

Abstract
Motile cilia are essential components of the mucociliary escalator and are central to respiratory-tract host defenses. Abnormalities in these evolutionarily conserved organelles cause primary ciliary dyskinesia (PCD). Despite recent strides characterizing the ciliome and sensory ciliopathies through exploration of the phenotype-genotype associations in model organisms, the genetic bases of most cases of PCD remain elusive. We identified nine related subjects with PCD from geographically dispersed Amish communities and performed exome sequencing of two affected individuals and their unaffected parents. A single autosomal-recessive nonsynonymous missense mutation was identified in HEATR2, an uncharacterized gene that belongs to a family not previously associated with ciliary assembly or function. Airway epithelial cells isolated from PCD-affected individuals had markedly reduced HEATR2 levels, absent dynein arms, and loss of ciliary beating. MicroRNA-mediated silencing of the orthologous gene in Chlamydomonas reinhardtii resulted in absent outer dynein arms, reduced flagellar beat frequency, and decreased cell velocity. These findings were recapitulated by small hairpin RNA-mediated knockdown of HEATR2 in airway epithelial cells from unaffected donors. Moreover, immunohistochemistry studies in human airway epithelial cells showed that HEATR2 was localized to the cytoplasm and not in cilia, which suggests a role in either dynein arm transport or assembly. The identification of HEATR2 contributes to the growing number of genes associated with PCD identified in both individuals and model organisms and shows that exome sequencing in family studies facilitates the discovery of novel disease-causing gene mutations.
Main Text
Dysfunction of cilia contributes to the pathophysiology of a growing number of syndromes that are collectively known as ciliopathies.1,2 Rapid progress in genetics has uncovered the basis of many rare syndromes involving the primary (sensory and nonmotile) cilium,2 but the genetic etiology of the prototypical disease of motor cilia, primary ciliary dyskinesia (PCD) (CILD1: MIM 244400), remains elusive for many individuals with PCD.1,3,4
PCD is a rare, genetically heterogeneous disorder that is usually inherited as an autosomal-recessive trait and typically presents with neonatal respiratory distress, sinopulmonary disease, otitis media, male infertility, and left-right laterality defects.3–6 Mutations in 14 genes are associated with PCD, but these account for fewer than half of all cases.3,4 Traditionally, genetic causes of PCD have been identified through sequencing of human homologs of genes discovered by screening for ciliary dysfunction in model organisms, including Chlamydomonas reinhardtii. Candidate genes found via this approach include proteins with roles in structural elements of the ciliary axoneme or in the preassembly of dynein arms.3,4,7–9 Once identified, a putative PCD-causing gene mutation is typically authenticated if it is a member of the ciliome, a collection of genes that code for proteins in the ciliary axoneme.10,11 However, recent identification of a cilia defect in the ortholog DNAAF3 (MIM 614566) in C. reinhardtii and zebrafish (Danio rerio) clearly showed that mutations in genes encoding nonciliary proteins also cause PCD.9
Amish communities present a unique population for genetic research owing to intermarriage, relative isolation, and detailed family recordkeeping.12,13 We identified nine related subjects (six males) with PCD in six subfamilies of geographically dispersed Amish communities (Figure 1A).14 Permission for research was obtained from the Washington University in St. Louis Human Research Protection Office (institutional review board). All subjects provided informed consent for diagnostic evaluation and genetic characterization. The diagnosis of PCD was considered on the basis of classic clinical features, which included chronic sinopulmonary disease, persistent neonatal hypoxemia, infertility, and laterality defects. Each subject had reduced nasal nitric oxide measurements and electron microscopy of the ciliary axonemes (Table 1) that showed absent outer dynein arms. In addition, most outer doublets lacked inner dynein arms, and a very small number (8.1%, range 7%–22%) assembled normal or truncated inner dynein arms. These data suggest a combined inner and outer dynein arm defect.
Figure 1.
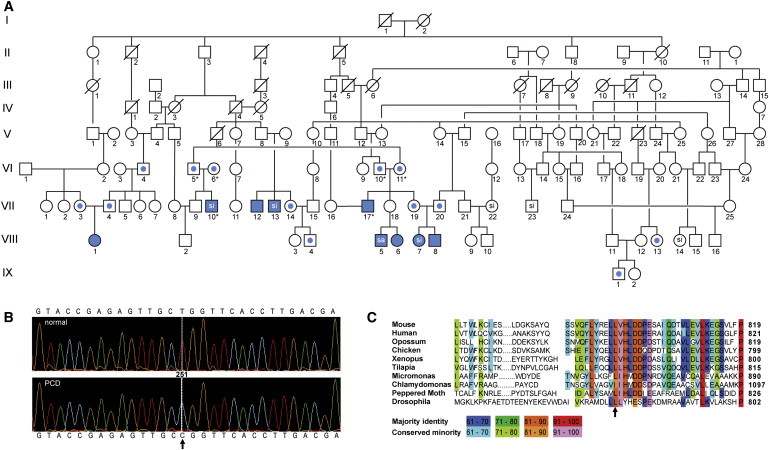
Family Pedigree and Genetic Analysis
(A) Pedigree of consanguineous kindred with related nuclear families in Amish communities from the midwestern United States. Solid symbols represent affected individuals; central dots represent heterozygous individuals. The following abbreviations are used: si, situs inversus totalis; sa, situs ambiguus. VIII-1 specifies the proband. Asterisks indicate subjects who underwent exome sequencing.
(B) Nucleotide sequences in affected HEATR2 region confirming exome sequencing results. HEATR2 mutation, NM_017802.3 (HEATR2): c.2384T>C, resulting in T-to-C transition at base position Chr7:819734 (hg19/GRCh37), is shown on the chromatograms for normal and PCD-affected subjects. Arrow indicates base change. This region does not have any reported nonsynonymous SNP or repeat elements. The HEATR2 variant allele segregated perfectly in the cohort and was homozygous mutant in all nine affected individuals.
(C) HEATR2 mutation leads to an amino acid change from leucine to proline, Leu795Pro, an amino acid conserved from humans to microalgae. Arrow indicates amino acid change.
Table 1.
Clinical Characteristics of Amish PCD-Affected Subjects with HEATR2 Mutant Alleles
| Subject | Age | Gender | Clinical Manifestations | Laterality | Ultrastructural Defect | Nasal NO (nl/min) |
|---|---|---|---|---|---|---|
| VII-12 | 36 | M | NRD, RS, CB, IN | SS | ODA-IDA | 1.2 |
| VII-13 | 40 | M | OM, RS, CB | SI | ODA-IDA | 1.6 |
| VII-10 | 20 | M | NRD, RS, CB, IN | SI | ODA-IDA | 4.2 |
| VII-17 | 28 | M | NRD, RS | SS | ODA-IDA | 3.6 |
| VIII-1 | 1.1 | F | NRD, RS | SS | ODA-IDA | 8.3 |
| VIII-7 | 8 | F | NRD, RS | SI | ODA-IDA | 11.6 |
| VIII-8 | 0.3 | M | NRD, RS | SI | ODA-IDA | 6.9 |
| VIII-6 | 0.3 | F | NRD, RS | SS | ODA-IDA | 7.5 |
| VIII-5 | 0.2 | M | CHD, NRD, OM | SA | ODA-IDA | 9.5 |
Normal nNO level > 100 nl/ml.
The following abbreviations are used: NO, nitric oxide; nl, nanoliter; M, male; F, female; NRD, neonatal respiratory distress or hypoxemia; RS, rhinosinusitus; CB, chronic bronchitis; IN, infertility; OM, chronic or recurrent otitis media; CHD, congenital heart disease; SS, situs solitus; SI, situs inversus totalis; SA, situs ambiguus; ODA, outer dynein arm; and IDA, inner dynein arm.
Sequencing of known candidate genes failed to identify a known PCD-causing mutation. With the use of GeneChip Genome-Wide Human SNP Array 6.0, DNA SNP analysis of nine affected and 13 unaffected family members identified homozygous regions on chromosome 12:88,373,816–88,974,238 and chromosome 13:48,516,791–49,056,026, which included candidate genes associated with ciliary or flagellar defects in humans and preclinical models, such as CEP290 (MIM 610142, centrosomal protein of 290 kDa), WDR51B (WD repeat-containing protein 51B), and RB1 (MIM 614041, retinoblastoma 1 tumor suppressor protein). We sequenced the exons for all genes within the regions of interest—C12orf50, C12orf29, CEP290, TMTC3, KITLG (MIM 184745), SUCLA2 (MIM 603921), NUDT15, MED4 (MIM 605718), ITM2B (MIM 117300), RB1, and LPAR6 (MIM 609239) —and no mutations were detected.
Exome capture and sequencing was performed with the Agilent SureSelect 38 Mb All Exon Hybridization Array on two affected children (VII-10 and VII-17), who were double first cousins, and their parents (VI-5, VI-6, VI-10, and VI-11) (Table 2). The SureSelect protocol was used to prepare libraries for paired-end sequencing (2 × 101 bp) on an Illumina HiSeq 2000 instrument. Exome sequencing identified an average of 15,508 total coding variants with greater than 5X coverage per exome, which were filtered against the 1000 Genomes Project and dbSNP build 132 for removal of known variants. Our filtering strategy only retained a variant if (1) it was not identified as a systematic artifact and (2) the variant was heterozygous in all four unaffected parents and homozygous in both affected offspring. We identified 56 sequence variants that segregated in an autosomal fashion, and from these, 55 variants were known and relatively common, with minor allele frequencies ranging from 4% to 69%. However, a rare, nonsynonymous variant in the HEATR2 also fit a model of autosomal-recessive inheritance (Table 2). This variant created a T-to-C transition on chromosome 7, base position 819734 (hg19/GRCh37), and changed a highly conserved amino acid in HEATR2 from leucine to proline (NM_017802.3 [HEATR2_i001]; p.Leu795Pro) (Figures 1B and 1C). Exome sequencing results were confirmed in the affected individuals, their parents, and a group of 27 related individuals that included seven additional PCD cases (Table S1 available online). Dideoxy sequencing of a 407 bp PCR fragment spanning chr7:819457–819863 used primers 5′TCCCGTGAGGTGTGGGTTTCTTAGG and 5′GGTCACACGGCTCAGCTACAGATGC. The HEATR2 variant allele (NM_017802.3 [HEATR2]: c.2384T>C) was homozygous in all nine affected individuals and heterozygous in the parents. These results were highly significant (Fisher's exact test, p = 2.6 × 10−8) for segregation of the variant allele in the 33 individuals genotyped. A search of the Exome Variant Server, which contains variants from 6,503 unrelated individuals including 4,300 European Americans, did not identify mutations in the HEATR2 sequence (chr7:819734). Moreover, the 1000 Genomes browser15 did not reveal variation at this position. Notably, despite genotype confirmation through additional resequencing and appropriate segregation of the HEATR2 variant, this region was not identified with homozygosity mapping,16 an approach that can have limitations when applied to inbred Amish kindred.17
Table 2.
Data Filtering Analysis
| Stepwise Filtering | VII-10 | VI-5 | VI-6 | VII-17 | VI-10 | VI-11 |
|---|---|---|---|---|---|---|
| Variants in coding regions with >5X coverage (n) | 14,936 | 14,969 | 16,280 | 15,418 | 15,735 | 15,707 |
| Variants after filtering against 1000 Genomes or dnSNP build 132 (n) | 1,196 | 1,477 | 1,560 | 1,638 | 1,745 | 1,738 |
| Variants after filtering against 51 exomes (n) | 892 | 1,061 | 909 | 1,121 | 1,230 | 1,228 |
| Variants that fit model (n) | 1 | 1 | 1 | 1 | 1 | 1 |
The total number of identified variants remaining after each step in the data filtering analysis is listed for each of the six exomes. Only one variant was novel, nonsynonymous, and heterozygous in all four parents and homozygous in both affected offspring.
HEATR2 is a member of a family of ten other uncharacterized HEAT-repeat-containing proteins in humans. The HEAT repeat is related to the armadillo/beta-catenin-like repeats and found in many other eukaryotic and prokaryotic proteins across a broad range of functional classes,18–21 including the four that give rise to the acronym HEAT (Huntingtin, elongation factor 3, PP2A, mTOR).22 None of the HEATR proteins have been previously linked to axonemal ultrastructure or have been related to other proteins associated with ciliary function. However, recent studies have implicated the HEAT-containing protein transportin (TNPO1, MIM 602901) in kinesin import into the cilium.23 Preliminary analyses showed that HEATR2 is a highly conserved gene and protein that is enriched in organisms with motile cilia and flagella (Figure 2A and Table S2).24 Furthermore, the amino acid modified by the observed variant is highly conserved in organisms with motile cilia and flagella (Figure 1C). We next sought to examine the function of its C. reinhardtii ortholog (Chlre4 gene model 525994; Phytozome v8.0 gene ID Cre09.g395500.t1). Artificial microRNA (amiRNA) directed to 20 bp sequences in the coding region25 were designed with the use of the Web MicroRNA Designer 3 website, cloned into pChlamiRNA3 vector,25 and verified via Sanger sequencing with established protocols.26 The inserts for hybridization were 5′CTAGTAAGTCAGTGGATGAGAAATGATCTCGCTGATCGGCACCATGGGGGTGGTGGTGATCAGCGCTATCATATCTCATCCACTGACTTG and 5′CTAGCAAGTCAGTGG ATGAGATATGATAGCGCTGATCACCACCACCCCCATGGTGCCGATCAGCGAGATCATTTCTCATCCACTGACTTA, which recognize bases 1441450–1441469 on chromosome 9 and are located in the terminal exon of the gene Cre09.g395500.t1. Two transformed strains with 8.8- and 4.6-fold reductions in mRNA levels in biological replicates were obtained. Flagella were isolated from cells grown on low-sulfur medium for 3 days in light and 2 days in the dark, then differentiated into gametes over 4 hr with the use of medium that lacks nitrogen, before deflagellation was induced through pH shock.27 Electron microscopy of htr2-silenced axonemes from these strains revealed missing outer dynein arms (Figure 2B) compared to wild-type C. reinhardtii axonemes. This ultrastructural defect was confirmed by immunoblot analyses of isolated silenced and wild-type flagella.29 The blots were incubated with primary antibodies ODA6 (DIC2) (1:1,000, a gift from D. Mitchell30), centrin (CNT1) (1:1,000, 20h5a, a gift from J. Salsibury31), IC138 (DIC4) (1:20,000, a gift from W. Sale32), and α-tubulin (1:1,000, DM1a, Sigma-Aldrich, St. Louis) for characterization of outer and inner dynein arm assembly. Antibodies were detected with enhanced chemiluminescnece,29 and band intensities from immunoblots were determined with the use of Photoshop software (Adobe Systems, San Jose, CA, USA), normalized, and reported as the fraction of α-tubulin. A markedly reduced level of the outer dynein arm intermediate chain, DIC2, was present compared to DIC4, a component of the inner dynein arm (Figure 2C). However, centrin, a protein found in a subset of inner dynein arms, was present and elevated in the amiRNA-silenced strain (Figure 2C). These findings are in line with the ultrastructural analysis by electron microscopy and suggest that htr2 plays a role in outer dynein arm assembly. To test this postulate, we assessed htr2 levels following deflagellation in wild-type cells. Expression of htr2 was increased 4.5-fold 20 min following deflagellation compared to pretreatment values, consistent with the well-known transcriptional upregulation of flagellar genes during cillogenesis.33
Figure 2.
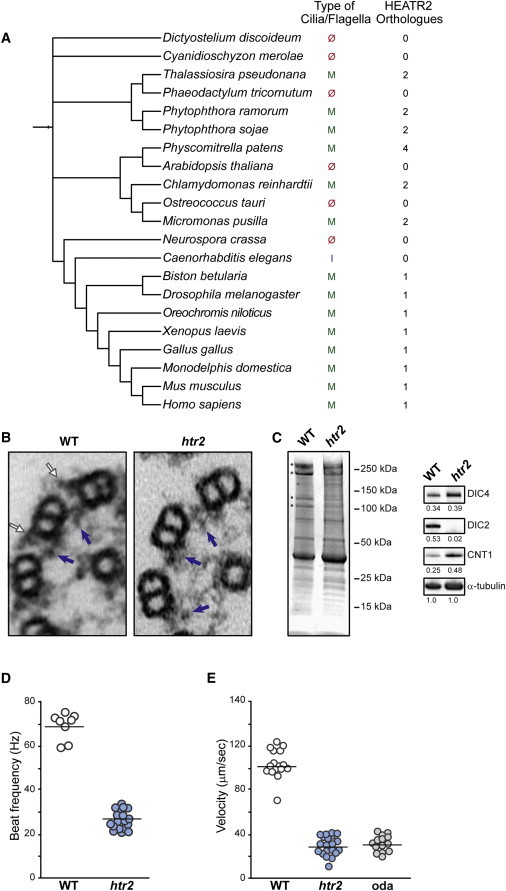
Ultrastructural and Functional Defects in Silenced C. reinhardtii
(A) Phylogenetic tree showing the relationship of HEATR2 among diverse organisms generated from iTOL v2.28 A BLAST search identified two to four proteins with similarity to HEATR2 in organisms that have motile cilia or flagella. In contrast, none of the 72 species that lack an orthologous HEATR2 have motile cilia or flagella, which suggests a conserved role for HEATR2-like proteins in the formation of motile cilia and flagella. The numeral on the right side of each species indicates the number of HEATR2-like proteins found in the relevant genome database.
(B) Ultrastructural appearance of outer microtubule doublets and dynein arms from C. reinhardtii flagella isolated from wild-type and amiRNA transformants with reduced HEATR2 mRNA. Outer dynein arms were absent in the transformant. Blue and white arrows indicate inner and outer dynein arms, respectively.
(C) Isolated axonemes from wild-type (WT) and HEATR2-silenced (htr2) strains were resolved by SDS-PAGE and stained with Coomassie blue. Protein molecular weights are shown on the right, and asterisks indicate bands with obvious differences between the preparations. The polypeptide at 250 kDa is probably a membrane protein that is often variable in axonemal preparations, whereas identities of polypeptides at 150 kDa are unknown. Immunoblots for structural proteins were normalized to α-tubulin, and values are indicated as fractions. DIC2 is a component of the outer dynein arms and is nearly absent in htr2 axonemes; however, DIC4 is a component of the inner dynein arms and is equivalent. CNT1 is a component of the inner dynein arms and is overrepresented in the axonemes from silenced strains, which may reflect their shorter length.
(D) Flagellar beat frequencies in wild-type uniflagellate (uni1-2) cells (WT) and uniflagellate double mutant (uni1-2:htr2) cells (htr2) (p < 0.0001).
(E) Swimming speeds of WT, htr2-silenced, and oda2 (oda, outer dynein arm mutant) cells (p < 0.0001).
To characterize the functional phenotype of the htr2-silenced strains, we performed motion analysis of swimming wild-type (CC-4430) and amiRNA transformant cells (amiRNA:htr2 and uni1-2;amiRNA:htr2 cells), using established methods.34,35 Swimming velocity was measured in biflagellate wild-type and htr2 algal cells, whereas rotation rate and beat frequency were measured in uniflagellate mutants (uni1-2) or double mutants (uni1-2;htr2). Rotation speed and beat frequency of uniflagellate cells were obtained from the mean slope and oscillation frequency of time series of angular displacement (Figure S1, Movies S1 and S2).35 Uniflagellate double mutant cells (uni1-2;htr2; n = 15) had a decreased beat frequency, 27.8 ± 3.8 Hz, and a slower rotation rate, 1.77 ± 0.63 revolutions per second (rps), compared to the htr2+ uniflagellate cells, at 69.7 ± 5.7 Hz (uni1-2; n = 8) (unpaired two-tailed Student's t test, p < 0.0001, Figure 2D) and 4.67 ± 0.57 rps, respectively (Figure S1). Moreover, biflagellate cells from silenced strains exhibited decreased cell velocities (29.7 ± 9.1 μm/s; n = 21 cells) compared to wild-type (103.7 ± 13.2 μm/s; n = 16 cells) (single-factor ANOVA, p < 0.0001, Figure 2E). These motion parameters in htr2-silenced cells corresponded closely to measured values in an oda2 mutant, which also has absent outer dynein arms.35
To elucidate the importance of HEATR2 in cilia assembly and its association with PCD, we evaluated the expression of HEATR2 in human multiciliated airway epithelial cells. Normal human lung, obtained from excess tissue of healthy lung donated for transplantation, was fixed and immunostained as previously described.29,36 Primary antibodies used included rabbit anti-HEATR2 (1:100, Sigma-Aldrich), mouse anti-acetylated α-tubulin (1:5,000, clone 6-11-B1, Sigma-Aldrich), and mouse anti-α-tubulin (1:4,000, clone B-5-1-2, Sigma-Aldrich) and were imaged with secondary antibodies conjugated to Alexa Fluor dyes (Life Technologies, Grand Island, NY, USA). Nuclei were stained with 1.5 μg/ml of DAPI (Vector Laboratories, Burlingame, CA, USA). Images were acquired with the use of epifluorescent microscopy with band-pass filter cubes optimized for the secondary dyes, interfaced with QCapture Pro software (QImaging, Surrey, BC, Canada), and adjusted globally with Photoshop software. HEATR2 was observed only in the cytoplasm of ciliated airway epithelial cells and was absent in cilia (Figures 3A and 3B). These findings are consistent with published proteomic analyses that did not include HEATR2 in cilia.10,11 Furthermore, within the cytoplasm, HEATR2 was observed in a diffuse punctate pattern (Figure 3B), which did not colocalize with endosomes, lysosomes, or basal bodies with the use of mouse anti-LAMP2 (1:200, Abcam, Cambridge, MA, USA), mouse anti-EEA1 (1:100, BD Biosciences, San Jose, CA, USA), and mouse anti-γ-tubulin (1:500, Clone Gtu-88, Sigma-Aldrich), respectively (Figure S2). The punctate pattern was also present in nasal epithelial cells recovered from healthy non-PCD-affected subjects (Figure 3C). To exclude secondary effects of chronic rhinosinusitis on HEATR2 levels in freshly collected samples from affected individuals,37 nasal epithelial cells were scraped from the inferior turbinate, expanded in culture, and then differentiated on semiporous supported membranes (Transwell, Corning, NY, USA) at an air-liquid interface.36 Preparations were maintained in culture for 4 to 10 weeks.36 Cultured nasal epithelial cells were harvested from membranes and immunostained as previously described.29,36 To assess HEATR2 levels in cultured cells, proteins were isolated and analyzed by immunoblot.29 The blots were incubated with the HEATR2 antibody (1:100 dilution) and detected with enhanced chemiluminescence.29 Cultured nasal epithelial cells from an individual with PCD (VIII-5 in Figure 1A) had markedly lower levels of HEATR2 compared to cells from non-PCD-affected normal subjects, as determined by both immunofluoresence and immunoblot analysis (Figures 3D and 3E). The actual fate of the mutant HEATR2 is unknown, though the Leu795Pro substitution is predicted to render the protein unstable.38 Prolonged immunoblot exposures showed that the size of the mutated HEATR2 was similar to that of the normal control (Figure S3).
Figure 3.
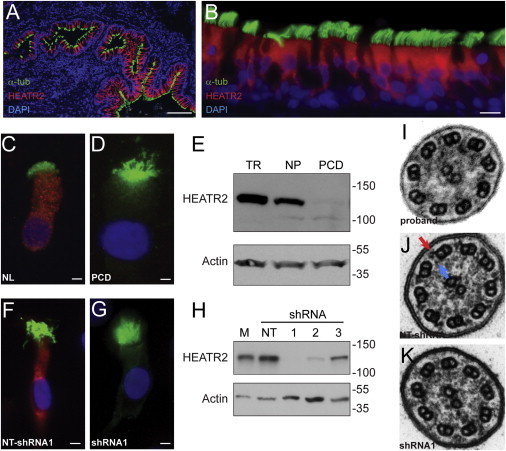
HEATR2 Localization in Ciliated Airway Epithelial Cells
(A and B) Photomicrographs of normal human lung section (scale bar = 100 μm) (A) and bronchial epithelium (B) following immunofluorescent staining for HEATR2, which reveals the cytoplasmic localization of the protein (HEATR2, red) only in ciliated cells (cilia marker, acetylated α-tubulin (α-tub), green; nuclei stained with DAPI, blue) (scale bar = 10 μm).
(C and D) Immunofluorescent staining of nasal epithelial cells cultured at an air-liquid interface from a healthy subject (NL) showing the presence of HEATR2 (C) and an individual with PCD showing a reduction of HEATR2 levels (D) (scale bar = 5 μm).
(E) Immunoblot analysis of tracheobronchial (TR) epithelial cells and nasal epithelia (NP) cells from a healthy subject and an individual with PCD (PCD) confirms the very low level of the mutant form of HEATR2.
(F and G) Immunoflourescent staining in representative human airway epithelial cells transfected with nontargeted, scrambled shRNA sequences (NT-shRNA) (F) and HEATR2-specific shRNA sequences (G) for HEATR2 (red) and acetylated α-tubulin (green) (scale bar = 5 μm).
(H) Immunoblot analyses of airway epithelial cells transfected with each of three different HEATR2-specific shRNA (1–3) or nontargeted shRNA (NT) sequences and nontransfected control cells (M).
(I–K) Ultrastructural appearance of cilium from the proband (I), and cilia from airway epithelial cells following transfection with (J) nontargeted and (K) HEATR2-targeted shRNA sequence 1. Blue and red arrows indicate inner and outer dynein arms, respectively.
We evaluated the effect of HEATR2 mutation on dynein arm function by examining cilia beat frequency. With a Nikon Ti-S inverted phase-contrast microscope enclosed in a customized environmental chamber maintained at 37°C, images were captured by a high-speed video camera and processed with the Sisson-Ammons Video Analysis system (Ammons Engineering, Mt. Morris, MI, USA).39 Five fields were captured for each sample, and whole fields were analyzed for generating a mean and SD. Simultaneous fluorescent images were obtained with a CCD Retiga-2000RV camera (QImaging) for determining cell viability through staining with calcein and ethidium homodimer-1 activity (Life Technologies). Live imaging showed that the nasal epithelial cells from a subject with a HEATR2 mutation had virtually immotile cilia, in contrast to those from healthy subjects (unpaired two-tailed Student's t test, p < 0.0001, n = 54 fields examined) (Figure S4, Movies S3 and S4).
To define the role of HEATR2 in differentiation, cilia assembly, and function of ciliated airway epithelial cells, we silenced HEATR2 expression with an RNAi approach in primary airway epithelial cells obtained from excess tracheal and bronchial tissue from lung donors. Human airway epithelial cells were transfected with nontargeted (scrambled) or HEATR2-specific small hairpin RNA (shRNA) sequences with the use of recombinant lentivirus that contained a cassette for conferring puromycin resistance. The shRNA-targeted sequences used were those generated by the RNAi Consortium (TRC) in the pLKO.1 hairpin RNA sequence transfer plasmid. shRNA sequences used were TRCN0000130727 (shRNA #1), TRCN0000129000 (shRNA #2), and TRCN0000146272 (shRNA #3) (obtained through Sigma-Aldrich). A nontargeted sequence with a yellow fluorescent protein reporter gene (gift from Y. Feng and G.D. Longmore, Washington University, St. Louis40) was used as a control sequence and for measuring transfection efficiency. For generating lentivirus for shRNA delivery, each vector was cotransfected with the pHR'8.2ΔR packaging and VSV-G envelope plasmids in HEK293T cells with the use of FuGENE 6 (Promega, Madison, WI, USA) according to the standard TRC protocol.41 Cultured, undifferentiated human airway epithelial cells (early passage) seeded onto supported Transwell membranes were incubated with medium containing freshly produced lentivirus with 10 μg/ml protamine sulfate for 24 hr. Cells were expanded for 4 to 6 days, then selected in puromycin (2.5 μg/ml) for an additional 5 days. Once confluent, an air-liquid interface condition was created to allow cell differentiation. HEATR2 expression was reproducibly inhibited by each of three different HEATR2-specific shRNA sequences in three independent preparations compared to cells transfected with the nontargeted shRNA sequence, as determined by immunoblot analysis and immunostaining (Figures 3F–3H). Each shRNA provided different degrees of HEATR2 inhibition; shRNA #1 and shRNA #2 were the most effective. Similar to the subject's nasal epithelial cells, cilia were present on the apical surface of preparations treated with each of the different shRNA sequences, which indicates that HEATR2 was not required for cilia formation (Figure S5), a finding that was analogous to our observations in the algal silenced strains. Moreover, the human airway epithelial cells had cilia with markedly reduced motility when assessed with high-speed video microscopy and compared to the preparations transfected with the control nontargeted sequences (unpaired two-tailed Student's t test, p < 0.0001, n = 54 fields) (Figure S5, Movies S5 and S6).39 Ultrastructural analysis of HEATR2 shRNA-silenced cells performed with the use of previously described protocols42 revealed absent outer dynein arms and truncated or possibly absent inner dynein arms (Figure 3K). These findings were similar to the axonemal defect observed in the proband (Figure 3I).
To better define the changes observed in the dynein arms, we immunostained ciliated cells with antibodies for DNAI1, an outer dynein arm polypeptide, using mouse anti-DNAI1 (1:5,000, gift from Lawrence Ostrowski, University of North Carolina, Chapel Hill, NC43), as well as DNAH7, an inner dynein arm polypeptide, using rabbit DNAH7 (1:50, Novus Biologicals, Littleton, CO, USA). DNAI1 was absent in ciliated airway epithelial cells from both the individual with PCD (Figure 4A) and HEATR2 shRNA-silenced cells (Figure S6). This lack of DNAI1 confirms the findings in the silenced C. reinhardtii and the ultrastructural studies in the human cells that the outer dynein arm does not assemble. However, the localization of the inner dynein arm protein DNAH7 to cilia was comparable in the normal and PCD airway epithelial cells (Figure 4B). This finding suggested that part or subsets of the inner dynein arms were still present, or that the protein was mislocalized elsewhere in the cilium. These data are consistent with our observation that silenced C. reinhardtii may have an abnormal distribution of inner dynein arms, reflected by altered centrin expression found in amiRNA-treated cells (Figure 2C).
Figure 4.
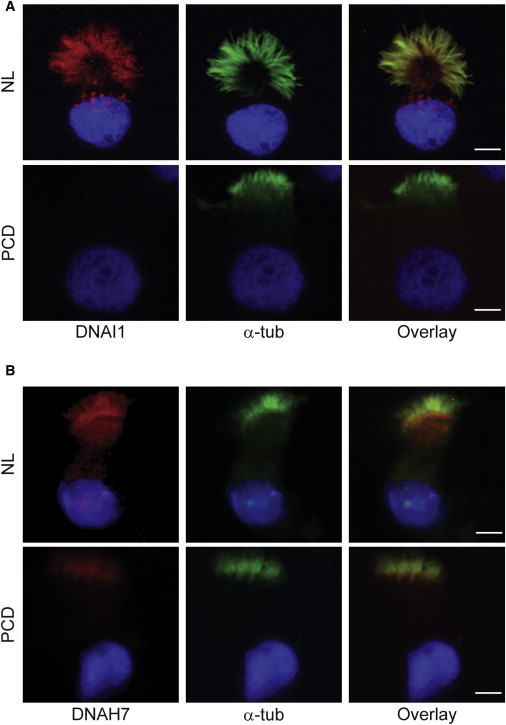
Expression and Localization of Specific Inner and Outer Dynein Arm Proteins in Ciliated Airway Epithelial Cells
(A) Immunofluorescent staining of nasal epithelial cells cultured at an air-liquid interface from a healthy subject (NL) showing the presence of outer dynein arm protein DNAI1 (red) and a subject with PCD (PCD) showing absence of HEATR2 expression.
(B) Expression of DNAH7 (red), an inner dynein arm protein, as seen in cultured nasal epithelial cells from an individual with PCD (PCD) and a healthy subject (NL). DNAH7 was expressed and localized to cilia in both normal and PCD airway epithelial cells. Cilia were identified by immunofluorescent staining with α-tubulin (green), and nuclei were stained with DAPI (blue). Scale bar = 5 μm.
By applying a whole-exome sequencing strategy to analyze the genome of several individuals with PCD from a large endogamous Amish family, we identified a previously uncharacterized gene, HEATR2, which encodes a highly conserved protein that is essential for the preassembly or stability of the axonemal dynein arms. Specifically, we found that the Leu795Pro substitution in HEATR2 reduced expression in ciliated airway epithelial cells in affected individuals to produce classic findings of PCD that include ultrastructural defects of the dynein arms and markedly dysmotile cilia. We confirmed that the function of HEATR2 is highly conserved by reproducing similar defects in dynein arm ultrastructure and markedly altered function in amiRNA-silenced C. reinhardtii flagella and human airway epithelial cilia. These studies and earlier proteomic analyses clearly establish that HEATR2 is not a structural protein in cilia and flagella, but its presence in the cytoplasm indicates a role in the preassembly of the dynein arms, similar to DNAAF1 (MIM 613190), DNAAF2 (MIM 612517), and DNAAF3, or in their transport to the basal bodies, such as ODA16.7–9 Significantly, the ciliary proteome44 did not predict HEATR2 involvement in ciliary biogenesis or assembly, and thus this tool has limitations for gene discovery in PCD. The precise mechanism by which HEATR2 interacts with other ciliary proteins and its involvement in motor dynein arm assembly remain unknown. Thus, HEATR2 joins the growing number of genes linked to PCD.
Acknowledgments
The authors thank Huawen Lin and Anne Sun for preliminary screens for HEATR2 amiRNA and Zhengyan Zhang for technical assistance with the C. reinhardtii silencing experiments. David Mitchell, Jeff Salsibury, Win Sale, Felix Y Feng, Greg Longmore, and Lawrence Ostrowski generously provided reagents for these studies. The authors also acknowledge Kimberly Burns, Wandy Beatty, and Marilyn Levy for assistance with electron microscopy; Milan Hazucha and Johnny Carson for their diagnostic expertise; Susan Minnix, Jane Quante, Kathryn Akers, Lu Huang, Michelle Farberman, Hauw Lie, Marianna Schmajuk, Adriana Lori, Michael Armstrong, and Hilda Metjian for their expert clinical and scientific support; and Cassie Mikols and Jian Xu from the Children's Discovery Institute Airway Epithelial Cell Core at Washington University School of Medicine for assistance with cell culture.
The authors were funded by the Children's Discovery Institute (T.E.D., P.V.B., S.L.B., S.K.D., and T.W.F.), the National Institutes of Health (NIH) awards HL082657 (T.W.F.), HL056244 (S.L.B.), GM32843 (S.K.D.), HL071798 (M.R.K., M.A.Z.), HL094976 (D.A.N., J.S.), HG005608 (D.A.N., J.S.), and HL083095 (A.C.P.), and the Ruth L. Kirschstein National Research Service Awards fellowship F32GM093598 (A.J.A.). In addition, this work was advanced by the Genetic Disorders of Mucociliary Clearance Consortium, HL096458 (M.A.Z., M.W.L., M.R.K., S.L.B., and T.W.F.), part of the NIH Rare Diseases Clinical Research Network, with programmatic support from the National Heart, Lung, and Blood Institute and the NIH Office of Rare Diseases Research. The views expressed do not necessarily reflect the official policies of the Department of Health and Human Services, nor does mention of trade names, commercial practices, or organizations imply endorsement by the U.S. government.
Supplemental Data
Web Resources
The URLs for the data presented herein are as follows:
1000 Genomes Browser, http://browser.1000genomes.org
Ciliary proteome database, http://v3.ciliaproteome.org/cgi-bin/index.php
Exome Variant Server, http://evs.gs.washington.edu/EVS
Interactive Tree of Life (iTOL) v2, http://itol.embl.de
MuStab for predicting mutant protein stability change, http://bioinfo.ggc.org/mustab
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Fliegauf M., Benzing T., Omran H. When cilia go bad: cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 2.Gerdes J.M., Davis E.E., Katsanis N. The vertebrate primary cilium in development, homeostasis, and disease. Cell. 2009;137:32–45. doi: 10.1016/j.cell.2009.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ferkol T.W., Leigh M.W. Ciliopathies: the central role of cilia in a spectrum of pediatric disorders. J. Pediatr. 2012;160:366–371. doi: 10.1016/j.jpeds.2011.11.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Leigh M.W., Pittman J.E., Carson J.L., Ferkol T.W., Dell S.D., Davis S.D., Knowles M.R., Zariwala M.A. Clinical and genetic aspects of primary ciliary dyskinesia/Kartagener syndrome. Genet. Med. 2009;11:473–487. doi: 10.1097/GIM.0b013e3181a53562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Afzelius B.A. A human syndrome caused by immotile cilia. Science. 1976;193:317–319. doi: 10.1126/science.1084576. [DOI] [PubMed] [Google Scholar]
- 6.Noone P.G., Leigh M.W., Sannuti A., Minnix S.L., Carson J.L., Hazucha M., Zariwala M.A., Knowles M.R. Primary ciliary dyskinesia: diagnostic and phenotypic features. Am. J. Respir. Crit. Care Med. 2004;169:459–467. doi: 10.1164/rccm.200303-365OC. [DOI] [PubMed] [Google Scholar]
- 7.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O'Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O'Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389. doi: 10.1038/ng.1106. S1–S2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Pazour G.J., Agrin N., Leszyk J., Witman G.B. Proteomic analysis of a eukaryotic cilium. J. Cell Biol. 2005;170:103–113. doi: 10.1083/jcb.200504008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ostrowski L.E., Blackburn K., Radde K.M., Moyer M.B., Schlatzer D.M., Moseley A., Boucher R.C. A proteomic analysis of human cilia: identification of novel components. Mol. Cell. Proteomics. 2002;1:451–465. doi: 10.1074/mcp.m200037-mcp200. [DOI] [PubMed] [Google Scholar]
- 12.Zariwala M.A., Omran H., Ferkol T.W. The emerging genetics of primary ciliary dyskinesia. Proc. Am. Thorac. Soc. 2011;8:430–433. doi: 10.1513/pats.201103-023SD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morton D.H., Morton C.S., Strauss K.A., Robinson D.L., Puffenberger E.G., Hendrickson C., Kelley R.I. Pediatric medicine and the genetic disorders of the Amish and Mennonite people of Pennsylvania. Am. J. Med. Genet. C. Semin. Med. Genet. 2003;121C:5–17. doi: 10.1002/ajmg.c.20002. [DOI] [PubMed] [Google Scholar]
- 14.Lie H., Zariwala M.A., Helms C., Bowcock A.M., Carson J.L., Brown D.E., 3rd, Hazucha M.J., Forsen J., Molter D., Knowles M.R. Primary ciliary dyskinesia in Amish communities. J. Pediatr. 2010;156:1023–1025. doi: 10.1016/j.jpeds.2010.01.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seelow D., Schuelke M. HomozygosityMapper2012—bridging the gap between homozygosity mapping and deep sequencing. Nucleic Acids Res. 2012;40(Web Server issue):W516–W520. doi: 10.1093/nar/gks487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pannain S., Weiss R.E., Jackson C.E., Dian D., Beck J.C., Sheffield V.C., Cox N., Refetoff S. Two different mutations in the thyroid peroxidase gene of a large inbred Amish kindred: power and limits of homozygosity mapping. J. Clin. Endocrinol. Metab. 1999;84:1061–1071. doi: 10.1210/jcem.84.3.5541. [DOI] [PubMed] [Google Scholar]
- 18.Andrade M.A., Petosa C., O'Donoghue S.I., Müller C.W., Bork P. Comparison of ARM and HEAT protein repeats. J. Mol. Biol. 2001;309:1–18. doi: 10.1006/jmbi.2001.4624. [DOI] [PubMed] [Google Scholar]
- 19.Neuwald A.F., Hirano T. HEAT repeats associated with condensins, cohesins, and other complexes involved in chromosome-related functions. Genome Res. 2000;10:1445–1452. doi: 10.1101/gr.147400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Park J.H., Aravind L., Wolff E.C., Kaevel J., Kim Y.S., Park M.H. Molecular cloning, expression, and structural prediction of deoxyhypusine hydroxylase: a HEAT-repeat-containing metalloenzyme. Proc. Natl. Acad. Sci. USA. 2006;103:51–56. doi: 10.1073/pnas.0509348102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Perry J., Kleckner N. The ATRs, ATMs, and TORs are giant HEAT repeat proteins. Cell. 2003;112:151–155. doi: 10.1016/s0092-8674(03)00033-3. [DOI] [PubMed] [Google Scholar]
- 22.Andrade M.A., Bork P. HEAT repeats in the Huntington's disease protein. Nat. Genet. 1995;11:115–116. doi: 10.1038/ng1095-115. [DOI] [PubMed] [Google Scholar]
- 23.Dishinger J.F., Kee H.L., Jenkins P.M., Fan S., Hurd T.W., Hammond J.W., Truong Y.N., Margolis B., Martens J.R., Verhey K.J. Ciliary entry of the kinesin-2 motor KIF17 is regulated by importin-beta2 and RanGTP. Nat. Cell Biol. 2010;12:703–710. doi: 10.1038/ncb2073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Powell S., Szklarczyk D., Trachana K., Roth A., Kuhn M., Muller J., Arnold R., Rattei T., Letunic I., Doerks T. eggNOG v3.0: orthologous groups covering 1133 organisms at 41 different taxonomic ranges. Nucleic Acids Res. 2012;40(Database issue):D284–D289. doi: 10.1093/nar/gkr1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Molnar A., Bassett A., Thuenemann E., Schwach F., Karkare S., Ossowski S., Weigel D., Baulcombe D. Highly specific gene silencing by artificial microRNAs in the unicellular alga Chlamydomonas reinhardtii. Plant J. 2009;58:165–174. doi: 10.1111/j.1365-313X.2008.03767.x. [DOI] [PubMed] [Google Scholar]
- 26.Lin H., Kwan A.L., Dutcher S.K. Synthesizing and salvaging NAD: lessons learned from Chlamydomonas reinhardtii. PLoS Genet. 2010;6:e1001105. doi: 10.1371/journal.pgen.1001105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Piperno G., Huang B., Luck D.J. Two-dimensional analysis of flagellar proteins from wild-type and paralyzed mutants of Chlamydomonas reinhardtii. Proc. Natl. Acad. Sci. USA. 1977;74:1600–1604. doi: 10.1073/pnas.74.4.1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Letunic I., Bork P. Interactive Tree Of Life v2: online annotation and display of phylogenetic trees made easy. Nucleic Acids Res. 2011;39(Web Server issue):W475–W478. doi: 10.1093/nar/gkr201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Pan J., You Y., Huang T., Brody S.L. RhoA-mediated apical actin enrichment is required for ciliogenesis and promoted by Foxj1. J. Cell Sci. 2007;120:1868–1876. doi: 10.1242/jcs.005306. [DOI] [PubMed] [Google Scholar]
- 30.Fowkes M.E., Mitchell D.R. The role of preassembled cytoplasmic complexes in assembly of flagellar dynein subunits. Mol. Biol. Cell. 1998;9:2337–2347. doi: 10.1091/mbc.9.9.2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Salisbury J.L., Baron A.T., Sanders M.A. The centrin-based cytoskeleton of Chlamydomonas reinhardtii: distribution in interphase and mitotic cells. J. Cell Biol. 1988;107:635–641. doi: 10.1083/jcb.107.2.635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hendrickson T.W., Perrone C.A., Griffin P., Wuichet K., Mueller J., Yang P., Porter M.E., Sale W.S. IC138 is a WD-repeat dynein intermediate chain required for light chain assembly and regulation of flagellar bending. Mol. Biol. Cell. 2004;15:5431–5442. doi: 10.1091/mbc.E04-08-0694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dutcher S.K. Flagellar assembly in two hundred and fifty easy-to-follow steps. Trends Genet. 1995;11:398–404. doi: 10.1016/s0168-9525(00)89123-4. [DOI] [PubMed] [Google Scholar]
- 34.Bayly P.V., Lewis B.L., Kemp P.S., Pless R.B., Dutcher S.K. Efficient spatiotemporal analysis of the flagellar waveform of Chlamydomonas reinhardtii. Cytoskeleton (Hoboken) 2010;67:56–69. doi: 10.1002/cm.20424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bayly P.V., Lewis B.L., Ranz E.C., Okamoto R.J., Pless R.B., Dutcher S.K. Propulsive forces on the flagellum during locomotion of Chlamydomonas reinhardtii. Biophys. J. 2011;100:2716–2725. doi: 10.1016/j.bpj.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.You Y., Richer E.J., Huang T., Brody S.L. Growth and differentiation of mouse tracheal epithelial cells: selection of a proliferative population. Am. J. Physiol. Lung Cell. Mol. Physiol. 2002;283:L1315–L1321. doi: 10.1152/ajplung.00169.2002. [DOI] [PubMed] [Google Scholar]
- 37.Al-Rawi M.M., Edelstein D.R., Erlandson R.A. Changes in nasal epithelium in patients with severe chronic sinusitis: a clinicopathologic and electron microscopic study. Laryngoscope. 1998;108:1816–1823. doi: 10.1097/00005537-199812000-00010. [DOI] [PubMed] [Google Scholar]
- 38.Teng S., Srivastava A.K., Wang L. Sequence feature-based prediction of protein stability changes upon amino acid substitutions. BMC Genomics. 2010;11(Suppl 2):S5. doi: 10.1186/1471-2164-11-S2-S5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sisson J.H., Stoner J.A., Ammons B.A., Wyatt T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 40.Feng Y., Nie L., Thakur M.D., Su Q., Chi Z., Zhao Y., Longmore G.D. A multifunctional lentiviral-based gene knockdown with concurrent rescue that controls for off-target effects of RNAi. Genomics Proteomics Bioinformatics. 2010;8:238–245. doi: 10.1016/S1672-0229(10)60025-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stewart S.A., Dykxhoorn D.M., Palliser D., Mizuno H., Yu E.Y., An D.S., Sabatini D.M., Chen I.S., Hahn W.C., Sharp P.A. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA. 2003;9:493–501. doi: 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Knowles M.R., Leigh M.W., Carson J.L., Davis S.D., Dell S.D., Ferkol T.W., Olivier K.N., Sagel S.D., Rosenfeld M., Burns K.A., Genetic Disorders of Mucociliary Clearance Consortium Mutations of DNAH11 in patients with primary ciliary dyskinesia with normal ciliary ultrastructure. Thorax. 2012;67:433–441. doi: 10.1136/thoraxjnl-2011-200301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ostrowski L.E., Yin W., Rogers T.D., Busalacchi K.B., Chua M., O'Neal W.K., Grubb B.R. Conditional deletion of dnaic1 in a murine model of primary ciliary dyskinesia causes chronic rhinosinusitis. Am. J. Respir. Cell Mol. Biol. 2010;43:55–63. doi: 10.1165/rcmb.2009-0118OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gherman A., Davis E.E., Katsanis N. The ciliary proteome database: an integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.