

Abstract
Cognitive deficits in AD correlate with progressive synaptic dysfunction and loss. The Rho family of small GTPases, including Rho, Rac, and Cdc42, has a central role in cellular motility and cytokinesis. Acetylcholinesterase inhibitor has been found to protect cells against a broad range of reagents-induced injuries. Present studies examined if the effect of HupA on neurite outgrowth in Aβ-treated neuronal cells executed via regulating Rho-GTPase mediated axon guidance relative gene expression. Affymetrix cDNA microarray assay followed by real-time RT-PCR and Western Blotting analysis were used to elucidate and analyze the signaling pathway involved in Aβ and HupA’s effects. The effects of Aβ and HupA on the neurite outgrowth were further confirmed via immunofluorescence staining. Aβ up-regulated the mRNA expressions of NFAT5, LIMK1, EPHA1, NTN4 and RAC2 markedly in SH-SY5Y cells. Co-incubation of Aβ and HupA reversed or decreased the changes of NFAT5, NTN4, RAC2, CDC42 and SEMA4F. HupA treated alone increased NFAT5, LIMK1, NTN4 significantly. Following qRT-PCR validation showed that the correlation of the gene expression ratio between microarray and qRT-PCR is significant. Western blot result showed that the change of CDC42 protein is consistent with the mRNA level while RAC2 is not. The morphological results confirmed that HupA improved, or partly reversed, the Aβ-induced damage of neurite outgrowth. The protective effect of HupA from Aβ induced morphological injury might be correlative to, at least partially, regulating the network of neurite outgrowth related genes.
Keywords: β-amyloid, axon guidance, neurite outgrowth, acetylcholinesterase inhibitor, huperzine A
Introduction
Alzheimer’s disease (AD) is the leading cause of dementia among the elderly and is characterized by accumulation of extracellular and vascular amyloid in the brain [1]. The key symptoms of AD are primarily caused by cholinergic dysfunction. A significant correlation has been found between a decrease in cortical cholinergic activity and the deterioration of mental test scores in patients with AD [1]. Cognitive deficits in AD correlate with progressive synaptic dysfunction and loss that may be initiated by soluble β-amyloid peptide and driven further by the accumulating neuropathological hallmarks, including intraneuronal neurofibrillary tangles, extracellular amyloid plaques, and neuron loss [1-3]. Both dystrophic neurites and dendritic spine loss are observed in AD and many mental retardation syndromes [3-8]. Soluble Aβ or Aβ oligomers correlates highly with synapse loss and the degree of dementia [9-17]. The evidence indicated that deregulation of Rho GTPase pathway is implicated in several pathological conditions, including neurodegen-erative disorders like AD [4,18]. The translocation of the GTPase to neurofibrillary tangles in dystrophic neuritis correlates with neuronal dystrophy reported in Alzheimer’s disease and AβPP overexpressing mice [19]. There is evidence that Rho GTPase activity regulates the formation of Aβ peptides during disease progression [20]. This pathology is characterized by a progressive loss in the number of dendritic spines, as well as by alterations in the synaptic efficacy and damage at the synaptic terminal [4,10]. Dendritic spines, major sites of synaptic contacts, are structurally reliant on the actin cytoskeleton. The dynamic regulation of actin polymerization is considered the main mechanism underlying morphological changes in dendritic spines. The Rho family of small GTPases, including Rho, Rac, and Cdc42, has a central role in cellular motility and cytokinesis due to its involvement in the regulation of actin cytoskeletal dynamics [21-25]. Rac/Cdc42 inhibits axon growth via the effector kinases p21-activated kinases (PAK) Rho or Rho-associated protein kinase (Rock) [18,26,27]. Previous studies demonstrated Aβ oligomers can also interfere with Rac and Cdc42 signaling and induce the loss of actin polymerization and of dendritic spines [20]. The evidence indicated that the β-site amyloid precursor protein cleaving enzyme 1 (BACE1) which is necessary to generate the Aβ peptide is play a central role in axon guidance [28,29].
Huperzine A (HupA), isolated from Chinese herb Huperzia serrata, is a potent, highly specific and reversible inhibitor of acetylcholinesterase [30]. It has been found to reverse or attenuate cognitive deficits in a broad range of animal models [31-33] and patients including aged subjects, patients with benign senescent forgetfulness, Alzheimer’s disease (AD) and vascular dementia (VD), with minimal peripheral cholinergic side effects compared with other AChEIs in use [30]. Besides the above mentioned AChE inhibiting effect, HupA possesses the ability, from our recent studies, to protect cells against hydrogen peroxide, β-amyloid protein (or peptide), glutamate, ischemia and staurosporine-induced cytotoxicity and apoptosis [31-35]. These protective effects are related to its ability to attenuate oxidative stress, regulate the expression of apoptotic proteins Bcl-2, Bax, P53 and caspase-3, protect mitochondria, and interfere with APP metabolism [30]. In addition to its AChE inhibition and antioxidation, the neuroprotective effect of HupA also involves other mechanisms, including targeting of the Wnt/β-catenin signaling pathway in AD brain [36]. It has been suggested that that AChE inhibitors are known to exert NGF-like activities by potentiating the neuritogenic effect of NGF [37]. Our previous studies also showed that HupA increased neurite outgrowth from undifferentiated PC12 cells [38]. As p75NTR interactors, it is well known that Rho GTPase family is involved in NGF signaling [39]. Recently, the study has shown that HupA treatment reduces synaptic deficits and Aβ-related pathological alterations, including Aβ deposition and oligomerization. This encourages us to explore the further mechanism. We presume that the effect of HupA on neurite outgrowth in Aβ-treated neuroblastoma might be executed via regulating Rho-GTPase mediated axon guidance relative gene expression. In order to elucidate and analyze the signaling pathway involved in HupA’s effects, an Affymetrix cDNA microarray assay followed by real-time RT-PCR, Western Blotting and immunofluorescence staining analysis were carried out. Expectedly, HupA reversed Aβ-induced changes in axon guidance relative gene expression, which might play an important role in its neuroprotective mechanism.
Material and methods
Cell culture and treatments
SH-SY5Y neuroblastoma cells, obtained from the American Type Culture Collection (ATCC), were maintained in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells were seeded into 100 mm dishes (2 × 104 cells per ml) in DMEM/F12 (1:1) medium, supplemented with 10% (v/v) fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin. Experiments were carried out 24-48 h after cells were seeded.
It has been well established that the truncated amyloidogenic peptide Aβ25-35 plays a key role in the pathogenesis of AD. Recently, Kaminsky et al [40] and Gulyaeva NV et al [41] presented analytical reviews on the proposed involvement of Aβ25-35 as a key player in pathogenesis of Alzheimer’s disease (AD). The authors provide a large body of evidence for the hypothesis that Aβ25-35 is a proxyholder for amyloidogenic peptides, using primarily the data of in vitro and in vivo experiments. Aβ25-35 peptide is effective on behavior, neuronal plasticity and cell death, as well as cholinergic system, and oxidative stress. Our previous reports are consistent with the hypothesis [30,34,35]. Based on the up-mentioned reports, amyloid β- peptide fragment 25-35 was used in the following experiments.
Active amyloid β peptide fragment 25-35 (Aβ25-35) was purchased from Sigma, dissolved in dH2O and aged by incubating in 37°C for 72 hours. Working solutions were diluted in phosphate-buffered saline (PBS) before using. Huperzine A (HupA), a colorless powder with m.p. 230 °C, and purity >99%, was dissolved and diluted in PBS. Pre-incubation with HupA was conducted 2 h before Aβ (10 μM) addition. HupA treated alone and PBS treated cells were used as controls. The cells were harvested for the further assays and detections after 24 h exposure of Aβ.
RNA extraction and cDNA microarray
Total RNA from the cells treated with PBS, HupA alone and Aβ with/without HupA co-treated was isolated using the RNeasy Mini Kit (Qiagen Inc., Valencia, CA) according to the manufacturer’s instructions with DNase I digestion to remove genomic DNA. cRNA is prepared according to Affymetrix protocols. Briefly, total RNA (10 μg) is first reverse transcribed using a T7-Oligo(dT) Promoter Primer in the first-strand cDNA synthesis reaction with One-Cycle cDNA Synthesis Kit (QIAGEN, Valencia, CA). Following RNase H-mediated second-strand cDNA synthesis (Two-Cycle cDNA Synthesis Kit), the double-stranded cDNA is purified and serves as a template in the subsequent in vitro transcription (IVT) reaction (MEGAscript® T7 Kit, Ambion, Inc., Austin, TX). The IVT reaction is carried out in the presence of T7 RNA Polymerase and a biotinylated nucleotide analog/ribonucleotide mix for complementary RNA (cRNA) amplification and biotin labeling. The biotinylated cRNA targets were then cleaned up (IVT cRNA Cleanup Kit), fragmented, and hybridized to GeneChip expression arrays.
Gene expression was measured by hybridization to Human Genome U133A (HG-U133A) Gene Chip DNA microarrays (Affymetrix, Santa Clara, CA, USA), containing 33,000 human genes to identify those that are uniquely expressed in the cells with different treatments. Differences in gene expression among groups were calculated with Affymetrix GeneChip® Operating Software (GCOS).
Real time RT-PCR
RNA was extracted using the TRIzol reagent. The concentration of nucleic acids was determined spectrophotometrically at 260 nm and 280 nm, taking into account the dilution factor. RQ1 RNase-free DNase (Promega) was used to remove relict of DNA which might interfere the output of PCR. For the PCR first strand synthesis was performed using the Omiscript® RT Kit (Qiagen Inc.). Real time PCR was performed in 0.2 ml thin wall PCR plates using the ABI PRISM 7000 Sequence Detection System and carried out with SYBR® Green PCR Core Reagents (TAKARA) according to the manufacturer’s instructions. The standard reaction mix consisted of SYBR Green supermix, forward and reverse primers at a final concentration of 5 pM each, 1 ng DNA template, DNase free water to give final volume of 20 μl. The mixture was kept in 50°C for 2 min and heated to 95°C for 10 min followed by 35 cycles with denaturation at 95°C for 15 s, annealing at 60°C for 1 min and extension at 72°C for 30 s. A group of genes were randomly selected to confirm the array results. Primer sequences used for real-time RT-PCR are listed in Table 1. Human GAPDH was used as reference gene. Relative quantification of genes expression was carried out by comparative Ct method according to manufacturer’s protocol. Briefly, the genes mRNA level was expressed in cycle threshold (Ct) value; the Ct values for each sample were averaged from duplicate. Differences between the mean Ct values of target genes and reference gene were calculated as ΔCtsample= Ct target-Ct gapdh for different treated groups, and that of the ΔCt for the control groups were set for calibrator (ΔCtcalibrator). Final results, the sample-calibrator ratio, expressed as N-fold differences of target genes expression in Aβ group compared with control, were determined as 2-(ΔCt sample - ΔCt calibrator).
Table 1.
Primer sequences used for real-time RT-PCR
| Gene bank ID | Gene title | Primer forward | Primer reverse |
|---|---|---|---|
| NM_001619 | ADRBK1 | CGATGAGGAGGACACAAAAGG | TCTGTCTCAGCGTTGATGGTG |
| NM_006464 | TGOLN2 | ACCTGATGCCAGATGCTATGG | GCAAGTCACGCCAAATCCAA |
| NM_002588 | PCDHGC3 | AGTGTTGGTGGAGGTTGTGGA | CACACTGAGCAAAGCGATGAC |
| NM_003255 | TIMP2 | TCATTCGTCTCCCGTCTTTG | GCACGGTTAAAAGACCAACGT |
| NM_001164 | A4 | GAAACGATGCTCTCATGCCAT | TCGTTTTCTGTGTTGGCTGG |
| NM_006732 | FOSB | AGCGAGCCGTTGAATTGGA | ACAGGCGGAGGAGAAAAGACA |
| NM_003790 | TNFRSF25 | TTGCAGAAGCCCTAAGTACGG | TTCGTGCTTCTTCGCCTTG |
| NM_003053 | SLC18A1 | GCATAGTCCCAACAGATCGGA | CTGGCATTTGGCAGCAAGA |
| NM_003381 | VIP | TGCTGATGGAGTTTTCACCAG | GACTGCATCTGAGTGACGTTTG |
| NM_020686 | ABAT | GCATCAGGAAATCGGATGGAG | TGCTTGGTGAACTCTGGCTCAT |
| NM_001909 | CTSD | CCCGACTTGCTGTTTTGTTCT | CCAATGCACGAAACAGATCTG |
| NM_005007 | NFKBIL1 | TTCCGAAGCCAGATTGAGACC | CCCCGAAGTTTCTTGCTTCTT |
| NM_001348 | DAPK3 | CGAGGAGTACTTCAGCAACACC | CGCCTTAATCCAGGAATGTTC |
| NM_001167942.1 | TNFAIP8L1 | GTTGGCTGGTTGGTTGCTGAT | AAAAGCTGCTGCCTCCCACTT |
| NM_005954 | MT3 | GTGTGGAGCACGTGGAGATAGT | TCATTCCTCCAAGGTCAGCAG |
| NM_005921.1 | MAP3K1 | TGCTAATTGACAGCACTGGTCA | TACCTCAGGTGCCATAAATGCA |
| NM_014841 | SNAP91 | GTTGTCATACTGTCTGTGCTGATGG | CAAACACCTTTCCAACCCAATATG |
| NM_003179 | SYP | GCCTGAGAAGAGTCGAGTGATATGG | CCGTTGGTTCTGTCCTCCTATTAAC |
| NM_021229 | NTN4 | ACTCCAGTCCTTTTCCATGCA | GCGAAGGTTGGTGATCTTCAG |
| NM_044472.2 | CDC42 | CTCCGGAAACTCAACCCAAA | GACGCAGAGGCTTTCAAACAG |
| NM_004263 | SEMA4F | CAGTGCTGAATGGTCCCTTCA | TGCCGGAGCTTCATGTTGTT |
| NM_002314 | LIMK1 | TGGTCCGCGAGAACAAGAAT | TTGCGGTCTGGCTTCTTGA |
| NM_002577 | PAK2 | AGGCTGTGCTGGATGTCCTAA | TGGCATTCAGTGCTGGTGTT |
| NM_002872 | RAC2 | CCTGTGGCGTTTCTTAGCAGA | GGATGCAGCACCTGCAAAT |
| NM_005232 | EPHA1 | ATCTTCACCACAGCCAGCGAT | CAACCGGTACCCATCCTCAAT |
| NM_006599 | NFAT5 | CACAGCGGTGGTGCTTAAAGA | TTTGCAGTCCTCAGTGCCACT |
| Z29373 | L1NCAM* | CCCTTTCGCCACAGTATGTCA | GCCGGAACATCCTCTCCTT AA |
| NM_014364 | GAPDH | TGACTTCAACAGCGACACCCA | CACCCTGTTGCTGTAGCCAAA |
| NM_001101.2 | ACTB | TTGTTACAGGAAGTCCCTTGCC | ATGCTATCACCTCCCCTGTGTG |
The neural cell adhesion molecule L1.
Western blotting
After incubation with PBS, Aβ, HupA alone or with Aβ for 24 h, cells were harvested, lysed in Passive Lysis Buffer containing protease inhibitors cocktail (Sigma P8430). Lysates were incubated for 30 min on ice, and then centrifuged at 10,000 X g for 10 min at 4°C. Protein concentration in the supernatant was determined by Coomassie blue protein binding method using protein quantification Kit-rapid (Sigma/Fluka) with bovine serum albumin as standard. The same amount of protein from different groups was used for immunoblotting. Samples were denatured in protein sample buffer (100 mM Tris-Cl pH 6.8, 4% SDS, 0.2% bromophenol blue, 20% glycerol, 20% H2O, 200 mM DTT) at 100°C for 10 min and loaded, separated on 10% SDS-polyacrylamide gels and transferred to nitrocellulose membranes in a Bio-Rad electrophoresis system. After blocking with TBST (Tris-buffered saline with 0.1% Tween) containing 5% non-fat milk, the membranes were kept at 4°C overnight with primary antibodies (1:5000 for β-ACTIN, 1:2000 for RAC1 and 1:1000 for RAC2 and CDC42, respectively), followed by HRP-conjugated second antibodies (1:5000 to 1:10000 dilution) at room temperature for 2 h. The target protein bands were detected using the ECL Western blotting detection system (Pierce) and autoradiography film (Fisher). The density of the RAC1, RAC2, CDC42 and β-ACTIN blotting bands were quantified by ImageJ software (1.37v, Wayne Rasband, National Institutes of Health, USA) as described [42].
Induction of neurite outgrowth
Undifferentiated PC12 rat pheochromocytoma cells were obtained from ATCC. The cells were maintained in DMEM media containing 10% (v/v) fetal bovine serum (FBS) (Gibco), 5% (v/v) horse serum (HS) (Gibco), 2mM glutamine (Gibco), 100 U/mL penicillin, and 100 μg/mL streptomycin (Gibco) in a humidified atmosphere of 95% air and 5% CO2 at 37°C. Cells (1×104 cells/well) were seeded in glass cover slips into a 12-well plate and cultured overnight. Subsequently the cells were washed twice with phosphate-buffered saline solution (PBS) and transferred to fresh serum-starved DMEM containing 1% HS, 0.5% FBS. After 2 h incubation with serum-starved DMEM, 100 ng/ml NGF(SinoBio, shanghai) was added to the cultures, which were incubated for 48 h. Cells were washed twice with PBS to remove NGF and replaced with 1μM Aβ1-42 (QiangYao, Shanghai) with or without Hup A for a further 48h. Pre-incubation with HupA (1 and 10 μ M) was conducted 2 hours before Aβ addition. Exposure to vehicle alone (culture medium) was used as the control. The cells were harvested for the further assays and detections after 48 h exposure of Aβ.
Immunofluorescence staining
PC12 cells cultured in glass cover slips were treated in serum-starved media for 2 days at 37°C under 95% air and 5% CO2. The cells were then washed with PBS and fixed with ice cold acetone for 30 min at -20°C. The cells were blocked with blocking buffer containing 5% BSA and 0.1% Triton X-100 for 30 min at RT. For double immunofluorescence staining, the cells were incubated with primary rabbit anti-MAP2 antibody (Abcam, 1:200) overnight at 4°C. Alexa Fluor 546 goat anti-rabbit IgG (H + L) (Invitrogen, 1:4000) was incubated at RT for 1 h. After rinsing with PBS and blocked with 5% BSA/PBS, cells were incubated with Alexa Fluor 488 conjugated anti-Tau-1 antibody (Millipore, 1:100) for 2h at RT. The nuclei were counterstained with VECTASHIELD® Mounting Medium with DAPI (1.5 μg/mL, Vector Laboratories, Inc.), and then the cells were photographed and analyzed with a fluorescent microscope (Nikon, ELWD 0.3, Japan). At least 3 independent immunofluorescence staining was performed to assess the reproducibility of the results.
Measurement of neurite outgrowth
Cell processes were defined as neurites when longer than the diameter of the cell body. The percentage of neurite-bearing cells in relation to the total number of cells was examined in 6-8 fields with an average of 15 cells per field under per treatment condition. After double immunofluorescence staining, neurites (dendrite and axon) formation was photographed and analyzed with a fluorescent microscope, and processes longer than one cell diameter were scored as neurites. Neurite length was determined by manually tracing the length of the longest neurite per cell using the ImageJ software (version 1.45s, NIH, USA) for neurite-bearing cells. At least 50 cells were randomly measured for each group.
Statistical analysis
All results are presented as mean ± SEM. Statistical analyses were performed with one-way ANOVA followed by least significant difference post hoc analysis or Duncan’s multiple-range test and t-test with threshold of p < 0.05. The results from individual experiments were averaged within each experimental group.
Results
Changes of axon guidance relative genes expression after the treatment of Aβ peptide with/without HupA
Using cDNA microarray, the gene expression profile was detected in Aβ with/without HupA and HupA alone treated human SH-SY5Y cells. Array data were verified by measuring the expression using quantitative RT-PCR. We randomly selected a group of genes to confirm the microarray data. PCR result showed that relative expression ratios of these genes determined by real-time quantitative RT-PCR were concordant with the expression levels observed in the corresponding microarray data. The correlation of the gene expression ratio between microarray ratio and qRT-PCR ratio is significant (P<0.0001). Interestingly, among more than 100 up or down regulated genes after the treatments above (data not shown), the genes involved in axon guidance were significantly regulated. RAC/Cdc42-PAK and RAC/Cdc42-ROCK pathway were affected which involved in axon attraction, axon repulsion and axon outgrowth (figure not shown). Aβ up-regulated the mRNA expressions of NFAT5, LIMK1, EPHA1, NTN4 and Rac2 markedly, but decreased Cdc42 expression. Co-incubation of Aβ and HupA reversed or decreased the changes of NFAT5, NTN4, RAC2, CDC42 and SEMA4F. HupA treated alone increased NFAT5, LIMK1, NTN4 significantly, but decreased RAC2, SEMA4F, PAK2 and LICAM (Table 2).
Table 2.
Aβ alters the axon guidance relative gene expression profile in SH-SY5Y cells
| GenBank ID | Symbol | Gene title | Fold changes | ||
|---|---|---|---|---|---|
|
| |||||
| Aβ | Aβ+HupA | HupA | |||
| NM_006599 | NFAT5 | Nuclear factor of activated T-cells 5, tonicity-responsive | 1.84 | 1.15 | 2.03 |
| NM_002314 | LIMK1 | LIM domain kinase 1 | 1.85 | 1.89 | 1.82 |
| NM_005232 | EPHA1 | EPH receptor A1 | 2.00 | 2.08 | 1.28 |
| NM_021229 | NTN4 | Netrin 4 | 3.85 | 2.14 | 2.00 |
| NM_002872 | RAC2 | small GTP binding protein Rac2 | 2.02 | 0.47 | 0.787 |
| NM_044472.2 | CDC42 | Cell division cycle 42 | 0.57 | 1.61 | 0.97 |
| NM_004263 | SEMA4F | Sema domain, immunoglobulin domain (Ig), transmembrane domain (TM) and short cytoplasmic domain | 0.87 | 2.16 | 0.76 |
| NM_002577 | PAK2 | p21 (CDKN1A)-activated kinase 2 | 0.82 | 0.85 | 0.52 |
| Z29373 | L1CAM* | neural cell adhesion molecule L1 | 0.94 | 1.15 | 0.56 |
The neural cell adhesion molecule L1.
Among the verified genes, axon guidance relative genes Cdc42, Rac2 and NFAT5 mRNA levels are coincidence with the array data. Rac2 and NFAT5 were up-regulated, while Cdc42 was down-regulated (Figure 1).
Figure 1.
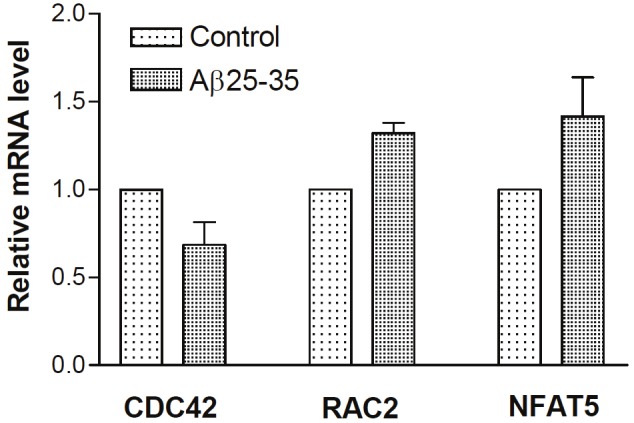
Aβ down-regulated CDC42 and up-regulated RAC2 and NFAT5 mRNA levels. SH-SY5Y cells were treated with PBS, Aβ, HupA alone or with Aβ for 24 h, respectively. Total RNA was extracted and analyzed by real-time RT-PCR and normalized to Human GAPDH mRNA levels. Shown are mean ± SEM of relative mRNA level.
Western blotting of Cdc42, RAC1 and RAC2
To confirm the evidence of microarray and qPCR, we further detected CDC42, RAC1 and RAC2 protein levels by Western blotting. The alteration of CDC42 protein is consistent with the mRNA regulation in Aβ and Aβ with HupA treated cells. However, the changes of RAC2 are opposite to that of the mRNA level (Figure 2). Treatment with Aβ or HupA alone did not affect RAC1 obviously, but co-incubation of HupA (1, 10 μM) and Aβ markedly increased RAC1 level. The effect of HupA treated alone up-regulated the CDC42 and RAC2 protein which is inconsistent with changes of the mRNA levels.
Figure 2.
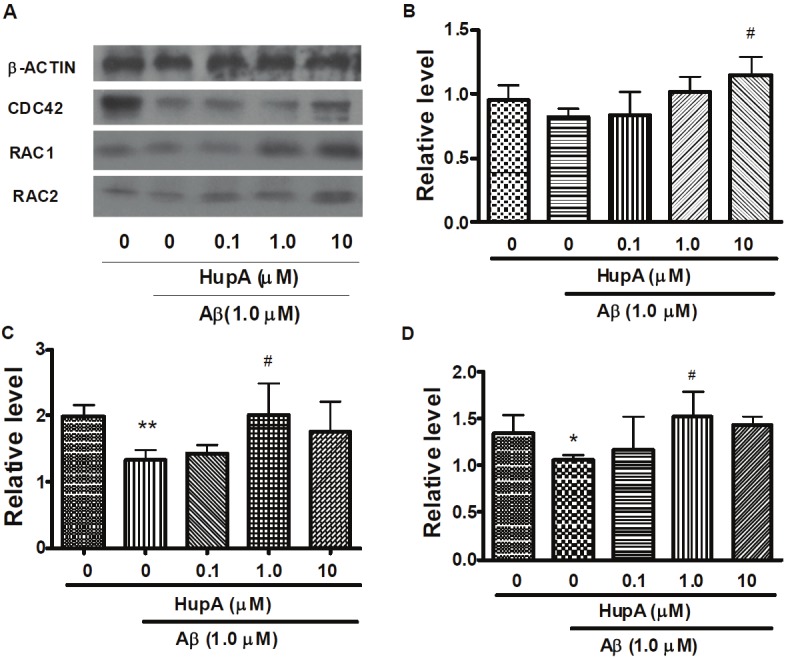
Western blotting analysis of CDC42, RAC1 and RAC2 protein levels in Aβ treated SH-SY5Y cells. After incubation with PBS, Aβ, HupA with Aβ for 24 h, SH-SY5Y cells were harvested and analyzed by western blot. Figure is the representative bands of β-ACTIN, CDC42, RAC1 and RAC2 Western blotting. A. Representative β-ACTIN, CDC42, RAC1 and RAC2 western blot bands. B, C and D. Immunoblots quantification of CDC42, RAC1 and RAC2, respectively. All bands were quantified and normalized by β-ACTIN. The data is expressed as mean ± SEM from three independent experiments. *p <0.05, **p <0.01 compared to control. #p < 0.05 compared to Aβ-treated group.
Double immunofluorescence staining and neurite outgrowth analysis of Aβ-treated differentiated PC12 cells
To further confirm if the phenotype was affected by the alterations of the above-mentioned genes expression, we next studied the effect of Aβ on the NGF-induced neurite outgrowth. PC12 cell is a cell line from pheochromocytoma of the rat adrenal medulla. It differentiates in response to nerve growth factor (NGF) with a dramatic change in phenotype, acquiring biochemical and morphological characteristics of sympathetic neurons. The elaboration of neurites occurs within 24-48 h of NGF exposure and continues for up to a week [43]. Thus, PC12 cells have been widely used as a model system in the investigation of neuronal differentiation in vitro, esp. in neurite outgrowth [44,45]. To determine the length of dendrite and axon, we performed double fluorescence staining (MAP-2 and Tau-1) followed by manually tracing the length analysis of NGF-induced PC12 cells. MAP-2 was used as a marker of dendrites, and Tau-1 as an axonal marker in neurons (Figure 3A and 4A). Both MAP-2 and Tau-1 were mainly localized in the cell body (Figure 3A and 4A). MAP-2 and Tau-1 positive neurites were almost undetectable in controls, but visualized in NGF-induced cells (Figure 3A). NGF (100 ng/ml) treatment induced neurite outgrowth in more than 60% of cells compared with control (Figure 3). Aβ treatment significantly reduced the number of neurite-bearing cells (Figure 4B) (reduced up to 35.63%, P<0.001) and shortened the neurite length (Figure 4A, C) compared to NGF control. These effects were markedly reversed by HupA (Figure 4A, B and C).
Figure 3.

NGF induced PC12 cells differentiation. PC12 cells were induced with 100ng/ml NGF for 48h. A. Cells were untreated (a-c), treated with 100ng/ml NGF (d-f) for 48 hours prior to fixation and staining with anti-MAP-2 (red) (a, d) or anti-Tau-1 (green) (b, e). Merged images revealed both most colocalized in the cell bodies (c, f). B. Neurite formation was examined under a fluorescent microscope after immunofluorescent staining, and processes longer than one cell diameter were scored as neurites. C. The longest length of neurites per cell was measured after immunofluorescent staining, and the mean value of neurite length was calculated. ***p <0.001. Scale bar is 100 μm.
Figure 4.
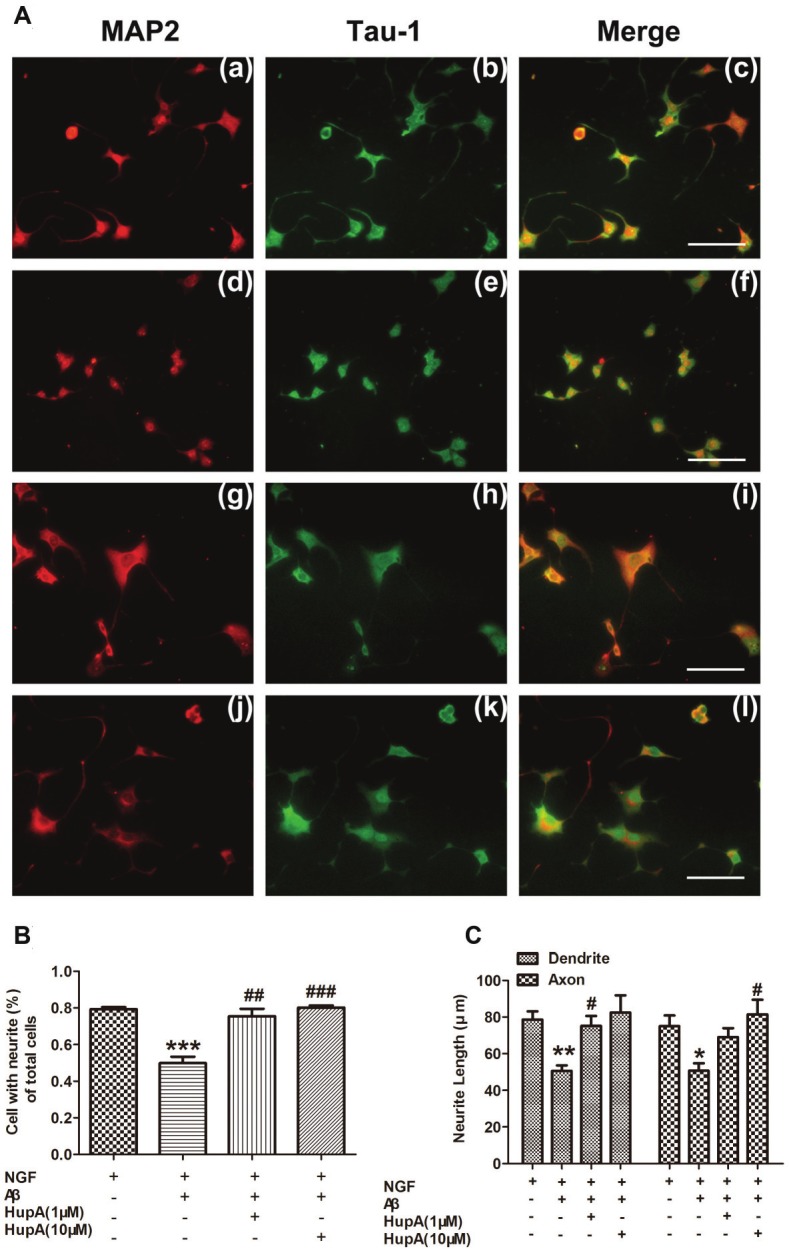
The effect ofHupA on the NGF-inducedneurite outgrowthof PC12 cells with Aβtreatment. Cells weretreated with NGF (a-c),NGF and 1 μM Aβ1-42(d-f), 1 μM (g-i) or 10μM (j-l) HupA, respectively.HupA was added2h before Aβ treatmentA. shows the representativeimages of immunofluorescentstainingunder different treatments:MAP-2 (red) (a,d, g, j), Tau-1 (green) (b,e, h, k) and merged imagesrevealed both (c, f, i, l). B. Neurite-bearingcells counting. Neuriteformation is measuredunder a fluorescent microscopeafter immunofluorescentstaining.The processes longerthan one cell diameterwere counted as neurites.C. The neuriteslength measurement.The longest length ofneurites was measuredafter immunofluorescentstaining, and themean value of neuritelength was calculated.Values are representedas means ±SEM. *p<0.05, **p <0.01 and ***p <0.001 for comparisonsbetween NGFtreatment and co-treatmentof NGF and Aβ; #p<0.05, ##p <0.01 and ###p <0.001 for comparisonsbetween NGFand Aβ co-treated groupand NGF, Aβ and HuperzineA co-treated group.Scale bar is 100 μm.
Discussion
The extension of axons and dendrites from the cell body is a complex and finely regulated process that requires the alteration of actin polymerization and depolymerization and tight dynamics of microtubules [46]. These events are under the control of various guidance cues and negative regulatory signals [47,48]. PAK and ROCK are both the family of serine/threonine protein kinases. PAK involves in regulating the actin-severing protein cofilin, the actin cytoskeleton, and dendritic function as downstream effectors of Rac1/Cdc42 [49,50]. By its kinase activity, ROCK regulates the activities of many target proteins (Rho, Rac, and Cdc42) which are involved in the regulation of cytoskeletal reorganization and then regulates dendrite initiation, growth, branching, spinogenesis, and spine maintenance [18,51,52].
The pheochromocytoma PC12 cell line has been used in both neurobiological [53] and neurotoxicological [53,54] studies as a model of neurite outgrowth. PC12 cells originate from chromaffin cells, whose differentiation into sympathetic neurons can also be induced by some factors including NGF, bFGF, and cAMP [55]. Previous reports studied that RhoA activity is correlated with neurite outgrowth in PC12 cells induced by cAMP [56], and Rab22 can promote nerve growth factor (NGF) signaling-dependent neurite outgrowth and gene expression in PC12 cells [57]. We described the method to quantify the number of neurites (dendrite and axon) bearing cells and neurite length after double immunofluorescence staining in PC12 cells using antibodies to Tau-1 as an axonal marker, and to MAP2 as a dendritic marker. Therefore the morphological changes are demonstrated in PC12 cells (Figure 3 and 4) to further confirm the result of microarray (Table 2), RT-PCR (Figure 1) and Western blotting (Figure 2). Our studies showed that Aβ regulates the genes expression involved in axon guidance in human neuroblastoma cells and inhibits NGF-induced neurite outgrowth in undifferentiated PC12 rat pheochromocytoma cells (Table 2, Figure 4), which is agree with some of previous reports [58-61]. Aβ up-regulated the mRNA expressions of NFAT5, LIMK1, EPHA1, NTN4 and Rac2 markedly, but decreased Cdc42 expression. SEMA4F, PAK2 and Neural cell adhension molecule L1 mRNA were not significantly regulated though a slight decrease was observed. All these factors are involved in the pathways in regulation of actin cytoskeleton and axon guidance pathways. Our further immunofluorescence staining showing the inhibition of neurite outgrowth by Aβ (Figure 4) confirmed the consequences of the genes expression alteration. Previous reports indicated that PAK protein and activity are markedly reduced in Alzheimer’s disease, and Aβ was directly involved in PAK signaling deficits [62]. Aβ-induced deficits showed an abnormal PAK activation and was accompanied by a rapid loss of F-actin and dendritic spines, which are the opposite responses compared to the normal activation of PAK by Rac/Cdc42 GTPase [4].
Even though a large number of reports have demonstrated the involvement of the Rho-ROCK pathway in the pathogenesis of several diseases [50], the pathogenesis of Rho GTPase pathways, esp. Rho-ROCK pathway, in AD development is still far less understood. In this study, Netrin4 (NTN4), the upstream factor of ROCK pathway was markedly up-regulated which suppose to activate ROCK signals. The result is agree with the previous report [63].
As a novel selective acetylcholinesterase inhibitor, HupA has been found, except for its potent AChE inhibition effect, to improve cognitive deficits in a broad range of animal models [33,64-67] and to protect cells against cytotoxicity and apoptosis induced by multiple pathogenic factors [30,32]. The mechanisms of these protective effects are still far less understood though we do find they related to attenuating oxidative stress [34,35,68,69], regulating the apoptotic proteins [31,33] and protecting mitochondria [70].
In this study, as shown in Table 2, Figure 2 and Figure 4, co-incubation of Aβ and HupA reversed or decreased the changes of NFAT5, NTN4, RAC2, CDC42 and SEMA4F. HupA itself increased NFAT5, LIMK1, NTN4 significantly, but decreased RAC2, SEMA4F, PAK2 and LICAM. As a consequence, the Aβ-induced neurite outgrowth damage was declined with a dose response manner. The data implied that HupA might protect neurons morphologically and functionally, at least partially, against Aβ-induced pathological process via regulating neurite outgrowth and synaptogenesis. Our previous studies indicated that NGF and NGF receptors involved in HupA’s non-cholinesterase inhibiting effects [30,32]. It has been well known that NGF is the first neurotrophin to promote neuronal survival and differentiation [71]. Upon NGF induced neurite outgrowth, the NGF receptor TrkA activates several small G proteins, including Ras, Rap1, and the Cdc42-Rac1-RhoA family [72]. P75 low-affinity NGF receptor p75NTR modulates, in a ligand-dependent fashion, the activity of intracellular proteins known to regulate actin assembly [39]. Aβ activates the RhoA GTPase by binding to p75 NTR leading to impair the initial steps of NGF signaling, therefore inactivation of RhoA GTPase can protect cultured hippocampal neurons against the noxious effects of Aβ [73]. Our previous data shown that HupA markedly increased the number of neurite-bearing cells with a significant up-regulation of NGF and P75 low-affinity NGF receptor [38,74]. From the study of hydrogen peroxide induced injury, the NGF and TrkA receptor mediate key events required for the neuroprotective actions of HupA. Among the downstream signaling events triggered by the action of NGF at the TrkA receptor, activation of the MAP/ERK kinase pathway may be particularly important for the ability of HupA to protect SH-SY5Y cells against oxidative stress [74]. The further data shown from present study agrees with the previous reports either from our study or the other’s. It is very likely that the effect of HupA on the regulation of axon guidance relative genes and proteins related to affecting NGF signaling.
In addition to inhibiting AChE activity, in our previous study, AChE mRNA expression and protein levels were significantly upregulated after treatment with HupA [38]. Accumulating evidence indicates that AChE may influence neurite outgrowth through a non-catalytic mechanism. Via protein-protein interactions, AChE’s effects on neurite outgrowth are not directly related to catalytic function but are nonetheless influenced by ligands with special structural features [75,76]. Now that HupA increased AChE mRNA and protein levels, though the AChE activity was significantly inhibited [38], the effect of HupA on neurite outgrowth relative genes and proteins may be associated with the level of AChE expression.
Acknowledgement
This work was supported by the grants (to Rui Wang) from National Natural Science Foundation of China 30572169, 81072627; Pujiang talent project of Shanghai (11PJ1402300); the 111 Project (Grant No. B07023) and the grants (to Dengshun Wang) from National Institutes of Health (NIH) grants AG025722 and AG029972, and an Alzheimer’s Association grant IIRG-08-90524. The authors are grateful to Professor Xi-can Tang (Shanghai Institute of Materia Medica, Chinese Academy of Sciences) for the advance and support.
References
- 1.Bartus RT, Dean RL 3rd, Beer B, Lippa AS. The cholinergic hypothesis of geriatric memory dysfunction. Science. 1982;217:408–414. doi: 10.1126/science.7046051. [DOI] [PubMed] [Google Scholar]
- 2.Hyman BT, Tanzi RE. Amyloid, dementia and alzheimers-diseae. Current Opinion in Neurology and Neurosurgery. 1992;5:88–93. [PubMed] [Google Scholar]
- 3.Scheff SW, Dekosky ST, Price DA. Quantitative assessment of cortical synaptic density in alzheimers-disease. Neurobiology of Aging. 1990;11:29–37. doi: 10.1016/0197-4580(90)90059-9. [DOI] [PubMed] [Google Scholar]
- 4.Ma QL, Yang FS, Calon F, Ubeda OJ, Hansen JE, Weisbart RH, Beech W, Frautschy SA, Cole GM. p21-activated kinase-aberrant activation and translocation in Alzheimer disease pathogenesis. Journal of Biological Chemistry. 2008;283:14132–14143. doi: 10.1074/jbc.M708034200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Oostra BA, Willemsen R. FMR1: A gene with three faces. Biochimica Et Biophysica Acta- General Subjects. 2009;1790:467–477. doi: 10.1016/j.bbagen.2009.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Steinbach P. Fragile X Syndrome. Medizinische Genetik. 2009;21:251–259. [Google Scholar]
- 7.Israely I, Costa RM, Silva AJ, Kosik KS, Liu X. Loss of delta - catenin changes synaptic composition and impairs plasticity and learning. Society for Neuroscience Abstract Viewer and Itinerary Planner. 2003;2003:Abstract No. 964.920. [Google Scholar]
- 8.Small DH. Network dysfunction in Alzheimer’s disease: does synaptic scaling drive disease progression? Trends in Molecular Medicine. 2008;14:103–108. doi: 10.1016/j.molmed.2007.12.006. [DOI] [PubMed] [Google Scholar]
- 9.Bate C, Gentleman S, Williams A. alpha-synuclein induced synapse damage is enhanced by amyloid-beta1-42. Mol Neurodegener. 2011;5:55. doi: 10.1186/1750-1326-5-55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bell KF, Claudio Cuello A. Altered synaptic function in Alzheimer’s disease. Eur J Pharmacol. 2006;545:11–21. doi: 10.1016/j.ejphar.2006.06.045. [DOI] [PubMed] [Google Scholar]
- 11.Zheng ZQ, Sabirzhanov B, Keifer J. Oligomeric Amyloid-beta Inhibits the Proteolytic Conversion of Brain-derived Neurotrophic Factor (BDNF), AMPA Receptor Trafficking, and Classical Conditioning. Journal of Biological Chemistry. 2010;285:34708–34717. doi: 10.1074/jbc.M110.150821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tampellini D, Capetillo-Zarate E, Dumont M, Huang ZY, Yu FM, Lin MT, Gouras GK. Effects of Synaptic Modulation on beta-Amyloid, Synaptophysin, and Memory Performance in Alzheimer’s Disease Transgenic Mice. Journal of Neuroscience. 2010;30:14299–14304. doi: 10.1523/JNEUROSCI.3383-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lacor PN, Buniel MC, Chang L, Fernandez SJ, Gong YS, Viola KL, Lambert MP, Velasco PT, Bigio EH, Finch CE, Krafft GA, Klein WL. Synaptic targeting by Alzheimer’s-related amyloid beta oligomers. Journal of Neuroscience. 2004;24:10191–10200. doi: 10.1523/JNEUROSCI.3432-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Lacor PN, Buniel MC, Furlow PW, Clemente AS, Velasco PT, Wood M, Viola KL, Klein WL. A beta oligomer-induced aberrations in synapse composition, shape, and density provide a molecular basis for loss of connectivity in Alzheimer’s disease. Journal of Neuroscience. 2007;27:796–807. doi: 10.1523/JNEUROSCI.3501-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Li SM, Hong SY, Shepardson NE, Walsh DM, Shankar GM, Selkoe D. Soluble Oligomers of Amyloid beta Protein Facilitate Hippocampal Long-Term Depression by Disrupting Neuronal Glutamate Uptake. Neuron. 2009;62:788–801. doi: 10.1016/j.neuron.2009.05.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shankar GM, Bloodgood BL, Townsend M, Walsh DM, Selkoe DJ, Sabatini BL. Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. Journal of Neuroscience. 2007;27:2866–2875. doi: 10.1523/JNEUROSCI.4970-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shankar GM, Li SM, Mehta TH, Garcia-Munoz A, Shepardson NE, Smith I, Brett FM, Farrell MA, Rowan MJ, Lemere CA, Regan CM, Walsh DM, Sabatini BL, Selkoe DJ. Amyloid-beta protein dimers isolated directly from Alzheimer’s brains impair synaptic plasticity and memory. Nature Medicine. 2008;14:837–842. doi: 10.1038/nm1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Benarroch EE. Rho GTPases - Role in dendrite and axonal growth, mental retardation, and axonal regeneration. Neurology. 2007;68:1315–1318. doi: 10.1212/01.wnl.0000259588.97409.8f. [DOI] [PubMed] [Google Scholar]
- 19.Huesa G, Baltrons MA, Gomez-Ramos P, Moran A, Garcia A, Hidalgo J, Frances S, Santpere G, Ferrer I, Galea E. Altered distribution of RhoA in Alzheimer’s disease and AbetaPP overexpressing mice. J Alzheimers Dis. 2010;19:37–56. doi: 10.3233/JAD-2010-1203. [DOI] [PubMed] [Google Scholar]
- 20.Antoine-Bertrand J, Villemure JF, Lamarche-Vane N. Implication of rho GTPases in neurodegenerative diseases. Curr Drug Targets. 2011;12:1202–1215. doi: 10.2174/138945011795906543. [DOI] [PubMed] [Google Scholar]
- 21.Chen LY, Rex CS, Babayan AH, Kramar EA, Lynch G, Gall CM, Lauterborn JC. Physiological Activation of Synaptic Rac > PAK (p-21 Activated Kinase) Signaling Is Defective in a Mouse Model of Fragile X Syndrome. Journal of Neuroscience. 2010;30:10977–10984. doi: 10.1523/JNEUROSCI.1077-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Viti S, Martino A, Musilli M, Fiorentini C, Diana G. The Rho GTPase activating CNF1 improves associative working memory for object-in-place. Behavioural Brain Research. 2010;212:78–83. doi: 10.1016/j.bbr.2010.03.049. [DOI] [PubMed] [Google Scholar]
- 23.Oh D, Han S, Seo J, Lee JR, Choi J, Groffen J, Kim K, Cho YS, Choi HS, Shin H, Woo J, Won H, Park SK, Kim SY, Jo J, Whitcomb DJ, Cho K, Kim H, Bae YC, Heisterkamp N, Choi SY, Kim E. Regulation of Synaptic Rac1 Activity, Long-Term Potentiation Maintenance, and Learning and Memory by BCR and ABR Rac GTPase-Activating Proteins. Journal of Neuroscience. 2010;30:14134–14144. doi: 10.1523/JNEUROSCI.1711-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Svitkina T, Lin WH, Webb DJ, Yasuda R, Wayman GA, Van Aelst L, Soderling SH. Regulation of the Postsynaptic Cytoskeleton: Roles in Development, Plasticity, and Disorders. Journal of Neuroscience. 2010;30:14937–14942. doi: 10.1523/JNEUROSCI.4276-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Narumiya S, Yasuda S. Rho GTPases in animal cell mitosis. Current Opinion in Cell Biology. 2006;18:199–205. doi: 10.1016/j.ceb.2006.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Hayashi K, Ohshima T, Mikoshiba K. Pak1 is involved in dendrite initiation as a downstream effector of Rac1 in cortical neurons. Molecular and Cellular Neuroscience. 2002;20:579–594. doi: 10.1006/mcne.2002.1144. [DOI] [PubMed] [Google Scholar]
- 27.Riento K, Ridley AJ. Rocks: Multifunctional kinases in cell behaviour. Nature Reviews Molecular Cell Biology. 2003;4:446–456. doi: 10.1038/nrm1128. [DOI] [PubMed] [Google Scholar]
- 28.Rajapaksha TW, Eimer WA, Bozza TC, Vassar R. The Alzheimer’s beta-secretase enzyme BACE1 is required for accurate axon guidance of olfactory sensory neurons and normal glomerulus formation in the olfactory bulb. Mol Neurodegener. 2011;6:88. doi: 10.1186/1750-1326-6-88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Cao L, Rickenbacher GT, Rodriguez S, Moulia TW, Albers MW. The precision of axon targeting of mouse olfactory sensory neurons requires the BACE1 protease. Sci Rep. 2012;2:231. doi: 10.1038/srep00231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang R, Yan H, Tang XC. Progress in studies of huperzine A, a natural cholinesterase inhibitor from Chinese herbal medicine. Acta Pharmacologica Sinica. 2006;27:1–26. doi: 10.1111/j.1745-7254.2006.00255.x. [DOI] [PubMed] [Google Scholar]
- 31.Wang R, Xiao XQ, Tang XC. Huperzine A attenuates hydrogen peroxide-induced apoptosis by regulating expression of apoptosis-related genes in rat PC12 cells. Neuroreport. 2001;12:2629–2634. doi: 10.1097/00001756-200108280-00009. [DOI] [PubMed] [Google Scholar]
- 32.Wang R, Tang XC. Neuroprotective effects of huperzine A - A natural cholinesterase inhibitor for the treatment of Alzheimer’s disease. Neurosignals. 2005;14:71–82. doi: 10.1159/000085387. [DOI] [PubMed] [Google Scholar]
- 33.Wang R, Zhang HY, Tang XC. Huperzine A attenuates cognitive dysfunction and neuronal degeneration caused by beta-amyloid protein-(1-40) in rat. European Journal of Pharmacology. 2001;421:149–156. doi: 10.1016/s0014-2999(01)01030-5. [DOI] [PubMed] [Google Scholar]
- 34.Xiao XQ, Wang R, Han YF, Tang XC. Protective effects of huperzine A on beta-amyloid (25-35) induced oxidative injury in rat pheochromocytoma cells. Neuroscience Letters. 2000;286:155–158. doi: 10.1016/s0304-3940(00)01088-0. [DOI] [PubMed] [Google Scholar]
- 35.Xiao XQ, Wang R, Tang XC. Huperzine A and tacrine attenuate beta-amyloid peptide-induced oxidative injury. Journal of Neuroscience Research. 2000;61:564–569. doi: 10.1002/1097-4547(20000901)61:5<564::AID-JNR11>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- 36.Wang CY, Zheng W, Wang T, Xie JW, Wang SL, Zhao BL, Teng WP, Wang ZY. Huperzine A activates Wnt/beta-catenin signaling and enhances the nonamyloidogenic pathway in an Alzheimer transgenic mouse model. Neuropsychopharmacology. 2011;36:1073–1089. doi: 10.1038/npp.2010.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Shigeta K, Ootaki K, Tatemoto H, Nakanishi T, Inada A, Muto N. Potentiation of nerve growth factor-induced neurite outgrowth in PC12 cells by a Coptidis Rhizoma extract and protoberberine alkaloids. Bioscience Biotechnology and Biochemistry. 2002;66:2491–2494. doi: 10.1271/bbb.66.2491. [DOI] [PubMed] [Google Scholar]
- 38.Tang LL, Wang R, Tang XC. Effects of huperzine A on secretion of nerve growth factor in cultured rat cortical astrocytes and neurite outgrowth in rat PC12 cells. Acta Pharmacologica Sinica. 2005;26:673–678. doi: 10.1111/j.1745-7254.2005.00130.x. [DOI] [PubMed] [Google Scholar]
- 39.Yamashita T, Tucker KL, Barde YA. Neurotrophin binding to the p75 receptor modulates Rho activity and axonal outgrowth. Neuron. 1999;24:585–593. doi: 10.1016/s0896-6273(00)81114-9. [DOI] [PubMed] [Google Scholar]
- 40.Kaminsky YG, Marlatt MW, Smith MA, Kosenko EA. Subcellular and metabolic examination of amyloid-beta peptides in Alzheimer disease pathogenesis: evidence for Abeta (25-35) Exp Neurol. 2010;221:26–37. doi: 10.1016/j.expneurol.2009.09.005. [DOI] [PubMed] [Google Scholar]
- 41.Gulyaeva NV, Stepanichev MY. A beta (25-35) as proxyholder for amyloidogenic peptides: In vivo evidence. Experimental Neurology. 2010;222:6–9. doi: 10.1016/j.expneurol.2009.12.019. [DOI] [PubMed] [Google Scholar]
- 42.Farrer M, Kachergus J, Forno L, Lincoln S, Wang DS, Hulihan M, Maraganore D, Gwinn-Hardy K, Wszolek Z, Dickson D, Langston JW. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Annals of Neurology. 2004;55:174–179. doi: 10.1002/ana.10846. [DOI] [PubMed] [Google Scholar]
- 43.Das KP, Freudenrich TM, Mundy WR. Assessment of PC12 cell differentiation and neurite growth: a comparison of morphological and neurochemical measures. Neurotoxicol Teratol. 2004;26:397–406. doi: 10.1016/j.ntt.2004.02.006. [DOI] [PubMed] [Google Scholar]
- 44.Khodagholi F, Tusi SK, Alamdary SZ, Amini M, Ansari N. 3-Thiomethyl-5,6-(dimethoxyphenyl)- 1,2,4-triazine improves neurite out growth and modulates MAPK phosphorylation and HSPs expression in H(2)O(2)-exposed PC12 cells. Toxicology in vitro : an international journal published in association with BIBRA. 2012;26:907–914. doi: 10.1016/j.tiv.2012.04.015. [DOI] [PubMed] [Google Scholar]
- 45.Labour MN, Banc A, Tourrette A, Cunin F, Verdier JM, Devoisselle JM, Marcilhac A, Belamie E. Thick collagen-based 3D matrices including growth factors to induce neurite outgrowth. Acta biomaterialia. 2012;8:3302–3312. doi: 10.1016/j.actbio.2012.05.015. [DOI] [PubMed] [Google Scholar]
- 46.Boukhelifa M, Parast MM, Valtschanoff JG, La- Mantia AS, Meeker RB, Otey CA. A role for the cytoskeleton-associated protein palladin in neurite outgrowth. Molecular Biology of the Cell. 2001;12:2721–2729. doi: 10.1091/mbc.12.9.2721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miller FD, Kaplan DR. Signaling mechanisms underlying dendrite formation. Current Opinion in Neurobiology. 2003;13:391–398. doi: 10.1016/s0959-4388(03)00072-2. [DOI] [PubMed] [Google Scholar]
- 48.Vaillant AR, Zanassi P, Walsh GS, Aumont A, Alonso A, Miller FD. Signaling mechanisms underlying reversible, activity-dependent dendrite formation. Neuron. 2002;34:985–998. doi: 10.1016/s0896-6273(02)00717-1. [DOI] [PubMed] [Google Scholar]
- 49.Ramakers GJA. Rho proteins, mental retardation and the cellular basis of cognition. Trends in Neurosciences. 2002;25:191–199. doi: 10.1016/s0166-2236(00)02118-4. [DOI] [PubMed] [Google Scholar]
- 50.Salminen A, Suuronen T, Kaarniranta K. ROCK, PAK, and Toll of synapses in Alzheimer’s disease. Biochemical and Biophysical Research Communications. 2008;371:587–590. doi: 10.1016/j.bbrc.2008.04.148. [DOI] [PubMed] [Google Scholar]
- 51.Govek EE, Newey SE, Van Aelst L. The role of the Rho GTPases in neuronal development. Genes & Development. 2005;19:1–49. doi: 10.1101/gad.1256405. [DOI] [PubMed] [Google Scholar]
- 52.Linseman DA, Loucks FA. Diverse roles of Rho family GTPases in neuronal development, survival, and death. Frontiers in Bioscience. 2008;13:657–676. doi: 10.2741/2710. [DOI] [PubMed] [Google Scholar]
- 53.Radio NM, Breier JM, Shafer TJ, Mundy WR. Assessment of chemical effects on neurite outgrowth in PC12 cells using high content screening. Toxicol Sci. 2008;105:106–118. doi: 10.1093/toxsci/kfn114. [DOI] [PubMed] [Google Scholar]
- 54.Zhaleh H, Azadbakht M, Pour AB. Effects of extracellular calcium concentration on neurite outgrowth in PC12 cells by staurosporine. Neurosci Lett. 2011;498:1–5. doi: 10.1016/j.neulet.2011.04.020. [DOI] [PubMed] [Google Scholar]
- 55.Sofroniew MV, Howe CL, Mobley WC. Nerve growth factor signaling, neuroprotection, and neural repair. Annu Rev Neurosci. 2001;24:1217–1281. doi: 10.1146/annurev.neuro.24.1.1217. [DOI] [PubMed] [Google Scholar]
- 56.Jeon CY, Moon MY, Kim JH, Kim HJ, Kim JG, Li Y, Jin JK, Kim PH, Kim HC, Meier KE, Kim YS, Park JB. Control of neurite outgrowth by RhoA inactivation. J Neurochem. 2012;120:684–698. doi: 10.1111/j.1471-4159.2011.07564.x. [DOI] [PubMed] [Google Scholar]
- 57.Wang L, Liang Z, Li G. Rab22 controls NGF signaling and neurite outgrowth in PC12 cells. Mol Biol Cell. 2011;22:3853–3860. doi: 10.1091/mbc.E11-03-0277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Magara F, Muller U, Li ZW, Lipp HP, Weissmann C, Stagljar M, Wolfer DP. Genetic background changes the pattern of forebrain commissure defects in transgenic mice underexpressing the beta-amyloid-precursor protein. Proceedings of the National Academy of Sciences of the United States of America. 1999;96:4656–4661. doi: 10.1073/pnas.96.8.4656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Zou YM. Axons find their way in the snow. Development. 2009;136:2135–2139. doi: 10.1242/dev.034686. [DOI] [PubMed] [Google Scholar]
- 60.Caltagarone J, Hamilton RL, Murdoch G, Jing Z, DeFranco DB, Bowser R. Paxillin and Hydrogen Peroxide-Inducible Clone 5 Expression and Distribution in Control and Alzheimer Disease Hippocampi. Journal of Neuropathology and Experimental Neurology. 2010;69:356–371. doi: 10.1097/NEN.0b013e3181d53d98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wilkinson B, Koenigsknecht-Talboo J, Grommes C, Lee CYD, Landreth G. Fibrillar beta-amyloid-stimulated intracellular signaling cascades require Vav for induction of respiratory burst and phagocytosis in monocytes and microglia. Journal of Biological Chemistry. 2006;281:20842–20850. doi: 10.1074/jbc.M600627200. [DOI] [PubMed] [Google Scholar]
- 62.Zhao LX, Ma QL, Calon F, Harris-White ME, Yang FS, Lim GP, Morihara T, Ubeda OJ, Ambegaokar S, Hansen JE, Weisbart RH, Teter B, Frautschy SA, Cole GM. Role of p21-activated kinase pathway defects in the cognitive deficits of Alzheimer disease. Nature Neuroscience. 2006;9:234–242. doi: 10.1038/nn1630. [DOI] [PubMed] [Google Scholar]
- 63.Petratos S, Li QX, George AJ, Hou X, Kerr ML, Unabia SE, Hatzinisiriou I, Maksel D, Aguilar MI, Small DH. The beta-amyloid protein of Alzheimers disease increases neuronal CRMP-2 phosphorylation by a Rho-GTP mechanism. Brain. 2008;131:90–108. doi: 10.1093/brain/awm260. [DOI] [PubMed] [Google Scholar]
- 64.Ou LY, Tang XC, Cai JX. Effect of huperzine A on working memory in reserpine- or yohimbine-treated monkeys. European Journal of Pharmacology. 2001;433:151–156. doi: 10.1016/s0014-2999(01)01500-x. [DOI] [PubMed] [Google Scholar]
- 65.Wang LM, Han YF, Tang XC. Huperzine A improves cognitive deficits caused by chronic cerebral hypoperfusion in rats. European Journal of Pharmacology. 2000;398:65–72. doi: 10.1016/s0014-2999(00)00291-0. [DOI] [PubMed] [Google Scholar]
- 66.Wang LS, Zhou J, Shao XM, Tang XC. Huperzine A attenuates cognitive deficits and brain injury in neonatal rats after hypoxia-ischemia. Brain Research. 2002;949:162–170. doi: 10.1016/s0006-8993(02)02977-3. [DOI] [PubMed] [Google Scholar]
- 67.Xiong ZQ, Cheng DH, Tang XC. Effects of huperzine A on nucleus basalis magnocellularis lesion-induced spatial working memory deficit. Acta Pharmacologica Sinica. 1998;19:128–132. [PubMed] [Google Scholar]
- 68.Xiao XQ, Yang JW, Tang XC. Huperzine A protects rat pheochromocytoma cells against hydrogen peroxide-induced injury. Neuroscience Letters. 1999;275:73–76. doi: 10.1016/s0304-3940(99)00695-3. [DOI] [PubMed] [Google Scholar]
- 69.Xiao XQ, Zhang HY, Tang XC. Huperzine A attenuates amyloid beta-peptide fragment 25-35-induced apoptosis in rat cortical neurons via inhibiting reactive oxygen species formation and caspase-3 activation. Journal of Neuroscience Research. 2002;67:30–36. doi: 10.1002/jnr.10075. [DOI] [PubMed] [Google Scholar]
- 70.Zhou J, Tang XC. Huperzine A attenuates apoptosis and mitochondria-dependent caspase-3 in rat cortical neurons. Febs Letters. 2002;526:21–25. doi: 10.1016/s0014-5793(02)03107-1. [DOI] [PubMed] [Google Scholar]
- 71.Levimontalcini R. The nerve growth-factor 35 years later. Science. 1987;237:1154–1162. doi: 10.1126/science.3306916. [DOI] [PubMed] [Google Scholar]
- 72.Huang EJ, Zang KL, Schmidt A, Saulys A, Xiang MQ, Reichardt LF. POU domain factor Brn-3a controls the differentiation and survival of trigeminal neurons by regulating Trk receptor expression. Development. 1999;126:2869–2882. doi: 10.1242/dev.126.13.2869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Chacon PJ, Garcia-Mejias R, Rodriguez-Tebar A. Inhibition of RhoA GTPase and the subsequent activation of PTP1B protects cultured hippocampal neurons against amyloid beta toxicity. Mol Neurodegener. 2011;6:14. doi: 10.1186/1750-1326-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Tang LL, Wang R, Tang XC. Huperzine A protects SHSY5Y neuroblastoma cells against oxidative stress damage via nerve growth factor production. European Journal of Pharmacology. 2005;519:9–15. doi: 10.1016/j.ejphar.2005.06.026. [DOI] [PubMed] [Google Scholar]
- 75.Brimijoin S, Koenigsberger C. Cholinesterases in neural development: New findings and toxicologic implications. Environmental Health Perspectives. 1999;107:59–64. doi: 10.1289/ehp.99107s159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Brimijoin S. Can cholinesterase inhibitors affect neural development? Environmental Toxicology and Pharmacology. 2005;19:429–432. doi: 10.1016/j.etap.2004.12.004. [DOI] [PubMed] [Google Scholar]