

Abstract
The HELLP syndrome is a pregnancy-associated disease inducing hemolysis, elevated liver enzymes, and low platelets in the mother. Although the HELLP symptoms occur in the third trimester in the mother, the origin of the disease can be found in the first trimester fetal placenta. A locus for the HELLP syndrome is present on chromosome 12q23 near PAH. Here, by multipoint nonparametric linkage, pedigree structure allele sharing, and haplotype association analysis of affected sisters and cousins, we demonstrate that the HELLP locus is in an intergenic region on 12q23.2 between PMCH and IGF1. We identified a novel long intergenic noncoding RNA (lincRNA) transcript of 205,012 bases with (peri)nuclear expression in the extravillous trophoblast using strand-specific RT-PCR complemented with RACE and FISH. siRNA-mediated knockdown followed by RNA-sequencing, revealed that the HELLP lincRNA activated a large set of genes that are involved in the cell cycle. Furthermore, blocking potential mutation sites identified in HELLP families decreased the invasion capacity of extravillous trophoblasts. This is the first large noncoding gene to be linked to a Mendelian disorder with autosomal-recessive inheritance.
Introduction
Preeclampsia (new-onset hypertension in pregnancy with proteinuria) and hemolysis, elevated liver enzymes, low platelets (HELLP) syndrome share a similar onset in the early placenta (1–3). Local placental dysfunction is followed by activation of compensatory pathways in the placenta (PlGF-sFLT1-endoglin axis) and mother (TGF-β–mediated NOS-dependent vasodilation) that ultimately trigger the appearance of systemic maternal symptoms (4–6). Genetically, the HELLP syndrome and preeclampsia are distinct entities (7). The familiar forms of preeclampsia involve the STOX1 gene (8). The HELLP gene is unknown, but 3 putative loci have been identified by genome-wide linkage analysis (7). Given the consistent observation in monozygotic parous twins that lack of concordancy for proteinuric hypertension is the rule rather than the exception, we searched these loci for the presence and identity of the HELLP gene, with the assumption that fetal (i.e., placental) contributions are essential for any genetic basis of HELLP (9).
Here we showed, by multipoint nonparametric linkage, pedigree structure allele sharing, and haplotype association analysis of affected sisters and cousins, that the HELLP locus resides in an intergenic region on chromosome 12q23.2 between PMCH and IGF1. By strand-specific RT-PCR complemented with rapid amplification of cDNA ends (RACE) and FISH, a novel long intergenic noncoding RNA (lincRNA) transcript with expression in the placental extravillous trophoblast was identified. By siRNA-mediated knockdown followed by RNA sequencing, the HELLP lincRNA was found to be involved in the cell cycle. By using morpholinos to block potential mutation sites, as identified in HELLP families, we observed a decreased invasion capacity of extravillous trophoblasts.
Results
Analysis of HELLP families reveals linkage to an intergenic region on 12q23.
Using the original cohort of families with the HELLP syndrome (n = 34; ref. 7), the 3 loci previously found to have nominal linkage were reanalyzed using additional microsatellite markers. The nonparametric lod scores for 12p12 and 20p12 decreased (from 1.55 to 1.08) and disappeared (from 1.70 to 0.37), respectively. The lod score for the 12q23 region increased from nominal to suggestive (i.e., from 2.1 to 2.37).
We subsequently tested 57 individuals (7 families with affected sib-pairs, 4 families with affected cousin-pairs, and 2 discordant monozygous twin sisters with their partners, of which 36 females were affected) with 26 microsatellite markers (D12S309–D12S395) in the 23.6-Mb region on 12q23. Nonparametric multipoint linkage analysis using Smnallele confirmed the 4-marker region between PAH and D12S1647 (lod and NPL scores >3; Figure 1A) and indicated recessive inheritance (in descending order: Smnallele, Spairs, Sall, Srobdom; ref. 10). Pedigree analysis narrowed this region to 2 minimal critical regions: D12S1607–PAH and D12S338–D123S317, each about 1 Mb. In the highly informative family 93113, in which 2 sisters married 2 brothers from an unrelated family, maximal allele sharing in all affected females from 2 generations (cousins and their mothers) was restricted to the first region near D12S1030 (Supplemental File 1; supplemental material available online with this article; doi: 10.1172/JCI65171DS1).
Figure 1. Identification of the HELLP locus on chromosome 12q23.2.

Multipoint nonparametric linkage analysis (A; see also Supplemental File 1), pedigree structure analysis with SNP/INDEL markers (B; see also Supplemental File 2), and HaploView analysis (C) defined the HELLP locus region as residing in a intergenic region of about 170 kb between C12orf48 and IGF1 on 12q23.2.
We found no mutations in the coding sequences of the 38 known and predicted genes within or near these 2 regions: DRAM, CCDC53, NUP37, C12orf48 (also referred to as PARPBP), PMCH, IGF1, PAH, ASCL1, C12orf42, BC041342, STAB2, NT5DC3, LOC253724 (also referred to as GNN), HSP90B1, TDG, GLT8D2, HCFC2, NFYB, TXNRD1, EID3, CHST11, BC030271, SLC41A2, C12orf45, APPL2, OCC-1, RFX4, ACACB, FOXN4, UBE3B, ANAPC7, CCDC63, RPH3A, OAS1, P/OKCL.4, OAS2, DDX54, and LHX5. Instead, using the 181 SNPs and insertion/deletion polymorphisms (INDEL) identified, we found the HELLP locus to reside in an intergenic region of 154 kb (chromosome 12: 101,114,674–101,268,434 bp; UCSC assembly NCBI36/hg18) between C12orf48 and IGF1 (Figure 1B and Supplemental File 2).
We confirmed this region by haplotype association analysis (HaploView) following deep sequencing. We tested 26 singletons (sisters and cousins) with 405 SNP markers. We took advantage of the placental genotype-maternal phenotype discrepancy characteristic for preeclampsia and HELLP (the maternal endophenotype is caused by the placental effector genotype, i.e., the genotype of the child born from the affected pregnancy), as this permitted case-control association analysis within the family cohort. Phenotypically affected females born from phenotypically affected mothers were genetically considered cases, as the disease genotypes of the former are homozygous or compound heterozygous; phenotypically affected females born from nonaffected mothers were considered controls, as the former are unaffected, heterozygous carriers. Using the “Solid Spine of LD” algorithm to evaluate blocks of linkage disequilibrium (LD) with a minimum D′ value of 0.8, the single block identified (chromosome 12: 101,150,849–101,320,921 bp) confirmed that the HELLP gene was present within the intergenic region between C12orf48 and IGF1 on 12q23.2 (Figure 1C), a finding that was confirmed to be significant by permutation testing (P = 0.0334; n = 5,000).
Discordant monozygotic twins confirm placental origin of the HELLP syndrome.
In the discordant monozygotic twin sister family, the HELLP linkage region presented as a cluster of minor alleles with heterozygous sharing between the affected twin sister and her partner, while completely absent in the partner of the nonaffected twin sister. Using 3 informative SNPs in this cluster, we observed complete agreement with the placental model: the presence of a fetal susceptibility gene, expressed in placenta cells in contact with maternal vessels, determined the maternal phenotype (Figure 2).
Figure 2. Parent-child segregation of 12q23.2 region in monozygotic twin sisters discordant for HELLP.

3 informative SNPs located within the HELLP region with linkage were analyzed for parent-child segregation in discordant, monozygotic twin sisters. Only 1 sister (patient 1) developed the HELLP syndrome during pregnancy. The other sister (patient 3) had 2 normal pregnancies. The minor alleles (red) were homozygous in the child born of affected patient 1 (patient A). In contrast, homozygosity for the major alleles (green) was seen in the children born of normal patient 3 (patients B and C). This pattern supports the presence of a placental susceptibility gene for the HELLP syndrome, in which the fetal genotype expressed in the placenta determines the maternal phenotype.
The HELLP transcript is a lincRNA expressed in extravillous trophoblasts.
We screened the intergenic region for transcription in SGHPL-5, a diploid cell line representative of first-trimester extravillous trophoblast, the fetal cell central in the etiology of preeclampsia and HELLP (11). We performed a complete analysis of the number, size, 5′- and 3′-ends, splicing pattern, and cellular location of the transcript(s) involved. This identified a single, unspliced large transcript with 5′-cap structure and 3′-polyA-tail of 205,012 bases (chromosome 12: 101,115,493–101,320,504 bp; UCSC assembly hg18) with no coding potential (CPC, –0.874346; ref. 12 and Figure 3). By FISH, expression was found to be nuclear, perinuclear, and cytoplasmic, both in vitro — in the extravillous trophoblast cell line (SGHPL-5) used for its identification (Figure 4, A–C) — and in vivo (i.e., first trimester placenta tissue), whereas no signal was detected using a sense probe as negative control (Figure 4D). In the anchoring villi of human first-trimester placenta tissue, (peri)nuclear expression was restricted to extravillous trophoblast cells and their precursors (i.e., column extravillous cytotrophoblasts; Figure 4E). In first-trimester placentabed showing a front of extravillous trophoblast invasion into the myometrium, invasive extravillous trophoblast cells actively involved in maternal spiral artery modification showed a distinct difference in subcellular localization: predominantly nuclear in the endovascular trophoblasts within maternal spiral arteries, while perinuclear and cytoplasmic in the interstitial trophoblast surrounding maternal spiral arteries (Figure 4, F and G). Comparison with the transcriptome assemblies of 17 adult tissues (in addition to placenta) in the human lincRNA catalog ( http://www.broadinstitute.org/genome_bio/human_lincrnas/) indicated that the HELLP lincRNA was preferentially, if not exclusively, expressed in the placenta (Supplemental File 3).
Figure 3. Location of the HELLP lincRNA on chromosome 12q23, identified by overlapping strand-specific PCR amplifications supplemented by RACE experiments to identify the 5′ and 3′ ends.

Figure 4. Localization of the HELLP lincRNA.

FISH showed nuclear (A), perinuclear (B), and cytoplasmic (C) localization in SGHPL-5 cells; (D) no signals were seen using a sense probe as negative control. (E) In first-trimester placental tissue, nuclear and perinuclear expression was found in the anchoring villous, consisting of column extravillous trophoblasts and villous cytotrophoblasts. (F) In first-trimester placentabed, localization was found to be nuclear in endovascular extravillous trophoblasts (arrows), whereas interstitial trophoblasts showed predominant perinuclear and cytoplasmic expression (arrowheads). (G) First-trimester placentabed showing the front (dotted line) of the extravillous trophoblast invasion into the myometrium. Inset: nuclear endovascular expression of the HELLP lincRNA (enlarged ×4.5). Scale bars: 5 μm (A–D); 30 μm (E and F); 100 μm (G).
Genome-wide RNA sequencing indicates involvement in the cell cycle.
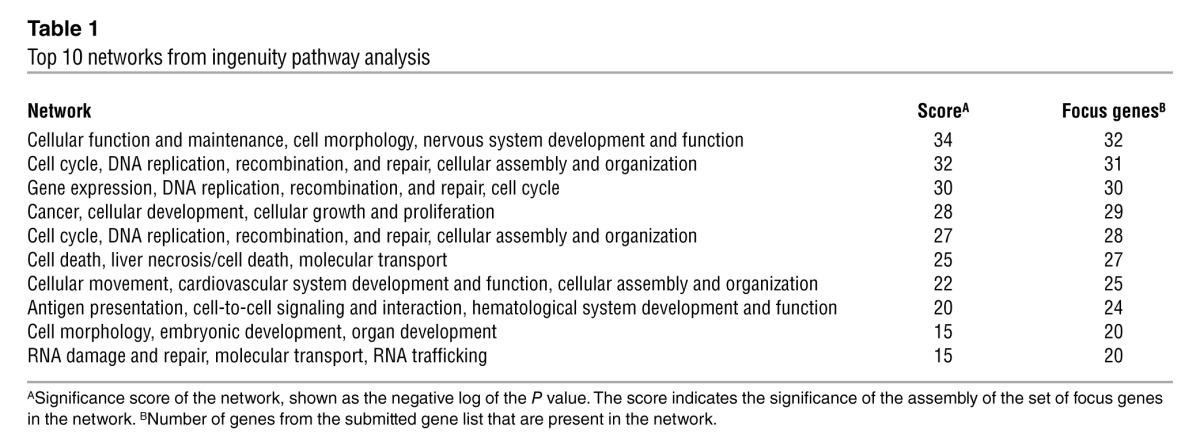
Given its unknown function, its large size, and the absence of additional landmarks preventing prioritization of regions for functional and mutational analyses, we performed genome-wide RNA-sequence (RNA-Seq) analysis after siRNA-mediated downregulation of the HELLP transcript in SGHPL-5 cells. The differential expression induced after HELLP transcript knockdown was analyzed using TopHat and Cufflinks software, calculating the significant differential expressions at the gene level. In this list, 3 significant gene_id hits corresponded with downregulation of the HELLP transcript on chromosome 12q23. These hits had q values (P value corrected for false discovery rate [FDR]) of 0.019, 0.030, and 0.031 and showed expression values in the control sample of 0.21, 0.12, and 0.15, respectively, whereas the siRNA-mediated knockdown sample value was 0 for all 3 hits. For subsequent validation analysis, the following selection criteria were used: q value threshold, <0.05; log2 fold change, ≥2; and a value of at least 1 of either the control sample or the siRNA-mediated knockdown sample. Furthermore, unannotated transcripts were excluded. By cross-checking the list of genes with the results obtained in an untransfected sample, the hits originating from transfection effects were omitted. This yielded 4 upregulated genes and 1,364 downregulated genes upon knockdown of the HELLP transcript. Validation of the RNA-Seq results was performed by quantitative RT-PCR assays on 14 transcripts, including all 4 upregulated genes, 5 downregulated genes with a q value of 0, a selection of 3 genes with q values between 0 and 0.05, and a gene that did not get through the additional selection criteria (GRB10) as a negative control. One of the upregulated genes, AZIN1, consists of 2 transcripts of which only 1 is upregulated, while the other is downregulated but with a log2 fold change of –1.47; therefore, this gene also did not get through the additional selection criteria. Quantitative RT-PCR (Supplemental File 4) showed that differential expression could be validated for genes with a q value below 0.01, with one exception; the upregulated gene MEG8 could not be validated with the opposite expression pattern observed likely to be caused and complicated by the 35 SNORD genes downstream of MEG8. The remaining transcripts — 1 with upregulated expression and 8 with downregulated expression after siRNA-mediated knockdown — could be validated. The gene list selected with the criteria used for validation analysis was accordingly adjusted to a q value threshold of <0.01. This validated set, excluding MEG8, consisted of 1,198 upregulated genes and 1 downregulated gene (Supplemental File 5) and was submitted to Ingenuity Pathway Analysis (IPA; see Methods). The top 10 networks from network analysis showed they were predominantly associated with the cell cycle (Table 1). Furthermore, function annotation analysis revealed significant increases in functions related to G1/S phase and cell death, whereas significant decreases were found for functions related to G2/M phase, cell survival, and migration (Supplemental File 6).
Table 1.
Top 10 networks from ingenuity pathway analysis
Blocking potential mutation sites decreases extravillous trophoblast invasion.
Although we have not yet identified all disease-causing mutations in the complete set of HELLP families, we identified a couple that segregate correctly with the disease phenotype in all generations with compound heterozygosity in the patients involved. These sequence variants, RW18-5 (C→T at position 101,120,459), HAPLO378 (A→G at position 101,319,702), and HAPLO215Rev (G→C at position 101,241,930), were not found in 200 control chromosomes of nonaffected individuals of similar race and therefore qualified as mutations (Figure 5A). Functional studies were performed using morpholinos to block the regions containing these sites. Validation of their experimental effect showed that blocking the different mutation sites using optimal morpholino concentrations (5 μM) seem to prevent HELLP lincRNA degradation, leading to a net increase in HELLP lincRNA levels (Figure 5B). Using the combination of HAPLO378 with HAPLO215Rev morpholinos (being compound heterozygotes in family 9265) with a concentration of 2.5 μM each did not show this effect as clearly. Single morpholinos at a concentration of 2.5 μM could also not show this increase in HELLP lincRNA levels (data not shown). If these sites are of functional importance and critical in the pathophysiology of the HELLP syndrome, the morpholino blocking effects should (a) be seen in trophoblast cells; (b) affect the expression of the same genes as identified by RNA-Seq, but in the opposite direction, as blocking shows an upregulation of the HELLP lincRNA instead of siRNA-mediated knockdown; (c) exhibit a greater effect when the mutations that segregate in the same patient are tested in combination. Finally, the transcriptional effects should be followed by a functional cellular effect, i.e., defective trophoblast invasion. This was exactly the situation observed when using the different morpholinos separately or in combination. The increase in HELLP lincRNA was followed by increased transcript levels of downstream effector genes when using the RW18-5 or HAPLO215Rev morpholino, while this was not observed for HAPLO378 (Figure 5C). In addition, while the individual effects of suboptimal doses (2.5 μM) of the morpholinos when tested separately were incomplete (data not shown), the effect was complete when HAPLO378 and HAPLO215Rev were tested in combination. Finally, we performed Matrigel invasion assays in the SGHPL-5 cells to test whether these transcriptional changes are accompanied by a phenotypic effect mimicking the primary clinical defect (i.e., decrease in trophoblast invasion). The results confirmed our findings: HAPLO378 did not have an effect on invasion, whereas RW18-5 significantly decreased the number of invaded cells, as did HAPLO378 and HAPLO215Rev in combination at 2.5 μM each. Furthermore, HAPLO215Rev showed a clear trend toward a decrease in invaded cells (P = 0.0645; Figure 5D).
Figure 5. Blocking potential mutation sites decreases extravillous trophoblast invasion.

(A) 3 potential mutations were investigated in detail. RW18-5 (C→T at position 101,120,459) occurred in family 9401; the mutation on the other allele has not yet been identified. HAPLO378 (A→G at position 101,319,702) and HAPLO215Rev (G→C at position 101,241,930) occurred as compound heterozygous mutations in family 9265. HAPLO378 was also present in family 93113. Women affected by HELLP are indicated by black symbols; children born of HELLP pregnancies are indicated by gray symbols. Potential mutagenic alleles are also shaded gray. (B) Morpholinos at a concentration of 5 μM blocking the potential mutagenic sites upregulated expression of the HELLP lincRNA in SGHPL-5 cells. (C) Upregulation of the HELLP lincRNA led to upregulation of downstream effector genes that showed downregulation after siRNA-mediated knockdown of the transcript when the RW18-5 or HAPLO215Rev morpholino was used at a concentration of 5 μM. The combination of HAPLO378 and HAPLO215Rev (2.5 μM each) also upregulated the downstream effector genes. (D) Invasion of SGHPL-5 cells significantly decreased when RW18-5 morpholinos were introduced, whereas HAPLO215Rev trended toward a significant decrease in invasion. HAPLO378 did not affect invasion. The combination of HAPLO215Rev and HAPLO378 (2.5 μM each) significantly decreased the amount of invaded cells. *P < 0.05; **P < 0.01.
Discussion
Here we show by multipoint nonparametric linkage, pedigree structure allele sharing, and haplotype association analysis of HELLP-affected sisters and cousins that the locus of the HELLP syndrome resides in an intergenic region on chromosome 12q23.2 between PMCH and IGF1. By strand-specific RT-PCR complemented with RACE experiments, we identified an unspliced noncoding RNA transcript of 205 kb in length located within this intergenic region. The transcript was furthermore found to be expressed in extravillous trophoblasts. In first-trimester placenta, the expression was predominantly nuclear in the endovascular extravillous trophoblasts, while interstitial extravillous trophoblasts mainly showed perinuclear and cytoplasmic expression. Results of whole-genome RNA-Seq indicated that the HELLP lincRNA is involved in 1 or more processes in the cell cycle, in which siRNA-mediated knockdown of the transcript leads to reduced activity of the G2/M phase of the cell cycle, while the G1/S phase shows an increase in activity.
By using morpholinos to block potential mutation sites as identified in HELLP families, we found that they were able to upregulate the expression of the HELLP lincRNA. This suggests that the morpholinos are blocking the breakdown of the transcript itself. Consistent with upregulated expression of the lincRNA, both the RW18-5 and especially the HAPLO215Rev morpholino were able to upregulate downstream effector genes found to be downregulated upon siRNA-mediated knockdown of the transcript. The combination of HAPLO378 and HAPLO215Rev morpholinos, as found in one of the HELLP families, showed similar effects. Finally, by using invasion assays, we found that morpholino RW18-5 and the combination of HAPLO378 with HAPLO215Rev significantly decreased the amount of invaded extravillous trophoblast cells, whereas HAPLO215Rev showed a trend toward a reduction in extravillous trophoblast invasion. This would fit the placental origin of the HELLP syndrome, in which extravillous trophoblasts show reduced invasion into the maternal decidua, caused by reduced differentiation of the extravillous trophoblasts from a proliferative toward an invasive phenotype. This concept was already indicated by the RNA-Seq results showing effects on the cell cycle, in which siRNA-mediated knockdown of the HELLP lincRNA led to decreased activity of the G2/M phase, whereas the G1/S phase showed increased activity.
In summary, we showed a region on chromosome 12q23 to be associated with the HELLP syndrome by genome-wide linkage analysis of families with the disease. The region contains a noncoding RNA transcript more than 205 kb in length. This lincRNA was localized in early placenta extravillous trophoblasts and, by genome-wide RNA analysis followed by pathway analysis, found to classify as an activator affecting a large set of genes involved in the cell cycle, in particular in activating the G2/M phase while deactivating the G1/S phase. Blocking potential mutation sites as identified in HELLP families decreased the invasion capacity of extravillous trophoblasts, consistent with the placental origin of the HELLP syndrome. lincRNAs have so far been implicated in chromatin remodeling, transcriptional control, and post-transcriptional processing (13–15). The next step is to identify the precise function of this lincRNA as a regulating noncoding RNA and its dysfunction in the HELLP syndrome.
Methods
Patient recruitment.
In our genetic studies, we included all HELLP families available to us that included at least 2 affected females (sisters or cousins). The original cohort of families (n = 67) with preeclampsia, eclampsia, the HELLP syndrome, or pregnancy-induced hypertension (PIH), recruited from 22 hospitals in the Netherlands and used for genome-wide linkage analysis, has been described elsewhere (7). We collected blood samples with informed consent of all affected females and their parents, when available. We collected blood samples from a monozygotic twin sister pair and their partners. The twin sister pair was discordant for the HELLP syndrome: only one developed the disease during pregnancy. The nonaffected twin sister had two normal pregnancies. We collected buccal swabs from all children available to us born from affected pregnancies and, if available, from nonaffected pregnancies from the same families.
In order for a family to be included, the clinical profiles of the affected females had to meet the strict criteria: (a) LDH ≥600 IU/l, ASAT ≥70 IU/l, ALAT ≥70 IU/l, and ≤100 platelets × 109/l; (b) at least 2 affected females in each family (single exception: monozygotic twin sisters); and (c) DNA available from at least 2 generations. For most families, DNA was available from 3 generations. All families were of mixed European descent, with no apparent relationship among families except for family 93113, in which 2 brothers married 2 sisters from an unrelated family. The daughters born from these couples both developed HELLP. These double–first cousins were informative for the fact that the IBD alleles shared were maximal within the minimal critical region. Ethics committee approval was secured from all participating hospitals. All of the participants or their legal representatives gave informed consent.
Multipoint linkage analysis with microsatellite markers.
We performed nonparametric allele-sharing linkage analysis with 26 microsatellite markers located on 12q23 between D12S309 and D12S395 on 53 individuals (35 affected females). The marker alleles in individual samples were analyzed by PCR with oligonucleotide primers labeled with 6-FAM or HEX. PCR product lengths were determined by capillary electrophoresis using ABI3100 or ABI3130XL genetic analyzer (Applied Biosystems) and analyzed using GeneScan or Genemapper (version 4.0; Applied Biosystems). DNA from Centre d’Etude du Polymorphisme Humain (CEPH) individual 1347/02 was considered the standard for DNA fragment lengths. Nonparametric lod scores were calculated using Allegro version 2 (16). Allele frequencies were obtained from the CEPH genotype database (version 10.0; www.cephb.fr). The order and position of the markers were derived from the Decode genetic map (17). Marker, linkage, and family data are shown in Supplemental File 1.
Identity-by-descent allele-sharing analysis with SNPs and INDEL polymorphism markers.
We designed primers using Primer 3 ( http://frodo.wi.mit.edu) or ExonPrimer ( http://ihg.gsf.de/ihg/ExonPrimer.html) to amplify exons of the 38 known and predicted genes in the regions with linkage. We analyzed exon fragments by cycle sequencing on an ABI3130XL Genetic Analyzer (Applied Biosystems). We analyzed the data by Sequencing Analysis (version 3.7; Applied Biosystems) or 4Peaks software ( http://www.mekentosj.com/science/4peaks). The SNPs and INDEL polymorphisms we identified were used for pedigree structure analysis of the regions with nonparametric lod scores >3 (Supplemental File 2).
Haplotype block analysis.
Tag SNPs were designed with Tagger (18) and sequenced as described above. See Supplemental File 7 for primer sequences. Case-control association analysis was performed for the familial cohort studied. Affected females born from affected mothers were considered cases (homozygous or compound heterozygous patients); affected females born from nonaffected mothers were considered controls (heterozygous carriers). We tested 26 singletons (sisters and cousins) with 405 SNP markers. We used Haploview version 4.2 (19) to evaluate blocks of LD using the “Solid Spine of LD” algorithm with a minimum D′ value of 0.8. The Solid Spine of LD method internal to Haploview defines a block when the first and last markers are in strong LD with all intermediate markers. Marker settings were as follows: HW P value, cutoff 1.0; minimum percentage genotype, 100; maximum number Mendelian errors, 1; minor allele frequency, 0.05.
Detailed transcript analysis.
From the extravillous trophoblast cell line SGHPL-5 (11), provided by J. Cartwright (St. George’s University of London, London, United Kingdom), RNA was isolated using the RNeasy mini kit (Qiagen) including on-column DNase treatment, and reverse transcription was performed using the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) using random hexamers as well as gene-specific reverse primers to confirm strand specificity. Overlapping PCRs covering the complete area between the genes PMCH and IGF1 was performed on cDNA using Platinum Taq DNA Polymerase High Fidelity (Invitrogen). To cover the complete transcript, 42 PCRs were needed yielding 2- to 7-kb PCR products. Confirmation of the correct products amplified was done by sequencing using BigDye Terminators and analyzed on an ABI3130XL genetic analyzer (Applied Biosystems). On the 5′ and 3′ ends of the transcript, where PCR products could no longer be amplified, 5′and 3′ RACE experiments were performed using the Generacer kit according to the manufacturer’s instructions (Invitrogen). The exact transcript boundaries were confirmed by sequencing. See Supplemental File 7 for primer sequences. The HELLP transcript sequence described herein was deposited in GenBank (accession no. JX088243).
Placental tissue collection.
First-trimester placentas were obtained at the time of elective termination of pregnancy. Informed consent was obtained from each patient, and collections were approved by the Ethical Committee of the VU University Medical Center and the Flevoziekenhuis Almere. Tissue was collected into ice-cold PBS. After extensive washing the placental tissues were submerged in Tissue-Tek and plated at –80°C.
FISH.
RNA FISH was carried out on SGHPL-5 cells and 5-μm-thin placental tissue cryostat sections, using oligonucleotide probes (Supplemental File 7) against the HELLP lincRNA labeled with digoxigenin (DIG) at the 5′ and 3′ end (Eurogentec), as described previously (20). Briefly, after fixation, slides were prehybridized for 30 minutes at 41°C (25°C below the Tm of the probe) in a humidified chamber with prehybridization buffer, then incubated with hybridization buffer containing an antisense or a sense probe with final concentrations of 100 ng/ml for 1 hour. Posthybridization washes with SSC buffer were done at 5°C higher than the hybridization temperature. Slides were incubated in blocking buffer (Roche) for 30 minutes, followed by anti-DIG/HRP antibody (Roche) preabsorbed at 1:500 dilution for another 30 minutes in a humidified chamber at room temperature. The signals were amplified using tyramide signal amplification (TSA, 1:50 dilution; PerkinElmer) for 5 minutes in a humidified chamber. Slides were dehydrated in ethanol and mounted in Vectashield containing DAPI nuclear stain (Vector Laboratories). Visualization was performed on a Leica DM5000B microscope.
siRNA-mediated knockdown.
The SGHPL-5 cell line was transfected with either scrambled siRNA or a combination of 4 siRNAs against the lincRNA evenly spread across the transcript (Supplemental File 7), using Lipofectamine RNAiMAX according to the manufacturer’s instructions (Invitrogen). siRNA knockdown was assessed by quantitative RT-PCR on RNA from transfected cells using primers and a probe (Supplemental File 7) specific for the transcript using the Taqman EZ RT-PCR kit (Applied Biosystems). The log2 fold change knockdown value was at least –5 in all transfections used.
RNA-Seq and pathway analysis after siRNA-mediated downregulation.
RNA-Seq was performed on 1 untransfected sample and 2 independent transfection couples consisting of a scrambled control sample and an siRNA-mediated knockdown sample as described above. Total RNA (1 μg/sample, DNase-treated, RIN ≥9.8) was subjected to a double round of poly-A mRNA purification, fragmented, and primed for cDNA library synthesis using the TruSeq RNA sample preparation kit (FC-122-1001). All procedures were done according to the manufacturer’s instructions (Illumina). Following validation (Agilent 2100 Bioanalyzer, DNA High Sensitivity) and normalization (AUC 200- to 500-bp fragments), samples were clustered (TruSeq paired-end cluster kit v3-cBot-HS, PE-401-3001) followed by paired-end sequencing (100 bp; TruSeq SBS kit v3-HS 200 cycles, FC-401-3001) on a HiSeq2000. To maximize coverage with inclusion of low-abundance transcripts (21, 22), each lane contained a single DNA library. Cluster densities were 623–970 K/mm2, q scores (≥Q30) 43.2%–80.5%, and FastQ output 44.44–69.17 gigabase for both forward and reverse reads. RNA-Seq reads were aligned to the preassembled reference genome (Illumina iGenome, data source UCSC assembly hg18; June 20, 2011) using Tophat (version 1.4.0) in combination with Bowtie (version 0.12.5) and SAMtools (version 0.1.18) using the default settings (23). Transcript assembly, abundance estimation (defined as fragments per kilobase of exon per million fragments mapped; FPKM), and differential expression was performed by sequential analysis of Tophat output (accepted_hits.bam). For this, transcripts were assembled using Cufflinks (version 1.3.0) under conditions (RABT assembly; ref. 24) permitting the identification of novel unannotated transcripts (transcripts.gtf) and with correction for fragment bias to account for biases in library preparation (25). The assemblies to be compared were merged (Cuffmerge), generating a transcript index (merged.gtf). Subsequently, differential analysis of significant changes in transcript expression, splicing and promoter use was performed (Cuffdiff) in the different transfection couples (scrambled vs. siRNA-mediated knockdown). All transfected samples were also compared with the untransfected sample. These data were checked for the occurrence of the HELLP lincRNA and downregulation of this transcript in the siRNA-mediated knockdown samples. The transfection couple of scrambled and siRNA-mediated knockdown samples with the highest difference was used in subsequent validation and pathway analysis in combination with the analyses with the untransfected sample to remove transfection-induced effects. Genes with significant differential expression were sorted by q value (P value corrected for FDR) and log2 (fold change). IPA (build 140500; Ingenuity Systems Inc.) was performed on the gene list with differentially expressed genes, including the additional selection criteria described above.
Validation experiments by quantitative RT-PCR.
Quantitative RT-PCR using gene expression assays (Applied Biosystems) for MEG8, GRB10, HMMR, USP1, NUF2, ERBB2IP, C12orf48, SMC2, RECQL, MRE11A, CRYAB, LOC388564, AZIN1 (transcript 1), and AZIN1 (transcript 2) were performed on an ABI7300. Normalization was done with gene expression assays for GAPDH.
Morpholino-mediated transcript block and Matrigel invasion assays.
Morpholinos blocking 3 different potential mutation sites (Supplemental File 7) and the standard negative control morpholino (Gene Tools) were delivered into the SGHPL-5 cell line using EndoPorter (Gene Tools) according to the manufacturer’s instructions. Efficient morpholino delivery was monitored using a fluorescein-labeled control. Cytotoxicity was observed at morpholino concentrations greater than 5 μM. RNA was isolated after 48 hours, followed by quantitative RT-PCR experiments for the HELLP lincRNA, HMMR, USP1, NUF2, ERBB2IP, C12orf48, SMC2, RECQL, and MRE11A and normalized for GAPDH, all as described above. Data were obtained from 3 independent experiments. In the invasion assays, morpholinos were delivered in serum-starved SGHPL-5 cells using EndoPorter. 50,000 cells were plated on 100 μl diluted Matrigel-coated (BD) 8.0-μm cell culture inserts, and the cells invaded for 48 hours. The insert membranes were fixed, covered in Vectashield with DAPI (Vector Laboratories), and coverslipped, after which the whole underside membrane was counted. Data were obtained from 6 independent experiments.
Statistics.
Unless otherwise indicated, statistical analyses were performed using Graphpad Prism software and a 1-sample t test with theoretical mean of 0. P values less than 0.05 were considered significant. Results are presented as mean ± SEM.
Study approval.
The studies performed on human samples were approved by the Ethical Committee of the VU University Medical Center and, in the case of placental tissues, also by the Ethical Committee of the Flevoziekenhuis Almere. Informed consent was obtained from all patients.
Supplementary Material
Acknowledgments
We are grateful for the collaboration of many colleagues and, most of all, for the contributions of the HELLP patients and their families. We further thank the donors and all participating staff of the Flevoziekenhuis Almere for the human placental tissue specimens used in this study. This work was supported by the Netherlands Organization for Scientific Research (NWO grants no. 950-10-612 and 91611177), European Union (SAFE LSHB-CT-2004-503243), and Foundation for Translational Research (STR).
Footnotes
Conflict of interest: The authors have declared that no conflict of interest exists.
Citation for this article: J Clin Invest. 2012;122(11):4003–4011. doi:10.1172/JCI65171.
See the related Commentary beginning on page 3837.
References
- 1.Baxter JK, Weinstein L. HELLP syndrome: the state of the art. Obstet Gynecol Surv. 2004;59(12):838–845. doi: 10.1097/01.ogx.0000146948.19308.c5. [DOI] [PubMed] [Google Scholar]
- 2.Sibai B, Dekker G, Kupferminc M. Pre-eclampsia. Lancet. 2005;365(9461):785–799. doi: 10.1016/S0140-6736(05)17987-2. [DOI] [PubMed] [Google Scholar]
- 3.Redman CW, Sargent IL. Latest advances in understanding preeclampsia. Science. 2005;308(5728):1592–1594. doi: 10.1126/science.1111726. [DOI] [PubMed] [Google Scholar]
- 4.Tjoa ML, van Vugt JM, Mulders MA, Schutgens RB, Oudejans CB, van Wijk IJ. Plasma placenta growth factor levels in midtrimester pregnancies. Obstet Gynecol. 2001;98(4):600–607. doi: 10.1016/S0029-7844(01)01497-1. [DOI] [PubMed] [Google Scholar]
- 5.Venkatesha S, et al. Soluble endoglin contributes to the pathogenesis of preeclampsia. Nat Med. 2006;12(6):642–649. doi: 10.1038/nm1429. [DOI] [PubMed] [Google Scholar]
- 6.Levine RJ, et al. Soluble endoglin and other circulating antiangiogenic factors in preeclampsia. N Engl J Med. 2006;355(10):992–1005. doi: 10.1056/NEJMoa055352. [DOI] [PubMed] [Google Scholar]
- 7.Lachmeijer AM, et al. A genome-wide scan for preeclampsia in the Netherlands. Eur J Hum Genet. 2001;9(10):758–764. doi: 10.1038/sj.ejhg.5200706. [DOI] [PubMed] [Google Scholar]
- 8.Van Dijk M, et al. Maternal segregation of the Dutch preeclampsia locus at 10q22 with a new member of the winged helix gene family. Nat Genet. 2005;37(5):514–519. doi: 10.1038/ng1541. [DOI] [PubMed] [Google Scholar]
- 9.Treloar SA, Cooper DW, Brennecke SP, Grehan MM, Martin NG. An Australian twin study of the genetic basis of preeclampsia and eclampsia. Am J Obstet Gynecol. 2001;184(3):374–381. doi: 10.1067/mob.2001.109400. [DOI] [PubMed] [Google Scholar]
- 10.McPeek MS. Optimal allele-sharing statistics for genetic mapping using affected relatives. Genet Epidemiol. 1999;16(3):225–249. doi: 10.1002/(SICI)1098-2272(1999)16:3<225::AID-GEPI1>3.0.CO;2-#. [DOI] [PubMed] [Google Scholar]
- 11.Choy MY, St Whitley G, Manyonda IT. Efficient, rapid and reliable establishment of human trophoblast cell lines using poly-L-ornithine. Early Pregnancy. 2000;4(2):124–143. [PubMed] [Google Scholar]
- 12.Kong L, et al. CPC: assess the protein-coding potential of transcripts using sequence features and support vector machine. Nucleic Acids Res. 2007;35(Web Server issue):W345–W349. doi: 10.1093/nar/gkm391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Moran VA, Perera RJ, Khalil AM. Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucleic Acids Res. 2012;40(14):6391–6400. doi: 10.1093/nar/gks296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guttman M, Rinn JL. Modular regulatory principles of large non-coding RNAs. Nature. 2012;482(7385):339–346. doi: 10.1038/nature10887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12(12):861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- 16.Gudbjartsson DF, Thorvaldsson T, Kong A, Gunnarsson G, Ingolfsdottir A. Allegro version 2. Nat Genet. 2005;37(10):1015–1016. doi: 10.1038/ng1005-1015. [DOI] [PubMed] [Google Scholar]
- 17.Kong A, et al. A high-resolution recombination map of the human genome. Nat Genet. 2002;31(3):241–247. doi: 10.1038/ng917. [DOI] [PubMed] [Google Scholar]
- 18.Gabriel SB, et al. The structure of haplotype blocks in the human genome. Science. 2002;296(5576):2225–2229. doi: 10.1126/science.1069424. [DOI] [PubMed] [Google Scholar]
- 19.Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21(2):263–265. doi: 10.1093/bioinformatics/bth457. [DOI] [PubMed] [Google Scholar]
- 20.De Planell-Saguer M, Rodicio MC, Mourelatos Z. Rapid in situ codetection of noncoding RNAs and proteins in cells and formalin-fixed paraffin-embedded tissue sections without protease treatment. Nat Protoc. 2010;5(6):1061–1073. doi: 10.1038/nprot.2010.62. [DOI] [PubMed] [Google Scholar]
- 21.Garber M, Grabherr MG, Guttman M, Trapnell C. Computational methods for transcriptome annotation and quantification using RNA-seq. Nat Methods. 2011;8(6):469–477. doi: 10.1038/nmeth.1613. [DOI] [PubMed] [Google Scholar]
- 22.Trapnell C, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol. 2010;28(5):511–515. doi: 10.1038/nbt.1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Trapnell C, Pachter L, Salzberg SL. TopHat: discovering splice junctions with RNA-Seq. Bioinformatics. 2009;25(9):1105–1111. doi: 10.1093/bioinformatics/btp120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Roberts A, Pimentel H, Trapnell C, Pachter L. Identification of novel transcripts in annotated genomes using RNA-Seq. Bioinformatics. 2011;27(17):2325–2329. doi: 10.1093/bioinformatics/btr355. [DOI] [PubMed] [Google Scholar]
- 25.Roberts A, Trapnell C, Donaghey J, Rinn JL, Pachter L. Improving RNA-Seq expression estimates by correcting for fragment bias. Genome Biol. 2011;12(3):R22. doi: 10.1186/gb-2011-12-3-r22. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.