

Abstract
Punctate palmoplantar keratodermas (PPKPs) are rare autosomal-dominant inherited skin diseases that are characterized by multiple hyperkeratotic plaques distributed on the palms and soles. To date, two different loci in chromosomal regions 15q22-15q24 and 8q24.13-8q24.21 have been reported. Pathogenic mutations, however, have yet to be identified. In order to elucidate the genetic cause of PPKP type Buschke-Fischer-Brauer (PPKP1), we performed exome sequencing in five affected individuals from three families, and we identified in chromosomal region 15q22.33-q23 two heterozygous nonsense mutations—c.370C>T (p.Arg124∗) and c.481C>T (p.Arg161∗)—in AAGAB in all affected individuals. Using immunoblot analysis, we showed that both mutations result in premature termination of translation and truncated protein products. Analyses of mRNA of affected individuals revealed that the disease allele is either not detectable or only detectable at low levels. To assess the consequences of the mutations in skin, we performed immunofluorescence analyses. Notably, the amount of granular staining in the keratinocytes of affected individuals was lower in the cytoplasm but higher around the nucleus than it was in the keratinocytes of control individuals. AAGAB encodes the alpha-and gamma-adaptin-binding protein p34 and might play a role in membrane traffic as a chaperone. The identification of mutations, along with the results from additional studies, defines the genetic basis of PPKP1 and provides evidence that AAGAB plays an important role in skin integrity.
Main Text
Palmoplantar keratodermas (PPKs) are familiar skin diseases characterized by epidermal hyperkeratosis of the palms and soles.1,2 Punctate palmoplantar keratoderma (PPKP) type I (PPKP1 [MIM 148600]), also called keratosis punctate palmoplantaris type Buschke-Fischer-Brauer, is a rare hereditary skin disease of the palms and soles and is characterized by multiple hyperkeratotic papules and central indentation irregularly distributed on the palms and soles (other palmoplantar keratoses have mostly diffuse hyperkeratinization). It was first described in 1910 by Buschke, Fischer,3 and Brauer.4 The inheritance pattern is mostly autosomal dominant, but simplex cases have also been described.5 Its incidence has not been extensively evaluated, but it is estimated as 1.17 per 100,000 individuals in Croatia.6 In mechanically irritated areas, confluent plaques can be found.5 The interfamilial and intrafamilial severity of the clinical picture shows broad variation. These lesions usually start to develop in early adolescence but also later in life.5 There have been reports in which PPK was associated with the development of both early- and late-onset malignancies, as well as squamous cell carcinomas.7–9
PPKP has previously been mapped to chromosomal region 15q22-15q24 for PPKP1 (MIM 148600)10 and region 8q24.13-8q24.21 for PPKP3 (MIM 101850),11 but the molecular basis of PPKP1 has not been identified yet. Recently, four different studies reported refinements of the critical region on chromosome 15 (Figure 1).12–15
Figure 1.
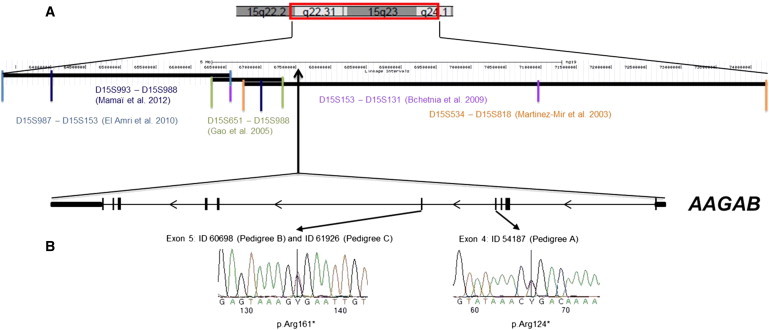
Linkage Intervals and Mutation Analysis of AAGAB
(A) In the upper panel, part of chromosome 15 shows the identified linkage intervals and the corresponding flanking microsatellites.10–15 The genomic positions of these markers are indicated in Mb. AAGAB resides within the region that was found by Martinez-Mir et al. (2003)10 and Bchetnia et al. (2009).13 In the lower panel is the genomic structure of AAGAB; ten exons are indicated as black bars. The noncoding parts of exons 1 and 10 are indicated with smaller bars.
(B) In the left panel is the c.481C>T transition in exon 5 from individual 60698 (pedigree B) and individual 61926 (pedigree C). In the right panel is the c.370C>T transition in exon 4 from individual 54187 (pedigree A).
Here, we describe the identification of pathogenic mutations in AAGAB, which encodes the alpha-and gamma-adaptin-binding protein p34. In addition, expression studies support the pathogenic role of AAGAB in the disease.
We identified three families composed of a total of 38 individuals (14 affected and 24 unaffected) available for further investigation. The disease seemed to be inherited in an autosomal-dominant mode. Two families are of Croatian origin, and one family is of German origin. All family members gave informed consent before participating, and experiments were performed under the approval of the institutional review board at the Ludwig Maximilians University of Munich; this approval also included adherence to the Declaration of Helsinki principles. Blood samples were taken from all 38 individuals. For establishing the phenotype, the palms and soles of all individuals participating in this study were clinically assessed. Keratoses on the soles were more severe than those on the palms, especially over pressure points (Figure 2). Histology by hematoxylin eosin (HE) staining showed a marked hyperkeratosis with focal parakeratosis and prominent hypergranulosis, which is consistent with PPKP1 (Figure S1B, available online). Phenotypic variability could be observed between, as well as within, families. To identify the genetic cause of PPKP1, we used exome-sequencing technology. We selected three cousins (54213, 54187, and 62727) from pedigree A and two index persons from pedigrees B (60698) and C (61926) for sequencing under the assumption that any gene harboring rare variants in all members of the three pedigrees might be a candidate for PPKP1. To minimize the proportion of alleles shared by descent, we selected distantly related members of pedigree A.
Figure 2.
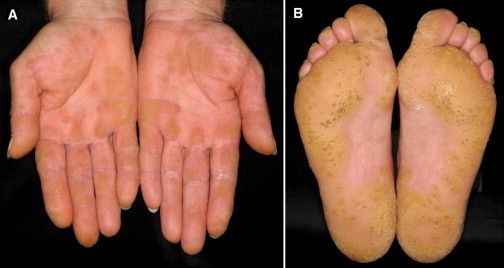
Clinical Picture
Disseminated hyperkeratotic papules on palms (A) and soles (B) of a 57-year-old index person of pedigree B.
Sequencing was performed as 100 bp paired-end runs on HiSeq2000 systems after in-solution enrichment of exonic sequences (SureSelect XT Human All Exon 50 Mb kit, Agilent). Read alignment was performed with the Burrows-Wheeler Aligner (v.0.5.8), and SAMtools (v.0.1.7) was used for detecting single-nucleotide variants and small insertions and deletions. We produced an average of 10.5 Gb of mappable sequence per sample, resulting in a mean read depth of 112, and greater than 88% of the target regions were covered at least 20 times. Variants were filtered for excluding those present in dbSNP132 and with an average heterozygosity greater than 0.02 and those present in more than 4 of 1,400 control exomes from individuals with other unrelated diseases.
This approach left only a single gene, AAGAB, which harbors a heterozygous nonsynonymous variant in all five affected individuals. The three investigated members of pedigree A showed at nucleotide position 370 a cytosine-to-thymine transition (c.370C>T) leading to a stop codon at amino acid position 124 (p.Arg124∗). In both pedigrees B and C, we identified at nucleotide position 481 a different nonsense mutation (c.481C>T) also leading to a stop codon at amino acid position 161 (p.Arg161∗) (Figure 1).
The variants were confirmed by Sanger sequencing and analyzed for cosegregation with the phenotype in all three pedigrees. Primer sequences and conditions for amplification for each variant are available upon request. All affected family members were found to show the mutation, whereas none of the unaffected individuals showed the mutation (Figure 3). We therefore concluded that the variants in AAGAB were indeed very likely to be causative for PPKP1 in pedigrees A, B, and C.
Figure 3.
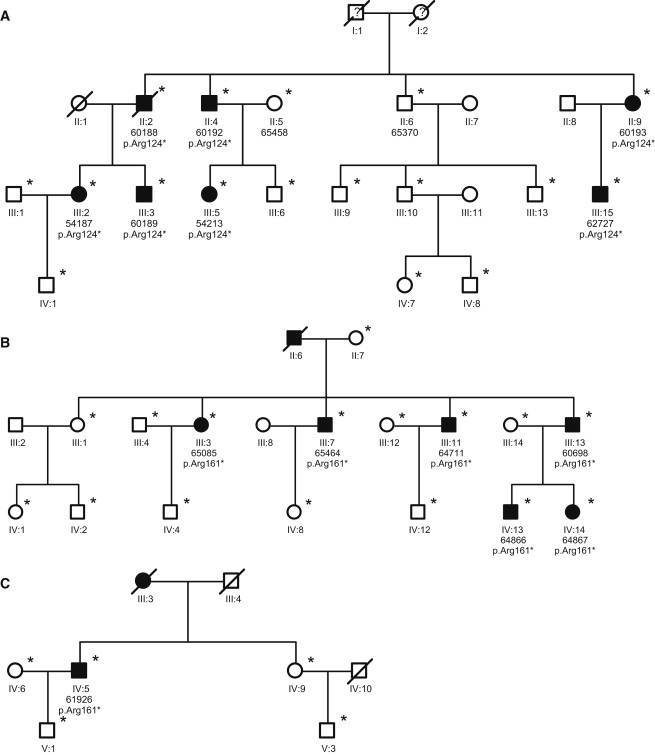
Pedigrees of Families A, B, and C
Pedigrees of families A (A), B (B), and C (C). Unaffected and affected family members are indicated by clear symbols and blackened symbols, respectively. Asterisks denote individuals genotyped for the variants in AAGAB. To maintain confidentiality, we have not shown genotypes of unaffected individuals. Diagonal bars through the symbols denote deceased individuals. Question marks in the symbols denote lacking information about the phenotype.
To determine whether variant c.481C>T, which was present in the affected members of pedigrees B and C, occurred on the same haplotype, we genotyped 21 individuals from both pedigrees by using oligonucleotide SNP arrays (HumanOmni2.5-Quad, Illumina). Haplotyping was performed with Merlin software.16 The haplotypes showing the c.481C>T variant are depicted in Table S1. The pedigrees shared a common haplotype across 1.2 Mb between markers SNP15-67469128 and SNP15-68728632. The fact that families B and C are of Croatian origin indicates that the affected individuals of family B and family C inherited the mutation by descent from a common ancestor.
AAGAB consists of ten exons with a coding sequence of 945 nucleotides. AAGAB has been described to be ubiquitously expressed by array analysis. We specifically showed by RT-PCR its expression in skin and an immortalized keratinocyte cell line (HaCaT cells), which is affected in PPKP1, supporting a role for AAGAB in the pathogenesis of PPKP1 (Figure S2).
Alpha-and gamma-adaptin-binding protein p34, whose subunit interacts with alpha and gamma adaptins might play a role in membrane trafficking. This interaction was mapped to these adaptins’ NH2-terminal domains, where the alpha and gamma adaptins show their highest homology; however, they are only 32% identical even at these domains. Another clue to the function of p34 comes from the observation that it can be coimmunoprecipitated with soluble adaptor complexes, both AP-1 and AP-2, although it is not enriched in purified clathrin-coated vesicles. This suggests that p34 might play a chaperone or chaperone-like role. Thus, it could act to prevent adaptors from coassembling with soluble clathrin or to participate in uncoating processes by supporting the removal of adaptors from the coated vesicles. p34 might also aid the recruitment of soluble adaptors onto the membrane. However, it is unlikely to be involved in the specificity of adaptor recruitment because it appears to interact equally well with both adaptor complexes.17
To assess the consequences of the mutations identified in AAGAB in the skin, we performed immunofluorescence analyses on the skin of affected and unaffected individuals, immunoblotting, and allele-specific PCR amplification.
For immunofluorescence analyses, normal human skin from the sole of the foot from four volunteers was used as a control. Biopsies of affected and unaffected areas of palm and sole skin were obtained from three affected individuals (54187, 60698, and 61926) from pedigrees A, B, and C. All was done with institutional approval and adherence to the Declaration of Helsinki from the Department of Dermatology at Ludwig Maximilians University Munich and Medical Faculty of the University of Heidelberg. Immediately after excision, tissues were snap frozen in liquid nitrogen and stored at −70°C.18
For immunofluorescence staining of AAGAB in normal and affected skin, we used three polyclonal antibodies raised against peptides of the unique AAGAB amino acid sequence composed of positions 126–214 (NBP1-87906), 224–300 (NBP1-87905), and the total recombinant protein (B01P) (Supplemental Data). In principle, all antisera showed staining patterns comparable to those from mice (B01P; used for all experiments) that give the strongest labeling (data shown).
In the normal palmoplantar skin used as a control, AAGAB showed granular staining in the cytoplasm, particularly of the stratum basale and the first suprabasal cell layers of the stratum spinosum. In upper layers of the stratum spinosum, cytoplasmic staining is increasingly lost and only detectable around a few keratinocyte nuclei (Figure S3A). Regularly, dermal vessels (“v” in Figures S3A–S3C) are also AAGAB-positive in all specimens.
From the affected persons, we investigated palm and sole sections of both affected (Figures S3B–S3B″) and unaffected (Figure S3C) skin areas. In affected regions, the cytoplasmic labeling was widely reduced and only rarely seen in the keratinocytes of the stratum basale and the lowermost stratum spinosum. The granular staining of nuclei was strongly enhanced and seen in the lower and middle layers of the stratum spinosum (Figures S3B–S3B″). This granular staining was clearly perinuclear and was best seen at higher magnifications (Figures S3B′–S3B″). Cytoplasmic and perinuclear granular staining have been observed previously.18–22 In these cases, such staining patterns were caused by keratin clumping of mutated and consequently defected keratin molecules.
The staining patterns of unaffected areas of affected persons (Figure S3C) were not significantly different than those of affected areas (Figure S3B), although the histology of these areas shows considerably lower stratification and cornification (Figure S1C). The fact that not all of the skin of the palms and soles is affected, although the mutated protein occurs in every cell, is not surprising because these observations have also been seen in other genetic skin diseases, such as focal ichthyoses or hyperkeratoses caused by keratin mutations.23–25
To further assess the consequences of the mutations, we performed immunoblot analyses. The nonsense mutations (c.370C>T and c.481C>T) identified in the three families are predicted to result in premature termination codons and truncated proteins. To examine both the wild-type and the truncated AAGAB, we cloned Taq polymerase-amplified PCR products in a eukaryotic expression vector by using the pcDNA3.1/V5-His TOPO TA Expression Kit (Invitrogen, Paisley, UK). The sequences of the primers used for cloning and sequencing of the respective constructs are available upon request. The protocols used for the cloning, cell culture, and immunoblot analysis have been described previously.26 COS7 cells were transiently transfected with AAGAB constructs, and cell lysates were obtained 30 hr after transfection. Protein blots (Figure 4A) stained with a monoclonal V5 antibody (Invitrogen, Paisley, UK) showed a signal of around 40 kDa (34 kDa without the tag), which corresponds to the size anticipated for the tagged wild-type protein. The tagged truncated proteins showed bands in their predicted size ranges of around 18 and 24 kDa (13.5 and 18 kDa, respectively, without the tag). The strong band for p.Arg161∗ indicates that the protein is stable and not degraded (Figure 4A). The faint band for p.Arg124∗ might indicate that the corresponding mRNA is mostly prone to mRNA decay (Figure 4A).
Figure 4.
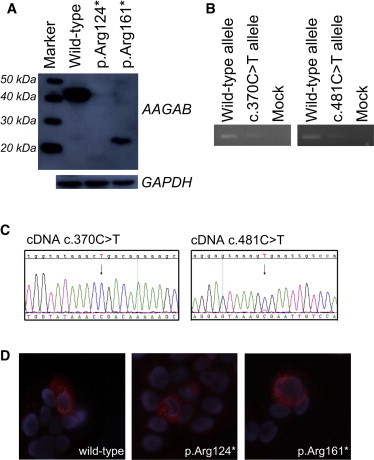
Immunoblot and Immuofluorescence Analysis of COS7 Cells Transiently Expressing Wild-Type and Mutant AAGAB and Allele-Specific PCR
(A) COS7 cells were transiently transfected with V5-tagged AAGAB wild-type or mutant (p.Arg124∗ or p.Arg161∗) constructs with the use of Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions and were harvested 30 hr after transfection. After lysis of cells in ripa buffer and sonication, lysates were subjected to electrophoresis and immunoblotting. We used monoclonal V5 antibody (R960-25, Invitrogen, 1:4.000) to detect AAGAB proteins. For normalization, we used a monoclonal GAPDH antibody (G8795, Sigma-Aldrich, 1:16.000). The wild-type AAGAB has a size of around 35 kDa, whereas the two truncated proteins have the anticipated sizes of 14 kDa and 18 kDa. However, we observe slightly larger bands, which might be due to the added tag and to glycosylation or phosphorylation of the protein.
(B) Allele-specific PCR was performed with cDNA that was generated from RNA extracted from the blood of affected individuals. We used the same forward primer (5′-ATGATGCTGTGAGATTTTATCCC-3′) for all reactions. The reverse primers used for studying AAGAB expression in individual 54187 were 5′-CACCATTCTTGAGCTTTTTGGCG-3′ for amplifying the wild-type allele and 5′-CACCATTCTTGAGCTTTTTGGCA-3′ for amplifying the mutant c.370C>T allele. The reverse primers used for studying AAGAB expression in individual 60698 were 5′- CATTCAGGGCTTGGACAATGCG-3′ for amplifying the wild-type allele and 5′-CATTCAGGGCTTGGACAATGCA-3′ for amplifying the c.481C>T mutant allele. We readily observed the wild-type allele in both cases, whereas the mutant allele was only present at reduced levels.
(C) Sequencing results of cDNA samples of affected individuals with gene-specific AAGAB primers. We could only detect a very small peak for the c.481C>T allele (arrow on the right), and we could not detect the c.370C>T allele at all (arrow on the left), indicating no presence of the respective mutant transcripts.
(D) Immunofluorescence analysis of transiently transfected HaCaT cells with the above-mentioned V5 antibody. No differences in staining were observed between cells expressing the wild-type (left) or the mutant (center = mutant p.Arg124∗; and right = mutant p.Arg161∗) AAGAB.
Because the immunoblot analysis is an overexpression assay and does not necessarily mirror the in vivo situation in affected individuals, we performed allele-specific PCR amplification of cDNA of affected individuals (Figure 4B) and obtained an mRNA transcript amount comparable to that in protein analyses.
To further test the possibility of mRNA decay, we also sequenced the respective cDNA samples of affected individuals (54187 and 60698) with gene-specific AAGAB primers. We could only detect a very small peak for the c.481C>T allele, whereas the allele showing the c.370C>T mutation was undetectable, indicating no presence of the respective mutant transcripts and thus the loss of the respective proteins (Figure 4C).
We then performed immunofluorescence analyses to determine the subcellular localization of the AAGAB proteins. Human embryonic kidney (HEK) 293 or HaCaT cells (an immortal human keratinocyte cell line) were transiently transfected with AAGAB wild-type or mutant constructs with the use of Lipofectamine 2000 (Invitrogen, Paisley, UK). Cells were fixed in ice-cold methanol acetone 1:1 for 5 min and then subjected to immunostaining with a V5 antibody. No differences in staining were observed between cells expressing the wild-type or the mutant AAGAB. Both proteins showed a normal distribution pattern in the cytoplasm (Figure 4D). However, a strong difference in the transfection efficiency between wild-type and mutants was obvious.
In summary, we identified two heterozygous nonsense mutations—c.370C>T (p.Arg124∗) and c.481C>T (p.Arg161∗)—in AAGAB. Analysis of mRNA of affected individuals revealed that the disease allele is either not detectable or only detectable at low levels, suggesting haploinsufficiency as a possible disease-causing mechanism. Interestingly, the granular staining that was observed in the cytoplasm in the skin of control individuals was widely reduced in the skin of affected individuals. AAGAB encodes the alpha-and gamma-adaptin-binding protein p34 and might play a role in membrane trafficking as a chaperone. Our findings define the genetic basis of PPKP and provide evidence that AAGAB has an important role in skin integrity.
AAGAB, whose function was nearly completely unknown, has thus been identified in processes of the skin, and its role as a putative chaperone has to be studied in the future.
Acknowledgments
The authors thank the individuals and their families for their participation in this study. We also thank R. Feldmann for technical assistance and J. Schweizer for helpful discussion. The work was supported by grants of the Wilhelm Sander-Stiftung, Munich (to L.L.), a scholarship for Excellence of the BGF (to K.G.), the German Ministry of Education and Research (01GS08160 and 01GR0802), and the European Commission 7th Framework Program (261123, GEUVADIS). R.C.B. is recipient of a Heisenberg Professorship from the German Research Foundation.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
ExonPrimer, http://ihg2.helmholtz-muenchen.de/ihg/ExonPrimer.html
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
UCSC Human Genome Browser, http://genome.cse.ucsc.edu/
References
- 1.Kelsell D.P., Stevens H.P. The palmoplantar keratodermas: Much more than palms and soles. Mol. Med. Today. 1999;5:107–113. doi: 10.1016/s1357-4310(98)01428-2. [DOI] [PubMed] [Google Scholar]
- 2.Braun-Falco M. Hereditary palmoplantar keratodermas. J. Dtsch. Dermatol. Ges. 2009;7:971–984. doi: 10.1111/j.1610-0387.2009.07058.x. quiz 984–985. [DOI] [PubMed] [Google Scholar]
- 3.Buschke A., Fischer W. Keratodermia mauculosa disseminate symmetrica palmaris and plantaris. Ikonographia Dermatologica. 1910;5l:183–192. [Google Scholar]
- 4.Brauer A. Über eine besondere Form des hereditären Keratoms (keratoderma disseminatum hereditarium palmare et plantare) Arch. Dermatol. Syph. 1913;114:211–236. [Google Scholar]
- 5.Emmert S., Küster W., Hennies H.C., Zutt M., Haenssle H., Kretschmer L., Neumann C. 47 individuals in 14 families with the rare genodermatosis keratosis puncatata palmopantaris Buschke-Fischer-Brauer. Eur. J. Dermatol. 2003;1:16–20. [PubMed] [Google Scholar]
- 6.Stanimirović A., Kansky A., Basta-Juzbasić A., Skerlev M., Beck T. Hereditary palmoplantar keratoderma, type papulosa, in Croatia. J. Am. Acad. Dermatol. 1993;29:435–437. doi: 10.1016/0190-9622(93)70207-a. [DOI] [PubMed] [Google Scholar]
- 7.Ena P., Cottoni F., Cerimele D., Saccabusi S., Retanda G. [Association of keratoderma punctata palmaris et plantaris with other morbid conditions (early grayness, carcinoma of the colon). Study of 3 families] G. Ital. Dermatol. Venereol. 1986;121:45–54. [PubMed] [Google Scholar]
- 8.Bennion S.D., Patterson J.W. Keratosis punctata palmaris et plantaris and adenocarcinoma of the colon. A possible familial association of punctate keratoderma and gastrointestinal malignancy. J. Am. Acad. Dermatol. 1984;10:587–591. doi: 10.1016/s0190-9622(84)80262-5. [DOI] [PubMed] [Google Scholar]
- 9.Stevens H.P., Kelsell D.P., Leigh I.M., Ostlere L.S., MacDermot K.D., Rustin M.H. Punctate palmoplantar keratoderma and malignancy in a four-generation family. Br. J. Dermatol. 1996;134:720–726. [PubMed] [Google Scholar]
- 10.Martinez-Mir A., Zlotogorski A., Londono D., Gordon D., Grunn A., Uribe E., Horev L., Ruiz I.M., Davalos N.O., Alayan O. Identification of a locus for type I punctate palmoplantar keratoderma on chromosome 15q22-q24. J. Med. Genet. 2003;40:872–878. doi: 10.1136/jmg.40.12.872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang X.J., Li M., Gao T.W., He P.P., Wei S.C., Liu J.B., Li C.R., Cui Y., Yang S., Yuan W.T. Identification of a locus for punctate palmoplantar keratodermas at chromosome 8q24.13-8q24.21. J. Invest. Dermatol. 2004;122:1121–1125. doi: 10.1111/j.0022-202X.2004.22507.x. [DOI] [PubMed] [Google Scholar]
- 12.Gao M., Yang S., Li M., Yan K.L., Jiang Y.X., Cui Y., Xiao F.L., Shen Y.J., Chen J.J., Liu J.B. Refined localization of a punctate palmoplantar keratoderma gene to a 5.06-cM region at 15q22.2-15q22.31. Br. J. Dermatol. 2005;152:874–878. doi: 10.1111/j.1365-2133.2005.06488.x. [DOI] [PubMed] [Google Scholar]
- 13.Bchetnia M., Charfeddine C., Kassar S., Hanchi I., Tounsi-Guettiti H., Rebai A., Osman A.D., Kubisch C., Abdelhak S., Boubaker S., Mokni M. Clinical, histological and genetic investigation of Buschke-Fischer-Brauer’s disease in Tunisian families. J. Dermatol. Sci. 2009;54:54–56. doi: 10.1016/j.jdermsci.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 14.El Amri I., Mamai O., Ghariani N., Denguezli M., Sriha B., Adala L., Saad A., Gribaa M., Nouira R. Étude clinique et génétique de la kératodermie palmopantaire de Buschke-Fischer-Brauer dans une famille tunisienne. Ann. Derm. Vénérol. 2010;137:269–275. doi: 10.1016/j.annder.2010.02.002. [DOI] [PubMed] [Google Scholar]
- 15.Mamaï O., Boussofara L., Adala L., Amara A., Ben Charfeddine I., Ghariani N., Sriha B., Denguezli M., Mili A., Belazreg T. Reduction of palmoplantar keratoderma Buschke-Fischer-Brauer locus to only 0.967Mb. J. Dermatol. Sci. 2012;67:210–212. doi: 10.1016/j.jdermsci.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 16.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 17.Page L.J., Sowerby P.J., Lui W.W.Y., Robinson M.S. Gamma-synergin: An EH domain-containing protein that interacts with gamma-adaptin. J. Cell Biol. 1999;146:993–1004. doi: 10.1083/jcb.146.5.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Uitto J., Richard G., McGrath J.A. Diseases of epidermal keratins and their linker proteins. Exp. Cell Res. 2007;313:1995–2009. doi: 10.1016/j.yexcr.2007.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lane E.B., McLean W.H.I. Keratins and skin disorders. J. Pathol. 2004;204:355–366. doi: 10.1002/path.1643. [DOI] [PubMed] [Google Scholar]
- 20.Joh G.Y., Traupe H., Metze D., Nashan D., Huber M., Hohl D., Longley M.A., Rothnagel J.A., Roop D.R. A novel dinucleotide mutation in keratin 10 in the annular epidermolytic ichthyosis variant of bullous congenital ichthyosiform erythroderma. J. Invest. Dermatol. 1997;108:357–361. doi: 10.1111/1523-1747.ep12286491. [DOI] [PubMed] [Google Scholar]
- 21.Math A., Frank J., Handisurya A., Poblete-Gutiérrez P., Slupetzky K., Födinger D., Winter D., Stingl G., Kirnbauer R. Identification of a de novo keratin 1 mutation in epidermolytic hyperkeratosis with palmoplantar involvement. Eur. J. Dermatol. 2006;16:507–510. [PubMed] [Google Scholar]
- 22.Bergman R., Khamaysi Z., Sprecher E. A unique pattern of dyskeratosis characterizes epidermolytic hyperkeratosis and epidermolytic palmoplantar keratoderma. Am. J. Dermatopathol. 2008;30:101–105. doi: 10.1097/DAD.0b013e3181614898. [DOI] [PubMed] [Google Scholar]
- 23.Korge B.P., Krieg T. The molecular basis for inherited bullous diseases. J. Mol. Med. 1996;74:59–70. doi: 10.1007/BF00196781. [DOI] [PubMed] [Google Scholar]
- 24.Corden L.D., McLean W.H. Human keratin diseases: Hereditary fragility of specific epithelial tissues. Exp. Dermatol. 1996;5:297–307. doi: 10.1111/j.1600-0625.1996.tb00133.x. [DOI] [PubMed] [Google Scholar]
- 25.Dereure O. [Keratin k6c mutations in focal palmoplantar keratoderma] Ann. Dermatol. Venereol. 2010;137:423–424. doi: 10.1016/j.annder.2010.03.018. [DOI] [PubMed] [Google Scholar]
- 26.Pasternack S.M., von Kügelgen I., Al Aboud K., Lee Y.A., Rüschendorf F., Voss K., Hillmer A.M., Molderings G.J., Franz T., Ramirez A. G protein-coupled receptor P2Y5 and its ligand LPA are involved in maintenance of human hair growth. Nat. Genet. 2008;40:329–334. doi: 10.1038/ng.84. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.