortality (18–20). Furthermore, a recent study demonstrated that infusion with a myristate-rich SFA cocktail rapidly induced LV hypertrophy in mice (21). These data suggest a role for dietary and plasma myristate in cardiac hypertrophy and dysfunction, but potential mechanisms remain unknown.
Thus, the literature has provided epidemiological links between dietary milk fat, the fatty acid myristate, and both cardiac dysfunction and diabetes in humans, while data in animals suggest a potential role for myristate in cardiac hypertrophy. Based on these findings, we hypothesized that the MFBD might induce a cardiac phenotype closely resembling human DbCM, with no requirement for genetic perturbations. Furthermore, we hypothesized that this phenotype would develop in a sphingolipid-dependent manner.
This report characterizes the cardiac phenotype of mice fed the MFBD, which is rich in SFAs but not UFAs, and implicates a specific SFA and sphingolipid metabolic route in the induction of hypertrophy and increased autophagy in response to lipid overload in cardiomyocytes.
Results
An MFBD induces cardiac hypertrophy and dysfunction.
Based on the documented connection between diets high in saturated fat and both insulin resistance and incidence of heart disease in humans, we hypothesized that the MFBD, which is high in SFAs and low in protective UFAs, would induce DbCM-like cardiac hypertrophy and dysfunction in wild-type mice. To test this hypothesis, mice were maintained on the MFBD, a traditional LBD, or an isocaloric low-fat control diet (CD) for fifteen weeks and then subjected to echocardiographic analyses. By 15 weeks of high-fat feeding, MFBD mice developed LV hypertrophy and functionally significant reductions in ejection fraction relative to mice fed the CD (Table 1). In contrast, LBD-fed mice maintained normal cardiac function and LV mass relative to CD-fed mice. These results suggested that a diet rich in saturated fat but not unsaturated fat promoted LV hypertrophy and diminished cardiac function.
Table 1.
Echocardiographic assessment of mice after 15 weeks of high-fat diet
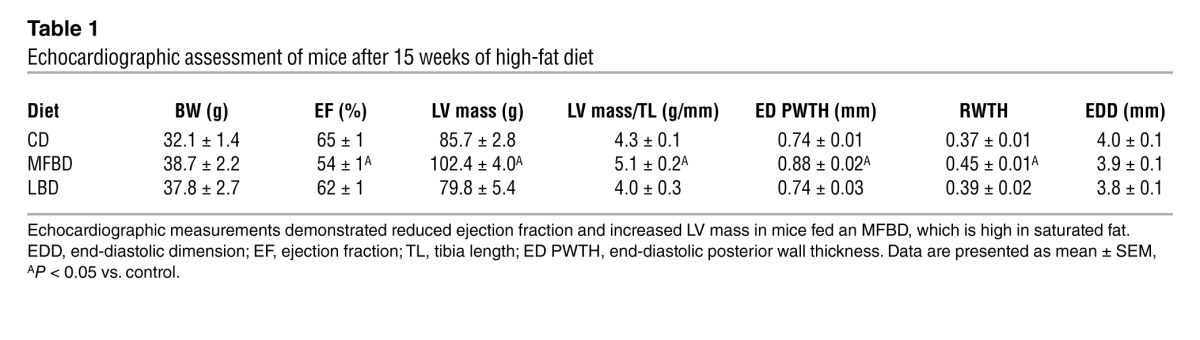
These findings led us to ask whether these observations constituted a pure DbCM phenotype, i.e., one independent of pressure overload and cardiac dilation. To determine this, blood pressure measurements, detailed echocardiographic studies, and histological assessments were performed on these mice after 18 weeks of high-fat feeding. Despite similar BWs, blood pressures, and end-diastolic diameters between high-fat diet groups (Figure 1A and Table 2), only MFBD mice displayed reduced ejection fraction, reduced early diastolic mitral annular velocity, increased end-diastolic posterior wall thickness, and LV hypertrophy (Table 2). Furthermore, only mice fed the MFBD displayed whole heart, LV, and RV hypertrophy and increased cardiomyocyte cross-sectional area (Figure 1, B and C). These findings indicate that the MFBD, but not the LBD or CD, induced concentric cardiac hypertrophy and reduced cardiac function in the absence of dilated cardiomyopathy or pressure overload.
Figure 1. An MFBD promotes sphingolipid-dependent cardiac hypertrophy.

Mice fed an MFBD for 18 weeks (A) did not experience greater obesity than mice on a traditional LBD, but they displayed (B) marked cardiac hypertrophy and (C) increased cardiomyocyte cross-sectional area. Myriocin treatment attenuated this hypertrophy without affecting weight gain. Black bars, control plus DMSO; light gray bars, milk fat plus DMSO; dark gray bars, milk fat plus myriocin; hatched bars, lard plus DMSO. All results are given as mean ± SEM. *P < 0.05 vs. control.
Table 2.
Echocardiographic assessment of mice after 18 weeks of high-fat diet
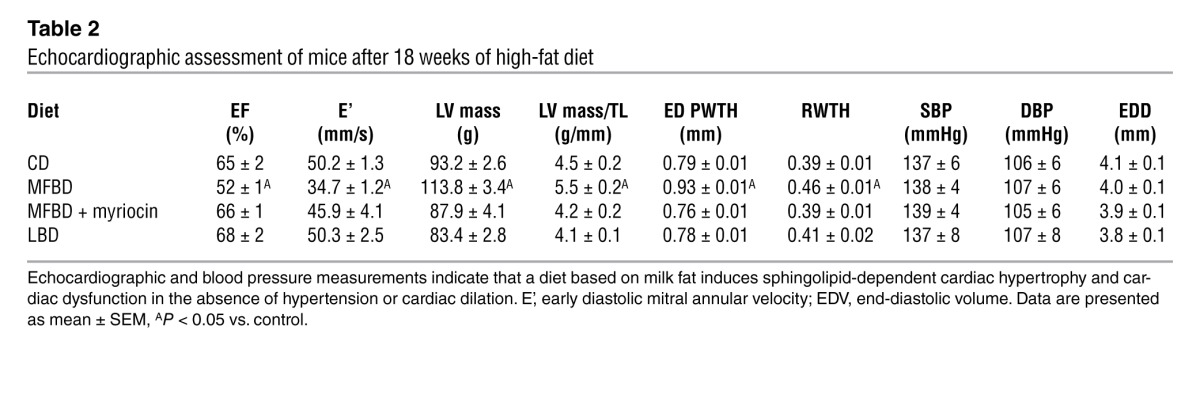
Cardiac outcomes in the MFBD are sphingolipid dependent.
Mechanistically, lipotoxicity has been implicated in the pathogenesis of DbCM (22). Previous studies in mice overexpressing a membrane-anchored lipoprotein lipase in the heart indicated that cardiac dysfunction, hypertrophy, and expression of heart failure markers were ameliorated by inhibition of de novo sphingolipid synthesis; however, the clinical relevance of this model to human DbCM may be limited (23). Moreover, only bulk sphingolipid synthesis has been implicated in these effects; the specific molecular species of sphingolipids and the enzymes that mediate them have remained undetermined.
To determine whether the cardiac remodeling observed in MFBD mice was dependent on sphingolipid synthesis, MFBD mice were treated with the de novo sphingolipid synthesis inhibitor myriocin and subjected to echocardiography and histological assessment. Strikingly, inhibition of de novo sphingolipid synthesis restored all measured echocardiographic parameters to normal levels in the MFBD mice (Table 2). Furthermore, myriocin treatment prevented LV and cardiomyocyte hypertrophy without reducing BW (Figure 1). These results dramatically highlight a role for sphingolipids in the pathogenesis of concentric cardiac hypertrophy and cardiac dysfunction in diet-induced obesity and DbCM.
Because attenuation of de novo sphingolipid synthesis prevented development of cardiomyopathy in the MFBD, we hypothesized that the cardiomyopathy phenotype may result from increases in bulk ceramide levels. To test this, ceramide and its precursor dihydroceramide were measured in hearts of mice fed the MFBD or the LBD. Surprisingly, total ceramide and dihydroceramide did not increase in the MFBD, but rather increased in the less cardiotoxic LBD, relative to control (Figure 2A). This perplexing result suggested a more subtle mechanism for sphingolipid-dependent lipotoxicity than merely a simple increase in bulk ceramide levels, as had previously been assumed.
Figure 2. An MFBD production of C14-ceramide.

(A) Total ceramide (Cer) and dihydroceramide (DHC) did not increase in mice fed an MFBD for 8 weeks, but (B) C14-ceramide increased specifically in the MFBD but not the LBD. All results are given as mean ± SEM. *P < 0.05 vs. control.
The term “ceramide” generally refers to any simple N-acylated sphingosine moiety; however, many different N-acyl chain lengths occur in ceramides, with the natural range extending from 12 to 26 carbons (24). Importantly, recent studies indicate that elevations of specific ceramide N-acyl chain lengths promote particular cellular and signaling outcomes in some systems (25, 26). In order to determine whether the MFBD induced changes in specific ceramides, individual ceramide species were quantified in hearts from mice after 8 weeks of high-fat feeding. This time point was chosen to minimize confounding effects of long-term insulin resistance, obesity, and inflammation. Strikingly, the MFBD specifically increased C14-ceramide (Figure 2B); similar results were obtained at later time points (data not shown). This finding was consistent with the lipid content of the MFBD, which contained significantly more myristate than either the CD or the LBD and promoted a 4-fold increase in plasma myristate levels (Supplemental Figure 1; supplemental material available online with this article; doi: 10.1172/JCI63888DS1). These results indicate that the MFBD induced a very precise readjustment of ceramide N-acyl chain length composition, possibly consequent to elevated plasma myristate levels.
Myristate induces hypertrophy through ceramide synthase 5.
Because of the high myristate content of the MFBD, we were led to ask whether exposure to myristate alone might induce hypertrophy in cardiomyocytes. To test this, we measured the effects of myristate treatment on cell size in isolated primary cardiomyocytes. Consistent with a specific role for myristate in cardiac hypertrophy, myristate treatment increased cell size in isolated cardiomyocytes (Figure 3A). Furthermore, as in the MFBD hearts, this hypertrophic response was completely attenuated by inhibition of de novo sphingolipid synthesis with myriocin. In contrast, treatment with palmitate, a fatty acid implicated in lipotoxicity in other tissues, did not induce cardiomyocyte hypertrophy. These findings suggest a specific role for myristate or its derivatives in the induction of sphingolipid-dependent cardiomyocyte hypertrophy.
Figure 3. Myristate induces hypertrophy in a sphingolipid- and CerS5-dependent manner.

(A) Myristate, but not palmitate, treatment induced hypertrophy in cardiomyocytes. This was prevented by inhibition of de novo sphingolipid synthesis with myriocin. Results are shown as percentage increase over BSA. (B) Myristate, but not palmitate, strongly induces C14-ceramide production in cardiomyocytes. (C) CerS5 and CerS6 are primarily responsible for synthesis of C14-ceramide. (D) siRNA-mediated knockdown of CerS5 is sufficient to prevent induction of hypertrophy by myristate. Results are given as percentage increase over control siRNA treated with BSA. For all panels, results are presented as mean ± SEM. *P < 0.05 vs. BSA; ***P < 0.005 vs. control BSA; #P < 0.05 vs. palmitate; †P < 0.005 vs. myristate DMSO.
The specific upregulation of C14-ceramide in the MFBD myocardium suggested that this metabolite, or the pathway that synthesizes it, may play a unique role in lipotoxic cardiac hypertrophy. Supporting this hypothesis, myristate treatment, which promoted cardiomyocyte hypertrophy, increased C14-ceramide levels 10-fold in isolated cardiomyocytes (Figure 3B). In contrast, palmitate treatment, which did not induce hypertrophy, had no effect on C14-ceramide levels. These findings led us to ask whether the pathway that produces C14-ceramide might be required for myristate-induced cardiomyocyte hypertrophy.
Ceramide synthesis occurs through N-acylation of a sphingoid base by one of 6 ceramide synthase (CerS) isoforms, each of which displays a unique profile of N-acyl chain length incorporation (Figure 3C) (27, 28). While both CerS5 and CerS6 synthesize C14-ceramide robustly in vitro (28), expression of CerS6 is very low in heart (28–30), suggesting CerS5 as the most likely candidate for cardiac C14-ceramide production. Thus, we tested to determine whether siRNA-mediated CerS5 knockdown was sufficient to prevent induction of cardiomyocyte hypertrophy by myristate. Indeed, it was found that CerS5 knockdown completely prevented lipotoxic cardiomyocyte hypertrophy in this system (Figure 3D). Thus, these findings implicate a specific CerS isoform in a component of the DbCM cardiac phenotype and lend credence to the hypothesis that the CerS5/C14-ceramide pathway is required for induction of lipotoxic cardiac hypertrophy.
The MFBD induces autophagy in a sphingolipid-dependent manner.
Numerous studies have identified autophagy as a key process that is upregulated in cardiac hypertrophy in response to pressure overload (31–36). Furthermore, sphingolipid-dependent autophagy has been demonstrated in numerous cell types, with the molecular mechanisms appearing to be specific to both the cell type and the mode of induction (reviewed in refs. 37–39). However, few studies have addressed the role of autophagy in DbCM in T2D, and none of them have directly tested the effect of lipid oversupply on cardiac autophagy.
To determine whether the MFBD increased autophagy, expression of the autophagy markers was measured in whole-heart lysate at 8 weeks. Indeed, mRNA levels of LC3B (Map1lc3b) and Beclin 1 (Becn1) were both strongly upregulated in the MFBD (Figure 4A). Furthermore, immunohistochemical and immunofluorescent staining demonstrated increased numbers of LC3B puncta in the LV of mice fed the MFBD, but not the LBD (Figure 4, B and C); moreover, this effect was attenuated in myriocin-treated mice, suggesting that autophagy in the MFBD mice occurred through a sphingolipid-dependent mechanism.
Figure 4. An MFBD promoted autophagy in cardiomyocytes.

(A) Myocardial mRNA levels of the autophagy markers Map1lc3b (LC3B) and Becn1 (Beclin 1) were upregulated by the MFBD but not by the LBD. (B) Immunohistochemical and (C) immunofluorescent staining revealed increased numbers of LC3B puncta (indicated by arrows in B and enclosed in circles in C) in the MFBD but not the LBD after 18 weeks of high-fat feeding. This effect was attenuated by myriocin treatment. Photographs were taken in a cross-sectional plane with a ×10 objective (B) and in a transverse plane using a ×60 objective (C). Results are presented as mean ± SEM. *P < 0.05 vs. control.
Myristate induces autophagy through ceramide synthase 5.
Because of the high myristate content of the MFBD and the confirmed role of exogenous myristate in the induction of cardiomyocyte hypertrophy in this system, we were led to ask whether exposure to myristate might also induce autophagy in cardiomyocytes. To test this, we treated isolated adult primary cardiomyocytes with myristate and measured levels of autophagy markers. Consistent with observations in whole heart, myristate treatment promoted overexpression of Becn1 and Atg7, another marker of autophagy, compared with BSA (Figure 5, A and B). In contrast, treatment with palmitate (a C16:0 fatty acid) did not induce expression of autophagy markers. These findings strongly paralleled those regarding the role of myristate in cardiomyocyte hypertrophy. Additionally, a prior study indicated that C14-ceramide and CerS5 increased under proautophagic conditions in cancer cells, although the potential mechanistic link was not investigated (40). Thus, we were led to ask whether CerS5 might also mediate induction of cardiomyocyte autophagy by myristate.
Figure 5. Myristate- and sphingolipid-dependent autophagy is mediated by CerS5 in cardiomyocytes.

Treatment with myristate, but not palmitate, increased mRNA levels of the autophagy markers (A) Becn1 and (B) Atg7 in isolated cardiomyocytes. (C) Overexpression of CerS5 in isolated cardiomyocytes resulted in an increase in Beclin 1 mRNA and protein. Immunoblot quantitation is normalized to actin and shown with a representative immunoblot. (D) siRNA-mediated knockdown of CerS5 reduced constitutive Beclin 1 expression and prevented DHS-mediated induction of Beclin 1 overexpression. Immunoblot quantitation is normalized to actin and shown with a representative immunoblot. (E) siRNA-mediated knockdown of CerS5 prevented induction of autophagy by myristate. Quantitation is paired with a representative immunoblot. For all representative immunoblots, lanes separated by white lines were run on the same gel but were noncontiguous. Results are presented as mean ± SEM. *P < 0.05 vs. control or BSA.
Previous studies have indicated that upregulation of BECN1 expression was sufficient to increase cardiac autophagy, both basally and in response to stress, while diminishing BECN1 expression attenuated stress-induced autophagy (34). Consistent with a role for CerS5 in regulation of cardiomyocyte autophagy in the present system, overexpression of CerS5 induced BECN1 expression in isolated cardiomyocytes, even in the absence of myristate treatment (Figure 5C). Furthermore, siRNA-mediated knockdown of endogenous CerS5 reduced basal expression of BECN1 (Figure 5D). To test directly the potential role of CerS5 in sphingolipid-mediated cardiomyocyte autophagy, we treated cells with the sphingoid base dihydrosphingosine (DHS), which serves as the immediate precursor of dihydroceramide. Because all CerS isoforms utilize DHS as a substrate, CerS5 knockdown would only prevent autophagy if that isoform were specifically required for this process. Supporting a role for a CerS-mediated sphingolipid pathway in cardiac autophagy, treatment with DHS induced BECN1 expression in isolated cardiomyocytes (Figure 5D). Strikingly, CerS5 knockdown completely prevented induction of BECN1 by DHS. This explicitly confirmed the requirement for this CerS isoform in sphingolipid-induced BECN1 expression in cardiomyocytes. Finally, to confirm the role for CerS5 in myristate-induced autophagy per se, myristate-treated cells were subjected to siRNA-mediated CerS5 knockdown. Consistent with the preceding results, CerS5 knockdown abrogated the induction of BECN1 expression by myristate treatment (Figure 5E). These findings demonstrated a requirement for CerS5 in sphingolipid-mediated induction of Beclin 1 protein and message levels in cardiomyocytes.
As suggestive as these results might be, they do not definitively demonstrate that myristate induces autophagy in cardiomyocytes. Indeed, it is possible that levels of autophagy mediators might be upregulated due to a defect in fusion of the autophagosome to the lysosome rather than increased autophagic flux. To test this possibility, autophagic flux was assessed in 2 complementary approaches, both of which utilized a GFP-LC3B fusion construct.
The first approach used the GFP-LC3B fusion construct to track LC3B lipidation and breakdown. The GFP-LC3B fusion protein is lipidated and processed similarly to native LC3B (41); however, the GFP moiety is more resistant to lysosomal proteases than the LC3B moiety, causing the free GFP protein to persist in the lysosome after breakdown of LC3B. Thus, levels of free GFP, detected by Western blot, can be used as a readout for breakdown of the inner membrane and contents of the autophagosome (41). Similarly to the results obtained with BECN1, it was found that overexpression of CerS5 in isolated cardiomyocytes increased expression of GFP-LC3B-I 4-fold (Figure 6A), while GFP-LC3B-II and free GFP expression increased 2- and 3-fold, respectively. Additionally, treatment with myristate increased levels of GFP-LC3B-II 7-fold and levels of free GFP 13-fold compared with BSA controls (Figure 6B). Finally, knockdown of CerS5 attenuated induction of all of these outcomes by myristate. Thus, these results indicate that myristate treatment stimulated lipidation and lysosomal degradation of LC3B, a finding consistent with increased autophagic flux.
Figure 6. Myristate promotes CerS5-dependent autophagic flux in cardiomyocytes.

Transfection with GFP-LC3B allowed visualization of autophagic puncta and tracking of autophagosome maturation and degradation in the lysosome. (A) Overexpression of CerS5 in isolated cardiomyocytes increased levels of GFP-LC3B-I, GFP-LC3B-II, and free GFP, indicating an increase in autophagy under these conditions, compared with empty vector. Immunoblot quantitations are presented with a representative immunoblot. White lines indicate that free GFP bands are shown with a shorter exposure of the same lanes from the same blot as the GFP-LC3B bands. (B) Treatment with myristate resulted in increased levels of GFP-LC3B-II and free GFP, indicating increased levels of mature autophagosomes and increased autophagic clearance. This effect was prevented by siRNA-mediated knockdown of CerS5. Immunoblot quantitations are presented with representative immunoblots; noncontiguous lanes, separated by white lines, are shown from the same gel. (C) Myristate treatment increased the number of GFP-labeled puncta in GFP-LC3B–expressing cells, suggesting increased numbers of autophagosomes; this effect was attenuated by CerS5 knockdown. All results are presented as mean ± SEM. *P < 0.05 vs. empty vector or control siRNA and BSA; ***P < 0.005 vs. control siRNA and BSA.
In a complementary approach, autophagosome dynamics were examined by fluorescent microscopy. To track autophagosome formation and fusion with the lysosome, GFP-LC3B–expressing cells were treated with myristate or BSA for 16 hours, loaded with the acidotropic dye LysoTracker Red, and examined by fluorescence microscopy over a 20-minute time course. This approach did not provide evidence of aberrant autophagosome accumulation and, rather, confirmed that fusion of autophagosomes to the lysosome, as indicated by fluorescent colocalization, is unimpaired in myristate-treated cells (Figure 7 and Supplemental Figures 5 and 6). Furthermore, it was revealed that total GFP-labeled puncta increased in myristate-treated cells, compared with those treated with BSA, and this increase was attenuated by CerS5 knockdown (Figure 6C). Taken together, all of these results demonstrate a requirement for CerS5 in induction of sphingolipid-mediated autophagic flux by myristate.
Figure 7. Myristate-treated cells do not display decreased autophagosome clearance.

After 16 hours of treatment with BSA (A and B) or myristate (C and D), GFP-LC3B–expressing cells were labeled with LysoTracker Red and observed by fluorescence microscopy for 20 minutes. Representative photomicrographs from the beginning and end of the time course (0 minutes and 20 minutes) are shown. Images were taken using a ×60 objective lens. In the panels presented, gamma settings of 0.7 for both red and green channels were used for all images. The heavily stained structure in the upper right quadrants of C and D is an adjacent cardiomyocyte undergoing cell death.
Autophagy is required for induction of hypertrophy by myristate in isolated cardiomyocytes.
While numerous reports have demonstrated an increase in autophagy in other hypertrophic cardiac conditions, it is still debated whether autophagy promotes or protects against cardiac hypertrophy in these systems (33–36, 42). To test the role for autophagy in regulation of cardiomyocyte hypertrophy, isolated cardiomyocytes were subjected to LC3B knockdown, which has been shown to impair autophagy (43). Strikingly, knockdown of LC3B prevented myristate-induced cardiomyocyte hypertrophy, suggesting a prohypertrophic role for autophagy in lipid overload (Figure 8). To confirm that this result was not due to an off-target effect of LC3B knockdown, cells were treated with 3-methyladenine, which inhibits autophagy via its action on type III phosphoinositide 3-kinases (44). Consistent with observations with LC3B knockdown, inhibition of autophagy by 3-methyladenine prevented myristate-induced cardiomyocyte hypertrophy (Supplemental Figure 8). These findings suggest that, at least in the context of cardiac lipid overload, increased autophagy is required for induction of cardiomyocyte hypertrophy.
Figure 8. Myristate induces hypertrophy in an autophagy-dependent manner.

Treatment with myristate induced hypertrophy in cardiomyocytes. This was prevented by knockdown of LC3B, which is a required component of the cellular autophagy machinery. Results are given as percentage increase over control siRNA treated with BSA. Results are presented as mean ± SEM. ***P < 0.005 vs. control BSA.
Discussion
Despite the considerable work that has been performed to tease apart the mechanisms of lipotoxic DbCM, to date, only general information has been published about the specific lipid mediators governing this process. In particular, although a few studies have implicated ceramides as a lipid class required for induction of cardiac lipotoxicity, the particular species involved have yet to be determined (23, 45–49). Additionally, the practical relevance of lipotoxicity to DbCM remained to be demonstrated, as all effective models of DbCM, to date, relied on genetic manipulations that disrupted cardiac lipid handling in a manner with no clinical parallels (ref. 2, reviewed in refs. 3, 4, 22).
The present study addresses both of these concerns. Here, we present a nontransgenic model of diet-induced obesity that robustly produced DbCM-like cardiac hypertrophy and functional impairment. Our findings revealed that an obesogenic diet based on milk fat, rather than lard, induced cardiac dysfunction, both gross and cellular hypertrophy, and increased autophagy in hearts of nontransgenic mice (Figures 1 and 4, and Tables 1 and 2). Furthermore, all of these outcomes were shown to be sphingolipid dependent. This diet, which was highly enriched in myristate (C14:0), also specifically potentiated production of the myristate-containing ceramide species C14-ceramide (Figure 2). These findings provided a compelling argument for further in vitro mechanistic work in isolated adult cardiomyocytes, where we were able to identify a specific pathway through which oversupply of an individual saturated fat induced cardiac hypertrophy. In isolated cells, myristate, but not palmitate (C16:0), was found to induce sphingolipid-dependent cardiomyocyte hypertrophy (Figure 3). Furthermore, myristate treatment potentiated C14-ceramide production, as was seen in the animal model. Subsequent to these observations, we identified a specific CerS isoform (CerS5) required for induction of hypertrophy by lipid overload (Figure 3).
This work also provided basic insight into the link between autophagy and lipotoxic cardiomyocyte hypertrophy. While numerous reports have demonstrated an increase in autophagy in other hypertrophic cardiac conditions, very few have examined cardiac autophagy in the context of T2D, and none have determined what effect lipid overload has on this pathway. Additionally, some controversy has surrounded the role for autophagy in the hypertrophic heart (31–36, 42). The debate centers on whether autophagy promotes or protects against cardiac hypertrophy in these systems. One major hypothesis unifies these 2 points of view, suggesting that autophagy is a “Goldilocks” phenomenon: too little is pathogenic; too much is pathogenic (50). In an isolated cardiomyocyte system, we demonstrated that oversupply of myristate, but not palmitate, promoted an increase in autophagy (Figures 5 and 6). Furthermore, we directly tested whether inhibition of autophagy would promote hypertrophy or prevent it. In fact, we found that fatty acid–induced hypertrophy was completely prevented by knockdown of LC3B or 3-methyladenine treatment, suggesting a pathogenic role for autophagy in cardiac lipid overload (Figure 8 and Supplemental Figure 8). Finally, we linked the induction of autophagy by myristate to the same CerS isoform implicated in the hypertrophic response (Figures 5 and 6).
Based on these findings, we propose a model in which a diet rich in myristate, which is a major constituent of milk fat–containing products such as butter and ice cream, induces cardiac hypertrophy through its effects on ceramide synthesis and composition. Namely, dietary myristate oversupply promotes ceramide synthesis via CerS5 and, consequently, increased autophagy. This increase in autophagy subsequently promotes hypertrophy through a mechanism that has yet to be identified.
Two major routes could potentially link autophagy and hypertrophy. The first mechanism would be mediated by conventional signaling pathways. While investigations remain ongoing, several potential connections have emerged through bioinformatic analyses. In particular, analysis of protein-protein interaction pathways has revealed links among 3 proteins of the autophagy pathway (i.e., Atg7, Atg12, and RB1CC1/FIP200) and the hypertrophy protein GSK3β via several routes, including FOXO1 and SIRT1 (S.B. Russo, unpublished data). Indeed, literature has already implicated FOXO1 and SIRT1 in regulation of both autophagy and hypertrophy in cardiomyocytes (51–54). Thus, it appears likely that autophagy and hypertrophy pathways may be subject to common regulators and could possibly engage in crosstalk.
The authors also hypothesize a second, less obvious mechanism by which upregulation of autophagy may be required for induction of hypertrophy. To develop the hypertrophic phenotype, cardiomyocytes undergo substantial subcellular remodeling of numerous components, including the mitochondria, sarcolemmal membrane, sarcoplasmic reticulum, and sarcomeres (55). Autophagy is a key process of cellular remodeling, and in particular, remodeling and recycling of the endoplasmic/sarcoplasmic reticulum and mitochondria (56, 57). On this basis, the authors speculate that, by altering the intracellular landscape via organelle turnover and by recycling intracellular components, induction of autophagy potentiates the organized remodeling prompted by prohypertrophic stimuli.
With regard to the mechanism linking C14-ceramide levels to autophagy, the authors of this study hypothesize that, in addition to upstream signaling roles, the effect of increased C14-ceramide may mediate membrane properties of the autophagosome and, thereby, the activity of membrane-resident proteins involved in autophagy. This notion is based on data from the literature that indicate that sphingolipid N-acyl chain lengths alter the biophysical properties of membranes, including curvature, fluidity, stability, and permeability (58–65). In particular, it has been shown that genetic ablation of CerS2 in mice promoted membrane fusion and budding (66). This was mechanistically attributed to alterations in lipid packing and membrane curvature, which occurred due to a shift toward shorter, unsaturated ceramides and a total ablation of C22- to C24-ceramide production. Furthermore, it has been shown that the activities of membrane-resident proteins are modulated by membrane biophysical properties and are sensitive to membrane lipid composition (67–69). In findings particularly relevant to this study, it was shown that membrane lipid composition and curvature were essential for targeting of Barkor/Atg14 to the budding autophagosome (70). Additionally, it has been shown that the proautophagic activity of BECN1 is mediated by its exit from sphingolipid-enriched membrane microdomains (so-called “lipid rafts”) in the context of traumatic brain injury (71). Thus, it seems plausible that increases in C14-ceramide may induce autophagy by promoting membrane budding and fusion and by altering localization or activity of membrane-resident proteins, including BECN1.
The diet model used in this study also represents an important technical advance in the study of lipotoxicity in DbCM. Specifically, previous studies of lipotoxic DbCM in mice have relied upon transgenic mouse models, including the ob/ob and db/db strains (reviewed in refs. 3, 4, 22). While these models closely mimic the DbCM phenotype, they do so by grossly perturbing cardiac metabolism and lipid handling. Thus, by their very nature, these studies confound any meaningful inference about roles for specific fatty acids and sphingolipid species in the pathophysiology of genotypically normal diabetic myocardium. Further complicating this effort, previously investigated diet-induced obesity models, which utilize high-fat feeding in wild-type mice, poorly reproduced the cardiac parameters of DbCM, including hypertrophy. This may be because the fatty acid profile of lard, which is the fat source for these diets, contains close to 60% combined oleic and linoleic acids, which have been shown to protect against lipotoxicity and might be expected to attenuate lipotoxic outcomes (11, 72).
In contrast, in the present study, feeding mice a diet based on milk fat, which is high in saturated fat and low in protective UFAs, robustly induced a DbCM phenotype (Tables 1 and 2). Furthermore, because this outcome was achieved in wild-type mice, meaningful conclusions could be drawn about the lipid species involved in this process. Thus, the present model represents a new and powerful tool to dissect the mechanisms underlying lipotoxic cardiomyopathy in a physiologically relevant context.
In conclusion, we present here what we believe is a new diet-based model of DbCM. This new model led us to define a specific role for C14-ceramide and CerS5 in promoting cardiac autophagy and subsequent hypertrophy of cardiomyocytes. Thus, this study implicates a specific ceramide species and CerS isoform in autophagy, hypertrophy, and cardiomyopathy resulting from lipid overload. Additionally, it provides a specific biochemical chain leading from dietary saturated fat to sphingolipid-mediated cardiac hypertrophy. Future studies aim to address the role for CerS5 in diet-induced obesity and insulin resistance in vivo as well as to identify the specific mechanisms by which myristate and C14-ceramide promote autophagy and hypertrophy.
Methods
Diets.
High-fat diets (60% calories from fat) and an isocaloric CD (10% calories from fat; TD.08810) were purchased from Harlan Laboratories Inc. High-fat diets were based on lard (TD.06414) or milk fat (TD.09766). The MFBD used anhydrous milk fat (also known as anhydrous butter oil) as its fat source; this substance is 99.8% fat and contains no detectible carbohydrate, including lactose. Both high-fat diets derived 18.4% of kcal from protein, 21.3% of kcal from carbohydrate, and 60.3% of kcal from fat. All components of the high-fat diets were identical, except for the substitution of anhydrous milk fat for lard. The CD derived 22.3% of kcal from protein, 60.9% of kcal from low-glycemic carbohydrate, and 16.8% of kcal from fat.
Animals.
Eight-week-old C57BL/6 male mice (Jackson Laboratory) were given water and chow ad libitum. For the study with time points of 8 and 16 weeks on the diets, 12 mice were placed in each group, and 6 mice were sacrificed per group per time point. For the study with time points of 15 and 18 weeks of feeding, 12 animals were assigned to each group. At the end of the study, at least 7 mice remained per group, with the exception of the lard group that received myriocin injections, of which only 5 survived. All mice from this study were sacrificed at 18 weeks. Metabolic characterization of these animals revealed a frank T2D phenotype in the MFBD-fed animals by 8 weeks. In particular, fasting plasma glucose levels in the MFBD mice exceeded 142% of control levels; this is consistent with and exceeds clinical diagnostic guidelines for T2D (73).
Echocardiography.
Mice underwent echocardiography to examine in vivo LV structure and function using a 40-MHz transducer and a Vevo 770 echocardiograph (VisualSonics). Three to six beats were averaged for each measurement. LV dimension and wall thickness were made at end-diastole and end-systole using American Society of Echocardiography criteria (74). Mean wall thickness was calculated as the average of interventricular septal wall thickness (IVS) and LV posterior wall thickness (LVPW). Relative wall thickness (RWTH) was calculated as the mean LV wall thickness divided by the LV internal dimension at end-diastole (LVIDd). LV mass was calculated as follows: LV mass = 0.8 × 1.05 × [(IVSd + LVPWd + LVIDd)3 – (LVIDd)3]. LV mass was normalized to BW (LV/BW ratio) and tibial length (LV/TL ratio). Ejection fraction (EF, %) was calculated as follows: EF = 100 × (LVEDV – LVESV)/LVEDV, where LV end-diastolic volume (LVEDV) and end-systolic volume (LVESV) were determined using Simpson’s method of discs. Both systolic (SBP) and diastolic blood pressure (DBP) were measured by using a standard, noninvasive, no-anesthesia, tail cuff pressure CODA system (Kent Scientific Corp). Five animals were analyzed per group at 15 and 18 weeks.
Histologic analysis.
At harvest, half of each heart was snap-frozen for biochemical analysis. The remaining half was submerged in 4% formaldehyde in PBS at 4°C overnight, transferred to 70% ethanol at 4°C, and paraffin embedded by the Histology Core at the MUSC. Paraffin sections were sliced to a thickness of 4 μM, deparaffinized as described in the Peroxidase Detection Reagent Pack (Thermo Scientific), and permeabilized with 0.5% Triton X-100. Sections were washed and blocked in 5% BSA in PBS, followed by incubation with LC3B antibody (MBL International) and anti-rabbit secondary antibody (Cell Signaling Technology) for light microscopy or Alexa Fluor 488 anti-rabbit secondary antibody (Cell Signaling Technology) for fluorescence microscopy. For fluorescence microscopy, cells were examined under a confocal microscope (Olympus FV10i), utilizing identical sensitivity and laser settings for all sections. Images were analyzed using FV10-ASW software (Olympus); identical thresholds and gamma settings were utilized for all images. For light microscopy, cells were stained as described in the Peroxidase Detection Reagent Pack and counterstained with H&E. Cross-sectional cell areas were quantified from images obtained at ×10 magnification, using Adobe PhotoShop 9.0 (Adobe Systems) on at least 10 fields from 3 animals per group, for a total of 335–475 cells analyzed per group. All cardiomyocytes in the proper plane of section were analyzed in each image field. Histograms of cell sizes, along with analyses of skewness and kurtosis, are provided in Supplemental Figure 2.
Cell culture.
Adult feline cardiomyocytes were isolated using a hanging heart preparation with enzymatic digestion, per approval of the Institutional Animal Care and Use Committee of the Ralph H. Johnson Veterans Affairs Medical Center, and cultured as described previously (75). Cardiomyocytes were plated on laminin-coated dishes at an initial density of 7.5 × 104 cells/ml. Cardiomyocytes for early studies were donated by Donald Menick (MUSC).
Free fatty acid treatment.
To prepare stock solutions of SFAs, medium was supplemented with 2% fatty acid–free BSA and either 1.5 mM myristate or 2.0 mM palmitate. Solutions were sonicated briefly, incubated for 15 minutes at 55°C, and cooled to 37°C. For studies in nontransfected cells, cells were incubated in 0.75 mM myristate or 1.0 mM palmitate for 16 hours. For experiments requiring transfection (described below), cells were treated with 0.1 mM myristate in order to minimize lipotoxicity. For experiments utilizing the de novo sphingolipid synthesis inhibitor myriocin or the autophagy inhibitor 3-methyladenine (Sigma-Aldrich), cells were pretreated with 1.0 μM myriocin, 2.5 mM 3-methyladenine, or vehicle for 30 minutes prior to addition of fatty acids. Inhibitors were allowed to remain in the medium for the duration of fatty acid treatment.
siRNA transfection and sphingoid base treatment.
Cardiomyocytes were transfected with siRNA oligonucleotides using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions. Custom feline siRNAs directed against CerS5 and LC3B were purchased from Invitrogen. AllStars Negative Control siRNA was from QIAGEN. At 24 hours after transfection, culture medium was supplemented with a final concentration of 2.5 μM C18-DHS (Matreya) or ethanol, or with 0.1 mM myristate or BSA. Cells were treated with DHS for a total of 3 hours or with myristate for a total of 16 hours.
DNA transfection.
Plasma DNA was transfected into cardiomyocytes using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s protocol. For CerS5 overexpression studies, cells were transfected with empty pcDNA3.0 or pcDNA3.0 containing the human CerS5 cDNA sequence (cloned and validated as described previously, ref. 76) and cultured for 24 hours. The CerS5 construct was provided by Anthony Futerman (Department of Biological Chemistry, Weizmann Institute of Science, Rehovot, Israel). For studies utilizing the GFP-LC3B construct, cells were cotransfected with pSELECT-GFP-LC3 (InvivoGen) and other plasmids or siRNAs, as indicated, and cultured for 24 hours prior to fatty acid treatment, as described above.
RNA isolation and quantitative real-time PCR analysis.
Total RNA was isolated with TRIzol (Invitrogen), according to the manufacturer’s protocol. cDNA was synthesized using SuperScript III (Invitrogen). Quantitative real-time PCR was performed using SYBR Green reagent (Bio-Rad) as described previously (10). Results were normalized to GAPDH. Primers for Map1lc3b, Becn1, Atg7, and GAPDH were obtained from SABiosciences. Feline Cers5 and Becn1 primers were ordered from Integrated DNA Technologies.
Analysis of cardiomyocyte area.
For analysis of isolated cardiomyocyte areas, cells were treated as indicated, fixed with 10% formalin, stained with H&E, and photographed and analyzed as described above. Mean cell area was determined from 10 fields each from a total of 5 to 6 replicates per group, isolated from a total of 2 separate animals, yielding a final total of 150–200 analyzed cells per treatment group. Cells demonstrating evidence of rounding (an indication of cell death) were excluded from analysis. Data are presented as mean ± SEM. Histograms of cell size, as well as analyses of kurtosis and skewness, are provided in Supplemental Figures 3, 4, and 7. Example photomicrographs showing cell size and morphology are provided in Supplemental Figure 9.
Western blotting.
Cells were washed twice with sterile filtered cold PBS, lysed with buffer containing 20 mM Tris (pH 7.5), 150 mM NaCl, 1 mM EDTA, 1 mM EGTA, 2.5 mM sodium pyrophosphate, 1 mM β-D-glycerophosphate, 1 mM sodium orthovanadate, 1% Triton X-100, and Complete Mini Protease Inhibitor (Roche), disrupted by freeze-thaw, and incubated on ice for 10 minutes. Insoluble material was pelleted by centrifugation at 4°C. Protein concentrations were determined using a Micro BCA Protein Assay Kit (Thermo-Fisher). Lysates were subjected to SDS-PAGE and immunoblotted as described previously (10). Membranes were incubated with antibodies against BECN1 (Cell Signaling Technology), GFP (Cell Signaling Technology), or actin (Sigma-Aldrich) and with anti-rabbit secondary antibody (Cell Signaling). Proteins were visualized by enhanced chemiluminescence. Images were quantitated using ImageJ software (NIH). Results were presented as mean ± SEM and shown with a representative immunoblot image.
Fluorescence microscopy of cardiomyocytes.
After transfection and fatty acid treatment (described above), half of the culture medium was removed and saved, and cells were preloaded for 30 minutes with 50 nM LysoTracker Red DND-99 (Invitrogen). The dye-containing medium was then removed and replaced with the dye-free aliquot, and cells were examined by confocal fluorescence microscopy using an Olympus FV10i microscope. To track autophagosome dynamics, images were taken every 5 minutes for 20 minutes at a magnification of ×60 with a zoom setting of ×3.9. Identical laser and sensitivity settings were utilized for all images. Images were analyzed using FV10-ASW software, as described above, using identical threshold and gamma settings for images presented in figures. For quantification of GFP-labeled puncta, cells were formalin fixed in the dark, rinsed in PBS, photographed at a magnification of ×60 with a zoom setting of ×2 and analyzed with FV10-ASW software, as described above. Puncta were counted for 25 cells per treatment group. Results are presented as mean ± SEM.
Lipidomic analysis.
Lipidomic profiling was performed by LC/MS by the Lipidomics Core Facility at MUSC, as described previously (77).
Statistics.
Data are expressed as mean ± SEM. Animal data were analyzed by 1-way ANOVA followed by Bonferroni’s t test. All other data were analyzed by 2-tailed, unpaired Student’s t test. For all experiments, P < 0.05 was considered statistically significant.
Study approval.
All animal protocols were in accordance with the Guide for the Care and Use of Laboratory Animals (NIH publication no. 85-23. Revised 1985) and were approved by the Institutional Animal Care and Use Committee at MUSC.
Supplementary Material
Acknowledgments
We thank Donald Menick for his invaluable support of this project in its early stages; Benjamin Addy and Santhosh Mani for cell culture and technical assistance; Wei Hu for technical assistance; and Yusuf Hannun for helpful comments. Imaging studies were performed using the Cell and Molecular Imaging Shared Resource (supported in part by Cancer Center Support Grant P30 CA138313 to the Hollings Cancer Center at MUSC), with help from Venkat Ramshesh. LC/MS lipid measurements were performed at the Lipidomics Core Facility at MUSC, which is supported by the COBRE in Lipidomics in Pathobiology (NIH P20RR017077). Support for this work was provided by grant number F30DK092125 from the National Institute of Diabetes and Digestive and Kidney Diseases (to S.B. Russo), merit awards from the Department of Veterans Affairs (to L.A. Cowart and M.R. Zile), and the NIH COBRE in Lipidomics and Pathobiology at MUSC (to L.A. Cowart).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):3919–3930. doi:10.1172/JCI63888.
References
- 1.Guha A, Harmancey R, Taegtmeyer H. Nonischemic heart failure in diabetes mellitus. Curr Opin Cardiol. 2008;23(3):241–248. doi: 10.1097/HCO.0b013e3282fcc2fa. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rijzewijk LJ, et al. Myocardial steatosis is an independent predictor of diastolic dysfunction in type 2 diabetes mellitus. J Am Coll Cardiol. 2008;52(22):1793–1799. doi: 10.1016/j.jacc.2008.07.062. [DOI] [PubMed] [Google Scholar]
- 3.Brice SE, Cowart LA. Sphingolipid metabolism and analysis in metabolic disease. Adv Exp Med Biol. 2011;721:1–17. doi: 10.1007/978-1-4614-0650-1_1. [DOI] [PubMed] [Google Scholar]
- 4.Bugger H, Abel ED. Rodent models of diabetic cardiomyopathy. Dis Model Mech. 2009;2(9–10):454–466. doi: 10.1242/dmm.001941. [DOI] [PubMed] [Google Scholar]
- 5.Wright JJ, et al. Mechanisms for increased myocardial fatty acid utilization following short-term high-fat feeding. Cardiovasc Res. 2009;82(2):351–360. doi: 10.1093/cvr/cvp017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Symons JD, et al. Contribution of insulin and Akt1 signaling to endothelial nitric oxide synthase in the regulation of endothelial function and blood pressure. Circ Res. 2009;104(9):1085–1094. doi: 10.1161/CIRCRESAHA.108.189316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Rennison JH, et al. Prolonged exposure to high dietary lipids is not associated with lipotoxicity in heart failure. J Mol Cell Cardiol. 2009;46(6):883–890. doi: 10.1016/j.yjmcc.2009.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ivanova M, et al. Activation of Akt kinase accompanies increased cardiac resistance to ischemia/reperfusion in rats after short-term feeding with lard-based high-fat diet and increased sucrose intake. Nutr Res. 2011;31(8):631–643. doi: 10.1016/j.nutres.2011.08.005. [DOI] [PubMed] [Google Scholar]
- 9.Chess DJ, Lei B, Hoit BD, Azimzadeh AM, Stanley WC. Effects of a high saturated fat diet on cardiac hypertrophy and dysfunction in response to pressure overload. J Card Fail. 2008;14(1):82–88. doi: 10.1016/j.cardfail.2007.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hu W, Bielawski J, Samad F, Merrill AH, JrCowart LA. Palmitate increases sphingosine-1-phosphate in C2C12 myotubes via upregulation of sphingosine kinase message and activity. J Lipid Res. 2009;50(9):1852–1862. doi: 10.1194/jlr.M800635-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hu W, Ross J, Geng T, Brice SE, Cowart LA. Differential regulation of dihydroceramide desaturase by palmitate versus monounsaturated fatty acids: implications for insulin resistance. J Biol Chem. 2011;286(19):16596–16605. doi: 10.1074/jbc.M110.186916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maedler K, Oberholzer J, Bucher P, Spinas GA, Donath MY. Monounsaturated fatty acids prevent the deleterious effects of palmitate and high glucose on human pancreatic beta-cell turnover and function. Diabetes. 2003;52(3):726–733. doi: 10.2337/diabetes.52.3.726. [DOI] [PubMed] [Google Scholar]
- 13.Rhee EP, et al. Lipid profiling identifies a triacylglycerol signature of insulin resistance and improves diabetes prediction in humans. J Clin Invest. 2011;121(4):1402–1411. doi: 10.1172/JCI44442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nestel PJ. Effects of dairy fats within different foods on plasma lipids. J Am Coll Nutr. 2008;27(6):735S–740S. doi: 10.1080/07315724.2008.10719751. [DOI] [PubMed] [Google Scholar]
- 15.Lovejoy JC, et al. Relationship of dietary fat and serum cholesterol ester and phospholipid fatty acids to markers of insulin resistance in men and women with a range of glucose tolerance. Metabolism. 2001;50(1):86–92. doi: 10.1053/meta.2001.19440. [DOI] [PubMed] [Google Scholar]
- 16.Kusunoki M, et al. Relationship between serum concentrations of saturated fatty acids and unsaturated fatty acids and the homeostasis model insulin resistance index in Japanese patients with type 2 diabetes mellitus. J Med Invest. 2007;54(3–4):243–247. doi: 10.2152/jmi.54.243. [DOI] [PubMed] [Google Scholar]
- 17.Ebbesson SO, et al. Individual saturated fatty acids are associated with different components of insulin resistance and glucose metabolism: the GOCADAN study. Int J Circumpolar Health. 2010;69(4):344–351. doi: 10.3402/ijch.v69i4.17669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yamagishi K, Nettleton JA, Folsom AR. Plasma fatty acid composition and incident heart failure in middle-aged adults: the Atherosclerosis Risk in Communities (ARIC) Study. Am Heart J. 2008;156(5):965–974. doi: 10.1016/j.ahj.2008.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu L, Nettleton JA, Bertoni AG, Bluemke DA, Lima JA, Szklo M. Dietary pattern, the metabolic syndrome, and left ventricular mass and systolic function: the Multi-Ethnic Study of Atherosclerosis. Am J Clin Nutr. 2009;90(2):362–368. doi: 10.3945/ajcn.2009.27538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Warensjo E, Sundstrom J, Vessby B, Cederholm T, Riserus U. Markers of dietary fat quality and fatty acid desaturation as predictors of total and cardiovascular mortality: a population-based prospective study. Am J Clin Nutr. 2008;88(1):203–209. doi: 10.1093/ajcn/88.1.203. [DOI] [PubMed] [Google Scholar]
- 21.Riquelme CA, et al. Fatty acids identified in the Burmese python promote beneficial cardiac growth. Science. 2011;334(6055):528–531. doi: 10.1126/science.1210558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.van de Weijer T, Schrauwen-Hinderling VB, Schrauwen P. Lipotoxicity in type 2 diabetic cardiomyopathy. Cardiovasc Res. 2011;92(1):10–18. doi: 10.1093/cvr/cvr212. [DOI] [PubMed] [Google Scholar]
- 23.Park TS, et al. Ceramide is a cardiotoxin in lipotoxic cardiomyopathy. J Lipid Res. 2008;49(10):2101–2112. doi: 10.1194/jlr.M800147-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mizutani Y, Kihara A, Igarashi Y. LASS3 (longevity assurance homologue 3) is a mainly testis-specific (dihydro)ceramide synthase with relatively broad substrate specificity. Biochem J. 2006;398(3):531–538. doi: 10.1042/BJ20060379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Senkal CE, Ponnusamy S, Bielawski J, Hannun YA, Ogretmen B. Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways. FASEB J. 2010;24(1):296. doi: 10.1096/fj.09-135087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hartmann D, et al. Long chain ceramides and very long chain ceramides have opposite effects on human breast and colon cancer cell growth. Int J Biochem Cell Biol. 2012;44(4):620–628. doi: 10.1016/j.biocel.2011.12.019. [DOI] [PubMed] [Google Scholar]
- 27.Stiban J, Tidhar R, Futerman AH. Ceramide synthases: roles in cell physiology and signaling. Adv Exp Med Biol. 2010;688:60–71. doi: 10.1007/978-1-4419-6741-1_4. [DOI] [PubMed] [Google Scholar]
- 28.Mizutani Y, Kihara A, Igarashi Y. Mammalian Lass6 and its related family members regulate synthesis of specific ceramides. Biochem J. 2005;390(pt 1):263–271. doi: 10.1042/BJ20050291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Laviad EL, et al. Characterization of ceramide synthase 2: tissue distribution, substrate specificity, and inhibition by sphingosine 1-phosphate. J Biol Chem. 2008;283(9):5677–5684. doi: 10.1074/jbc.M707386200. [DOI] [PubMed] [Google Scholar]
- 30.Grosch S, Schiffmann S, Geisslinger G. Chain length-specific properties of ceramides. Prog Lipid Res. 2012;51(1):50–62. doi: 10.1016/j.plipres.2011.11.001. [DOI] [PubMed] [Google Scholar]
- 31.Hein S, et al. Progression from compensated hypertrophy to failure in the pressure-overloaded human heart: structural deterioration and compensatory mechanisms. Circulation. 2003;107(7):984–991. doi: 10.1161/01.CIR.0000051865.66123.B7. [DOI] [PubMed] [Google Scholar]
- 32.Kostin S, et al. Myocytes die by multiple mechanisms in failing human hearts. Circ Res. 2003;92(7):715–724. doi: 10.1161/01.RES.0000067471.95890.5C. [DOI] [PubMed] [Google Scholar]
- 33.Nakai A, et al. The role of autophagy in cardiomyocytes in the basal state and in response to hemodynamic stress. Nat Med. 2007;13(5):619–624. doi: 10.1038/nm1574. [DOI] [PubMed] [Google Scholar]
- 34.Zhu H, et al. Cardiac autophagy is a maladaptive response to hemodynamic stress. J Clin Invest. 2007;117(7):1782–1793. doi: 10.1172/JCI27523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ferdous A, Battiprolu PK, Ni YG, Rothermel BA, Hill JA. FoxO, autophagy, and cardiac remodeling. J Cardiovasc Transl Res. 2010;3(4):355–364. doi: 10.1007/s12265-010-9200-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cao DJ, et al. Histone deacetylase (HDAC) inhibitors attenuate cardiac hypertrophy by suppressing autophagy. Proc Natl Acad Sci U S A. 2011;108(10):4123–4128. doi: 10.1073/pnas.1015081108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zheng W, et al. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim Biophys Acta. 2006;1758(12):1864–1884. doi: 10.1016/j.bbamem.2006.08.009. [DOI] [PubMed] [Google Scholar]
- 38.Lavieu G, et al. Is autophagy the key mechanism by which the sphingolipid rheostat controls the cell fate decision? Autophagy. 2007;3(1):45–47. doi: 10.4161/auto.3416. [DOI] [PubMed] [Google Scholar]
- 39.Ponnusamy S, et al. Sphingolipids and cancer: ceramide and sphingosine-1-phosphate in the regulation of cell death and drug resistance. Future Oncol. 2010;6(10):1603–1624. doi: 10.2217/fon.10.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Spassieva SD, Mullen TD, Townsend DM, Obeid LM. Disruption of ceramide synthesis by CerS2 down-regulation leads to autophagy and the unfolded protein response. Biochem J. 2009;424(2):273–283. doi: 10.1042/BJ20090699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klionsky DJ, et al. Guidelines for the use and interpretation of assays for monitoring autophagy in higher eukaryotes. Autophagy. 2008;4(2):151–175. doi: 10.4161/auto.5338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gottlieb RA, Mentzer RM. Autophagy during cardiac stress: joys and frustrations of autophagy. Annu Rev Physiol. 2010;72:45–59. doi: 10.1146/annurev-physiol-021909-135757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujita N, et al. An Atg4B mutant hampers the lipidation of LC3 paralogues and causes defects in autophagosome closure. Mol Biol Cell. 2008;19(11):4651–4659. doi: 10.1091/mbc.E08-03-0312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Petiot A, Ogier-Denis E, Blommaart EF, Meijer AJ, Codogno P. Distinct classes of phosphatidylinositol 3′-kinases are involved in signaling pathways that control macroautophagy in HT-29 cells. J Biol Chem. 2000;275(2):992–998. doi: 10.1074/jbc.275.2.992. [DOI] [PubMed] [Google Scholar]
- 45.Ussher JR, et al. Inhibition of de novo ceramide synthesis reverses diet-induced insulin resistance and enhances whole body oxygen consumption. Diabetes. 2010;59(10):2453–2464. doi: 10.2337/db09-1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Watson ML, Coghlan M, Hundal HS. Modulating serine palmitoyl transferase (SPT) expression and activity unveils a crucial role in lipid-induced insulin resistance in rat skeletal muscle cells. Biochem J. 2009;417(3):791–801. doi: 10.1042/BJ20081149. [DOI] [PubMed] [Google Scholar]
- 47.Yang G, Badeanlou L, Bielawski J, Roberts AJ, Hannun YA, Samad F. Central role of ceramide biosynthesis in body weight regulation, energy metabolism, and the metabolic syndrome. Am J Physiol Endocrinol Metab. 2009;297(1):E211–E224. doi: 10.1152/ajpendo.91014.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Soltys CL, Buchholz L, Gandhi M, Clanachan AS, Walsh K, Dyck JR. Phosphorylation of cardiac protein kinase B is regulated by palmitate. Am J Physiol Heart Circ Physiol. 2002;283(3):H1056–H1064. doi: 10.1152/ajpheart.00275.2002. [DOI] [PubMed] [Google Scholar]
- 49.Zhou YT, et al. Lipotoxic heart disease in obese rats: implications for human obesity. Proc Natl Acad Sci U S A. 2000;97(4):1784–1789. doi: 10.1073/pnas.97.4.1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xie M, Morales CR, Lavandero S, Hill JA. Tuning flux: autophagy as a target of heart disease therapy. Curr Opin Cardiol. 2011;26(3):216–222. doi: 10.1097/HCO.0b013e328345980a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sengupta A, Molkentin JD, Yutzey KE. FoxO transcription factors promote autophagy in cardiomyocytes. J Biol Chem. 2009;284(41):28319–28331. doi: 10.1074/jbc.M109.024406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ni YG, et al. Foxo transcription factors blunt cardiac hypertrophy by inhibiting calcineurin signaling. Circulation. 2006;114(11):1159–1168. doi: 10.1161/CIRCULATIONAHA.106.637124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hariharan N, Maejima Y, Nakae J, Paik J, Depinho RA, Sadoshima J. Deacetylation of FoxO by Sirt1 plays an essential role in mediating starvation-induced autophagy in cardiac myocytes. Circ Res. 2010;107(12):1470–1482. doi: 10.1161/CIRCRESAHA.110.227371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Planavila A, Iglesias R, Giralt M, Villarroya F. Sirt1 acts in association with PPARalpha to protect the heart from hypertrophy, metabolic dysregulation, and inflammation. Cardiovasc Res. 2011;90(2):276–284. doi: 10.1093/cvr/cvq376. [DOI] [PubMed] [Google Scholar]
- 55.Dhalla NS, Saini-Chohan HK, Rodriguez-Leyva D, Elimban V, Dent MR, Tappia PS. Subcellular remodelling may induce cardiac dysfunction in congestive heart failure. Cardiovasc Res. 2009;81(3):429–438. doi: 10.1093/cvr/cvn281. [DOI] [PubMed] [Google Scholar]
- 56.Yorimitsu T, Klionsky DJ. Autophagy: molecular machinery for self-eating. Cell Death Differ. 2005;12(suppl 2):1542–1552. doi: 10.1038/sj.cdd.4401765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Iglewski M, Hill JA, Lavandero S, Rothermel BA. Mitochondrial fission and autophagy in the normal and diseased heart. Curr Hypertens Rep. 2010;12(6):418–425. doi: 10.1007/s11906-010-0147-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dupuy F, Fanani ML, Maggio B. Ceramide N-acyl chain length: a determinant of bidimensional transitions, condensed domain morphology, and interfacial thickness. Langmuir. 2011;27(7):3783–3791. doi: 10.1021/la105011x. [DOI] [PubMed] [Google Scholar]
- 59.Nybond S, Bjorkqvist YJ, Ramstedt B, Slotte JP. Acyl chain length affects ceramide action on sterol/sphingomyelin-rich domains. Biochim Biophys Acta. 2005;1718(1–2):61–66. doi: 10.1016/j.bbamem.2005.10.009. [DOI] [PubMed] [Google Scholar]
- 60.MeghaSawatzki P, Kolter T, Bittman R, London E. Effect of ceramide N-acyl chain and polar headgroup structure on the properties of ordered lipid domains (lipid rafts). Biochim Biophys Acta. 2007;1768(9):2205–2212. doi: 10.1016/j.bbamem.2007.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nyholm TK, Grandell PM, Westerlund B, Slotte JP. Sterol affinity for bilayer membranes is affected by their ceramide content and the ceramide chain length. Biochim Biophys Acta. 2010;1798(5):1008–1013. doi: 10.1016/j.bbamem.2009.12.025. [DOI] [PubMed] [Google Scholar]
- 62.Holopainen JM, Brockman HL, Brown RE, Kinnunen PK. Interfacial interactions of ceramide with dimyristoylphosphatidylcholine: impact of the N-acyl chain. Biophys J. 2001;80(2):765–775. doi: 10.1016/S0006-3495(01)76056-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Goni FM, Alonso A. Effects of ceramide and other simple sphingolipids on membrane lateral structure. Biochim Biophys Acta. 2009;1788(1):169–177. doi: 10.1016/j.bbamem.2008.09.002. [DOI] [PubMed] [Google Scholar]
- 64.Sot J, Aranda FJ, Collado MI, Goni FM, Alonso A. Different effects of long- and short-chain ceramides on the gel-fluid and lamellar-hexagonal transitions of phospholipids: a calorimetric, NMR, and x-ray diffraction study. Biophys J. 2005;88(5):3368–3380. doi: 10.1529/biophysj.104.057851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Montes LR, Ruiz-Arguello MB, Goni FM, Alonso A. Membrane restructuring via ceramide results in enhanced solute efflux. J Biol Chem. 2002;277(14):11788–11794. doi: 10.1074/jbc.M111568200. [DOI] [PubMed] [Google Scholar]
- 66.Silva LC, et al. Ablation of ceramide synthase 2 strongly affects biophysical properties of membranes. J Lipid Res. 2012;53(3):430–436. doi: 10.1194/jlr.M022715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cybulski LE, de Mendoza D. Bilayer hydrophobic thickness and integral membrane protein function. Curr Protein Pept Sci. 2011;12(8):760–766. doi: 10.2174/138920311798841681. [DOI] [PubMed] [Google Scholar]
- 68.Ibarguren M, et al. End-products diacylglycerol and ceramide modulate membrane fusion induced by a phospholipase C/sphingomyelinase from Pseudomonas aeruginosa. Biochim Biophys Acta. 2010;1798(1):59–64. doi: 10.1016/j.bbamem.2009.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Nishio M, et al. Overexpressed GM1 suppresses nerve growth factor (NGF) signals by modulating the intracellular localization of NGF receptors and membrane fluidity in PC12 cells. J Biol Chem. 2004;279(32):33368–33378. doi: 10.1074/jbc.M403816200. [DOI] [PubMed] [Google Scholar]
- 70.Fan W, Nassiri A, Zhong Q. Autophagosome targeting and membrane curvature sensing by Barkor/Atg14(L). Proc Natl Acad Sci U S A. 2011;108(19):7769–7774. doi: 10.1073/pnas.1016472108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bigford GE, Alonso OF, Dietrich D, Keane RW. A novel protein complex in membrane rafts linking the NR2B glutamate receptor and autophagy is disrupted following traumatic brain injury. J Neurotrauma. 2009;26(5):703–720. doi: 10.1089/neu.2008.0783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Maedler K, Spinas GA, Dyntar D, Moritz W, Kaiser N, Donath MY. Distinct effects of saturated and monounsaturated fatty acids on beta-cell turnover and function. Diabetes. 2001;50(1):69–76. doi: 10.2337/diabetes.50.1.69. [DOI] [PubMed] [Google Scholar]
- 73.American Diabetes Association Diagnosis and classification of diabetes mellitus. Diabetes Care. 2006;29(suppl 1):S43–S48. [PubMed] [Google Scholar]
- 74.Ikonomidis JS, et al. Accelerated LV remodeling after myocardial infarction in TIMP-1-deficient mice: effects of exogenous MMP inhibition. Am J Physiol Heart Circ Physiol. 2005;288(1):H149–H158. doi: 10.1152/ajpheart.00370.2004. [DOI] [PubMed] [Google Scholar]
- 75.Kent RL, Mann DL, Urabe Y, Hisano R, Hewett KW, Loughnane M. Contractile function of isolated feline cardiocytes in response to viscous loading. Am J Physiol. 1989;257(5 pt 2):H1717–H1727. doi: 10.1152/ajpheart.1989.257.5.H1717. [DOI] [PubMed] [Google Scholar]
- 76.Riebeling C, Allegood JC, Wang E, Merrill AH, Jr, Futerman AH. Two mammalian longevity assurance gene (LAG1) family members, trh1 and trh4, regulate dihydroceramide synthesis using different fatty acyl-CoA donors. J Biol Chem. 2003;278(44):43452–43459. doi: 10.1074/jbc.M307104200. [DOI] [PubMed] [Google Scholar]
- 77.Bielawski J, Szulc ZM, Hannun YA, Bielawska A. Simultaneous quantitative analysis of bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Methods. 2006;39(2):82–91. doi: 10.1016/j.ymeth.2006.05.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.