

Abstract
Neurofibromatosis type 1 (NF-1, Recklinghausen disease) is the most common hereditary multitumor syndrome with an incidence at birth of approximately 1:3000. However, the significant variation in the expression of the disease not infrequently precludes early diagnosis. As a consequence of non-familiarity with their frequency and wide clinicopathological spectrum, gastrointestinal manifestations of NF-1 are seldom thought of in routine clinical practice and might thus be significantly under-recognized. Their heterogeneous spectrum ranges from localized microscopic proliferative lesions of autonomic nerves and interstitial cells of Cajal and diffuse microscopic ganglio/neuro/fibromatosis to grossly recognizable mass-forming neurofibromas and gastrointestinal stromal tumors (GIST). Furthermore, neuroendocrine neoplasms, particularly of the periampullary duodenum seem to be quite characteristic of this disease. Based on our own experience and the available literature, this review summarizes and discusses the clinicopathological spectrum of gastrointestinal manifestations of NF-1 including putative proliferative precursor lesions with emphasis on the differential diagnostic aspects of these disorders and their molecular pathogenesis. In addition, this review underlines the great value of specific gastrointestinal findings in uncovering undiagnosed or missed NF-1 cases.
Keywords: NF-1, Recklinghausen disease, GIST, Gastrointestinal stromal tumor, Neurofibromatosis, Neuroendocrine tumor
Introduction
Neurofibromatosis type 1 (NF-1; OMIM #162200) or Recklinghausen disease represents one of the most common autosomal dominant hereditary tumor syndromes with an incidence of 1:3000 births and a prevalence of 1:4-5.000 [1]. The disorder is caused by alterations in the NF-1 gene, a tumor suppressor gene located on chromosome 17q11.2 [2]. NF-1 encodes neurofibromin, a cytoplasmic protein which controls cellular proliferation by inactivating the p21 RAS and the MAP kinase pathway [2]. Alterations in the NF-1 gene usually represent nonsense, frame shift or splice mutations and deletions but rarely translocations may be encountered as well.
NF-1 is inherited as autosomal dominant with complete penetrance and variable expression [1]. The NF1 gene has one of the highest/new mutation rates in humans. Because of this high mutation susceptibility, approximately 50% of NF-1 patients have no family history of the disorder [1]. The clinical presentation varies greatly depending on the nature of the mutation, the time at which the mutation occurs, and the presence of additional molecular alterations in other related genes [1]. In particular, some patients may have the segmental form of the disease, thus presenting with somatic mosaicism reflected in lesions restricted to a specific part (dermatome) of the body [3,4]. Because of the large size of the NF1 gene and lack of mutation hot spots, mutation analysis is usually not practicable as an initial tool for identifying NF-1. Therefore, the diagnosis of NF-1 is based on the presence of two or more of the major clinical criteria established by the National Institutes of Health (NIH) Consensus Development Conference in 1988 [1] (Table 1).
Table 1.
Diagnostic criteria of neurofibromatosis type 1/Recklinghausen’s disease*
| Six or more café au lait macules (>0.5 cm in children or > 1.5 cm in adults). |
| Two or more cutaneous or subcutaneous neurofibromas or one plexiform neurofibroma. |
| Axillary or inguinal freckling. |
| Optic pathway glioma |
| Two or more Lisch nodules (iris hamartomas seen on slit lamp examination) |
| One first-degree relative with NF-1 |
| Two or more Lisch nodules |
| Bony dysplasia (sphenoid wing dysplasia, bowing of long bone +/- pseudoarthrosis) |
two or more criteria are needed for diagnosis (from ref. 1).
The frequency of intra-abdominal (gastrointestinal or retroperitoneal) manifestations of NF-1 varied greatly in previous studies ranging from 5%-25% [5-8]. This variation probably reflects the poverty of symptoms related to intraabdominal manifestations of NF-1 (only 5% are symptomatic). Gastrointestinal manifestations of NF-1 usually arise during midlife or later; generally later than the appearance of the cutaneous manifestations of the disease. In a previous study from a large specialized center, intra-abdominal (gastrointestinal or retroperitoneal) manifestations necessitating surgical intervention occurred in 2.5% of patients followed up regularly at a median age of 40 years [7,8]. However, other studies reported a higher incidence approaching 25% [9,10]. Early diagnosis of abdominal manifestations of NF-1 is necessary for appropriate treatment to avoid serious organic complications related to tumor mass. This review summarizes the most important gastrointestinal and related intra-abdominal lesions occurring in NF-1 patients including two illustrative cases from our files that clearly demonstrate under-recognition of this disorder during routine clinical practice.
Gastrointestinal findings suggestive of undiagnosed NF-1
Two illustrative cases from our files
Case one
A middle-aged man without a family history of NF-1 presented with non-specific abdominal symptoms. Imaging studies showed an upper abdominal mass associated with the jejunum. At laparotomy, a 4.5 cm mass arising from the jejunum was removed via a segment resection (Figure 1A). A small nodule was seen a few centimeters from the large mass and submitted for frozen section to rule out metastatic disease and showed typical histology of a small GIST of the bowel wall (very low risk tumor). Histological examination of the large mass showed an intermediate risk GIST with spindle cell morphology and numerous skeinoid fibers (Figure 1B). Randomly sampled regional lymph nodes contained within the resected mesenteric fat showed a minute focus of well differentiated neuroendocrine tumor confined to one lymph node. A primary neuroendocrine tumor could not be found on imaging investigations. The tumor was negative for somatostatin by immunohistochemistry. Both GISTs showed a wild type sequence for KIT exon 9, 11, 13, 17 and PDGFRA exon 12, 14 and 18. The findings were judged as highly suspicious for NF-1 and a further dermatological assessment has been recommended. This displayed multiple large café-au-lait spots on the thigh and back. One of a few cutaneous nodules detected on his forehead and neck region was excised and was histologically neurofibroma. A working diagnosis of NF-1 was made and the patient was referred to genetic counseling. This case illustrates the significant under recognition of NF-1 features as this patient was seen by several physicians who did not give any attention to his subtle cutaneous changes. The case also clearly points out the important value of gastrointestinal findings in identifying undiagnosed NF-1. Thus, the presence of multifocal wild type GIST in the small bowel, particularly if concurrent to neuroendocrine tumor as in this case should be judged as highly suspicious for NF-1 and should be recommended as a further criterion in the list of diagnostic findings for NF-1.
Figure 1.

A: Large duodenal GIST from a patient with undiagnosed NF-1. B: Histology showed a spindle cell GIST with multiple eosinophilic collagen deposits between tumor cells (skeinoid fibers) (H&E-stain, original magnification x400). Note nuclear variation typical of NF1-GIST.
Case two
An elderly man was diagnosed with a duodenal tumor after an episode of gastrointestinal bleeding. A paraffin block was received for mutational assessment for KIT and PDGFRA mutations for potential therapeutic option. No detailed clinical data were supplied to our pathology department. Histological examination showed an epithelial neoplasm infiltrating the mucosa, submucosa and muscularis propria of the duodenum with tubule formation and strong expression of somatostatin (Figure 2A). The adjacent duodenal wall showed a minute focus of interstitial cell of Cajal hyperplasia forming a 1 mm GIST that co-expressed CD117 and CD34 (Figure 2B). The findings were considered typical of NF-1 and further clinical information was sought. This uncovered that the patient has numerous cutaneous neurofibromas and has several surgical procedures for large metastatic GISTs. Again, this case clearly illustrates that the combination of somatostatinoma and GIST should be considered highly pathognomonic for NF-1 and this might be of great value in recognizing this disorder.
Figure 2.

A: duodenal somatostatinoma stained strongly with somatostatin (original magnification x200). B: diffuse interstitial cell of Cajal hyperplasia and a minute microscopic GIST focus in the duodenal wall surrounding somatostatinoma in A (CD117, original magnification x400).
Review of GI manifestations of NF-1 from the English literature
True neurogenic neoplasms of the digestive system in NF-1 patients
Peripheral nerve sheath tumors of different histological subtypes may develop in the gastrointestinal organs of NF-1 patients (Table 2). However, compared to the cutaneous manifestations, neurogenic tumors are relatively uncommon in the gastrointestinal tract. They may occur at any site from the esophagus to the anorectum and in the associated peritoneal and mesenteric soft tissues [5-7] (Figure 3). According to previous reports, benign tumors significantly outnumbered their malignant counterpart. Symptoms are generally non-specific and included abdominal pain, bleeding and symptoms related to the presence of a tumor mass (obstruction, jaundice, etc.). In a previous series [7], neurofibroma was the most frequent manifestation and affected mainly the small bowel, retroperitoneum and less frequently the colon. Visceral neurofibroma may show any of the subtypes commonly observed at peripheral cutaneous and somatic soft tissue sites including localized, diffuse and plexiform variants [7,11].
Table 2.
Reported gastrointestinal manifestations in NF-1
| 1. True neurogenic neoplasms |
| Solitary neurofibroma |
| Diffuse or plexiform neurofibroma |
| Gastric schwannoma (single case reported) |
| Diffuse mucosal/submucosal neurofibromatosis |
| Ganglioneuromatosis |
| Gangliocytic paraganglioma |
| Malignant peripheral nerve sheath tumor (very rare) |
|
|
| 2. Interstitial cell of Cajal lesions |
| Multifocal clinical gastrointestinal stromal tumors (GISTs) |
| Minute incidental GIST tumorlets (usually non-gastric) |
| Microscopic diffuse or multifocal interstitial cell of Cajal hyperplasia |
| Motility disorders related to Cajal cell lesions |
|
|
| 3. Neuroendocrine tumors |
| Carcinoid tumors at any gastrointestinal location |
| Periampullary somatostatinoma |
| Rarely, insulinoma and gastrinoma |
|
|
| 4. Miscellaneous neoplasms and lesions |
| Adenocarcinoma at different gastrointestinal sites |
| Vasculopathy |
Figure 3.
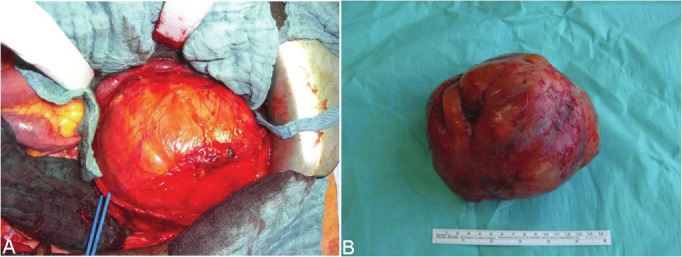
B: A large retroperitoneal neurofibroma in NF-1 during surgical mobilization. No other gastrointestinal lesions have been detected during surgery in this patient. A: The resected tumor was surrounded by a smooth thin capsule. Histology revealed typical neurofibroma without evidence of malignancy.
On the other hand, gastrointestinal schwannomas usually occur sporadically unrelated to NF-1 and NF-2. Although recent studies have shown that digestive schwannomas are histologically and molecularly more related to plexiform neurofibroma than peripheral soft tissue schwannoma [12], gastrointestinal schwannoma is not known to occur regularly in the setting of NF-1. To date, only a single case of well documented digestive schwannoma occurred in the stomach of a patient with a known NF-1 [11]. The patient had a synchronous diffuse neurofibroma of the vermiform appendix and a microscopic gastric gastrointestinal stromal tumor (GIST) [11]. However, a majority of gastrointestinal neoplasms reported as NF-1-associated schwannomas preceding the establishment of current diagnostic criteria probably represented GISTs. Both sporadic and NF-1-associated variants of malignant peripheral nerve sheath tumor (MPNST) are quite uncommon in the gastrointestinal tract. Again, there exist only very rare well documented cases of this sarcoma type in the gastrointestinal tract as opposed to a plethora of presumable MPNSTs reported in the pre-KIT era; a majority if not all of them were probably GISTs. Retroperitoneal/peritoneal MPNSTs related to NF-1 were seen in two patients in one previous study [7].
So-called gastrointestinal neurofibromatosis is a heterogeneous group of disorders
NF-1 patients may present with a diffuse neurofibromatous proliferation expanding the lamina propria mucosae and the submucosa of the gastrointestinal tract. This may either take the form of a pure neurofibromatosis or be admixed with a ganglioneuromatous component (Figure 4). Historically, the term gastrointestinal neurofibromatosis has been applied to a group of clinicopathologically and genetically heterogeneous group of conditions, some of them could be well characterized in recent years. However, because of the lacking diagnostic criteria during the decades preceding the discovery of KIT role in GISTs, some of reported cases in the literature are difficult to categorize precisely. One familial example of these lesions has been shown to represent a manifestation of a germline PDGFRA mutation [13]. A recently reported case featured both fibrous tumors and epithelioid GISTs in a patient with PDGFRA germline mutation [14]. These fibrous tumors occurring in the setting of PDGFRA germline mutation have to be separated from the so-called gastrointestinal neurofibromatosis. Isolated intestinal neurofibromatous proliferations (IINP) in patients without clinical evidence of a syndromic disease represent a further member of this heterogeneous group of lesions [15]. However, isolated intestinal neurofibromatous proliferations differ from NF-1-associated gastrointestinal neurofibromatosis by the mere absence of further manifestation of NF-1. Currently, it remains obscure, whether such lesions represent a segmental or localized visceral form of NF-1 limited to the gastrointestinal tract or a distinct disease entity [16]. Furthermore, gastrointestinal ganglioneuromatosis may represent a manifestation of either NF-1 or multiple endocrine neoplasia type 2b [17,18]. Similar findings have also been reported in patients with juvenile and adenomatous colonic polyposis and as isolated diseases. Thus, close monitoring and clinical follow-up of these patients is mandatory to exclude any other syndromic disorder before rendering a diagnosis of sporadic isolated intestinal neurofibromatous proliferations (Table 3).
Figure 4.
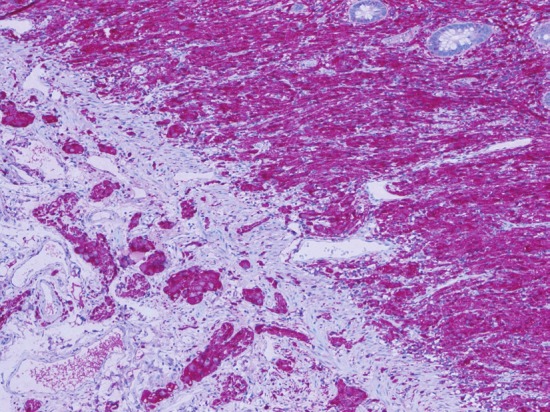
Diffuse intestinal ganglio/neurofibromatosis in NF-1 highlighted by protein S100 stain (original magnification x200).
Table 3.
Disorders presenting with diffuse ganglio/neurofibromatous proliferation in the gastrointestinal tract
| 1. NF-1-associated gastrointestinal neurofibromatosis. |
| 2. NF-1-asscoaited gastrointestinal ganglioneuromatosis. |
| 3. Gastrointestinal ganglioneuromatosis associated with MEN-2b. |
| 4. Ganglioneuromatosis/plexiform neurofibroma limited to the gastrointestinal tract (segmental NF-1?). |
| 5. Ganglioneuromatous polyposis related to juvenile and hamartomatous polyposis syndromes. |
| 6. Ganglioneuromatous polyposis unrelated to a known syndromic disorder. |
| 7. Isolated intestinal neurofibromatous proliferations (carefully exclude subtle features of NF-1). |
Periampullary neoplasms in NF-1 patients
Tumors arising in the ampulla of Vater and the periampullary duodenum are among the most common and characteristic gastrointestinal manifestations of NF-1. In a previous review, 12 of 28 (43%) periampullary somatostatinomas reported in the literature occurred in patients with NF-1 as opposed to only 6% of their pancreatic counterparts [19]. Two thirds of somatostatinomas occurred in the pancreas while only one third affected the duodenum. The symptoms related to ampullary somatostatinoma varied from absent to significant tumor-related symptoms including obstructive jaundice, cholangitis, pancreatitis, hemorrhage, and duodenal obstruction. Although the tumors usually stain strongly for somatostatin by immunohistochemistry, symptoms of the somatostatinoma syndrome (diarrhea, diabetes mellitus, dyspepsia, and cholelithiasis) are uncommon [20]. Twenty-five percent of the cases metastasized to regional lymph nodes, but liver metastases were not frequent. Of note, none of the MEN1-associated somatostatinomas of the duodenum or the pancreas metastasized in contrast to NF-1-associated and sporadic somatostatinomas [20].
The combination of GIST and somatostatinoma in NF-1 is rare (7 cases) [19,21,22]. Although a majority of NF-1 associated neuroendocrine neoplasms are ampullary somatostatinomas, other endocrine tumors including gastrinoma [23], insulinoma [24] and gangliocytic paraganglioma [22] have been rarely reported. Furthermore, Costi et al described a case of periampullary adenocarcinoma in a patient with NF-1 and multiple gastroduodenal and intestinal GISTs [25]. They reviewed the literature and found further 11 reports on epithelial non-endocrine neoplasms of the ampullary region. These neoplasms affected women and men equally at a mean age of 47 yrs. Their patient had also evidence of Islet cell microadenomatosis of the pancreas [25].
NF-1-associated gastrointestinal stromal tumor (GISTs)
Gastrointestinal neoplasms resembling leiomyomas in NF-1 patients were first reported by Lukash & Johnson [26]. More recently, GISTs have been increasingly documented as an abdominal manifestation of the disease. GISTs were detected in 25% of NF-1 patients at autopsy [9,10]. Thus, they represent the most common gastrointestinal manifestation of NF-1. In the large series of GISTs documented at the Armed Forces Institute of Pathology (AFIP, Washington, D.C.) NF-1 associated GISTs represented 1.5% of all GISTs from different gastrointestinal sites [27]. However, taken by site, NF-1 associated GISTs represented 6%, 4% and 0.06% of duodenal, jejuno-ileal and gastric GISTs at the AFIP respectively. The mean age at GIST diagnosis was 49 years compared to a mean age of 56 years for small bowel GISTs in general. Most NF-1 associated GISTs present as small asymptomatic lesions with low mitotic activity and they generally follow a benign clinical course. Histologically, they display spindle cell morphology with numerous brightly eosinophilic PAS-positive pathological collagen fibers that have been referred to as skeinoid fibers based on their ultrastructural similarity to “skein of yarn” [28] (Figure 1B). However, skeinoid fibers are not pathognomonic to NF-1 associated GIST and are common in sporadic small bowel GIST as well. In a series of 45 patients, five patients developed metastatic disease and died of tumor (all had a tumor size >5 cm or a mitotic rate >5/50 HPFs, or both) [27]. Similar to KIT/PDGFRA wild type sporadic GISTs, NF-1-associated GISTs show a variable but generally incomplete response to the tyrosine kinase inhibitor imatinib mesylate [29,30]. From a differential diagnostic point of view, multiple GISTs in NF-1 should be distinguished from sporadic multifocal GIST caused by distinct somatic KIT or PDGFRA mutations [31], multifocal wild type GIST of unknown pathogenesis [32] and familial GIST syndromes resulting from germline KIT [33] and PDGFRA [14] mutations.
Molecular pathogenesis of GISTs and other gastrointestinal neoplasms in NF-1
With rare exceptions, GISTs in the setting of NF-1 do not harbor mutations in KIT or PDGFRA (wild type) [27,34]. Of 120 patients analyzed in previous series (reviewed in Mussi et al [30]), 7 showed KIT and 3 PDGFRA mutations. (Total mutation rate: 8%). However, the significance of these mutations and their nature (primary or secondary) remains unclear. Despite their molecular profiles, NF-1 associated GISTs are uniformly KIT positive by immunohistochemistry. A recent study demonstrated that NF-1 associated GISTs have an intact mitochondrial expression of the succinate dehydrogenase subunits (SDHB), thus being similar to their sporadic KIT/PDGFRA-mutated counterpart (so-called type 1 GISTs or SDHB-positive GISTs) and unrelated to the pediatric-type and Carney triad-associated GISTs that uniformly lack this feature (so-called type 2 or SDHB-deficient GISTs) [35,36].
The NF1 gene product, neurofibromin, is a member of the GAP family of ras regulatory proteins that functions as a tumor suppressor by negatively regulating the ras pathway. Thus, beiallelic NF-1 inactivation results in constitutive ras activation thereby increasing the downstream mitogenic signaling through the MAP kinase cascade [37]. Interestingly, gain-of-function mutations of KIT also result in constitutive activation of downstream signaling pathways including the ras-MAP kinase cascade. Accordingly, it is likely that activation of the ras-MAP kinase cascade may be a common pathogenetic mechanism in both sporadic and NF-1-associated GISTs [37]. Mitotic recombination has been postulated as an alternative pathogenesis of GIST in NF-1 [38]. Somatic inactivation of the wild-type NF-1 allele has been suggested as a probable molecular event underlying GIST development in NF-1 patients. Thus, inactivation of neurofibromin may represent an alternate mechanism to activate the MAP-kinase pathway, while the JAK-STAT3 and PI3K-AKT pathways were found less activated in NF1-related GIST compared to sporadic GISTs [37]. Array comparative genomic hybridization (aCGH) experiments on NF1-related GISTs showed similar chromosomal aberrations as seen in sporadic GISTs [37]. Additionally, loss of heterozygosity (LOH) at 14q and 22q may contribute to the relatively early phase of tumor development of NF-1 GIST [39]. Similar to GISTs, the exact molecular pathogenesis of epithelial and other gastrointestinal neoplasms in NF-1 patients remains largely obscure. In islet1-Cre conditional knockout mice loss of NF-1 resulted in gastric epithelial hyperplasia with appearance of atypical epithelial cells [40]. The authors illustrated that the epithelial hyperplasia was not due to an autonomous effect of NF-1 and that possibly other types of cells (probably stromal fibroblasts), after losing NF-1, stimulate the growth of epithelial cells through a paracrine activity.
Proliferative mesenchymal precursor lesions in the gastrointestinal tract in NF-1: a hint to the GIST histogenesis
The association of GISTs with the myenteric plexus of Auerbach has been suggested since the publication by Herrera in 1984 based on electron microscopic observations [41]. Later, it became evident that tumor growth in GISTs detected at an early stage in patients with NF-1 is closely associated with the nerve fibers of the Auerbach plexus [42]. The current concept of GIST histogenesis argues for a tumor origin from interstitial cells of Cajal or their non-committed precursor mesenchymal cells [43]. Interstitial cell of Cajal hyperplasia in NF-1 is common in resection specimens for GISTs [27]. It may present either as minute nodules or forms a longitudinal band along the plane of the Auerbach plexus between the internal and external muscle layer (Figure 5A, B). While the interstitial cell of Cajal hyperplasia is believed to be a polyclonal proliferation as a direct consequence of constitutive KIT activation caused by germline mutations in KIT in familial GIST syndromes [33], it is conceivable that interstitial cell of Cajal hyperplasia in NF-1 might be secondary to NF-1 haploinsufficiency [37]. The subsequent loss of heterozygosity at NF-1 and accumulation of additional chromosomal alterations would explain the emergence of gross GIST in the background of microscopic interstitial cell of Cajal hyperplasia in NF-1. Thus, loss of heterozygosity at NF-1 is probably necessary, but not sufficient for tumor development. Interstitial cell of Cajal hyperplasia has been implicated in dysmotility in cases of familial GIST syndromes and in intestinal neuronal dysplasia [44]. Gastrointestinal dysmotility occasionally observed in NF-1 patients might be related to the proliferative lesions of interstitial cells of Cajal and associated myenteric plexus hyperplasia and their interference with the physiological function of neurotransmission in the myenteric plexus [45].
Figure 5.

A: Diffuse interstitial cell of Cajal hyperplasia forming a thick layer encasing autonomic nerves of the Auerbach plexus adjacent to a larger GIST nodule (upper right). B: minute nodular focus of hyperplastic Cajal cells forming a microscopic tumorlet (lower right) from same patient (CD117 stain, original magnification x200).
NF-1-associated vasculopathies
Several vascular lesions have been described in NF-1 including renal artery stenosis, arterial aneurysms, and arteriovenous fistulae. The renal arteries are most frequently affected. NF-1-associated vasculopathies have been classified into pure intimal type, advanced intimal type, intimal aneurysmal, and nodular [46]. Schwann cell proliferative lesions within vascular walls and superimposed degenerative changes and fibrosis have been implicated in the pathogenesis of these vasculopathic changes [47]. However, it is currently believed that intimal smooth muscle rather than a Schwann cell proliferation leads to the development of these lesions. It has been suggested that NF-1 gene dysregulation may have a major role in vascular smooth muscle proliferation. A recent case showed vasculopathic changes in vessels within and in the immediate vicinity of a periampullary somatostatinoma associated with NF-1 [48]. The authors speculated that somatostatinoma may have exerted a paracrine effect on local vessels leading to the observed vascular pathology.
Coexistence of GIST and neuroendocrine neoplasms
As illustrated above, co-existence of ampullary somatostatin-producing neuroendocrine tumor (somatostatinoma) and GIST in the gastrointestinal tract represents an almost pathognomonic feature of NF-1. Phaeochromocytoma is another rare intra-abdominal manifestation of NF-1, affecting approximately 1-3% of patients and it may coexist with GIST [49]. It is necessary not to confuse coexistence of adrenal phaeochromocytoma and small bowel or even gastric GIST with other rare hereditary and non-hereditary disorders featuring both GIST and paraganglioma or other endocrine tumors (Table 4). In this setting, demonstration of an intact granular mitochondrial expression of SDHB by immunohistochemistry practically rules out the possibility of Carney triad, Carney-Stratakis syndrome and other rare disorders caused by germline mutation in the SDH genes [36]. It is important in this context to recognize that carcinoid tumors may rarely occur in patients with sporadic GISTs without a known underlying genetic disorder. In a previous extensive review of the literature, carcinoid tumors constituted 3% of all secondary neoplasms detected in patients with GISTs [50].
Table 4.
Disorders featuring GIST and Neuroendocrine tumors (NET)
| Disease | Associated gastrointestinal and abdominal tumors | Gene/s affected | Mode of inheritance |
|---|---|---|---|
| NF-1 | Multiple GIST, NET (adrenal, ampulla, pancreas and other sites) | NF-1 | Autosomal dominant |
| Multiple endocrine neoplasia type 1 & 2 | Multifocal NETs, very rare cases of GISTs were reported in MEN-1 &MEN-2 patients | MEN-1 or RET | Autosomal dominant |
| Carney triad | Multiple gastric GIST and extra-adrenal paraganglioma | Unknown | Non-heritable |
| Carney-Stratakis syndrome | Familial GIST and multiple paragangliomas | SDH A,B,C,D | Autosomal dominant |
| Von Hippel Lindau disease | Renal cell carcinoma, endocrine pancreas tumors, one case of GIST reported | vHL | Autosomal dominant |
| Miscellaneous | GIST at different sites and various NET types (carefully exclude underlying syndromes) | Unknown | Sporadic, non-hereditary, etiology unknown |
Coexistence of GIST and peripheral nerve sheath tumors
The concurrence of gastrointestinal benign peripheral nerve sheath tumors with GISTs seems to be unique. In a previous review [11], three patients with NF-1 had benign peripheral nerve sheath tumors and minute incidental (n=2) or large (n=1) gastric GISTs. Of the remaining three patients without known NF-1, one elderly patient had colonic schwannoma coexisting with large epithelioid gastric GIST [51]. Another report concerned a 61-yr man with sigmoid granular cell tumor followed by ileal GIST 4 months later, but no mention was made of NF-1 [52]. Another patient had a large gastric GIST and an incidental reticular schwannoma in the jejunum, but data on possible NF-1 were not conclusive [11]. In a series of NF-1 patients who have been closely followed up, synchronous gastric GIST and jejunal neurofibroma were detected in one patient [7]. However, the gastric GIST showed high grade features and lacked KIT and PDGFRA expression making a sarcoma more likely [7]. Other than this case, we are not aware of any report documenting concurrent GIST and MPNST in the gastrointestinal tract. Thus, with a few rare exceptions, coexistence of a benign peripheral nerve sheath tumor in the gastrointestinal tract and GIST seems to be unique and should alert to the possibility of an underlying undiagnosed NF-1.
Resume
Gastrointestinal manifestations of NF-1 are generally under-recognized by both clinicians and pathologists. However, following the establishment of well defined and reproducible diagnostic criteria for GISTs, smooth muscle neoplasms and true neurogenic (Schwann cell) neoplasms in the gastrointestinal tract in the KIT era, gastrointestinal manifestations of NF-1 are becoming increasingly recognized and may have a substantial impact on the diagnosis and recognition of this disorder. Coexistence of GIST and peripheral nerve sheath tumors in the gastrointestinal tract and/or GIST and somatostatinoma or ampullary/periampullary neuroendocrine neoplasm represents a typical and almost pathognomonic feature of NF-1 and should be considered a surrogate marker for identifying affected patients.
References
- 1.Ferner RE. Neurofibromatosis 1 and neurofibromatosis 2: a twenty first century perspective. Lancet Neurol. 2007;6:340–51. doi: 10.1016/S1474-4422(07)70075-3. [DOI] [PubMed] [Google Scholar]
- 2.Gottfried ON, Viskochil DH, Couldwell WT. Neurofibromatosis Type 1 and tumorigenesis: molecular mechanisms and therapeutic implications. Neurosurg Focus. 2010;28:E8. doi: 10.3171/2009.11.FOCUS09221. [DOI] [PubMed] [Google Scholar]
- 3.Messiaen L, Vogt J, Bengesser K, Fu C, Mikhail F, Serra E, Garcia-Linares C, Cooper DN, Lazaro C, Kehrer-Sawatzki H. Mosaic type-1 NF1 microdeletions as a cause of both generalized and segmental neurofibromatosis type-1 (NF1) Hum Mutat. 2011;32:213–9. doi: 10.1002/humu.21418. [DOI] [PubMed] [Google Scholar]
- 4.Ruggieri M, Huson SM. The clinical and diagnostic implications of mosaicism in the neurofibromatoses. Neurology. 2001;56:1433–43. doi: 10.1212/wnl.56.11.1433. [DOI] [PubMed] [Google Scholar]
- 5.Fuller CE, Williams GT. Gastrointestinal manifestations of type 1 neurofibromatosis (von Recklinghausen’s disease) Histopathology. 1991;19:1–11. doi: 10.1111/j.1365-2559.1991.tb00888.x. [DOI] [PubMed] [Google Scholar]
- 6.Heuschkel R, Kim S, Korf B, Schneider G, Bousvaros A. Abdominal migraine in children with neurofibromatosis type 1: a case series and review of gastrointestinal involvement in NF1. J Pediatr Gastroenterol Nutr. 2001;33:149–54. doi: 10.1097/00005176-200108000-00010. [DOI] [PubMed] [Google Scholar]
- 7.Basile U, Cavallaro G, Polistena A, Giustini S, Orlando G, Cotesta D, Petramala L, Letizia C, Calvieri S, De Toma G. Gastrointestinal and retroperitoneal manifestations of type 1 neurofibromatosis. J Gastrointest Surg. 2010;14:186–94. doi: 10.1007/s11605-009-0940-5. [DOI] [PubMed] [Google Scholar]
- 8.Cavallaro G, Basile U, Polistena A, Giustini S, Arena R, Scorsi A, Zinnamosca L, Letizia C, Calvieri S, De Toma G. Surgical management of abdominal manifestations of type 1 neurofibromatosis: experience of a single center. Am Surg. 2010;76:389–96. [PubMed] [Google Scholar]
- 9.Ghrist TD. Gastrointestinal involvement in neurofibromatosis. Arch Intern Med. 1963;112:357–362. doi: 10.1001/archinte.1963.03860030111011. [DOI] [PubMed] [Google Scholar]
- 10.Zöller ME, Rembeck B, Odén A, Samuelsson M, Angervall L. Malignant and benign tumors in patients with neurofibromatosis type 1 in a defined Swedish population. Cancer. 1997;79:2125–31. [PubMed] [Google Scholar]
- 11.Agaimy A, Märkl B, Kitz J, Wünsch PH, Arnholdt H, Füzesi L, Hartmann A, Chetty R. Peripheral nerve sheath tumors of the gastrointestinal tract: a multicenter study of 58 patients including NF1-associated gastric schwannoma and unusual morphologic variants. Virchows Arch. 2010;456:411–22. doi: 10.1007/s00428-010-0886-8. [DOI] [PubMed] [Google Scholar]
- 12.Lasota J, Wasag B, Dansonka-Mieszkowska A, Karcz D, Millward CL, Ryś J, Stachura J, Sobin LH, Miettinen M. Evaluation of NF2 and NF1 tumor suppressor genes in distinctive gastrointestinal nerve sheath tumors traditionally diagnosed as benign schwannomas: A study of 20 cases. Lab Invest. 2003;83:1361–71. doi: 10.1097/01.lab.0000087591.29639.e3. [DOI] [PubMed] [Google Scholar]
- 13.de Raedt T, Cools J, Debiec-Rychter M, Brems H, Mentens N, Sciot R, Himpens J, de Wever I, Schöffski P, Marynen P, Legius E. Intestinal neurofibromatosis is a subtype of familial GIST and results from a dominant activating mutation in PDGFRA. Gastroenterology. 2006;131:1907–12. doi: 10.1053/j.gastro.2006.07.002. [DOI] [PubMed] [Google Scholar]
- 14.Carney JA, Stratakis CA. Stromal, fibrous, and fatty gastrointestinal tumors in a patient with a PDGFRA gene mutation. Am J Surg Pathol. 2008;32:1412–20. doi: 10.1097/PAS.0b013e31816250ce. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carter JE, Laurini JA. Isolated intestinal neurofibromatous proliferations in the absence of associated systemic syndromes. World J Gastroenterol. 2008;14:6569–71. doi: 10.3748/wjg.14.6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hirata K, Kitahara K, Momosaka Y, Kouho H, Nagata N, Hashimoto H, Itoh H. Diffuse ganglioneuromatosis with plexiform neurofibromas limited to the gastrointestinal tract involving a large segment of small intestine. J Gastroenterol. 1996;31:263–267. doi: 10.1007/BF02389528. [DOI] [PubMed] [Google Scholar]
- 17.Shekitka KM, Sobin LH. Ganglioneuromas of the gastrointestinal tract. Relation to Von Recklinghausen disease and other multiple tumor syndromes. Am J Surg Pathol. 1994;18:250–7. [PubMed] [Google Scholar]
- 18.Thway K, Fisher C. Diffuse ganglioneuromatosis in small intestine associated with neurofibromatosis type 1. Ann Diagn Pathol. 2009;13:50–4. doi: 10.1016/j.anndiagpath.2007.06.001. [DOI] [PubMed] [Google Scholar]
- 19.Tanaka S, Yamasaki S, Matsushita H, Ozawa Y, Kurosaki A, Takeuchi K, Hoshihara Y, Doi T, Watanabe G, Kawaminami K. Duodenal somatostatinoma: a case report and review of 31 cases with special reference to the relationship between tumor size and metastasis. Pathol Int. 2000;50:146–52. doi: 10.1046/j.1440-1827.2000.01016.x. [DOI] [PubMed] [Google Scholar]
- 20.Garbrecht N, Anlauf M, Schmitt A, Henopp T, Sipos B, Raffel A, Eisenberger CF, Knoefel WT, Pavel M, Fottner C, Musholt TJ, Rinke A, Arnold R, Berndt U, Plöckinger U, Wiedenmann B, Moch H, Heitz PU, Komminoth P, Perren A, Klöppel G. Somatostatin-producing neuroendocrine tumors of the duodenum and pancreas: incidence, types, biological behavior, association with inherited syndromes, and functional activity. Endocr Relat Cancer. 2008;15:229–41. doi: 10.1677/ERC-07-0157. [DOI] [PubMed] [Google Scholar]
- 21.Klein A, Clemens J, Cameron J. Periampullary neoplasms in von Recklinghausen’s disease. Surgery. 1989;106:815–9. [PubMed] [Google Scholar]
- 22.Relles D, Baek J, Witkiewicz A, Yeo CJ. Periampullary and duodenal neoplasms in neurofibromatosis type 1: two cases and an updated 20-year review of the literature yielding 76 cases. J Gastrointest Surg. 2010;14:1052–61. doi: 10.1007/s11605-009-1123-0. [DOI] [PubMed] [Google Scholar]
- 23.Lee WS, Koh YS, Kim JC, Park CH, Joo YE, Kim HS, Cho CK, Choi SK, Rew JS, Kim SJ. Zollinger-Ellison syndrome associated with neurofibromatosis type 1: a case report. BMC Cancer. 2005;5:85. doi: 10.1186/1471-2407-5-85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Perren A, Wiesli P, Schmid S, Montani M, Schmitt A, Schmid C, Moch H, Komminoth P. Pancreatic endocrine tumors are a rare manifestation of the neurofibromatosis type 1 phenotype: molecular analysis of a malignant insulinoma in a NF-1 patient. Am J Surg Pathol. 2006;30:1047–51. doi: 10.1097/00000478-200608000-00018. [DOI] [PubMed] [Google Scholar]
- 25.Costi R, Caruana P, Sarli L, Violi V, Roncoroni L, Bordi C. Ampullary adenocarcinoma in neurofibromatosis type 1. Case report and literature review. Mod Pathol. 2001;14:1169–74. doi: 10.1038/modpathol.3880454. [DOI] [PubMed] [Google Scholar]
- 26.Lukash WM, Jolmson BB. Gastrointestinal neoplasms in von Recklinghausen’s disease. South Med J. 1969;62:1237. doi: 10.1097/00007611-196910000-00019. [DOI] [PubMed] [Google Scholar]
- 27.Miettinen M, Fetsch JF, Sobin LH, Lasota J. Gastrointestinal stromal tumors in patients with neurofibromatosis 1: a clinicopathologic and molecular genetic study of 45 cases. Am J Surg Pathol. 2006;30:90–6. doi: 10.1097/01.pas.0000176433.81079.bd. [DOI] [PubMed] [Google Scholar]
- 28.Schaldenbrand JD, Appelman HD. Solitary solid stromal gastrointestinal tumors in von Recklinghausen’s disease with minimal smooth muscle differentiation. Hum Pathol. 1984;15:229–32. doi: 10.1016/s0046-8177(84)80184-7. [DOI] [PubMed] [Google Scholar]
- 29.Lee JL, Kim JY, Ryu MH, Kang HJ, Chang HM, Kim TW, Lee H, Park JH, Kim HC, Kim JS, Kang YK. Response to imatinib in KIT- and PDGFRA-wild type gastrointestinal stromal associated with neurofibromatosis type 1. Dig Dis Sci. 2006;51:1043–6. doi: 10.1007/s10620-006-8003-1. [DOI] [PubMed] [Google Scholar]
- 30.Mussi C, Schildhaus HU, Gronchi A, Wardelmann E, Hohenberger P. Therapeutic consequences from molecular biology for gastrointestinal stromal tumor patients affected by neurofibromatosis type 1. Clin Cancer Res. 2008;14:4550–5. doi: 10.1158/1078-0432.CCR-08-0086. [DOI] [PubMed] [Google Scholar]
- 31.Agaimy A, Dirnhofer S, Wünsch PH, Terracciano LM, Tornillo L, Bihl MP. Multiple sporadic gastrointestinal stromal tumors (GISTs) of the proximal stomach are caused by different somatic KIT mutations suggesting a field effect. Am J Surg Pathol. 2008;32:1553–9. doi: 10.1097/PAS.0b013e31817587ea. [DOI] [PubMed] [Google Scholar]
- 32.Agaimy A, Märkl B, Arnholdt H, Wünsch PH, Terracciano LM, Dirnhofer S, Hartmann A, Tornillo L, Bihl MP. Multiple sporadic gastrointestinal stromal tumours arising at different gastrointestinal sites: pattern of involvement of the muscularis propria as a clue to independent primary GISTs. Virchows Arch. 2009;455:101–8. doi: 10.1007/s00428-009-0803-1. [DOI] [PubMed] [Google Scholar]
- 33.Chen H, Hirota S, Isozaki K, Sun H, Ohashi A, Kinoshita K, O’Brien P, Kapusta L, Dardick I, Obayashi T, Okazaki T, Shinomura Y, Matsuzawa Y, Kitamura Y. Polyclonal nature of diffuse proliferation of interstitial cells of Cajal in patients with familial and multiple gastrointestinal stromal tumours. Gut. 2002;51:793–6. doi: 10.1136/gut.51.6.793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kinoshita K, Hirota S, Isozaki K, Ohashi A, Nishida T, Kitamura Y, Shinomura Y, Matsuzawa Y. Absence of c-kit gene mutations in gastrointestinal stromal tumours from neurofibromatosis type 1 patients. J Pathol. 2004;202:80–5. doi: 10.1002/path.1487. [DOI] [PubMed] [Google Scholar]
- 35.Gill AJ, Chou A, Vilain R, Clarkson A, Lui M, Jin R, Tobias V, Samra J, Goldstein D, Smith C, Sioson L, Parker N, Smith RC, Sywak M, Sidhu SB, Wyatt JM, Robinson BG, Eckstein RP, Benn DE, Clifton-Bligh RJ. Immunohistochemistry for SDHB divides gastrointestinal stromal tumors (GISTs) into 2 distinct types. Am J Surg Pathol. 2010;34:636–44. doi: 10.1097/PAS.0b013e3181d6150d. [DOI] [PubMed] [Google Scholar]
- 36.Wang JH, Lasota J, Miettinen M. Succinate Dehydrogenase Subunit B (SDHB) Is Expressed in Neurofibromatosis 1-Associated Gastrointestinal Stromal Tumors (Gists): Implications for the SDHB Expression Based Classification of Gists. J Cancer. 2011;2:90–3. doi: 10.7150/jca.2.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maertens O, Prenen H, Debiec-Rychter M, Wozniak A, Sciot R, Pauwels P, De Wever I, Vermeesch JR, de Raedt T, De Paepe A, Speleman F, van Oosterom A, Messiaen L, Legius E. Molecular pathogenesis of multiple gastrointestinal stromal tumors in NF1 patients. Hum Mol Genet. 2006;15:1015–23. doi: 10.1093/hmg/ddl016. [DOI] [PubMed] [Google Scholar]
- 38.Stewart DR, Corless CL, Rubin BP, Heinrich MC, Messiaen LM, Kessler LJ, Zhang PJ, Brooks DG. Mitotic recombination as evidence of alternative pathogenesis of gastrointestinal stromal tumours in neurofibromatosis type 1. J Med Genet. 2007;44:e61. doi: 10.1136/jmg.2006.043075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yamamoto H, Tobo T, Nakamori M, Imamura M, Kojima A, Oda Y, Nakamura N, Takahira T, Yao T, Tsuneyoshi M. Neurofibromatosis type 1-related gastrointestinal stromal tumors: a special reference to loss of heterozygosity at 14q and 22q. J Cancer Res Clin Oncol. 2009;135:791–8. doi: 10.1007/s00432-008-0514-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lin L, Chen J, Richardson JA, Parada LF. Mice lacking neurofibromin develop gastric hyperplasia. Am J Physiol Gastrointest Liver Physiol. 2009;297:G751–61. doi: 10.1152/ajpgi.00007.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Herrera GA, Pinto de Moraes H, Grizzle WE, Han SG. Malignant small bowel neoplasm of enteric plexus derivation (plexosarcoma). Light and electron microscopic study confirming the origin of the neoplasm. Dig Dis Sci. 1984;29:275–84. doi: 10.1007/BF01296263. [DOI] [PubMed] [Google Scholar]
- 42.Walsh NM, Bodurtha A. Auerbach’s myenteric plexus. A possible site of origin for gastrointestinal stromal tumors in von Recklinghausen’s neurofibromatosis. Arch Pathol Lab Med. 1990;114:522–5. [PubMed] [Google Scholar]
- 43.Hirota S. Gastrointestinal stromal tumors: their origin and cause. Int J Clin Oncol. 2001;6:1–5. doi: 10.1007/pl00012072. [DOI] [PubMed] [Google Scholar]
- 44.Jeng YM, Mao TL, Hsu WM, Huang SF, Hsu HC. Congenital interstitial cell of cajal hyperplasia with neuronal intestinal dysplasia. Am J Surg Pathol. 2000;24:1568–72. doi: 10.1097/00000478-200011000-00016. [DOI] [PubMed] [Google Scholar]
- 45.Wu JF, Chen HL, Peng SS, Yu TW, Lee WT, Chang MH. Neurofibromatosis type I and intestinal neuronal dysplasia type B in a child: report of one case. Acta Paediatr Taiwan. 2003;44:232–4. [PubMed] [Google Scholar]
- 46.Salyer WR, Salyer DC. The vascular lesions of neurofibromatosis. Angiology. 1974;25:509–10. doi: 10.1177/000331977402500803. [DOI] [PubMed] [Google Scholar]
- 47.Hamilton SJ, Friedman JM. Insights into the pathogenesis of neurofibromatosis 1 vasculopathy. Clin Genet. 2000;58:341–4. doi: 10.1034/j.1399-0004.2000.580501.x. [DOI] [PubMed] [Google Scholar]
- 48.Chetty R, Vajpeyi R. Vasculopathic changes, a somatostatin-producing neuroendocrine carcinoma and a jejunal gastrointestinal stromal tumor in a patient with type 1 neurofibromatosis. Endocr Pathol. 2009;20:177–81. doi: 10.1007/s12022-009-9083-1. [DOI] [PubMed] [Google Scholar]
- 49.Kramer K, Hasel C, Aschoff AJ, Henne-Bruns D, Wuerl P. Multiple gastrointestinal stromal tumors and bilateral pheochromocytoma in neurofibromatosis. World J Gastroenterol. 2007;13:3384–7. doi: 10.3748/wjg.v13.i24.3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Agaimy A, Wünsch PH, Sobin LH, Lasota J, Miettinen M. Occurrence of other malignancies in patients with gastrointestinal stromal tumors. Semin Diagn Pathol. 2006;23:120–9. doi: 10.1053/j.semdp.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 51.Miettinen M, Shekitka KM, Sobin LH. Schwannomas in the colon and rectum: a clinicopathologic and immunohistochemical study of 20 cases. Am J Surg Pathol. 2001;25:846–55. doi: 10.1097/00000478-200107000-00002. [DOI] [PubMed] [Google Scholar]
- 52.Ince AT, Yavuzer D, Kiliç G, Kendir T, Demirtürk P. Coincidental occurrence of granular cell tumor and gastrointestinal stromal tumor in a patient. Turk J Gastroenterol. 2008;19:135–6. [PubMed] [Google Scholar]