

Abstract
Defects of mitochondrial protein synthesis are clinically and genetically heterogeneous. We previously described a male infant who was born to consanguineous parents and who presented with severe congenital encephalopathy, peripheral neuropathy, myopathy, and lactic acidosis associated with deficiencies of multiple mitochondrial respiratory-chain enzymes and defective mitochondrial translation. In this work, we have characterized four additional affected family members, performed homozygosity mapping, and identified a homozygous splicing mutation in the splice donor site of exon 2 (c.504+1G>A) of RMND1 (required for meiotic nuclear division-1) in the affected individuals. Fibroblasts from affected individuals expressed two aberrant transcripts and had decreased wild-type mRNA and deficiencies of mitochondrial respiratory-chain enzymes. The RMND1 mutation caused haploinsufficiency that was rescued by overexpression of the wild-type transcript in mutant fibroblasts; this overexpression increased the levels and activities of mitochondrial respiratory-chain proteins. Knockdown of RMND1 via shRNA recapitulated the biochemical defect of the mutant fibroblasts, further supporting a loss-of-function pathomechanism in this disease. RMND1 belongs to the sif2 family, an evolutionary conserved group of proteins that share the DUF155 domain, have unknown function, and have never been associated with human disease. We documented that the protein localizes to mitochondria in mammalian and yeast cells. Further studies are necessary for understanding the function of this protein in mitochondrial protein translation.
Main Text
Resulting from mitochondrial or nuclear DNA defects, disorders of mitochondrial protein synthesis have been reported in a heterogeneous group of individuals presenting predominantly with prenatal- or congenital-onset lethal encephalopathies associated with combined deficiencies of respiratory-chain and oxidative-phosphorylation (OXPHOS) enzymes.1–3 Mutations in nuclear genes have been described in eight mitochondrial transfer-RNA-modifying factors,4–13 three mitochondrial ribosomal proteins,14–16 and three mitochondrial elongation factors.17–22 Additional mutations have been documented in the gene encoding the translational activator of cytochrome c oxidase (COX) I (TACO1) (MIM 612958)23 and in C12orf65 (encoded by C12orf65 [MIM 613541]), a protein belonging to the family of mitochondrial class I peptide release factors.24
In 2008, we described an 18-day-old male infant (individual VI-1 in Figure 1) who was born to consanguineous parents from Saudi Arabia and who presented with severe neonatal encephaloneuromyopathy and lactic acidosis.1 At birth, the infant was unresponsive but was successfully resuscitated and intubated. He had few spontaneous limb movements and required mechanical ventilation. Examination revealed prominent tongue fasciculations, bilateral equinus foot deformities, and profound hypotonia of the arms and legs. Tendon reflexes were absent. He developed myoclonic jerks and died at the age of 18 months. A muscle biopsy when he was 24 days old showed severe COX (complex IV) deficiency. Fibroblasts showed a generalized and severe defect of mitochondrial protein synthesis, reductions of all mitochondrial-translated products, normal mitochondrial transcript levels, reduction in the steady-state levels of complexes I, IV, and V, and a normal complex II (encoded entirely by nuclear genes).1 We have now characterized four additional affected family members and have performed homozygosity mapping in the family (Figure 1). Three affected infants (VI-3, VI-7, and VI-8) similarly presented with severe neonatal encephaloneuromyopathy at birth and showed lethargy, respiratory failure, profound floppiness, hyporeflexia or areflexia, equinus deformities, lactic acidosis, and death in the first year of life, whereas the fifth affected individual (VI-9) was stillborn and did not have skeletal deformities (Table S1, available online). Deficiencies in the activity of multiple mitochondrial respiratory-chain enzymes were confirmed in individual VI-3 fibroblasts (Table 1).
Figure 1.
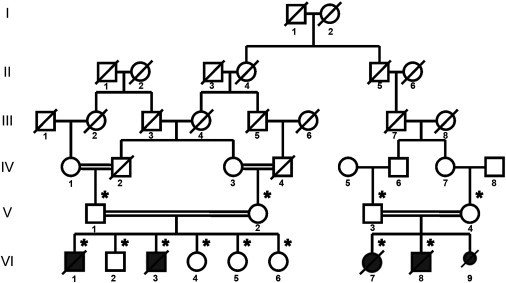
Pedigree of the family
The following symbols are used: black symbols, affected individuals; open symbols, unaffected relatives; small black symbols, stillborns; diagonal lines, deceased persons; and asterisks, DNA available.
Table 1.
Activity of Mitochondrial Respiratory-Chain Enzymes in VI-3 and Control Fibroblasts after Transient Transfection with Empty Vector or with a Construct Encoding RMND1 Isoform 1
| Complex | VI-3 Fibroblasts | Control Fibroblasts | VI-3 with Empty Vector | Control with Empty Vector | VI-3 with RMND1 | Control with RMND1 |
|---|---|---|---|---|---|---|
| IV/CS | 0.06 ± 0.002 | 0.69 ± 0.02 | 0.11 ± 0.07 | 0.59 ± 0.44 | 0.14 ± 0.08 | 0.5 ± 0.41 |
| II+III/CS | 0.05 ± 0.001 | 0.11 ± 0.03 | 0.02 ± 0.01 | 0.05 ± 0.02 | 0.03 ± 0.02 | 0.04 ± 0.02 |
| I+III/CS | 1.1 ± 0.88 | 2.59 ± 0.07 | 0.98 ± 0.79 | 1.75 ± 0.77 | 1.46 ± 0.59 | 1.80 ± 1.29 |
Activity is expressed in μmol/min/mg protein and is normalized to citrate synthase. Values represent the mean ± standard deviation of at least three different experiments and three different transfections. The following abbreviations are used: complex IV, cytochrome c oxidase (COX); complexes II+III, succinate cytochrome c reductase; complexes I+III, nicotinamide dehydrogenase (NADH) cytochrome c reductase; and CS, citrate synthase.
Informed consent for analyses of biological samples was obtained under a protocol approved by the institutional review board at Columbia University Medical Center. Genomic DNA was purified from blood according to standard procedures. To identify areas of shared homozygosity among affected individuals, we performed a high-density genome-wide genotyping of four affected and eight unaffected relatives by using the Illumina 660W-Quad gene chip array (Illumina, San Diego, CA), which features over 650,000 markers across the genome. Clustering, normalization, and genotype calls were performed with the dedicated GenomeStudio 2010.3 Genotyping Module (Illumina). SNP genotypes were analyzed in PLINK software for standard quality controls.25 Genome-wide homozygosity mapping, which we conducted with the Homozygosity Mapper program by using default parameters and restricting the analysis to areas of homozygosity shared between all affected individuals, identified a single region of homozygosity shared identically by descent in all affected individuals (Figure S1A). This region spanned <1Mb between markers rs519861 and rs926777 in 6q25 and included only seven coding genes (Figure S1B). We sequenced all exons of the seven genes included in the candidate locus by dideoxy DNA sequencing. All affected individuals were homozygous for a G-to-A transition (c.504+1G>A) in the canonical splice donor site of exon 2 of the long isoform of RMND1 (RefSeq accession number NM_017909.2) (Figure 2). Parents were carriers, four unaffected siblings were found to be heterozygous, and two were homozygous for the wild-type allele. This variant was absent in 210 control alleles, as well as in dbSNP 135 and the 1000 Genomes Project. Primers used for sequencing RMND1 are listed in Table S2.
Figure 2.
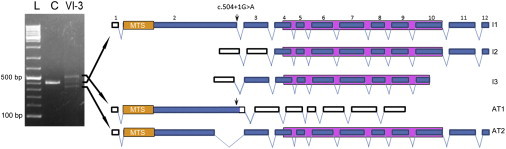
Schematic Representation of RMND1 Wild-Type and Aberrant Transcripts
RMND1 has four splicing variants, of which only three encode proteins: ENST00000367303 (12 exons, isoform 1 [I1]), ENST00000367303 (11 exons, isoform 2 [I2]), and ENST00000444024 (9 exons, isoform 3 [I3]). All three coding variants contain a DUF155 domain indicated by a purple box. Coding regions are represented by blue boxes, and noncoding regions are represented by white boxes. The longest transcript has a predicted N-terminal 33 amino acid mitochondrial targeting sequence (MTS). On the left is agarose gel showing the PCR product of RMND1 cDNA from control (C) and VI-3 fibroblasts. “L” indicates the 100 bp DNA ladder. Splice-site mutation c.504+1G>A (arrow) produces two aberrant transcripts (AT1 and AT2) and reduces the amount of wild-type transcript (middle band). The longer transcript, AT1, includes the insertion of 88 nucleotides after exon 2 and generates a premature stop at codon 171. In the shorter variant, AT2, a cryptic splice site in exon 2 is activated and results in an in-frame deletion of 75 nucleotides and a protein 25 amino acids shorter than the wild-type. RNA was extracted by the PureLink RNA Mini Kit (Ambion, Austin, TX) and reverse transcribed into cDNA with the VILO RT-PCR kit (Invitrogen, Grand Island, NY). RT-PCR of exons 2–7 was performed, and the RT-PCR products were electrophoresed in 2% agarose gels.
To evaluate RMND1 mRNA and protein in fibroblasts from individual VI-3 and controls, we performed real-time PCR (RT-PCR) and immunoblot analyses. RT-PCR analysis of RMND1 showed three transcripts expressed from the mutant allele: (1) the wild-type (31% ± 1.5), encoding a 449 amino acid protein; (2) a longer transcript (40% ± 3.5) that has 88 additional nucleotides and that leads to the insertion of a premature stop codon at codon 171, 54 amino acids upstream of the start of the only known functional domain of the protein (amino acids 225–404); and (3) a shorter variant (32% ± 2.5), in which a cryptic splice site in exon 2 is activated and produces an in-frame deletion of 75 nucleotides (25 amino acids) (Figure 2). In addition, quantitative RT-PCR (qRT-PCR) with a probe that detects all three splice variants showed mildly reduced RMND1 expression relative to that of the β-actin (ACTB) transcript in individual VI-3 compared with the control (Figure 3A). In accordance with the marked reduction of the wild-type transcript, immunoblots showed decreased levels of the 52 kDa band corresponding to the expected RMND1 long isoform and a 28 kDa band in VI-3 fibroblasts compared with control fibroblasts (Figure 3B–3C).
Figure 3.
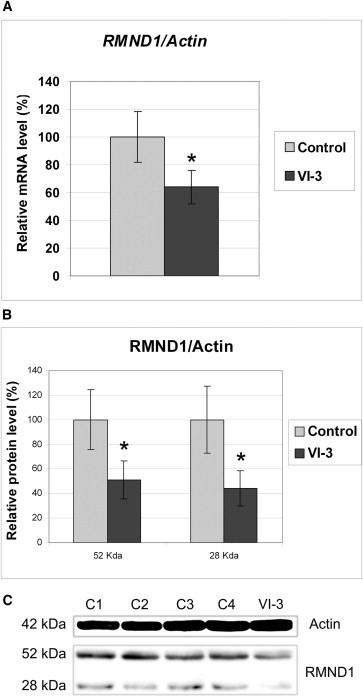
RMND1 Is Partially Reduced in VI-3 Fibroblasts
(A) qRT-PCR was performed for characterizing the expression level of RMND1 mRNA in VI-3 fibroblasts with the use of TaqMan Assays for RMND1 and ACTB (β-actin) transcripts (Applied Biosystems). Values are expressed as percentages of controls. Data are represented as the mean ± standard deviation and are the results of at least three different experiments. The asterisk (∗) indicates a Student’s t test p < 0.05.
(B and C) Immunoblot for measuring the level of RMND1 in VI-3 fibroblasts. Proteins were extracted, and concentrations were measured with the BCA Protein Assay Kit (Pierce). Five micrograms of protein was electrophoresed in a SDS-12%-PAGE gel, transferred to Immun-Blot PVDF membranes (Biorad, Hercules, CA, USA), and probed with rabbit polyclonal RMND1 antibody (product #HPA031399, Sigma-Aldrich, St. Louis, MO) at a 1:1,000 dilution. A mouse monoclonal actin antibody (Sigma-Aldrich) was used at a 1:10,000 dilution. Protein-antibody interaction was detected with peroxidase-conjugated goat anti-mouse IgG antibody (1:5,000) (Sigma-Aldrich) with SuperSignal chemiluminiscence detection kit (Thermo Fisher Scientific, Waltham, MA). Quantitation of the bands was performed by densitometric analysis with the National Institutes of Health ImageJ software package (version 1.45). Values are expressed as percentages of controls. Data are represented as the mean ± standard deviation and are the results of at least three different experiments. The asterisks (∗) indicate a Student’s t test p < 0.05. The following abbreviations are used: C1–C4, controls; and actin, β-actin.
To confirm the pathogenicity of the mutation, we overexpressed RMND1 in VI-3 and control fibroblasts. Efficiency of overexpression was confirmed by qRT-PCR and immunoblots (Figure S2). Biochemical activities of mitochondrial respiratory-chain enzymes, as well as COX and succinate dehydrogenase (SDH) cytochemistry, were performed as described.26
In mutant fibroblasts, the biochemical activities of mitochondrial enzymes were greater after RMND1 cDNA overexpression than after empty-vector transfection (Table 1). Specifically, we observed enhanced activities of complexes I+III (55.7% ± 5.7 after empty-vector transfection and 90.7% ± 5.7 after RMND1 transfection) and IV (16.3% ± 4.7 after empty vector transfection and 23.3% ± 7.1 after RMND1 transfection) (Figure 4A). Increased COX activity without change in baseline normal SDH activity was confirmed by cytochemistry (Figure S3).
Figure 4.
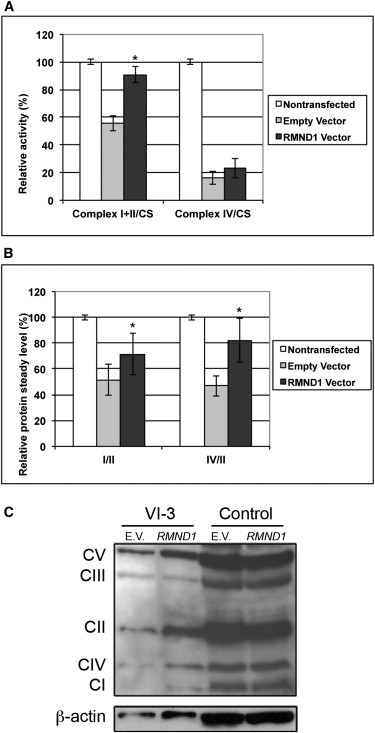
Complementation Analysis in VI-3 and Control Fibroblasts
After transient transfection with a construct encoding wild-type RMND1, mutant cells showed increased biochemical activities of mitochondrial respiratory-chain enzymes normalized to citrate synthase (CS) (A) and an increase in the steady-state level of mitochondrial OXPHOS subunits (B and C). Values are represented as the mean ± standard deviation, reflect the results of at least three different transfection experiments, and are expressed as percentages of the untransfected control. The asterisks (∗) indicate a Student’s t test p < 0.05.
Quantitative evaluation of the steady-state level of respiratory-chain enzyme subunits by immunoblot analyses with the Total OXPHOS Complexes Detection Kit cocktail of antibodies (MitoSciences, Eugene, OR, USA) revealed decreased amounts of mitochondrial-encoded OXPHOS subunits that increased after transfection with wild-type RMND1 cDNA. Specifically, after RMND1 overexpression, as well as after normalization to SDH as a marker of mitochondrial mass, there were significant increases in levels of complex I (51.1% ± 8.0 after empty-vector transfection and 71.5% ± 17 after RMND1 transfection) and complex IV (47% ± 8.0 after empty-vector transfection and 82% ± 16 after RMND1 transfection) in absolute values (Figures 4B–4C).
To further investigate the function of RMND1, we used shRNA-mediated knockdown of the protein. The clone with the lowest levels of RMND1 transcript and protein (about 60% of wild-type levels) was chosen for measuring respiratory-chain and OXPHOS activities, steady-state level, and synthesis (Figure 5).
Figure 5.
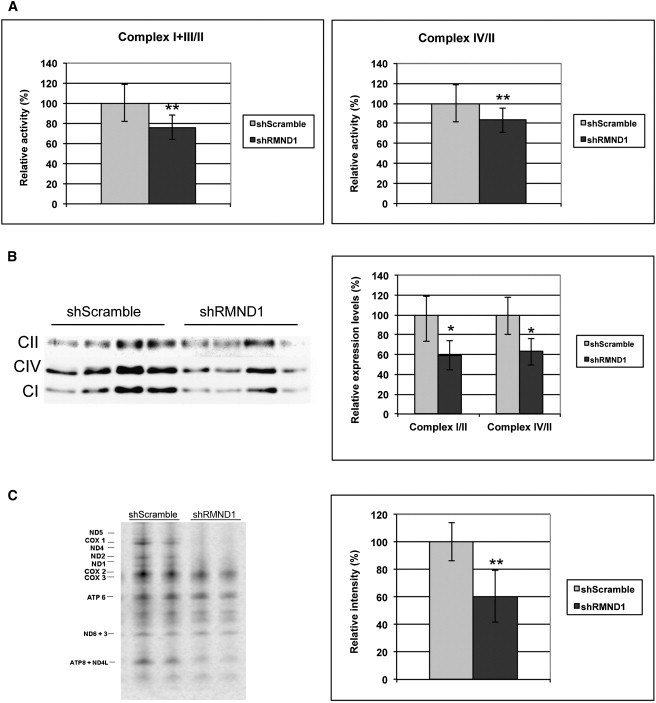
Effects of RMND1 Knockdown in HeLa Cells
HeLa cells were cultured in DMEM with 10% fetal bovine serum (FBS) until they were 70%–80% confluent. Transfection with a scramble shRNA-pLKO plasmid (negative control) and a RMND1-specific TRC shRNA-pLKO plasmid construct (TRCN0000135730, Sigma Aldrich) was mediated by Lipofectamine 2000 (Invitrogen). Five hours after transfection, cells were selected with Puromycin in DMEM 2% FBS, and transfected clones were individually expanded in DMEM 10% FBS. RMND1 knockdown was assessed in 30 clones by qRT-PCR and immunoblot analysis.
(A) Activity of mitochondrial respiratory-chain enzymes.
(B and C) Immunoblot analysis of the steady-state levels of mitochondrial respiratory enzymes (B) and pulse labeling with [35S] methionine of mitochondrial proteins (C). A total of 5 × 105 cells cultured in 10 cm culture plates were washed in methionine-free DMEM and subsequently incubated in the same medium supplemented with 15% dialyzed FBS, 1.2 mM sodium pyruvate, and glucose for 30 min. Cytosolic protein synthesis was inhibited by the addition of emetine (0.1 μg/μl) for 7 min at 37°C. Mitochondrial proteins were labeled with 50 μCi [35S]-methionine Redivue (Amersham Biosciences, Piscataway, NJ) in methionine-free medium and incubated for 1 hr at 37°C. After treatment, cells were incubated for 10 min in DMEM supplemented with 10% FBS and were collected by scraping. Protein aliquots (15 μg per sample) were electrophoresed in a 4%–12% Bis-Tris polyacrylamide gradient gel for 4 hr at 85V. The gel was dried for 60 min at 80°C and analyzed with a phosphorimager. The seven subunits of complex I (ND), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated at the left. Values are expressed as percentages of the control and are represented as the mean ± standard deviation. One asterisk (∗) indicates a Student’s t test p < 0.05; ∗∗ indicates a Student’s t test p < 0.01.
To assess mitochondrial translation, pulse labeling of cell cultures from shRNA and control clones was performed as described1 with slight modifications. Consistent with our observations in mutant fibroblasts, as compared to wild-type cells, those with 60% depletion of RMND1 showed decreased activities of complexes I+III (75%) and IV (83%), reduced levels of complexes I (59%) and IV (62%), and diminished mitochondrial proteins synthesis (60%) (Figures 5A–5C).
To assess the potential role of RMND1 in the assembly of ribosomal subunits, we measured 12S and 16S rRNA levels by qRT-PCR. We did not detect any differences in 12S and 16S rRNAs among mutant skin fibroblasts, RMND1-depleted HeLa cells, or corresponding wild-type cells (data not shown).
To confirm the localization of RMND1 in the mitochondria of mammalian cells, we performed immunohistochemistry and immunoblots. Fluorescence images, collected and analyzed by a laser-scanning confocal microscope showed that the fusion protein colocalizes with mitochondria (Figure 6A). An immunoblot of isolated cytosolic fraction, endoplasmic reticulum (ER), crude mitochondria, pure mitochondria, and ER + mitochondria from HeLa cells showed a 52 kDa band in all fractions and a 28 kDa band in total cell lysate and in all mitochondrial-containing fractions (Figure 6B). The 28 kDa band was absent in both the cytosol and ER fractions (Figure 6B). An immunoblot with YFP antibody (1:1000, 632380, Clontech) of whole 239T cell extracts transfected with a construct encoding YFP-RMND1 fusion protein confirmed the specificity of the two bands, corresponding to the expected long RMND1 isoform (52 kDa) and to the presumably cleaved mitochondrial protein (28 kDa) (Figure 6C).
Figure 6.
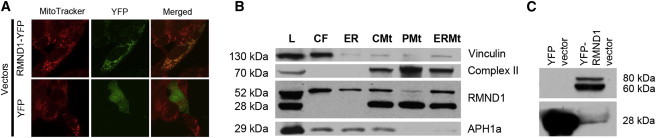
RMND1 Localization in Mammalian Cells by Immunohistochemistry and Quantitation by Immunoblot Analysis
(A) RMND1 cDNA was amplified from a commercial vector containing the human full-length cDNA with the use of a BamHI-recognition-site-integrated forward primer 5′-AGG ATCCGCCATGCCAGCCACACTCCTCAGAGCCG-3′ and AgeI-recognition-site-integrated reverse primer 5′-CGACCGGTGATTTCATGGTTGGAAGGTGTG-3′. PCR product was digested with BamHI and AgeI and cloned into a pEYFP expression vector in-frame and upstream of the YFP coding sequence. Fidelity of the fusion RMND1-YFP construct in the expression vector was verified by sequencing, and 2.5 μg was used for transiently transfecting human embryonic kidney (HEK) 293T cells as described above. Culture media were supplemented with 0.1 μM MitoTracker Red (Invitrogen) for 30 min before the cells were fixed with 4% formalin PBS for the detection of mitochondria.
(B) Purification of mitochondria and cellular fractions was performed as previously described,27 and immunoblot analysis was performed with primary antibodies anti-RMND1 (1:1,000, Sigma), anti-Complex II (1:5,000, MitoScience, Abcam, Cambridge, MA), anti-APH1a (1:1,000, Abcam), and anti-Vinculin (1:5,000, Sigma-Aldrich, St. Louis, MO) and either peroxidase-conjugated anti-rabbit (1:2,000) or peroxidase-conjugated anti-mouse IgG (1:5,000) as a secondary antibody (Sigma-Aldrich). Cellular fractions were isolated from HeLa cells (25 μg of protein from the total lysate [L]), and equal amounts (6 μg) from each fraction (cytoplasmic fraction [CF], endoplasmatic reticulum [ER], crude mitochondria [CMt], pure mitochondria [PMt], and ER + mitochondria [ERMt]) were loaded onto the gel. The known molecular masses (in kDa) of proteins used for confirming cell-fraction enrichment are indicated in the left margin of the gel.
(C) Immunoblot analysis of total HEK 293T cell extracts transfected with YFP-RMND1 fusion protein with the antibody against green fluorescent protein confirmed the specificity of the two bands detected by the RMND1 antibody.
We generate a disrupted version of Saccharomyces cerevisiae rmnd1 (RMND1 ortholog) to investigate mitochondrial function.28,29 Mitochondrial fractionation and intramitochondrial localization of Ydr282cp with an antibody against the HA epitope were performed as described.30 An immunoblot with the HA epitope antibody detected a ∼50 kDa protein (Figure S4).
In accordance with data on the ydr282c mutant strain in the yeast genome collection, yeast containing the ydr282c-null allele showed a normal phenotype, including normal growth in respiration-dependent glycerol medium and mitochondrial protein synthesis (Figure S5).
Mitochondrial protein synthesis is a complex and still poorly understood process requiring a number of initiation, elongation, and termination factors, all of which are encoded by nuclear genes together with mtDNA-encoded ribosomal and transfer RNA.31 Defects in 18 genes directly or indirectly involved in mitochondrial protein synthesis have been described.1,4–25 In muscle and fibroblasts from an individual with consanguineous parents and a fatal infantile-onset encephalomyopathy with lactic acidosis, we previously demonstrated evidence of a mitochondrial protein synthesis by documenting severe COX deficiency, partially reduced biochemical activities of complexes I and V, normal SDH activity, and decreases of all mitochondrial-translation products with normal levels of mitochondrial transcripts.1
Here, we report four additional affected relatives, homozygosity mapping, and identification of a homozygous splice-site mutation in the gene encoding RMND1 in all four affected individuals. We have shown that the c.504+1G>A splice donor mutation reduces the wild-type transcript and produces two aberrant transcripts. One alters the reading frame of the protein and produces a prematurely truncated protein with loss of the DUF155 domain. The other is predicted to produce a protein truncated by 25 amino acids. The fact that the truncated protein was absent in an immunoblot with an antibody that recognizes the carboxy-terminus of RMND1 suggests that this aberrant polypeptide is unstable and degraded. These data, together with the results of the RNA interference of the wild-type transcript—showing reduced mitochondrial protein synthesis in partially depleted cells—indicate that the disease is caused by loss, rather than toxic gain, of function. We did not find mutations in RMND1 in ten other individuals with documented defects of mitochondrial protein synthesis,2 suggesting that mutations in RMND1 might be rare.
According to Uniprot, RMND1 has at least three isoforms produced by alternative splicing: isoform 1, the longest one (449 amino acids), is localized to mitochondria;32 isoform 2 (lacking amino acids 1–211); and isoform 3 (containing alternative amino acids 205–208 DAAN>GTSS and missing amino acids 209–449). However, in addition to the predicted 52 kDa full-length RMND1, immunoblot analysis revealed a shorter protein (28 kDa), which was abundant in purified mitochondrial fractions; therefore, we postulate that the 28 kDa RMND1 is the cleaved mitochondrial protein. We sublocalized the yeast ortholog, Ydr282c, to the inner mitochondrial membrane with the C terminus facing the intermembrane space.
RMND1 belongs to the sif2 family, an evolutionary conserved family of proteins that have unknown function and that share the DUF155 domain. rmnd1 is conserved in species down to yeast. The S. cerevisiae ortholog, YDR282C, has no known functions but has been reported to interact with the functional ortholog of the human Nieman Pick C1 protein, which is involved in sphingolipid metabolism.33 Although we have shown that the yeast rmnd1 ortholog is a mitochondrial protein, the fact that yeast null for rmnd1 are viable in glycerol medium indicates that respiration is not severely affected. Therefore, either the protein is not involved in yeast mitochondrial protein synthesis or its function overlaps with those of other proteins in yeast. In fact, divergent functions in yeast and humans have been observed for other mitochondrial proteins, including TACO1, which is required for human COX I mitochondrial-translation activation, whereas the yeast ortholog does not serve this function.23 Although COX deficiency is prominent in the affected individual’s muscle and fibroblasts, the involvement of other respiratory-chain enzymes and the generalized mitochondrial-translation defect in mutant fibroblasts and in RMND1 knockdown cells indicate that RMND1 is not a COX-subunit-specific translational activator.
Intriguingly, several RMND1 orthologs seem to be involved in cell division,34 and mitochondrial ribosomal proteins have been shown to be involved in cell-cycle regulation.35 However, against the notion that RMND1 is a structural ribosomal protein are the normal levels of 12S and 16S rRNA in mutant fibroblasts and in RMND1-depleted HeLa cells because mutations in mitochondrial small-subunit ribosomal proteins have been shown to decrease 12S rRNA.14,15
In conclusion, we have identified a human mutation affecting a DUF155 protein in a family with a severe autosomal-recessive mitochondrial-translation defect. Further studies are necessary for understanding the function of RMND1 and elucidating its role in mitochondrial protein synthesis.
Acknowledgments
We are grateful to all of the affected individuals and relatives and Ali Gharavi for their collaborations. The work was partially supported by the Marriott Mitochondrial Disorder Clinical Research Fund. C.M.Q. is supported by National Institute of Health (NIH) grant K23 HD065871 from the Eunice Kennedy Shriver National Institute of Child Health & Human Development (NICHD). M.H. is supported by NIH grants R01 HD057543 and R01 HD056103 from the NICHD and the Office of Dietary Supplements, as well as U54NS078059 from the National Institute of Neurological Disorders and Stroke and the NICHD, and a grant from the Muscular Dystrophy Association. M.H.B. and C.C.F.B. are supported by Fundacão Amparo a Pesquisa São Paulo. S.S.C. is supported by the American Heart Association Scientist Development Grant 0930151N and by the American Society of Nephrology Carl W. Gottschalk Research Scholar Grant.
Supplemental Data
Web Resources
The URLs for data presented are as follows:
Ensembl, www.ensembl.org
HomozygosityMapper, http://www.homozygositymapper.org
Human MitoCarta, http://www.broadinstitute.org/pubs/MitoCarta/human.mitocarta.html
MitoProt: Prediction of Mitochondrial Targeting Sequences, http://ihg.gsf.de/ihg/mitoprot.html
NCBI dbSNP, www.ncbi.nlm.nih.gov/snp
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
UniProt: The Universal Protein Resource, www.ebi.ac.uk/uniprot
Saccharomyces Genome Database, www.yeastgenome.org
References
- 1.Ferreiro-Barros C.C., Tengan C.H., Barros M.H., Palenzuela L., Kanki C., Quinzii C., Lou J., El Gharaby N., Shokr A., De Vivo D.C. Neonatal mitochondrial encephaloneuromyopathy due to a defect of mitochondrial protein synthesis. J. Neurol. Sci. 2008;275:128–132. doi: 10.1016/j.jns.2008.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kemp J.P., Smith P.M., Pyle A., Neeve V.C., Tuppen H.A., Schara U., Talim B., Topaloglu H., Holinski-Feder E., Abicht A. Nuclear factors involved in mitochondrial translation cause a subgroup of combined respiratory chain deficiency. Brain. 2011;134:183–195. doi: 10.1093/brain/awq320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rötig A. Human diseases with impaired mitochondrial protein synthesis. Biochim. Biophys. Acta. 2011;1807:1198–1205. doi: 10.1016/j.bbabio.2011.06.010. [DOI] [PubMed] [Google Scholar]
- 4.Bykhovskaya Y., Casas K., Mengesha E., Inbal A., Fischel-Ghodsian N. Missense mutation in pseudouridine synthase 1 (PUS1) causes mitochondrial myopathy and sideroblastic anemia (MLASA) Am. J. Hum. Genet. 2004;74:1303–1308. doi: 10.1086/421530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fernandez-Vizarra E., Berardinelli A., Valente L., Tiranti V., Zeviani M. Nonsense mutation in pseudouridylate synthase 1 (PUS1) in two brothers affected by myopathy, lactic acidosis and sideroblastic anaemia (MLASA) J. Med. Genet. 2007;44:173–180. doi: 10.1136/jmg.2006.045252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Riley L.G., Cooper S., Hickey P., Rudinger-Thirion J., McKenzie M., Compton A., Lim S.C., Thorburn D., Ryan M.T., Giegé R. Mutation of the mitochondrial tyrosyl-tRNA synthetase gene, YARS2, causes myopathy, lactic acidosis, and sideroblastic anemia—MLASA syndrome. Am. J. Hum. Genet. 2010;87:52–59. doi: 10.1016/j.ajhg.2010.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scheper G.C., van der Klok T., van Andel R.J., van Berkel C.G., Sissler M., Smet J., Muravina T.I., Serkov S.V., Uziel G., Bugiani M. Mitochondrial aspartyl-tRNA synthetase deficiency causes leukoencephalopathy with brain stem and spinal cord involvement and lactate elevation. Nat. Genet. 2007;39:534–539. doi: 10.1038/ng2013. [DOI] [PubMed] [Google Scholar]
- 8.Isohanni P., Linnankivi T., Buzkova J., Lönnqvist T., Pihko H., Valanne L., Tienari P.J., Elovaara I., Pirttilä T., Reunanen M. DARS2 mutations in mitochondrial leucoencephalopathy and multiple sclerosis. J. Med. Genet. 2010;47:66–70. doi: 10.1136/jmg.2009.068221. [DOI] [PubMed] [Google Scholar]
- 9.Edvardson S., Shaag A., Kolesnikova O., Gomori J.M., Tarassov I., Einbinder T., Saada A., Elpeleg O. Deleterious mutation in the mitochondrial arginyl-transfer RNA synthetase gene is associated with pontocerebellar hypoplasia. Am. J. Hum. Genet. 2007;81:857–862. doi: 10.1086/521227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zeharia A., Shaag A., Pappo O., Mager-Heckel A.M., Saada A., Beinat M., Karicheva O., Mandel H., Ofek N., Segel R. Acute infantile liver failure due to mutations in the TRMU gene. Am. J. Hum. Genet. 2009;85:401–407. doi: 10.1016/j.ajhg.2009.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Steenweg M.E., Ghezzi D., Haack T., Abbink T.E., Martinelli D., van Berkel C.G., Bley A., Diogo L., Grillo E., Te Water Naudé J. Leukoencephalopathy with thalamus and brainstem involvement and high lactate ‘LTBL’ caused by EARS2 mutations. Brain. 2012;135:1387–1394. doi: 10.1093/brain/aws070. [DOI] [PubMed] [Google Scholar]
- 12.Bayat V., Thiffault I., Jaiswal M., Tétreault M., Donti T., Sasarman F., Bernard G., Demers-Lamarche J., Dicaire M.J., Mathieu J. Mutations in the mitochondrial methionyl-tRNA synthetase cause a neurodegenerative phenotype in flies and a recessive ataxia (ARSAL) in humans. PLoS Biol. 2012;10:e1001288. doi: 10.1371/journal.pbio.1001288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pierce S.B., Chisholm K.M., Lynch E.D., Lee M.K., Walsh T., Opitz J.M., Li W., Klevit R.E., King M.C. Mutations in mitochondrial histidyl tRNA synthetase HARS2 cause ovarian dysgenesis and sensorineural hearing loss of Perrault syndrome. Proc. Natl. Acad. Sci. USA. 2011;108:6543–6548. doi: 10.1073/pnas.1103471108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Miller C., Saada A., Shaul N., Shabtai N., Ben-Shalom E., Shaag A., Hershkovitz E., Elpeleg O. Defective mitochondrial translation caused by a ribosomal protein (MRPS16) mutation. Ann. Neurol. 2004;56:734–738. doi: 10.1002/ana.20282. [DOI] [PubMed] [Google Scholar]
- 15.Saada A., Shaag A., Arnon S., Dolfin T., Miller C., Fuchs-Telem D., Lombes A., Elpeleg O. Antenatal mitochondrial disease caused by mitochondrial ribosomal protein (MRPS22) mutation. J. Med. Genet. 2007;44:784–786. doi: 10.1136/jmg.2007.053116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Galmiche L., Serre V., Beinat M., Assouline Z., Lebre A.S., Chretien D., Nietschke P., Benes V., Boddaert N., Sidi D. Exome sequencing identifies MRPL3 mutation in mitochondrial cardiomyopathy. Hum. Mutat. 2011;32:1225–1231. doi: 10.1002/humu.21562. [DOI] [PubMed] [Google Scholar]
- 17.Coenen M.J., Antonicka H., Ugalde C., Sasarman F., Rossi R., Heister J.G., Newbold R.F., Trijbels F.J., van den Heuvel L.P., Shoubridge E.A., Smeitink J.A. Mutant mitochondrial elongation factor G1 and combined oxidative phosphorylation deficiency. N. Engl. J. Med. 2004;351:2080–2086. doi: 10.1056/NEJMoa041878. [DOI] [PubMed] [Google Scholar]
- 18.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 19.Smeitink J.A., Elpeleg O., Antonicka H., Diepstra H., Saada A., Smits P., Sasarman F., Vriend G., Jacob-Hirsch J., Shaag A. Distinct clinical phenotypes associated with a mutation in the mitochondrial translation elongation factor EFTs. Am. J. Hum. Genet. 2006;79:869–877. doi: 10.1086/508434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Smits P., Antonicka H., van Hasselt P.M., Weraarpachai W., Haller W., Schreurs M., Venselaar H., Rodenburg R.J., Smeitink J.A., van den Heuvel L.P. Mutation in subdomain G’ of mitochondrial elongation factor G1 is associated with combined OXPHOS deficiency in fibroblasts but not in muscle. Eur. J. Hum. Genet. 2011;19:275–279. doi: 10.1038/ejhg.2010.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vedrenne V., Galmiche L., Chretien D., de Lonlay P., Munnich A., Rötig A. Mutation in the mitochondrial translation elongation factor EFTs results in severe infantile liver failure. J. Hepatol. 2012;56:294–297. doi: 10.1016/j.jhep.2011.06.014. [DOI] [PubMed] [Google Scholar]
- 22.Valente L., Tiranti V., Marsano R.M., Malfatti E., Fernandez-Vizarra E., Donnini C., Mereghetti P., De Gioia L., Burlina A., Castellan C. Infantile encephalopathy and defective mitochondrial DNA translation in patients with mutations of mitochondrial elongation factors EFG1 and EFTu. Am. J. Hum. Genet. 2007;80:44–58. doi: 10.1086/510559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weraarpachai W., Antonicka H., Sasarman F., Seeger J., Schrank B., Kolesar J.E., Lochmüller H., Chevrette M., Kaufman B.A., Horvath R., Shoubridge E.A. Mutation in TACO1, encoding a translational activator of COX I, results in cytochrome c oxidase deficiency and late-onset Leigh syndrome. Nat. Genet. 2009;41:833–837. doi: 10.1038/ng.390. [DOI] [PubMed] [Google Scholar]
- 24.Antonicka H., Ostergaard E., Sasarman F., Weraarpachai W., Wibrand F., Pedersen A.M., Rodenburg R.J., van der Knaap M.S., Smeitink J.A., Chrzanowska-Lightowlers Z.M., Shoubridge E.A. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am. J. Hum. Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.DiMauro S., Servidei S., Zeviani M., DiRocco M., DeVivo D.C., DiDonato S., Uziel G., Berry K., Hoganson G., Johnsen S.D. Cytochrome c oxidase deficiency in Leigh syndrome. Ann. Neurol. 1987;22:498–506. doi: 10.1002/ana.410220409. [DOI] [PubMed] [Google Scholar]
- 27.Stone S.J., Vance J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- 28.Hill J.E., Myers A.M., Koerner T.J., Tzagoloff A. Yeast/E. coli shuttle vectors with multiple unique restriction sites. Yeast. 1986;2:163–167. doi: 10.1002/yea.320020304. [DOI] [PubMed] [Google Scholar]
- 29.Rothstein R.J. One-step gene disruption in yeast. Methods Enzymol. 1983;101:202–211. doi: 10.1016/0076-6879(83)01015-0. [DOI] [PubMed] [Google Scholar]
- 30.Barros M.H., Johnson A., Gin P., Marbois B.N., Clarke C.F., Tzagoloff A. The Saccharomyces cerevisiae COQ10 gene encodes a START domain protein required for function of coenzyme Q in respiration. J. Biol. Chem. 2005;280:42627–42635. doi: 10.1074/jbc.M510768200. [DOI] [PubMed] [Google Scholar]
- 31.Smits P., Smeitink J., van den Heuvel L. Mitochondrial translation and beyond: Processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Costanzo M., Baryshnikova A., Bellay J., Kim Y., Spear E.D., Sevier C.S., Ding H., Koh J.L., Toufighi K., Mostafavi S. The genetic landscape of a cell. Science. 2010;327:425–431. doi: 10.1126/science.1180823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhou X., Li Q., Chen X., Liu J., Zhang Q., Liu Y., Liu K., Xu J. The Arabidopsis RETARDED ROOT GROWTH gene encodes a mitochondria-localized protein that is required for cell division in the root meristem. Plant Physiol. 2011;157:1793–1804. doi: 10.1104/pp.111.185827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yoo Y.A., Kim M.J., Park J.K., Chung Y.M., Lee J.H., Chi S.G., Kim J.S., Yoo Y.D. Mitochondrial ribosomal protein L41 suppresses cell growth in association with p53 and p27Kip1. Mol. Cell. Biol. 2005;25:6603–6616. doi: 10.1128/MCB.25.15.6603-6616.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.