

Abstract
About half of people with trisomy 21 have a congenital heart defect (CHD), whereas the remainder have a structurally normal heart, demonstrating that trisomy 21 is a significant risk factor but is not causal for abnormal heart development. Atrioventricular septal defects (AVSD) are the most commonly occurring heart defects in Down syndrome (DS), and ∼65% of all AVSD is associated with DS. We used a candidate-gene approach among individuals with DS and complete AVSD (cases = 141) and DS with no CHD (controls = 141) to determine whether rare genetic variants in genes involved in atrioventricular valvuloseptal morphogenesis contribute to AVSD in this sensitized population. We found a significant excess (p < 0.0001) of variants predicted to be deleterious in cases compared to controls. At the most stringent level of filtering, we found potentially damaging variants in nearly 20% of cases but fewer than 3% of controls. The variants with the highest probability of being damaging in cases only were found in six genes: COL6A1, COL6A2, CRELD1, FBLN2, FRZB, and GATA5. Several of the case-specific variants were recurrent in unrelated individuals, occurring in 10% of cases studied. No variants with an equal probability of being damaging were found in controls, demonstrating a highly specific association with AVSD. Of note, all of these genes are in the VEGF-A pathway, even though the candidate genes analyzed in this study represented numerous biochemical and developmental pathways, suggesting that rare variants in the VEGF-A pathway might contribute to the genetic underpinnings of AVSD in humans.
Introduction
Congenital heart defects (CHD) are the most common form of birth defect, occurring in nearly 1 in 100 live births.1 There is clear evidence for a significant genetic contribution to most congenital heart malformations; however, our understanding of the specific genes involved in the majority of CHD is limited. On the other hand, our knowledge of the genetic control of heart development has grown exponentially over the past few decades, yielding a wealth of information about the genes responsible for the exquisitely orchestrated cascade of events that result in the formation of the vertebrate four-chambered heart.2 Clearly, defects in any number of these genes could lead to a heart malformation, yet numerous studies to date have failed to show a significant contribution from some of the most promising candidate genes to the cause of heart defects in humans.3–5 However, studies of CHD are often done with phenotypically heterogeneous cohorts, which might dilute the power to detect specific associations with genetic variants.
Atrioventricular septal defect (AVSD [MIM 606215]), also known as common atrioventricular (AV) canal or endocardial cushion defect, is a clinically significant congenital heart malformation. A recent study from the National Center on Birth Defects and Developmental Disabilities found that the birth incidence for nonsyndromic AVSD is 0.83/10,000 live births,6 and the estimated incidence for all AVSD is 3–5/10,000 live births.7 AVSD is found most often in children with Down syndrome (DS [MIM 190685]), who account for approximately 65% of all AVSD cases.8 Among those with DS, about 18% have a complete AVSD.9 Thus, compared to the euploid population, individuals with DS have about a 2,000-fold-increased risk of AVSD. Although having three copies of chromosome 21 genes certainly contributes to this risk, the increased gene dosage itself is not sufficient to cause the defect. Indeed, half or more of those with DS have a structurally normal heart. Clearly, other factors in addition to trisomy 21 are required for causing AVSD in children with DS.
The identification of gene mutations and potentially damaging rare variants associated with familial AVSD suggests that the risk factors for AVSD in DS might include variants in heart-development genes that are not necessarily on chromosome 21.10–12 To date, the gene most frequently associated with AVSD is CRELD1 (MIM 607170). Missense mutations in CRELD1 were originally identified in a small cohort of simplex AVSD cases,12 with subsequent identification of additional mutations in individuals with DS-associated AVSD.13 One of those mutations, c.985C>T (p.Arg329Cys, RefSeq accession number NM_001077415.2), is recurrent and has been identified in multiple unrelated individuals with AVSD.12,13 Numerous additional studies have shown that CRELD1 mutations are specifically associated with the AVSD phenotype and have been found in several different ethnic and racial groups.3,14–17 It is estimated that approximately 5% of individuals with nonsyndromic AVSD carry a CRELD1 missense mutation. Incomplete penetrance has been demonstrated for many of the mutations, indicating that deleterious CRELD1 mutations function as risk factors for AVSD but can be maintained in the general population as benign variants.
Missense mutations in GATA4 (MIM 600576) have also been shown to cause cardiac septal defects in humans, including AVSD in rare families with autosomal-dominant inheritance of CHD.11,18 Mutations in GATA4 are infrequently associated with simplex AVSD cases,16,19,20 although inheritance from unaffected parents indicates that additional risk factors are required for causing the defect. Rare variants in ALK2 (ACVR1 [MIM 102576]) have also been associated with CHD, including one case of an individual with DS and a partial AVSD, otherwise known as an ostium primum atrial septal defect.21 Additional ALK2 variants have been identified in individuals within the AVSD clinical spectrum, although these have been found in only a very small percentage of cases analyzed.22 Functional analyses support a role for these uncommon variants contributing to the risk of heart defects. Additional rare variants in other genes have been identified in a small number of AVSD cases; however, their pathogenicity remains uncertain.23 One mutation in TBX5 (MIM 601620) was identified in an individual with AVSD and Holt-Oram syndrome (MIM 142900).24 Somatic mutations in TBX5 and NKX2.5 (MIM 600584) have been identified in formalin-fixed hearts from deceased individuals with AVSD and other congenital heart malformations, including individuals with DS and AVSD,25,26 suggesting a potentially pathogenic role for nongermline events in CHD. However, other studies of fresh-frozen tissue from similarly malformed hearts failed to replicate these findings.27,28 Overall, the mutations associated with AVSD have been found in only a small proportion of affected individuals, indicating involvement of additional, as-yet-unidentified genes.
To advance our knowledge of the genetic basis for AVSD, we have employed the strategy of studying individuals with DS as a population sensitized to the actions of additional genetic risk factors, including genetic variants on chromosome 21 and on other chromosomes. Our premise is that the 2,000-fold increase in risk for developing an AVSD due to trisomy 21 unmasks additional modifiers, making it possible to detect them in a smaller cohort than for the euploid population. We hypothesize that rare genetic variants, incompletely penetrant on a euploid background, act synergistically with trisomy 21 to increase risk for AVSD. Under this model, we focus on rare nonsynonymous coding variants as being the most likely disease candidates, an approach that has been successful in identifying causative genes for Mendelian disorders via whole-exome sequencing.29
Subjects and Methods
Study Participants
Study participants were recruited from centers across the United States under protocols approved by the institutional review boards for each participating center and with informed consent from a custodial parent for each subject. Recruitment and enrollment methods have been extensively documented through several earlier studies.9,30,31 Individuals enrolled in the study had a diagnosis of full trisomy 21, with the vast majority confirmed by a karyotype. Individuals with partial or mosaic trisomy 21 were not enrolled. A single cardiologist (K.J.D.) reviewed medical records and classified cases as individuals with DS who had a complete, balanced AVSD documented by echocardiogram (DS+AVSD) and controls as individuals with DS who had a documented structurally normal heart (DS without CHD). Individuals with patent ductus arteriosus and patent foramen ovale were allowed as controls. The majority of the samples included in this sequencing analysis were self-identified via maternal questionnaire as non-Hispanic whites (white), although a small subset of non-Hispanic black (black) and Hispanic white (Hispanic) individuals were also included. Specifically, of the 141 cases, 111 were white, 25 were black, and 5 were Hispanic. For the 141 controls, 113 were white, 23 were black, and 5 were Hispanic.
Candidate Genes
The 26 candidate genes resequenced in this study and the rationale for inclusion are shown in Table S1 (available online). In general, candidate genes for mutation analyses were selected on the basis of evidence that disruption of gene function could result in AVSD. Genes involved in multiple pathways and developmental processes were included.
Sequencing
Genomic DNA for all samples was extracted from low-passage lymphoblastoid cell lines. Sequencing was performed via the traditional Sanger method from PCR amplicons targeted to encompass coding regions of candidate genes and the surrounding exon-intron boundaries, 3′ and 5′ untranslated regions (UTRs), and ∼2 kb of intergenic sequence up- and downstream of the transcription start and endpoints. Eighteen of the genes were resequenced in 141 cases and 141 controls through the University of Washington National Heart, Lung, and Blood Institute (NHLBI) Resequencing and Genotyping Service, D. Nickerson, Director. Eight genes were resequenced in at least 100 of the same cases, all white, through the Oregon Clinical Translational Research Institute (OCTRI) at Oregon Health & Science University, and variants were genotyped in 100 white controls. One gene, CRELD1, was resequenced in 135 white cases and controls. Samples resequenced by OCTRI were a subset of the same samples resequenced by the University of Washington NHLBI Resequencing Center, with some additional samples included in the CRELD1 study. Variants identified in this study will be deposited into the National Center for Biotechnology Information Short Genetic Variations database (dbSNP) in accordance with NHLBI policy.
Variant Classification
Variants were classified as noncoding or coding, synonymous, nonsynonymous, nonsense, insertion, deletion, splicing, and frameshift. Previously documented variants were identified through dbSNP, and the reported allele frequencies were compared to the allele frequencies in this study. Variants were further classified as case-specific (variants found only in cases), control-specific (variants found only in controls), or nonspecific (variants found in both cases and controls).
Bioinformatic Analyses
We used the MutPred web application tool to classify missense variants according to predicted likelihood of being deleterious to the protein product.32 MutPred is a supervised method based on protein-structure- and function-derived attributes, as well as evolutionary information that provides a computed general score (g) indicating the probability that a given variant is damaging and/or disease-associated versus neutral through the use of human-disease and neutral alleles for training. In addition, MutPred predicts the molecular cause of damage to the protein on the basis of a gain or loss of structural and functional properties and provides a p value (p) for each structural or functional prediction. Hypotheses that specified structural and functional properties of the protein are affected can then be generated on the basis of a combination of the general score and the p value. Mutations are binned according to the relevant hypothesis. Actionable hypotheses are assigned to variants with scores of g > 0.5 and p < 0.05. Confident hypotheses refer to variants with g > 0.75 and p < 0.05. Very confident hypotheses refer to variants with g > 0.75 and p < 0.01. We analyzed all case- and control-specific missense variants using these parameters to determine which variants were the most likely to be associated with disease.
In addition, case- or control-associated variants were evaluated for the potential to disrupt splicing through alteration of conserved sequences at intron-exon boundaries and intronic branchpoint consensus sequences.
The nonspecific variants were further analyzed for determination of whether there were variants that were statistically overrepresented in cases or controls.
Lastly, case- or control-associated variants in the 5′ UTR were evaluated for potential disruption of transcription factor binding sites via a recently developed method for predicting disease-associated functional variants within gene-regulatory regions.33
Statistical Methods
The variants found in both cases and controls were analyzed for determining whether any variants were statistically overrepresented in cases or controls. Case- and control-specific variants were binned or collapsed in several ways, and comparisons were made between the case and control groups. Missense variants were binned on the basis of the three levels of confidence for hypotheses (i.e., actionable, confident, and very confident). Potential splicing variants and potential gene-regulatory variants were also binned.
p values for comparison of total numbers of variants in cases versus controls were calculated with Fisher’s exact test, reporting two-sided p values. We used the simplifying assumption that each gene was sequenced in exactly 141 cases and 141 controls in order to perform tests across genes and pathways; sensitivity analysis to variations in this assumption (e.g., assuming 120 individuals instead of 141) made essentially no difference in the p values.
Two-tailed Student’s t tests were used for determining whether the transcriptional activity from GATA5 substitutions in luciferase assays was significantly different from wild-type; p values <0.05 were considered significant.
Expression Constructs
Case-specific genetic variants in GATA5 (MIM 611496, RefSeq NM_080473.4) with general scores of g > 0.5—c.8A>G (p.Gln3Arg), c.424T>C (p.Tyr142His), and c.477C>G (p.Phe159Leu)—were introduced into cDNA clones (OriGene Technologies, Rockville, MD, USA) with a QuikChange XL Site-Directed Mutagenesis kit (Stratagene, La Jolla, CA, USA) according to manufacturer’s instructions. The single-base mutations and the integrity of all constructs were confirmed through sequencing of both strands.
Dual Luciferase Assay
The luciferase assay was performed with the Dual-Luciferase Reporter Assay System (Promega, Madison, WI, USA) with the use of the pLightSwitch-VEGFA promoter plasmid (pLS-VEGFA), which has the human VEGF-A (MIM 192240) promoter cloned upstream of the luciferase gene. COS-7 cells were cotransfected with the expression vectors for wild-type or mutant GATA5, pLS-VEGFA, and a Renilla luciferase control vector, pGL4.73 (hRluc/SV40). Empty vector was used separately as a control, and the Renilla expression served as an internal standard for transfection efficiency. Cell extracts were prepared 48 hr after transfection and assayed for luciferase activity. Luciferase-activity values were corrected for Renilla activity, and the results were reported as ± SEM, obtained from three separate transfection assays.
Results
Resequencing of the 26 candidate genes identified a total of 2,221 variants, of which 502 were in coding regions and 1,719 were in noncoding sequences. All variants were classified according to the nature of the alteration and binned into DS with AVSD case-specific, DS with no CHD control-specific, or nonspecific groups.
Noncoding Variants
Noncoding single-nucleotide variants (SNVs), insertions, or deletions were identified in all 26 candidate genes, most frequently in introns. None of the variants identified in introns altered the consensus sequences for known splicing elements (splice junctions and branchpoint-defining sequences). With the exception of case- or control-specific rare variants, none of the variants in introns had a significant association with cases or controls. The case-specific and control-specific intronic variants were not considered further in this study because of the uncertainty in assessing functional significance.
The 5′ UTR regions for each gene were analyzed for variants with the potential to alter transcription factor binding sites. Case- or control-specific SNVs in the 5′ UTR were uncommon but did occur in ACVR1 (one case), CITED2 (MIM 602937) (two in controls), COL6A3 (MIM 120250) (one case), CRELD1 (two in cases), CTGF (MIM 121009) (one control), CYR61 (MIM 602369) (two in cases), FGF2 (MIM 134920) (one case), ROCK1 (MIM 601702) (two in cases, four in controls), and SHH (MIM 600725) (one case). None of these rare variants were found in more than one individual, and none were reported in dbSNP. Also, none of these 5′ UTR variants showed any indication of changing predicted transcription factor recognition sites, and they were therefore of unknown consequence. All other 5′ UTR SNVs were found in both cases and controls and had no significant association with either group. Details regarding those SNVs can be found in Table S2.
Coding Variants
Coding variants included numerous previously documented SNVs, small insertions or deletions, and frameshift variants. None of the previously documented variants showed significant overrepresentation in cases or controls. Consequently, we focused on the newly identified variants for additional analyses.
Only one nonsense SNV was identified; c.3832G>T (p.Glu1278∗), a truncating mutation in exon 30 of COL18A1 (MIM 120328, RefSeq NM_130444.2), was identified in a single control sample. We hypothesize that this truncating variant in COL18A1, a gene located on chromosome 21, results in a normal disomic level of expression of this gene by eliminating expression from the one trisomic truncating allele.
A previously unreported deletion was also identified in COL18A1. The 10 bp frameshift deletion at the 3′ end of exon 33, c.4067_4068del, was detected in 69 individuals; 25 cases were heterozygous for the deletion compared to 41 controls (p = 0.03). One case and two controls were homozygous for the deletion. The variant encodes a truncated protein of 1,441 amino acids, with 85 substituted amino acids at the C terminus of the predicted protein product, disrupting the triple-helical (collagenous) domain, and terminating prior to the NC1 domain that includes the coding region for endostatin. Although seen in nearly 25% of our study cohort, this isoform has not been previously reported. The significant overrepresentation in controls compared to cases suggests a potentially protective effect.
COL6A3 had a previously undocumented 3 bp deletion in exon 40, c.8877_8879delTGC (RefSeq NM_004369.2). The deletion removes a single alanine residue from an alanine-rich region of the protein. Out of the 282 individuals resequenced for this gene, 24 cases and 26 controls were heterozygous for the deletion (p = 0.87).
Another undocumented 3 bp deletion, c.693_695delAGA (RefSeq NM_007341.2), was identified in exon 6 of SH3BGR (MIM 602230) and had similar rates of heterozygous insertion in 89 cases and 96 controls (p = 0.57). The deletion removes a glutamic acid residue from the protein sequence. Approximately 50% of the amino acids encoded by this exon are glutamic acid residues, so it appears unlikely that the loss of one glutamic acid residue will have an impact on the protein.
A 3 bp duplication was detected in exon 1 of CRELD2 (MIM 607171) in a single case. The duplication, c.129_131dupCAG (RefSeq NM_001135101.1), adds a glutamine residue to the CRELD domain, which is a glutamine-rich region and is not predicted to have any functional consequence.
The majority of undocumented coding variants identified in this study were single-nucleotide missense variants. A total of 238 nonsynonymous SNVs were identified in 21 of the 26 resequenced genes. Five of the genes, BMP2 (MIM 112261), BMP5 (MIM 112265), GATA4, HEY2 (MIM 604674), and WNT9A (MIM 602863), had no missense variants in either cases or controls, suggesting that these genes generally have a low tolerance for variation, at least on the genetic background of trisomy 21.
All missense SNVs were analyzed for indications that they were common variants. Documented SNPs found in dbSNP were further evaluated for evidence of overrepresentation in cases compared to controls. There were 63 previously documented SNVs. None were found to have a significant bias in allele representation. There were 175 SNVs that were not found in dbSNP, which were also evaluated for allelic bias in cases compared to controls. Fifty-eight variants were found exclusively in cases or in controls; 34 were case-specific and 24 were control-specific. The remaining 117 SNVs were nonspecific and did not show any significant allelic association with cases compared to controls.
Potentially Damaging Variants
Altogether, 13 of 26 resequenced genes had variants in either cases or controls that were potentially damaging according to MutPred analyses (g > 0.5). A total of 34 case-specific missense SNVs were identified in nine genes, all of which were heterozygous changes (Table 1). Nine of these were recurrent, i.e., found in more than one unrelated affected individual. The other 25 missense variants were identified in one individual each. No individual carried more than one of these case-specific variants. In all, 48 individual cases carried putatively deleterious case-specific SNVs.
Table 1.
Case-Specific Missense Variants Predicted to be Deleterious
| Gene | Nucleotide Change | Amino Acid Change | Frequencya(Race)b | General Score (Confidence Level)c | Structure or Function Hypotheses (Probability Scores) |
|---|---|---|---|---|---|
|
COL6A1d |
c.350T>C | p.Val117Ala | 2/141 (W) | 0.776 (VC) | loss of helix (p = 0.0076), gain of loop (p = 0.0079), loss of stability (p = 0.0292), gain of disorder (p = 0.0353), gain of ubiquitination at p.Lys121 (p = 0.04441) |
| c.2614C>T | p.Arg872Trp | 1/141 (B) | 0.790 (C) | loss of methylation at p.Arg872 (p = 0.0207), loss of disorder (p = 0.0259), gain of catalytic residue at p.Arg872 (p = 0.0467) | |
| c.2304G>C | p.Gln768His | 1/141 (W) | 0.537 (A) | gain of sheet (p = 0.0016), loss of helix (p = 0.0017), gain of loop (p = 0.024) | |
| c.2170G>A | p.Ala724Thr | 1/141 (W) | 0.542 (NP) | NP | |
|
COL6A2d |
c.2558G>A | p.Arg853Gln | 1/141 (W) | 0.869 (C) | gain of ubiquitination at p.Glu851 (p = 0.0354) |
| c.316G>A | p.Glu106Lys | 5/141 (W) | 0.759 (C) | gain of methylation at p.Glu106 (p = 0.0122) | |
| c.2528G>A | p.Arg843Gln | 1/141 (W) | 0.635 (NP) | NP | |
| c.2182G>A | p.Val728Met | 1/141 (W) | 0.545 (NP) | NP | |
|
COL6A3 |
c.8209A>C | p.Lys2737Gln | 1/141 (W) | 0.650 (A) | loss of ubiquitination at p.Lys2737 (p = 0.0283), loss of sheet (p = 0.0315), loss of methylation at Lys2737 (p = 0.0355) |
| c.1216C>T | p.Arg406Cys | 1/141 (W) | 0.643 (A) | gain of sheet (p = 0.0344), loss of helix (p = 0.0376) | |
| c.3445C>T | p.Arg1149Trp | 1/141 (W) | 0.625 (A) | loss of disorder (p = 0.0107) | |
| c.4117G>A | p.Ala1373Thr | 1/141 (H) | 0.543 (A) | gain of phosphorylation at p.Ala1373 (p = 0.0412) | |
| c.8236G>A | p.Glu2746Lys | 1/141 (W) | 0.513 (A) | gain of ubiquitination at p.Glu2746 (p = 0.0258), loss of sheet (p = 0.0315), gain of methylation at p.Glu2746 (p = 0.0427) | |
| c.7873G>T | p.Asp2625Tyr | 1/141 (W) | 0.897 (NP) | NP | |
| c.3191G>A | p.Arg1064Gln | 1/141 (W) | 0.834 (NP) | NP | |
| c.176G>A | p.Arg59Gln | 1/141 (W) | 0.707 (NP) | NP | |
| c.4813A>G | p.Ile1605Val | 1/141 (B) | 0.568 (NP) | NP | |
|
COL18A1d |
c.4786G>A | p.Gly1596Arg | 1/141 (B) | 0.810 (VC) | gain of MoRFe binding (p = 0.0081) |
| c.331G>A | p.Gly111Arg | 1/141 (B) | 0.835 (C) | loss of helix (p = 0.0167), gain of sheet (p = 0.0266), gain of MoRFe binding (p = 0.0311) | |
| c.5156C>T | p.Ser1719Leu | 1/141 (B) | 0.603 (A) | loss of disorder (p = 0.0152) | |
| c.1051C>T | p.Arg351Cys | 1/141(W) | 0.640 (NP) | NP | |
| c.2635G>A | p.Asp879Asn | 2/141 (W,B) | 0.590 (NP) | NP | |
| c.142C>G | p.Pro48Ala | 4/141 (W, W, B, H) | 0.578 (NP) | NP | |
| c.1777G>A | p.Val593Met | 1/141 (W) | 0.554 (NP) | NP | |
|
CRELD1 |
c.985C>T | p.Arg329Cys | 2/135 (W) | 0.860 (validated) | NP; biochemical analysis shows misfolding |
| c.1240G>A | p.Glu414Lys | 1/135 (W) | 0.798 (VC) | gain of methylation (p = 0.016), gain of MoRF binding (p = 4 × 10−04) | |
| CRELD2 | c.287C>T | p.Ala96Val | 3/100 (W) | 0.643 (NP) | NP |
|
FBLN2 |
c.3116T>C | p.Ile1039Thr | 1/141 (W) | 0.696 (A) | loss of stability (p = 0.0211) |
| c.2749G>A | p.Gly917Ser | 1/141 (W) | 0.577 (NP) | NP | |
|
FRZB |
c.299T>C | p.Glu100Ser | 1/141 (W) | 0.543 (A) | gain of disorder (p = 0.0078) |
| c.695G>A | p.Arg232Gln | 2/141 (W) | 0.634 (NP) | NP | |
| GATA5 | c.8A>G | p.Gln3Arg | 2/141 (W) | 0.712 (C; validated) | gain of MoRFe binding (p = 8 × 10−04), gain of methylation (p = 0.0283); transcription assay shows gain of function |
| c.424T>C | p.Tyr142His | 1/141(W) | 0.743 (A) | gain of disorder (p = 0.0409) | |
| c.477C>G | p.Glu159Leu | 2/141(W, B) | 0.754 (NP) | NP | |
The cDNA positions for the nucleotide changes are based on the following reference sequences: COL6A1, NM_001848.2; COL6A2, NM_001849.3; COL6A3, NM_004369.2; COL18A1, NM_130444.2; CRELD1, NM_001077415.2; CRELD2, NM_001135101.1; FBLN2, NM_001004019.1; FRZB, NM_001463.2; and GATA5, NM_080473.4.
Frequency is the number of individuals in which each variant was identified over the number of total cases resequenced for that gene.
Race codes are as follows: W, white; B, black; and H, Hispanic.
Confidence levels are as follows: A, actionable hypotheses; C, confident hypotheses; VC, very confident hypotheses; and NP, none predicted.
Gene is located on chromosome 21 (trisomic for this population).
Gain of MoRF binding represents gain of molecular recognition factor binding (interaction with other molecules is enhanced).
Ten genes had potentially damaging nonsynonymous variants that occurred exclusively in controls (Table 2). Six of the ten genes were the same as those that harbored case-specific damaging missense changes. There were a total of 24 potentially damaging missense SNVs in controls, each occurring only once. No control individual carried more than one of the potentially damaging variants. Of the genes with unique missense SNVs found in both cases and controls, all but one had more potentially damaging variants in the cases. The exception was FBLN2 (MIM 135821), which had two potentially damaging variants in cases and four in controls.
Table 2.
Control-Specific Missense Variants Predicted to be Deleterious
| Gene | Nucleotide Change | Amino Acid Change | Frequencya(Race)b | General Score (Confidence Level)c | Structure or Function Hypotheses (Probability Scores) |
|---|---|---|---|---|---|
| CITED2 | c.792G>T | p.Gln264His | 1/141 (W) | 0.512 (NP) | NP |
| COL6A1d | c.3001A>C | p.Ser1001Arg | 1/141 (W) | 0.568 (NP) | NP |
|
COL6A2d |
c.3029T>G | p.Phe1010Cys | 1/141 (W) | 0.814 (NP) | NP |
| c.1129C>T | p.Arg377Cys | 1/141 (B) | 0.566 (A) | loss of MoRFe binding (p = 0.0068), loss of methylation at Arg377 (p = 0.0139) | |
| c.1336G>A | p.Asp446Asn | 1/141 (B) | 0.511 (NP) | NP | |
|
COL6A3 |
c.2180A>C | p.Tyr727Ser | 1/141 (W) | 0.769 (C) | gain of disorder (p = 0.0248) |
| c.1688A>G | p.Asp563Gly | 1/141 (W) | 0.760 (NP) | NP | |
| c.4090G>A | p.Val1364Met | 1/141 (B) | 0.686 (NP) | NP | |
| c.2666G>A | p.Arg889His | 1/141 (W) | 0.601 (NP) | NP | |
| c.4561G>A | p.Glu1521Lys | 1/141 (H) | 0.569 (A) | gain of MoRFe binding (p = 0.0014), gain of methylation at Glu1521 (p = 0.0144), loss of ubiquitination at Lys1518 (p = 0.0272) | |
| c.6263C>T | p.Pro2088Leu | 1/141 (B) | 0.516 (A) | gain of MoRFe binding (p = 0.043) | |
| c.2794G>T | p.Ala932Ser | 1/141 (W) | 0.515 (A) | gain of disorder (p = 0.0218) | |
|
COL18A1d |
c.373G>A | p.Val125Ile | 1/141 (B) | 0.714 (NP) | NP |
| c.1264C>T | p.Arg422Cys | 1/141 (H) | 0.554 (NP) | NP | |
| c.3637C>T | p.Arg1213Trp | 1/141 (W) | 0.610 (A) | loss of methylation at Arg1213 (p = 0.007) | |
| CTGF | c.172C>G | p.Arg58Gly | 1/141 (W) | 0.709 (NP) | NP |
|
FBLN2 |
c.2770A>C | p.Asn924His | 1/141 (W) | 0.909 (NP) | NP |
| c.2630C>T | p.Thr877Met | 1/141 (W) | 0.776 (NP) | NP | |
| c.2658C>A | p.Asn886Lys | 1/141 (B) | 0.615 (C) | gain of methylation at Asn886 (p = 0.0045), gain of catalytic residue at Asn886 (p = 0.007), gain of ubiquitination at Asn886 (p = 0.0452) | |
| c.3467C>T | p.Ala1156Val | 1/141 (W) | 0.610 (NP) | NP | |
| GATA5 | c.83C>T | p.Ala28Val | 1/141 (B) | 0.707 (NP) | NP |
| TBX20 | c.461C>G | p.Pro154Arg | 1/141 (B) | 0.718 (C) | gain of MoRFe binding (p = 3e-04), loss of ubiquitination at Lys158 (p = 0.0395) |
| VTN | c.728T>C | p.Ile243Thr | 1/141 (W) | 0.718 (NP) | NP |
| c.1267G>A | p.Asp423Asn | 1/141 (B) | 0.532 (C) | loss of sheet (p = 0.0126), loss of loop (p = 0.0374) | |
The cDNA positions for the nucleotide changes are based on the following reference sequences: CITED2, NM_006079.3; COL6A1, NM_001848.2; COL6A2, NM_001849.3; COL6A3, NM_004369.2; COL18A1, NM_130444.2; CTGF, NM_001901.2; FBLN2, NM_001004019.1; GATA5, NM_080473.4; TBX20, NM_001077653.1; and VTN, NM_000638.3.
Frequency is the number of individuals in which each variant was identified over the number of total cases resequenced for that gene.
Race codes are as follows: W, white; B, black; and H, Hispanic.
Confidence levels are as follows: A, actionable hypotheses; C, confident hypotheses; VC, very confident hypotheses; and NP, none predicted.
Gene is located on chromosome 21 (trisomic for this population).
Gain of MoRF binding represents gain of molecular recognition factor binding (interaction with other molecules is enhanced).
Sixteen variants affecting the triple-helical domains of the four collagen genes analyzed were not scored because MutPred does not provide reliable predictions for those sequences. These amino acid substitutions all occurred in variable positions of the triple-helical domains. Importantly, none altered the conserved glycine residue of the Gly-X-Y consensus sequence (X is any amino acid; Y is hydroxyl-proline or hydroxyl-lysine). These are known hot spots for disease-associated mutations in collagen genes, because the glycine residue is the only amino acid compatible with triple-helical formation.34 Because the variants identified were in the X position, they are considered probably benign.
Overall, 24 individual controls carried putatively deleterious control-specific SNVs compared to the 48 individual cases that carried damaging case-specific mutations (p = 0.0016).
Examination of Case- and Control-Specific Missense Variants by Hypothesis Binning
One of the valuable aspects of MutPred is its ability to generate hypotheses regarding the functional consequences of mutations and assign a probability score to these predicted properties. Variants can therefore be classified by combination of the general score and property scores. MutPred combined scores are binned into three categories: actionable hypotheses (g > 0.5, p < 0.05), confident hypotheses (g > 0.75, p < 0.05), and very confident hypotheses (g > 0.75, p < 0.01). Using these criteria, we further filtered case- and control-specific variants, eliminating those that did not fall into one of these categories. At this level of filtering, 15 variants were eliminated from the case-specific group, leaving 19 high-probability variants among 26 of the 141 cases (18.4% of cases) in eight different genes (Table 3). Fifteen variants were removed from the control-specific group, leaving only nine high-probability variants among 141 control individuals (6.4% of controls) and six genes. This resulted in the elimination of COL6A1 (MIM 120220) and GATA5 from the control-specific variant list, because there were only case-specific high-probability variants in those genes when binned by these criteria (Table 4). Accordingly, there was a significant excess of variants predicted to be deleterious in cases (26/141) compared to controls (9/141) (p = 0.0013).
Table 3.
Case-Specific Variants with Actionable Hypotheses
| Gene | Protein Variant | Frequencya(Race)b | General Score (Confidence Level)c | Structure or Function Hypotheses (Probability Scores) |
|---|---|---|---|---|
|
COL6A1d |
p.Val117Ala | 2/141 (W) | 0.776 (VC) | loss of helix (p = 0.0076), gain of loop (p = 0.0079), loss of stability (p = 0.0292), gain of disorder (p = 0.0353), gain of ubiquitination at Lys121 (p = 0.04441) |
| p.Arg872Trp | 1/141 (B) | 0.790 (C) | loss of methylation at Arg872 (p = 0.0207), loss of disorder (p = 0.0259), gain of catalytic residue at Arg872 (p = 0.0467) | |
| p.Gln768His | 1/141 (W) | 0.537 (A) | gain of sheet (p = 0.0016), loss of helix (p = 0.0017), gain of loop (p = 0.024) | |
|
COL6A2d |
p.Arg853Gln | 1/141 (W) | 0.869 (C) | gain of ubiquitination at Lys851 (p = 0.0354) |
| p.Glu106Lys | 5/141 (W) | 0.759 (C) | gain of methylation at Glu106 (p = 0.0122) | |
|
COL6A3 |
p.Lys2737Gln | 1/141 (W) | 0.650 (A) | loss of ubiquitination at Lys2737 (p = 0.0283), loss of sheet (p = 0.0315), loss of methylation at Lys2737 (p = 0.0355) |
| p.Arg406Cys | 1/141 (W) | 0.643 (A) | gain of sheet (p = 0.0344), loss of helix (p = 0.0376) | |
| p.Arg1149Trp | 1/141 (W) | 0.625 (A) | loss of disorder (p = 0.0107) | |
| p.Ala1373Thr | 1/141 (H) | 0.543 (A) | gain of phosphorylation at Ala1373 (p = 0.0412) | |
| p.Glu2746Lys | 1/141 (W) | 0.513 (A) | gain of ubiquitination at Glu2746 (p = 0.0258), loss of sheet (p = 0.0315), gain of methylation at Glu2746 (p = 0.0427) | |
|
COL18A1d |
p.Gly1596Arg | 1/141 (B) | 0.810 (VC) | gain of MoRFe binding (p = 0.0081) |
| p.Gly111Arg | 1/141 (B) | 0.835 (C) | loss of helix (p = 0.0167), gain of sheet (p = 0.0266), Gain of MoRF binding (p = 0.0311) | |
| p.Ser1719Leu | 1/141 (B) | 0.603 (A) | loss of disorder (p = 0.0152) | |
|
CRELD1 |
p.Arg329Cysb | 2/135 (W) | 0.860 (NP) | NP; biochemical analysis shows misfolding |
| p.Glu414Lys | 1/135 (W) | 0.798 (VC) | gain of methylation (p = 0.016), gain of MoRF binding (p = 4 × 10−04) | |
| FBLN2 | p.Ile1039Thr | 1/141 (W) | 0.696 (A) | loss of stability (p = 0.0211) |
| FRZB | p.Phe100Ser | 1/141 (W) | 0.543 (A) | gain of disorder (p = 0.0078) |
| GATA5 | p.Gln3Argb | 2/141 (W) | 0.712 (C) | gain of MoRFe binding (p = 8 × 10−04), gain of methylation (p = 0.0283); transcription assay shows gain of function |
| p.Tyr142His | 1/141(W) | 0.743 (A) | gain of disorder (p = 0.0409) | |
Frequency is the number of individuals in which each variant was identified over the number of total cases resequenced for that gene.
Race codes are as follows: W, white; B, black; and H, Hispanic.
Confidence levels are as follows: A, actionable hypotheses; C, confident hypotheses; VC, very confident hypotheses; and NP, none predicted.
Gene is located on chromosome 21 (trisomic for this population).
Gain of MoRF binding represents gain of molecular recognition factor binding (interaction with other molecules is enhanced).
Table 4.
Control-Specific Variants with Actionable Hypotheses
| Gene | Protein Variant | Frequencya(Race)b | General Score (Confidence Level)c | Structure or Function Hypotheses (Probability Scores) |
|---|---|---|---|---|
| COL6A2d | p.Arg377Cys | 1/141 (B) | 0.566 (A) | loss of MoRFe binding (p = 0.0068), loss of methylation at Arg377 (p = 0.0139) |
|
COL6A3 |
p.Tyr727Ser | 1/141 (W) | 0.769 (C) | gain of disorder (p = 0.0248) |
| p.Glu1521Lys | 1/141 (H) | 0.569 (A) | gain of MoRFe binding (p = 0.0014), gain of methylation at Glu1521 (p = 0.0144), loss of ubiquitination at Lys1518 (p = 0.0272) | |
| p.Pro2088leu | 1/141 (B) | 0.516 (A) | gain of MoRFe binding (p = 0.043) | |
| p.Ala932Ser | 1/141 (W) | 0.515 (A) | gain of disorder (p = 0.0218) | |
| COL18A1d | p.Arg1213Trp | 1/141 (W) | 0.610 (A) | loss of methylation at Arg1213 (p = 0.007) |
| FBLN2 | p.Asn886Lys | 1/141 (B) | 0.615 (C) | gain of methylation at Asn886 (p = 0.0045), gain of catalytic residue at Asn886 (p = 0.007), gain of ubiquitination at Asn886 (p = 0.0452) |
| TBX20 | p.Pro154Arg | 1/141 (B) | 0.718 (C) | gain of MoRFe binding (p = 3 × 10−04), loss of ubiquitination at Lys158 (p = 0.0395) |
| VTN | p.Asp423Asn | 1/141 (B) | 0.532 (C) | loss of sheet (p = 0.0126), loss of loop (p = 0.0374) |
Frequency is the number of individuals in which each variant was identified over the number of total cases resequenced for that gene.
Race codes are as follows: W, white; B, black; and H, Hispanic.
Confidence levels are as follows: A, actionable hypotheses; C, confident hypotheses; VC, very confident hypotheses; and NP, none predicted.
Gene is located on chromosome 21 (trisomic for this population).
Gain of MoRF binding represents gain of molecular recognition factor binding (interaction with other molecules is enhanced).
Examination of SNVs by Race and/or Ethnic Group
The case and control cohorts were largely non-Hispanic white, but there were some black (25 cases, 23 controls) and Hispanic (5 cases, 5 controls) individuals included in the resequencing. We removed variants that were exclusive to black and Hispanic samples from consideration to determine whether those genotypes had a significant impact on the data. Recurrent rare variants that were identified in whites and other race and/or ethnic groups were retained in the data set. For missense variants with a general score g > 0.5, there were case-specific variants in nine genes, with a total of 28 variants in 42 of the 111 white cases, whereas control-specific variants were found in eight genes, with a total of 13 variants in 13 of the 113 white controls (p < 0.0001). For missense variants that met the more stringent criteria of actionable, confident, or very confident hypotheses (Table S3), there were case-specific variants in eight genes, with 14 variants found in 21 different individuals. Control-specific variants were found in only two genes, COL6A3 and COL18A1, with three variants in three individuals (Table S4), although these two genes have both case- and control-specific variants at this level of filtering. In summary, in the total data set there was a significant excess of deleterious missense variants in nearly 19% (21/111) of white cases, but only 2.6% (3/113) of controls (p < 0.0001). Consequently, we conclude that the small amount of race and/or ethnic group heterogeneity did not falsely inflate the finding that there is an excess of deleterious mutations in cases versus controls.
Inheritance of Recurrent Variants
The highest-probability case-specific variants included four recurrent variants, in COL6A1, COL6A2 (MIM 120240), CRELD1, and GATA5, which were each identified in more than one unrelated individual (Table 3). DNA from biological parents was resequenced for determination of whether the variants were inherited or occurred spontaneously. All of the recurrent variants were inherited as a single copy from a parent in all cases except one. The COL6A2 c.316G>A (p.Glu106Lys) variant appears to have occurred as a de novo mutation in one individual, but was inherited from a parent in the other four cases. There was no bias as to the gender of the parent of origin for inherited variants.
Alteration of GATA5 Activity by Functional Variants
GATA5 activity was measured for the three case-specific amino acid substitutions, p.Gln3Arg, p.Tyr142His, and p.Phe159Leu, with the use of a luciferase reporter assay with the luciferase gene under the control of the VEGF-A promoter. VEGF-A is a known transcriptional target of GATA5 and an important regulator of AV cushion morphogenesis.35 The most significant functional-hypothesis score was the case-specific variant, GATA5 c.8A>G (p.Gln3Arg), which was identified in two unrelated AVSD cases. The functional prediction indicated enhanced binding of the p.Arg3 substitution to its DNA targets (p = 8 × 10−04). The p.Arg3 substitution showed significantly more activity 48 hr after transfection, with a >2-fold increase in the signal from the VEGF luciferase reporter compared to wild-type (Figure 1). The relative activity of p.Arg3 compared to p.Gln3 correlated well with the MutPred prediction of increased molecular recognition factor (MoRF) binding by p.Arg3. In comparison, the recurrent p.Phe159Leu substitution showed significantly lower activity compared to wild-type GATA5. Although this c.477C>G (p.Phe159Leu) variant had the highest general score of the GATA5 variants, it did not trigger functional hypotheses by MutPred. The p.Tyr142His substitution showed activity levels that trended higher than wild-type but did not reach statistical significance.
Figure 1.
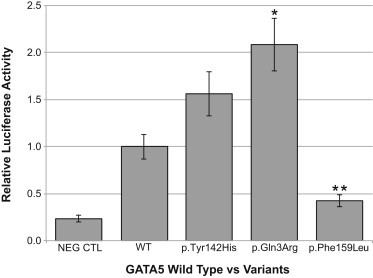
The Transcriptional Activation Ability of GATA5 Variants as Measured with a Luciferase Assay
COS-7 cells were transfected with VEGF-A promoter-reporter construct and either wild-type GATA5 construct (WT) or one of the three GATA5 mutant constructs, as described in Subjects and Methods. Cells were harvested 48 hr posttransfection, and luciferase activity was measured. Reporter-gene activity was quantitated as luciferase activity relative to WT, which was normalized to a relative luciferase activity value of 1.0. NEG CTRL is the empty vector. The bars for each GATA5 mutant construct are identified by the protein variant. Each bar represents a mean ± SEM of three individual experiments. ∗p = 0.004 and ∗∗p = 0.0009 (two-tailed Student’s t test).
Discussion
The search for genetic modifiers of risk for CHD is often challenged by the lack of availability of sufficiently large and phenotypically homogeneous study cohorts. Benson and Martin recently estimated that a sample size of 5,000 would be required for a 1:1 case-control study to detect a 40% increase in risk for a trait that occurs in 1% of the population.36 To address this problem, we chose to study individuals with DS, a sensitized population with a single highly penetrant risk factor for heart defects. The 2,000-fold increase in risk for developing an AVSD due to trisomy 21 should make it possible to detect disease-associated genetic variants in a smaller cohort than for the euploid population. However, even in a sensitized population that is selected to be homogenous, the ability to predict which variants are the most likely to confer risk of disease is of paramount importance. Given the prevalence of rare variants in the human genome, the potential for spurious association is high, so analysis requires robust predictions of functional consequences of variants.
MutPred was selected for bioinformatic evaluation of missense changes on the basis of a recent comparison between several computational methods that demonstrated its superior performance in making accurate predictions compared to other commonly used algorithms.37 MutPred has the added advantage of providing functional hypotheses with probability scores, which aids in prioritizing follow-up studies. Using a carefully phenotyped, sensitized population to identify genetic risk factors for AVSD and rigorous evaluation of genetic variants, we show that there is an excess of potentially damaging missense variants in DS+AVSD cases compared to DS without CHD controls. In our most stringent analysis, including filtering on the basis of strict functional criteria and limiting the analysis to white non-Hispanic individuals, we found highly probable deleterious missense variants in 15% of cases and none in controls (p < 0.0001). It was notable that the two genes, COL6A3 and COL18A1, with actionable and confident variants in controls, also had variants in cases, suggesting a potential lack of specificity for AVSD. If we consider genes that have only case- or control-specific variants at this level of filtering, it would remove all of the genes in the control-specific group but leave six genes in the case-specific group, with 10 variants in 17 individuals. These variants were restricted to six out of the 26 genes investigated, COL6A1, COL6A2, CRELD1, FBLN2, FRZB (MIM 605083), and GATA5. Inclusion of all variants in these genes with a significant general score (g > 0.5) resulted in a total of 17 missense variants in 25 different individuals, or 18% of all cases. Other genes, including CRELD2, COL6A3, and COL18A1, also had more potentially deleterious variants in cases compared to controls. Some of these variants could well be pathogenic but will be of lower priority for follow-up studies. In addition, most of the COL18A1 variants were found in black or Hispanic individuals, which might reflect specific variation in those populations. Control-specific missense mutations with significant general scores were also identified, albeit at a much lower rate than case-specific mutations. Many of these occurred in genes that also harbored case-specific variants, but some genes had variants exclusively in controls, including CITED2, CTGF, TBX20 (MIM 606061), and VTN (MIM 193190). Control-specific variants could represent protective alleles, although the low frequency might also indicate spurious association.
This study indicates that a significant proportion of AVSD risk in our study cohort might be attributable to rare, highly deleterious variants in six genes: COL6A1, COL6A2, CRELD1, FBLN2, FRZB, and GATA5. With the exception of CRELD1, all of the high-probability mutations are in genes that have not been previously associated with AVSD, although COL6A1 and COL6A2 have been implicated in CHD in DS.38–41 The recurrence of case-specific variants in the COL6A1, COL6A2, CRELD1, and GATA5 genes in unrelated individuals provides additional support for the premise that these genes are involved in AVSD etiology, especially considering that there were no recurrent variants specific to controls. One of those variants, the c.985C>T (p.Arg329Cys) mutation in CRELD1, has been associated with AVSD in studies across different syndromic and nonsyndromic populations.12,13 It had a highly significant general score (g = 0.860), but no predicted structure or function hypotheses in MutPred. However, we showed previously that the p. Arg329Cys substitution, which we predicted would disrupt the disulfide bonding pattern of an epidermal growth factor (EGF) domain, does indeed affect protein folding.12 Further, a null allele of Creld1, which has no heart or other phenotype on a disomic background in a mouse model, greatly increases the occurrence of CHD in a mouse model of DS.42
In this study we identified a previously unreported variant in GATA5, c.8A>G (p.Gln3Arg), in two individuals. This change increased activation of transcription, consistent with the highly probable hypothesis that this amino acid change would result in stronger MoRF binding (p = 8 × 10−04) predicted by MutPred. The dramatic increase in GATA5 activation of the VEGF-A promoter indicates the potential for the p.Arg3 substitution to affect AV cushion morphogenesis by increasing VEGF-A expression, which is known to contribute to the cause of CHD in mouse models.43,44 Another newly identified recurrent GATA5 variant, c.477C>G (p.Phe159Leu), significantly decreased GATA5 activity to less than half that of wild-type. Decreased VEGF-A expression has also been associated with CHD in mouse models, indicating that this is also a pathogenic substitution, and that changes that result in significant increases or decreases in GATA5 activity can contribute to the etiology of AVSD. There are also recurrent variants in COL6A1 and COL6A2. The c.316G>A (p.Glu106Lys) case-specific variant in COL6A2 was found in five unrelated individuals. Collectively, in this study alone we found potentially damaging recurrent variants in 10% of cases across four genes, and none in controls (p = 0.0002).
Recurrence of damaging mutations is consistent with our premise that genetic risk factors for AVSD are present at a low frequency in the general population. These variants are incompletely penetrant and benign in the general population but contribute to AVSD susceptibility on a sensitive background.42 Trisomy 21 confers substantial risk for CHD but still requires additional “hits” to elicit the defect. Our data indicates that those hits occur as missense changes in a subset of heart-development genes. We propose that those same variants confer risk of AVSD in the non-DS population and that the defect occurs when a sufficient number of factors combine to breach the disease threshold. This is supported by our previous studies showing that the CRELD1 p.Arg329Cys mutation is associated with both DS-related and nonsyndromic AVSD and is inherited from an unaffected parent,12,13 as well as our finding that the other recurrent case-specific variants presented here are also inherited from unaffected parents.
One finding of particular interest is the apparent involvement of COL6A1 and COL6A2 in DS-related AVSD. These genes are located together on human chromosome 21q22.3 and are therefore trisomic in DS. COL6A1 and COL6A2 encode two of the three chains for the heterotrimeric type VI collagen protein. Variation in these genes has been implicated in the etiology of CHD in DS through genetic association studies, although only common variants were previously considered.38–41 However, the implications of genetic association between variants in collagen VI genes and AVSD have never been fully realized. In addition, abnormal expression of type VI collagen protein has been shown in DS fetal hearts and correlates with morphological defects of AV endocardial-derived structures.45 This has driven further speculation that genetic variation in the collagen VI genes is somehow associated with AVSD. Most recently, overexpression of COL6A2 and DSCAM (MIM 602523) from an exogenous α-MHC promoter in transgenic mouse hearts has been shown to disrupt heart development in doubly transgenic mice.46 In spite of all of the suggestive data, this is compelling evidence that rare missense variants in COL6A1 and COL6A2 are specifically associated with AVSD.
Paradoxically, mutations in COL6A1, COL6A2, and COL6A3 are known to cause two skeletal muscular dystrophies: Bethlem myopathy (MIM 158810) and Ullrich congenital muscular dystrophy (MIM 254090). Bethlem myopathy is thought to be autosomal dominant, although many cases occur sporadically as a result of de novo mutations. Ullrich myopathy is generally autosomal recessive, with heterozygous carriers being asymptomatic. Cardiomyopathies and arrhythmias are common in individuals with Bethlem myopathy, but septal malformations have not been reported.47 The mutations found in these muscle diseases are usually deletions, exon skipping, missplicing, or substitutions for the conserved glycine residues in the triple-helical domains, although a few other missense variants have also been associated with the disease.48 By contrast, the potentially deleterious variants identified in this study were found exclusively in the coding regions for the N-terminal and C-terminal globular domains of COL6A1 and COL6A2. The C-terminal globular domains are required for initial monomer assembly into heterotrimers from the chains encoded by COL6A1, COL6A2, and COL6A3. The N-terminal globular domains participate in the higher-ordered assembly of collagen VI microfibrils following folding of the heterotrimers. The AVSD-associated COL6A1 and COL6A2 variants were inherited from self-reported phenotypically normal parents, demonstrating that these are not dominant skeletal myopathy mutations, but might be incompletely penetrant AVSD risk alleles that are pathogenic on the sensitized trisomy 21 background. If there is an incompletely penetrant or very mild skeletal phenotype associated with these mutations, we might not detect it in the parents. It is also possible that these variants could exacerbate the hypotonia commonly found in children with DS. However, we do not have information about the course of hypotonia in these individuals, so that cannot be assessed. It is also possible that these variants do not affect skeletal muscle but are instead specific to the heart. Pleiotropy for collagen VI gene mutations could be due to differences in the nature of the variants found in heart defects compared to skeletal muscle abnormalities. Further study of these variants will significantly advance our understanding of collagen VI biology. In addition, COL6A1 and COL6A2 are known to be quite variable in their composition in terms of polymorphic missense variants, so the potential for intragenic allelic interactions is significant. However, we did not find any specific associations between case- or control-specific rare variants and nonspecific SNVs, suggesting that COL6A1 and COL6A2 intragenic allelic interactions are not significant in DS.
Although we have focused here on missense variants, it is notable that there are other variants of interest that deserve follow-up consideration. In particular, the rare case-specific SNVs in the 5′ UTRs of several of the candidate genes are of unknown consequence but certainly could have the potential to alter regulation of gene expression. Additionally, the 10 bp deletion in exon 33 of COL18A1, which is predicted to truncate the protein, warrants further investigation. The overrepresentation in controls compared to cases (p < 0.03) suggests a protective effect. The role of type XVIII collagen in heart development is poorly understood; therefore, investigation of this variant could be informative in terms of both the pathogenesis of AVSD and heart development in general.
Pathway analysis led to the unexpected finding that all six genes harboring purportedly deleterious case-specific variants are associated in some way with VEGF-A (Table 5). VEGF-A is potent mitogen that is a known regulator of AV valvuloseptal morphogenesis, wherein it is critical for the formation of the AV endocardial cushions and helps guide the morphogenesis of those primordial structures into the AV valves.52,53 In animal models, both increased and decreased levels of VEGF-A expression during heart development are associated with CHD, including AVSD.43,44,54,55 Consequently, deleterious mutations in genes that alter VEGF-A expression or are responsive to VEGF-A have a clear developmental link to AVSD, suggesting a mechanism by which the variants identified in this study could contribute to the pathogenesis of AVSD. However, the effect of variation in these genes might extend to other aspects of AV valve development. Although development of the AV endocardial cushions is critical to AV septation, there is also a significant contribution from the myocardium and from both endothelium- and nonendothelium-derived mesenchyme. The dorsal mesenchymal protrusion (DMP) is a structure arising from the venous pole of the heart that is critical to proper AV septation. Indeed, study of the Tc1 mouse model for DS has led to speculation that AVSD in DS results from an abnormality in the DMP.56,57 Although there is no established developmental role for our six genes harboring AVSD-associated variants in the development of the DMP, it is possible that they play unknown roles in the formation of that structure.
Table 5.
Relationship of AVSD Candidate Genes to VEGF-A
| Gene | Relationship |
|---|---|
| COL6A1 | expression is responsive to VEGF-A49 |
| COL6A2 | expression is responsive to VEGF-A49 |
| CRELD1 | modulates cell response to VEGF-A (C.L.M., unpublished data) |
| FBLN2 | downregulates VEFG-A50 |
| FRZB | upstream regulator of VEGF-A expression51 |
| GATA5 | transcriptional regulation of VEGF-A expression35 |
In contrast, other genes in our study have direct links to the DMP, which is derived from the second heart field (SHF). Our study includes five genes central to the SHF regulatory network: SHH, BMP4 (MIM 112262), GATA4, TBX1 (MIM 602054), and TBX20. None of these candidate genes had any potentially damaging variants in our study cohort, and, in general, they showed substantially less genetic variation than many of the other candidate genes. Of course, this does not rule out involvement of impaired DMP development in AVSD, and the absence of genetic variation in these genes suggests that alterations to their protein products might not be tolerated in DS.
We propose that most or all variants that show additive effects with trisomy 21 to interfere with heart development are the same variants that combine to produce AVSD in all individuals affected with this CHD. Our study demonstrates that the recurrent variants identified in 10% of cases are present as benign variants in the general population and are therefore available to confer risk when acting in concert with additional factors. The sensitized DS background provides an increased signal for these variants, which could be difficult to identify in euploid individuals with nonfamilial CHD. In this study, no one individual had more than one deleterious rare variant in any of these genes, which might reflect the very large effect of trisomy 21. The absence of the predisposing effect of trisomy 21 would require that a greater number of risk factors, each of smaller effect size, come together to produce the AVSD phenotype. This is supported by the significantly lower risk to euploid individuals, indicating that there is no single AVSD risk factor of large effect size in the euploid population. We anticipate that all six of the genes harboring AVSD-associated variants identified in this study will likewise be associated with AVSD in other cohorts, as has been seen for the recurrent CRELD1 missense mutation, p.Arg329Cys, which has been identified in nonsyndromic AVSD, DS-associated AVSD, and AVSD in heterotaxy syndrome (MIM 606217).
Discovery of AVSD-associated genetic variants in this study provides insight into the identity of other probable modifiers. We anticipate that additional mutations in VEGF-A pathway genes will eventually be implicated in the pathogenesis of AVSD. In addition, we envision that allelic interactions between the genes identified and variation in interacting proteins will have the potential to further modify risk but also to potentially influence phenotype and outcomes. This might be particularly true for AVSD, given that there is evidence that DS-associated AVSD, which tends to be a common AV canal balanced between the left and right ventricles, is anatomically distinct from most euploid AVSD.58 In addition, complete AVSD is more often an isolated cardiac malformation in DS than in euploid cases.59 It is possible that the presence of an extra copy of chromosome 21 drives development toward that specific outcome, and the presence of additional risk factors more greatly influences risk of abnormal heart development than the precise nature of the defect. Likewise, outcomes might be differentially affected depending on the presence or absence of trisomy 21. In one study, fetuses with DS and AVSD had a better survival rate than those with nonsyndromic AVSD, including a higher rate of successful surgical repair.60
Although there are many possible developmental routes to AVSD, as indicated by mouse models, the findings in this study suggest that the VEGF-A pathway could play a predominant role in the cause of AVSD in DS. However, candidate-gene studies are by nature biased; thus, additional studies are required to determine the global role of the VEGF-A pathway in AVSD. It is thanks to the families with DS that variations in these genes that contribute to the most frequent human congenital anomaly are coming to light.
Acknowledgments
This work was supported in part by NHLBI, R01HD083300 (R.H.R., S.L.S., and C.L.M.), T32 HL094294 (C.A.), the American Heart Association Predoctoral Fellowship Program (A.E.L.), the National Institute for Child Health and Development, F32 HD046337 (L.J.H.B.), the Western Undergraduate Exchange Program, the Portland State University Laurels Scholarship, the Rose Tucker Charitable Trust, the Throckmorton Scholarship Fund (K.E.), Children’s Healthcare of Atlanta Cardiac Research Committee, the National Center for Research Resources (NCRR), UL1RR024140 (OCTRI), and the technical assistance of the General Clinical Research Center at Emory University (National Institutes of Health/NCRR M01 RR00039). Resequencing services were provided by the University of Washington Department of Genome Sciences under U.S. Federal Government contract number N01-HV-48194 from the NHLBI. The authors thank Robert Glanville for critical reading of the manuscript.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
National Center for Biotechnology Information Short Genetic Variations database (dbSNP), http://www.ncbi.nlm.nih.gov/projects/SNP/
MutPred, http://mutpred.mutdb.org/
Online Mendelian Inheritance in Man (MIM), http://www.omim.org
References
- 1.Hoffman J.I., Kaplan S. The incidence of congenital heart disease. J. Am. Coll. Cardiol. 2002;39:1890–1900. doi: 10.1016/s0735-1097(02)01886-7. [DOI] [PubMed] [Google Scholar]
- 2.Srivastava D., Olson E.N. A genetic blueprint for cardiac development. Nature. 2000;407:221–226. doi: 10.1038/35025190. [DOI] [PubMed] [Google Scholar]
- 3.Posch M.G., Perrot A., Schmitt K., Mittelhaus S., Esenwein E.M., Stiller B., Geier C., Dietz R., Gessner R., Ozcelik C., Berger F. Mutations in GATA4, NKX2.5, CRELD1, and BMP4 are infrequently found in patients with congenital cardiac septal defects. Am. J. Med. Genet. A. 2008;146A:251–253. doi: 10.1002/ajmg.a.32042. [DOI] [PubMed] [Google Scholar]
- 4.Esposito G., Butler T.L., Blue G.M., Cole A.D., Sholler G.F., Kirk E.P., Grossfeld P., Perryman B.M., Harvey R.P., Winlaw D.S. Somatic mutations in NKX2–5, GATA4, and HAND1 are not a common cause of tetralogy of Fallot or hypoplastic left heart. Am. J. Med. Genet. A. 2011;155A:2416–2421. doi: 10.1002/ajmg.a.34187. [DOI] [PubMed] [Google Scholar]
- 5.Liu H., Dai L., Mao M., Wang X., Hua Y., Xie L. Absence of association between length variation of an intronic region in the NFATC1 gene and congenital heart defects in a Han Chinese population. DNA Cell Biol. 2012;31:88–91. doi: 10.1089/dna.2011.1286. [DOI] [PubMed] [Google Scholar]
- 6.Hartman R.J., Riehle-Colarusso T., Lin A., Frías J.L., Patel S.S., Duwe K., Correa A., Rasmussen S.A., National Birth Defects Prevention Study Descriptive study of nonsyndromic atrioventricular septal defects in the National Birth Defects Prevention Study, 1997-2005. Am. J. Med. Genet. A. 2011;155A:555–564. doi: 10.1002/ajmg.a.33874. [DOI] [PubMed] [Google Scholar]
- 7.Reller M.D., Strickland M.J., Riehle-Colarusso T., Mahle W.T., Correa A. Prevalence of congenital heart defects in metropolitan Atlanta, 1998-2005. J. Pediatr. 2008;153:807–813. doi: 10.1016/j.jpeds.2008.05.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ferencz C., Neill C.A., Boughman J.A., Rubin J.D., Brenner J.I., Perry L.W. Congenital cardiovascular malformations associated with chromosome abnormalities: an epidemiologic study. J. Pediatr. 1989;114:79–86. doi: 10.1016/s0022-3476(89)80605-5. [DOI] [PubMed] [Google Scholar]
- 9.Freeman S.B., Bean L.H., Allen E.G., Tinker S.W., Locke A.E., Druschel C., Hobbs C.A., Romitti P.A., Royle M.H., Torfs C.P. Ethnicity, sex, and the incidence of congenital heart defects: a report from the National Down Syndrome Project. Genet. Med. 2008;10:173–180. doi: 10.1097/GIM.0b013e3181634867. [DOI] [PubMed] [Google Scholar]
- 10.Sheffield V.C., Pierpont M.E., Nishimura D., Beck J.S., Burns T.L., Berg M.A., Stone E.M., Patil S.R., Lauer R.M. Identification of a complex congenital heart defect susceptibility locus by using DNA pooling and shared segment analysis. Hum. Mol. Genet. 1997;6:117–121. doi: 10.1093/hmg/6.1.117. [DOI] [PubMed] [Google Scholar]
- 11.Garg V., Kathiriya I.S., Barnes R., Schluterman M.K., King I.N., Butler C.A., Rothrock C.R., Eapen R.S., Hirayama-Yamada K., Joo K. GATA4 mutations cause human congenital heart defects and reveal an interaction with TBX5. Nature. 2003;424:443–447. doi: 10.1038/nature01827. [DOI] [PubMed] [Google Scholar]
- 12.Robinson S.W., Morris C.D., Goldmuntz E., Reller M.D., Jones M.A., Steiner R.D., Maslen C.L. Missense mutations in CRELD1 are associated with cardiac atrioventricular septal defects. Am. J. Hum. Genet. 2003;72:1047–1052. doi: 10.1086/374319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maslen C.L., Babcock D., Robinson S.W., Bean L.J., Dooley K.J., Willour V.L., Sherman S.L. CRELD1 mutations contribute to the occurrence of cardiac atrioventricular septal defects in Down syndrome. Am. J. Med. Genet. A. 2006;140:2501–2505. doi: 10.1002/ajmg.a.31494. [DOI] [PubMed] [Google Scholar]
- 14.Guo Y., Shen J., Yuan L., Li F., Wang J., Sun K. Novel CRELD1 gene mutations in patients with atrioventricular septal defect. World J. Pediatr. 2010;6:348–352. doi: 10.1007/s12519-010-0235-7. [DOI] [PubMed] [Google Scholar]
- 15.Kusuma L., Dinesh S.M., Savitha M.R., Krishnamurthy B., Narayanappa D., Ramachandra N.B. A maiden report on CRELD1 single-nucleotide polymorphism association in congenital heart disease patients of Mysore, South India. Genet Test Mol Biomarkers. 2011;15:483–487. doi: 10.1089/gtmb.2010.0246. [DOI] [PubMed] [Google Scholar]
- 16.Sarkozy A., Esposito G., Conti E., Digilio M.C., Marino B., Calabrò R., Pizzuti A., Dallapiccola B. CRELD1 and GATA4 gene analysis in patients with nonsyndromic atrioventricular canal defects. Am. J. Med. Genet. A. 2005;139:236–238. doi: 10.1002/ajmg.a.31018. [DOI] [PubMed] [Google Scholar]
- 17.Zatyka M., Priestley M., Ladusans E.J., Fryer A.E., Mason J., Latif F., Maher E.R. Analysis of CRELD1 as a candidate 3p25 atrioventicular septal defect locus (AVSD2) Clin. Genet. 2005;67:526–528. doi: 10.1111/j.1399-0004.2005.00435.x. [DOI] [PubMed] [Google Scholar]
- 18.Hirayama-Yamada K., Kamisago M., Akimoto K., Aotsuka H., Nakamura Y., Tomita H., Furutani M., Imamura S., Takao A., Nakazawa M., Matsuoka R. Phenotypes with GATA4 or NKX2.5 mutations in familial atrial septal defect. Am. J. Med. Genet. A. 2005;135:47–52. doi: 10.1002/ajmg.a.30684. [DOI] [PubMed] [Google Scholar]
- 19.Rajagopal S.K., Ma Q., Obler D., Shen J., Manichaikul A., Tomita-Mitchell A., Boardman K., Briggs C., Garg V., Srivastava D. Spectrum of heart disease associated with murine and human GATA4 mutation. J. Mol. Cell. Cardiol. 2007;43:677–685. doi: 10.1016/j.yjmcc.2007.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tomita-Mitchell A., Maslen C.L., Morris C.D., Garg V., Goldmuntz E. GATA4 sequence variants in patients with congenital heart disease. J. Med. Genet. 2007;44:779–783. doi: 10.1136/jmg.2007.052183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Joziasse I.C., Smith K.A., Chocron S., van Dinther M., Guryev V., van de Smagt J.J., Cuppen E., Ten Dijke P., Mulder B.J., Maslen C.L. ALK2 mutation in a patient with Down’s syndrome and a congenital heart defect. Eur. J. Hum. Genet. 2011;19:389–393. doi: 10.1038/ejhg.2010.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smith K.A., Joziasse I.C., Chocron S., van Dinther M., Guryev V., Verhoeven M.C., Rehmann H., van der Smagt J.J., Doevendans P.A., Cuppen E. Dominant-negative ALK2 allele associates with congenital heart defects. Circulation. 2009;119:3062–3069. doi: 10.1161/CIRCULATIONAHA.108.843714. [DOI] [PubMed] [Google Scholar]
- 23.Lagendijk A.K., Smith K.A., Bakkers J. Genetics of congenital heart defects: a candidate gene approach. Trends Cardiovasc. Med. 2010;20:124–128. doi: 10.1016/j.tcm.2010.10.003. [DOI] [PubMed] [Google Scholar]
- 24.Borozdin W., Bravo Ferrer Acosta A.M., Bamshad M.J., Botzenhart E.M., Froster U.G., Lemke J., Schinzel A., Spranger S., McGaughran J., Wand D. Expanding the spectrum of TBX5 mutations in Holt-Oram syndrome: detection of two intragenic deletions by quantitative real time PCR, and report of eight novel point mutations. Hum. Mutat. 2006;27:975–976. doi: 10.1002/humu.9449. [DOI] [PubMed] [Google Scholar]
- 25.Reamon-Buettner S.M., Borlak J. Somatic NKX2-5 mutations as a novel mechanism of disease in complex congenital heart disease. J. Med. Genet. 2004;41:684–690. doi: 10.1136/jmg.2003.017483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Reamon-Buettner S.M., Hecker H., Spanel-Borowski K., Craatz S., Kuenzel E., Borlak J. Novel NKX2-5 mutations in diseased heart tissues of patients with cardiac malformations. Am. J. Pathol. 2004;164:2117–2125. doi: 10.1016/S0002-9440(10)63770-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Draus J.M., Jr., Hauck M.A., Goetsch M., Austin E.H., 3rd, Tomita-Mitchell A., Mitchell M.E. Investigation of somatic NKX2-5 mutations in congenital heart disease. J. Med. Genet. 2009;46:115–122. doi: 10.1136/jmg.2008.060277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Salazar M., Consoli F., Villegas V., Caicedo V., Maddaloni V., Daniele P., Caianiello G., Pachón S., Nuñez F., Limongelli G. Search of somatic GATA4 and NKX2.5 gene mutations in sporadic septal heart defects. Eur. J. Med. Genet. 2011;54:306–309. doi: 10.1016/j.ejmg.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 29.Cooper G.M., Shendure J. Needles in stacks of needles: finding disease-causal variants in a wealth of genomic data. Nat. Rev. Genet. 2011;12:628–640. doi: 10.1038/nrg3046. [DOI] [PubMed] [Google Scholar]
- 30.Freeman S.B., Torfs C.P., Romitti P.A., Royle M.H., Druschel C., Hobbs C.A., Sherman S.L. Congenital gastrointestinal defects in Down syndrome: a report from the Atlanta and National Down Syndrome Projects. Clin. Genet. 2009;75:180–184. doi: 10.1111/j.1399-0004.2008.01110.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Locke A.E., Dooley K.J., Tinker S.W., Cheong S.Y., Feingold E., Allen E.G., Freeman S.B., Torfs C.P., Cua C.L., Epstein M.P. Variation in folate pathway genes contributes to risk of congenital heart defects among individuals with Down syndrome. Genet. Epidemiol. 2010;34:613–623. doi: 10.1002/gepi.20518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li B., Krishnan V.G., Mort M.E., Xin F., Kamati K.K., Cooper D.N., Mooney S.D., Radivojac P. Automated inference of molecular mechanisms of disease from amino acid substitutions. Bioinformatics. 2009;25:2744–2750. doi: 10.1093/bioinformatics/btp528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhao Y., Clark W.T., Mort M., Cooper D.N., Radivojac P., Mooney S.D. Prediction of functional regulatory SNPs in monogenic and complex disease. Hum. Mutat. 2011;32:1183–1190. doi: 10.1002/humu.21559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engel J., Prockop D.J. The zipper-like folding of collagen triple helices and the effects of mutations that disrupt the zipper. Annu. Rev. Biophys. Biophys. Chem. 1991;20:137–152. doi: 10.1146/annurev.bb.20.060191.001033. [DOI] [PubMed] [Google Scholar]
- 35.Reiter J.F., Alexander J., Rodaway A., Yelon D., Patient R., Holder N., Stainier D.Y. Gata5 is required for the development of the heart and endoderm in zebrafish. Genes Dev. 1999;13:2983–2995. doi: 10.1101/gad.13.22.2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Benson D.W., Martin L.J. Complex story of the genetic origins of pediatric heart disease. Circulation. 2010;121:1277–1279. doi: 10.1161/CIR.0b013e3181d98516. [DOI] [PubMed] [Google Scholar]
- 37.Thusberg J., Olatubosun A., Vihinen M. Performance of mutation pathogenicity prediction methods on missense variants. Hum. Mutat. 2011;32:358–368. doi: 10.1002/humu.21445. [DOI] [PubMed] [Google Scholar]
- 38.Baptista M.J., Fairbrother U.L., Howard C.M., Farrer M.J., Davies G.E., Trikka D., Maratou K., Redington A., Greve G., Njølstad P.R., Kessling A.M. Heterotrisomy, a significant contributing factor to ventricular septal defect associated with Down syndrome? Hum. Genet. 2000;107:476–482. doi: 10.1007/s004390000395. [DOI] [PubMed] [Google Scholar]
- 39.Davies G.E., Howard C.M., Farrer M.J., Coleman M.M., Bennett L.B., Cullen L.M., Wyse R.K., Burn J., Williamson R., Kessling A.M. Genetic variation in the COL6A1 region is associated with congenital heart defects in trisomy 21 (Down’s syndrome) Ann. Hum. Genet. 1995;59:253–269. doi: 10.1111/j.1469-1809.1995.tb00746.x. [DOI] [PubMed] [Google Scholar]
- 40.Davies G.E., Howard C.M., Farrer M.J., Coleman M.M., Cullen L.M., Williamson R., Wyse R.K., Kessling A.M. Unusual genotypes in the COL6A1 gene in parents of children with trisomy 21 and major congenital heart defects. Hum. Genet. 1994;93:443–446. doi: 10.1007/BF00201672. [DOI] [PubMed] [Google Scholar]
- 41.Davies G.E., Howard C.M., Gorman L.M., Farrer M.J., Holland A.J., Williamson R., Kessling A.M. Polymorphisms and linkage disequilibrium in the COL6A1 and COL6A2 gene cluster: novel DNA polymorphisms in the region of a candidate gene for congenital heart defects in Down’s syndrome. Hum. Genet. 1993;90:521–525. doi: 10.1007/BF00217452. [DOI] [PubMed] [Google Scholar]
- 42.Li H., Cherry S., Klinedinst D., DeLeon V., Redig J., Reshey B., Chin M.T., Sherman S.L., Maslen C.L., Reeves R.H. Genetic modifiers predisposing to congenital heart disease in the sensitized Down syndrome population. Circ. Cardiovasc. Genet. 2012;5:301–308. doi: 10.1161/CIRCGENETICS.111.960872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dor Y., Camenisch T.D., Itin A., Fishman G.I., McDonald J.A., Carmeliet P., Keshet E. A novel role for VEGF in endocardial cushion formation and its potential contribution to congenital heart defects. Development. 2001;128:1531–1538. doi: 10.1242/dev.128.9.1531. [DOI] [PubMed] [Google Scholar]
- 44.Kumar S.D., Yong S.K., Dheen S.T., Bay B.H., Tay S.S. Cardiac malformations are associated with altered expression of vascular endothelial growth factor and endothelial nitric oxide synthase genes in embryos of diabetic mice. Exp. Biol. Med. (Maywood) 2008;233:1421–1432. doi: 10.3181/0806-RM-186. [DOI] [PubMed] [Google Scholar]
- 45.Gittenberger-de Groot A.C., Bartram U., Oosthoek P.W., Bartelings M.M., Hogers B., Poelmann R.E., Jongewaard I.N., Klewer S.E. Collagen type VI expression during cardiac development and in human fetuses with trisomy 21. Anat. Rec. A Discov. Mol. Cell. Evol. Biol. 2003;275:1109–1116. doi: 10.1002/ar.a.10126. [DOI] [PubMed] [Google Scholar]
- 46.Grossman T.R., Gamliel A., Wessells R.J., Taghli-Lamallem O., Jepsen K., Ocorr K., Korenberg J.R., Peterson K.L., Rosenfeld M.G., Bodmer R., Bier E. Over-expression of DSCAM and COL6A2 cooperatively generates congenital heart defects. PLoS Genet. 2011;7:e1002344. doi: 10.1371/journal.pgen.1002344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Finsterer J., Stöllberger C. Primary myopathies and the heart. Scand. Cardiovasc. J. 2008;42:9–24. doi: 10.1080/14017430701854953. [DOI] [PubMed] [Google Scholar]
- 48.Lampe A.K., Bushby K.M. Collagen VI related muscle disorders. J. Med. Genet. 2005;42:673–685. doi: 10.1136/jmg.2002.002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Weston G.C., Haviv I., Rogers P.A. Microarray analysis of VEGF-responsive genes in myometrial endothelial cells. Mol. Hum. Reprod. 2002;8:855–863. doi: 10.1093/molehr/8.9.855. [DOI] [PubMed] [Google Scholar]
- 50.Law E.W., Cheung A.K., Kashuba V.I., Pavlova T.V., Zabarovsky E.R., Lung H.L., Cheng Y., Chua D., Lai-Wan Kwong D., Tsao S.W. Anti-angiogenic and tumor-suppressive roles of candidate tumor-suppressor gene, Fibulin-2, in nasopharyngeal carcinoma. Oncogene. 2012;31:728–738. doi: 10.1038/onc.2011.272. [DOI] [PubMed] [Google Scholar]
- 51.Person A.D., Garriock R.J., Krieg P.A., Runyan R.B., Klewer S.E. Frzb modulates Wnt-9a-mediated beta-catenin signaling during avian atrioventricular cardiac cushion development. Dev. Biol. 2005;278:35–48. doi: 10.1016/j.ydbio.2004.10.013. [DOI] [PubMed] [Google Scholar]
- 52.Armstrong E.J., Bischoff J. Heart valve development: endothelial cell signaling and differentiation. Circ. Res. 2004;95:459–470. doi: 10.1161/01.RES.0000141146.95728.da. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Lambrechts D., Carmeliet P. Sculpting heart valves with NFATc and VEGF. Cell. 2004;118:532–534. doi: 10.1016/j.cell.2004.08.022. [DOI] [PubMed] [Google Scholar]
- 54.Hallaq H., Pinter E., Enciso J., McGrath J., Zeiss C., Brueckner M., Madri J., Jacobs H.C., Wilson C.M., Vasavada H. A null mutation of Hhex results in abnormal cardiac development, defective vasculogenesis and elevated Vegfa levels. Development. 2004;131:5197–5209. doi: 10.1242/dev.01393. [DOI] [PubMed] [Google Scholar]
- 55.Montano M.M., Doughman Y.Q., Deng H., Chaplin L., Yang J., Wang N., Zhou Q., Ward N.L., Watanabe M. Mutation of the HEXIM1 gene results in defects during heart and vascular development partly through downregulation of vascular endothelial growth factor. Circ. Res. 2008;102:415–422. doi: 10.1161/CIRCRESAHA.107.157859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Dunlevy L., Bennett M., Slender A., Lana-Elola E., Tybulewicz V.L., Fisher E.M., Mohun T. Down’s syndrome-like cardiac developmental defects in embryos of the transchromosomic Tc1 mouse. Cardiovasc. Res. 2010;88:287–295. doi: 10.1093/cvr/cvq193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Benson D.W., Sund K.L. Looking down the atrioventricular canal. Cardiovasc. Res. 2010;88:205–206. doi: 10.1093/cvr/cvq302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mahle W.T., Shirali G.S., Anderson R.H. Echo-morphological correlates in patients with atrioventricular septal defect and common atrioventricular junction. Cardiol. Young. 2006;16(Suppl 3):43–51. doi: 10.1017/s1047951106000758. [DOI] [PubMed] [Google Scholar]
- 59.Friedberg M.K., Kim N., Silverman N.H. Atrioventricular septal defect recently diagnosed by fetal echocardiography: echocardiographic features, associated anomalies, and outcomes. Congenit. Heart Dis. 2007;2:110–114. doi: 10.1111/j.1747-0803.2007.00082.x. [DOI] [PubMed] [Google Scholar]
- 60.Berg C., Kaiser C., Bender F., Geipel A., Kohl T., Axt-Fliedner R., Krapp M., Knöpfle G., Herberg U., Breuer J. Atrioventricular septal defect in the fetus—associated conditions and outcome in 246 cases. Ultraschall Med. 2009;30:25–32. doi: 10.1055/s-2008-1027438. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.