

Abstract
CaBPs are a family of Ca2+-binding proteins related to calmodulin and are localized in the brain and sensory organs, including the retina and cochlea. Although their physiological roles are not yet fully elucidated, CaBPs modulate Ca2+ signaling through effectors such as voltage-gated Cav Ca2+ channels. In this study, we identified a splice-site mutation (c.637+1G>T) in Ca2+-binding protein 2 (CABP2) in three consanguineous Iranian families affected by moderate-to-severe hearing loss. This mutation, most likely a founder mutation, probably leads to skipping of exon 6 and premature truncation of the protein (p.Phe164Serfs∗4). Compared with wild-type CaBP2, the truncated CaBP2 showed altered Ca2+ binding in isothermal titration calorimetry and less potent regulation of Cav1.3 Ca2+ channels. We show that genetic defects in CABP2 cause moderate-to-severe sensorineural hearing impairment. The mutation might cause a hypofunctional CaBP2 defective in Ca2+ sensing and effector regulation in the inner ear.
Introduction
Hearing loss is a common sensory disorder that can significantly impact quality of life. The estimated incidence of congenital hearing loss in developed countries is 1 in 650–1,000, and the majority of cases are attributable to genetic factors.1 Genetic hearing loss can be part of a syndrome or the only detectable disease manifestation (nonsyndromic). Autosomal-recessive nonsyndromic hearing loss (arNSHL) is the most common type of inherited hearing loss and is genetically heterogeneous; 61 genes and >100 loci have been reported (Hereditary Hearing Loss Homepage). The fact that genetic hearing loss is exceptionally heterogeneous can be explained by the complexity of the auditory system, which involves coordination of multiple processes involving different parts of the ear and nervous system. A defect in any part of this complex chain of events can lead to hearing impairment or deafness.
Most families segregating arNSHL typically have a prelingual, bilateral, severe-to-profound hearing loss. An exception is found with mutations in TECTA (MIM 602574) and STRC (MIM 606440); these mutations cause prelingual moderate-to-severe hearing loss that is usually most pronounced in the mid-frequency range.2–4 Recently, we identified a locus associated with arNSHL on 11q12.3-11q13.3 (DFNB93) in an Iranian family that also presents a similar moderate-to-severe hearing-loss phenotype.5 This is the third arNSHL-associated locus associated with a milder phenotype.
CaBP2 is a member of a subfamily of Ca2+ binding proteins with high similarity to calmodulin (CALM1).6 Like calmodulin, CaBP family members interact with and regulate effectors such as voltage-gated Cav channels in a Ca2+-dependent manner.7,8 In the cochlea, Ca2+ influx through Cav1.3 (L-type) Ca2+ channels into hair cells triggers synaptic transmission to spiral ganglion neurons that convey the auditory information to the brain.9,10 Ca2+-binding proteins (CABPs) localized in hair cells might modulate presynaptic Ca2+ influx, e.g., by antagonizing calmodulin-mediated Ca2+-dependent inactivation (CDI) of Cav1.3 channels.11,12 Here, we report that a mutation in CABP2 (MIM 607314) is the cause of DFNB93 moderate-to-severe hearing loss and reveal a role for CaBP2 in the mammalian auditory system.
Material and Methods
Clinical Investigation
Pure-tone audiograms and speech-reception thresholds were obtained from all individuals. Pure-tone audiometry was performed with air conduction at 250, 500, 1,000, 2,000, 4,000 and 8,000 Hz, and bone conduction was performed at 250, 500, 1,000, 2,000, and 4,000 Hz. In addition, transient-evoked otoacoustic emissions (TEOAEs) were recorded in all affected individuals and click-evoked broadband (100–3,000 Hz) auditory brainstem response (ABR) measurements were done on a subset of affected individuals. All affected members underwent comprehensive ophthalmological examinations, including a visual acuity test, a color vision test, cover test, retinoscopy, autorefractors, slit-lamp examination, pupil dilation, and a visual field test. To investigate cardiovascular problems, we recorded electrocardiograms and echocardiographs on most of the affected individuals.
All Iranian families and 100 Iranian controls were ascertained by the Cellular and Molecular Research Center, Shahrekord, Iran. The three families affected by mutations in CABP2 are of Iranian ethnicity. Families Sh10 and Sh11 live in the Fars province in the south of Iran, and the He family is from the Chaharmahal va Bakhtiari province in the southwest of Iran. All medical examinations of the Sh10, Sh11, and He families were performed in a local clinical center.
An additional 24 unrelated Belgian and Dutch individuals who show a specific moderate-to-severe hearing-loss profile were collected through the Centre of Medical Genetics, Antwerp and Radboud University Nijmegen Medical Centre.
Fourteen unrelated families affected by Usher syndrome were collected through the Radboud University Medical Centre. Five families were diagnosed with Usher syndrome type I (USH1 [MIM 276900]), five families were diagnosed with Usher syndrome type II (USH2 [MIM 276901]), and four families were diagnosed with atypical Usher syndrome. Per family, DNA was available from the parents, one to three affected individuals, and one to three unaffected individuals. These families could so far not be explained by two mutated alleles of the nine known genes in which mutations cause Usher syndrome; these genes are MYO7A (MIM 276903), USH1C (MIM 605242), CDH23 (MIM 605516), PCDH15 (MIM 605514), USH1G (MIM 607696), USH2A (MIM 608400), GPR98 (MIM 602851), DFNB31 (MIM 607928), and CLRN1 (MIM 606397).
This study was carried out according to the guidelines of the ethical committees of the University of Antwerp and Shahrekord University of Medical Sciences. Informed consent was obtained from all individuals.
Target Enrichment and Next-Generation Sequencing
We developed a custom SureSelect DNA capture array (1M) (Agilent Technologies, Santa Clara, USA) that contains all gene exons and noncoding RNAs in the complete 9.6 Mb DFNB93 region. Gene exons and noncoding RNA coordinates were selected on the basis of the Ensembl genome database (hg19/GRCh37 draft).5 In addition, we added 1,000 bp to the 5′ UTR of each gene and 50 bp to the 5′ end and 20 bp to the 3′ end of each exon to include the promoter region and to detect possible splice-site mutations. We then filtered the final selected region (1.3 Mb) for repeats (UCSC RepeatMasker) to exclude the development of probes that capture nonspecific regions in the genome. The custom DNA capture array was designed with a 20× probe tiling.
DNA from affected member V:14 of the DFNB93-affected family (Sh10; Figure 1) was fragmented with a Covaris S220 instrument (Covaris, Woburn, USA). Subsequently, the sample was prepared for sequencing according to the SureSelect DNA Capture Array protocol (Agilent Technologies, Santa Clara, USA). In short, universal adaptor nucleotides were ligated, and the library was amplified before hybridization to the array (65°C for 65 hr). Hybridized molecules were eluted, amplified, and quantified with quantitative PCR (qPCR) and diluted to a final concentration of 10 nM. Enriched DNA fragments were then sequenced on an Illumina Genome Analyzer, GAIIx (Illumina, San Diego, USA) with the use of 1× 72 bp reads.
Figure 1.
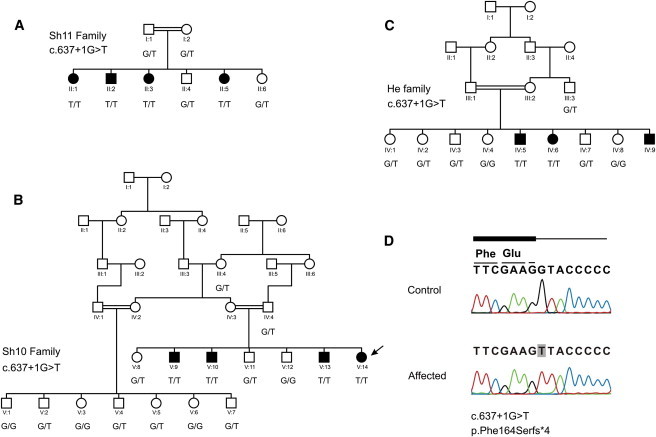
Pedigrees
(A–C) Pedigrees and segregation of the c.637+1G>T (p.Phe164Serfs∗4) mutation in Iranian families Sh11 (A), Sh10 (B), and He (C).
(D) Sequencing results of the c.637+1G>T mutation. The affected individual sequenced with next-generation sequencing is indicated with an arrow. Nomenclature was based on NM_016366.2 and NP_057450.2.
Data Analysis
NextGENe (SoftGenetics, State College, USA), a workbench for computational analysis of genomic data, was used for quality control, data manipulation, alignment to the human genome, and detection of indels and variants. Because sequencing revealed a great number of homozygous variants, we next used the program ANNOVAR13 to perform a stepwise filtering approach to screen the identified variants in order to select those likely to be implicated in the disorder. First, variants present in the 1000 Genome Project Full Project Phase 1 (November 2010 release) or in dbSNP131 were excluded. Second, variants were annotated and screened for nonsynonymous variants, nonsense mutations, frameshift mutations, splice-site mutations, and coding indels.
Sanger DNA Sequencing
Primers surrounding possibly damaging variants identified by next-generation sequencing and surrounding all exons of CABP2 were designed with primer3 software.14 PCR was carried out under standard conditions, and direct sequencing of the PCR product was performed on an ABI 3130xl sequencer (Applied Biosystems, Foster City, USA).
Genotyping
Linkage to the DFNB93 region and refinement of the region were done by genotyping microsatellite markers and SNPs. Information for microsatellite markers was taken from the NCBI STS database. Markers CABP2M4, CABP2M5, and CABP2M8 are not in this database and were developed on the basis of the human genome sequence (NCBI build 37.2) (primers are available upon request). In short, PCR was carried out under standard conditions. PCR products were fluorescently labeled, and capillary electrophoresis and pattern visualization were performed with an ABI 3130xl automatic DNA sequencer (Applied Biosystems, Foster City, USA). SNPs were genotyped by direct sequencing of the PCR product as described in the previous section.
Exon Trapping
Using genomic DNA from an affected and control individual, we used PCR to amplify a fragment containing 105 bp of CABP2 intron 5, 148 bp of CABP2 exon 6, and 622 bp of CABP2 intron 6 (with wild-type [WT] and mutant [MT] 5′ donor sites), and we cloned it into the pSPL3 exon trapping vector (Life technologies, Carlsbad, USA) by using BamHI and EcoRI restriction enzymes (Fermentas, St. Leon-Rot, Germany). COS-7 cells were seeded in 60 mm dishes and transfected with 2 μg plasmid DNA (WT, MT, and empty pSPL3) by the use of FuGENE 6 transfection reagent (Roche, Basel, Switzerland). All transfections were performed in duplicates. Total RNA was extracted 24 hr after transfection with the use of Trizol reagent (Invitrogen, Carlsbad, USA), RNA was DNase treated with the DNA-free kit (Ambion, Austin, USA), and cDNA was made by means of reverse transcriptase with the SA2 primers and Superscript III Reverse Transcriptase (Invitrogen, Carlsbad, USA). Subsequently, PCR was used for amplifying cDNA with pSPL3 primers SD6 and SA2 and was followed by a nested PCR reaction with dUSD2 and dUSA4 primers (Life technologies, Carlsbad, USA). Products were visualized with agarose-gel electrophoresis. Bands were cut out and purified by the QiaEX gel extraction kit II (QIAGEN, Hilden, Germany) before sequencing. Sequencing was performed on an ABI 3130xl sequencer (Applied Biosystems, Foster City, USA). The results were confirmed by repeated independent PCR and sequencing reactions.
CaBP2 Constructs, Purification, and Ca2+-Binding Assays
Before amplifying CaBP2 from the cDNA clone IRAKp961K1413Q (ImaGenes GmbH, Berlin, Germany) with oligonucleotides CABP2O1 and CABP2O2, we removed a 4 nt insertion after exon 2 of the original cDNA to obtain the sequence as reported in RefSeq NM_016366.2 by means of overlap PCR. For this, two fragments of CaBP2 were amplified (primer: fwd1, rev1; fwd2, rev2) and fused to one construct by overlap PCR. Overlap PCR was also used for introducing a premature stop codon (p.Phe164∗), resulting in a construct comparable to the result of exon skipping evoked by the CABP2 c.637+1G>T mutation (primers: fwd1, rev2; p.Phe164∗, rev3, fwd3). All CaBP2 constructs were subcloned into a pET28a vector (EMD Biosciences, San Diego, CA) with restriction sites BamHI and NotI such that an N-terminal His6-tag was fused to all CaBP2 constructs. All constructs were verified by DNA sequencing.
Protein purification was performed as described earlier.15 In brief, the E. coli strain Rosetta 2 (DE3) (Agilent Technologies) was transformed with pET28a containing cDNA for His6-CaBP2-wt or His6-CaBP2-p.Phe164∗, and 100 ml of bacterial overnight culture was inoculated in 5 l of 2YT medium (BD Biosciences, Franklin Lakes, NJ) and grown at 30°C. Overexpression was induced by the addition of 1 mM isopropyl β-D-1-thiogalactopyranoside at an optical density (at 600 nm) of 0.6 and incubation at 16°C for 15–20 hr. Cells were harvested by centrifugation, resuspended, and opened up with a Branson 250 sonicator (Branson, Danbury, CT). After centrifugation at 21,280 g, the His6-tagged proteins were affinity purified from the supernatant with a HisTrap FF crude column (GE Healthcare, Munich, Germany). The proteins were eluted from the column, and the His6-tag was cleaved by the incubation of the protein solution with 1 U thrombin per 1 ml protein solution overnight at 4°C. Next, we added 5 mM of EDTA for chelation of Ca2+ and then reduced the volume of the protein solution in a Vivaspin 20 (10,000 MWCO) concentrator (Sartorius Stedim Biotech, Göttingen, Germany). This protein fraction was loaded on a gel-filtration column (Superdex 200 10/300, GE Healthcare) and run at 0.5 ml/min in buffer (pH 7.4) pretreated with Chelex100 (Bio-Rad, Munich, Germany). Only the fractions corresponding to the monomers of CaBP2-wt or CaBP2-p.Phe164∗ were collected for Ca2+-binding assays, and the volume was further reduced in a Vivaspin 20 concentrator (Sartorius Stedim Biotech). Both proteins were readily purified in the absence and presence of Ca2+. However, Ca2+ enhanced the fraction of monomeric CaBP2 and was therefore present until the gel filtration for the experiments shown in the manuscript. Isothermal titration calorimetry (ITC) was performed with a VP-ITC isothermal titration calorimeter (MicroCal/GE Healthcare, Munich, Germany) at 20°C with 210–250 s of spacing time between injections for allowing complete temperature equilibration with the reference cell. CaBP2-wt or CaBP2-p.Phe164∗ proteins (110–120 μM) were titrated with a buffer solution containing 5 mM Ca2+ with the following parameters: one injection with 2 μl, 50 more injections with 5 μl, and a stirring speed of 300 rpm. A blank titration was performed with the same parameters but without protein in the sample cell, i.e., Ca2+-containing buffer was injected into Ca2+-free buffer.
Analysis of CaBP2 Regulation of Cav1.3 in Transfected Cells
Human embryonic kidney (HEK) cells transformed with SV40 T-antigen (HEK 293T cells) were maintained in DMEM with 10% fetal bovine serum (Invitrogen, CA), 50 U/ml penicillin, and 50 μg/ml streptomycin at 37°C in a humidified atmosphere with 5% CO2. Cells were grown to ∼80% confluence and transfected with FuGENE 6 (Roche, Basel, Switzerland) according to the manufacturer’s protocol. Cells were transfected with cDNAs encoding rat Cav1.3 α1 subunit (1 μg from D. Lipscombe at Brown University; GenBank accession number AF370010), β2a (0.5 μg; RefSeq accession number NM_053581), and α2δ (0.5 μg; GenBank M21948), as well as with 0.06, 0.006, or 0.0006 μg WT or p.Phe164∗ GFP-CaBP2 or 0.06 μg pEGFP-N1 vector (for Cav1.3 alone). The human WT and MT CaBP2 proteins were cloned into NheI and BamHI sites of the pEGFP-N1 vector (Clontech, CA) and verified by DNA sequencing before use.
Whole-cell patch-clamp recordings were performed at room temperature 36–60 hr after transfection. Extracellular recording solutions contained 150 mM Tris, 1 mM MgCl2, and 10 mM CaCl2. Intracellular recording solutions contained 140 mM N-methyl-d-glucamine, 10 mM HEPES, 2 mM MgCl2, 2 mM Mg-ATP, and 5 mM EGTA. The pH of extracellular and intracellular recording solutions was adjusted to 7.3 with methanesulfonic acid. These conditions, including the use of relatively high external Ca2+ and internal EGTA, were used for maximizing the Ca2+ currents and are typical of those used for characterizing Ca2+-dependent inactivation of Cav1.3 in transfected HEK 293T cells.11 Reagents used for electrophysiological recordings were obtained from Sigma-Aldrich (St. Louis, MO). Electrode resistances in the recording solutions were typically 2–4 MΩ. Currents were recorded with an EPC-10 patch-clamp amplifier driven by Patch-Master software (HEKA Electronics, Lambrecht/Pfalz, Germany). Leak and capacitive transients were subtracted according to a P8 protocol. Data were analyzed with routines written in IGOR Pro software (WaveMetrics, Lake Oswego, OR). Averaged data represent the means ± the standard error of the mean [SEM]. Statistical differences between groups were determined by a student’s t test (inactivation data) or ANOVA (IV data) with Sigma Plot software (Systat Software, San Jose, CA). Current-voltage curves were fit with the following function: g(V − E) / [1 + exp[(V − V1/2) / k] + b], where g is the maximum conductance, V is the test potential, E is the apparent reversal potential, V1/2 is the potential of half activation, k is the slope factor, and b is the baseline.
For immunoblotting, HEK 293T cells (36 hr after transfection) were homogenized in 1 ml of ice-cold standard RIPA lysis buffer containing Complete Mini Protease Inhibitor Cocktail (Roche) and were solubilized at 4°C for 30 min. Insoluble material was removed by centrifugation at 600 × g for 5 min, and the supernatants were used immediately or aliquoted and stored at −80°C. Equal amounts (20 μg) of proteins were resolved by SDS-PAGE and were transferred to nitrocellulose. Immunoblotting was performed with rabbit polyclonal antibodies against CaBP2 (UW92 1:2,000)6,16 and was followed by HRP-conjugated anti-rabbit antibodies (1:3,000; Amersham Biosciences, NJ) and enhanced chemiluminescent detection reagents (GE Healthcare, WI).
Results
Next-Generation Sequencing of the DFNB93 Region Identifies a Splice-Site Mutation in CABP2
The DFNB93 region contains more than 300 annotated and hypothetical genes, and several genes are expressed in the mouse and human inner ear. Because there are many strong candidate genes in the region,5 we sequenced all genes and noncoding genes in this region by using a custom DNA capture array to identify the disease-causing mutation in one affected individual from the family. The final target region on the array included 1.1Mb for a total of 449,007 probes, and contained 85% of the original target area. After sequencing, reads were aligned to the human genome, and 27.7% of the reads were on target. Mean coverage in the target area was 97.6×.
After the identified homozygous variants were filtered through the 1000 Genomes Project November 2010 release and dbSNP131, 47 previously unreported variants remained and included two exonic mutations, one splicing mutation, six nontranslated mutations, 16 intergenic (downstream or upstream) mutations, and 22 intronic mutations. The two exonic variants included one nonsynonymous variant, c.1379A>G (p.Glu460Gly) (RefSeq NM_003626.2 [NP_003617.1]), in PPFIA1 (MIM 611054) and synonymous variant c.174G>A (p.Pro58Pro) (RefSeq NM_033036.2 [NP_149025.1]) in GAL3ST3 (MIM 608234). The splice-site variant, c.637+1G>T (RefSeq NM_016366.2), was located at the 5′ donor site of intron 6 of CABP2 (Figure 1 and Figure S1, available online).
The variants in PPFIA1 and CABP2 were subsequently validated by Sanger DNA sequencing, which only confirmed the splicing variant in CABP2. Both variants were present in 100% of the reads, although the variant in PPFIA1 only had 5× coverage compared to the 34× coverage of CABP2—this might explain the lack of confirmation of the first variant. Next, we checked the inheritance of the CABP2 variant in the entire Sh10 family (Figure 1) and screened an additional 100 random Iranian controls to ensure that the variant is not a frequent polymorphism. The mutation was not detected in any of the controls, and inheritance was consistent with hearing loss in the family.
Several prediction programs (Human splicing finder v.2.4.1, NNSPLICE 0.9, and Netgene 2)17–19 indicate that the c.637+1G>T mutation in CABP2 causes a loss of the functional donor site at this position. Loss of the 5′ donor site will most likely result in exon skipping of exon 6 or the use of cryptic splice sites.
CABP2 c.637+1G>T Leads to Exon Skipping
Because we could not detect CABP2 mRNA expression in human blood or saliva, we evaluated the effect of the c.637+1G>T CABP2 mutation on splicing by an exon-trapping system. We cloned exon 6 of CABP2 and the surrounding introns (with WT and MT c.637+1G>T 5′ donor sites) into the exon-trapping vector pSPL3 (Figure 2A). The multiple cloning site of pSPL3 is located within the intron of a minigene and is flanked by functional splice donor (sD) and splice acceptor (sA) sites. If the cloned DNA contains an exon with functional sD and sA sites, splicing will occur between the vector and insert sequences. If regular minigene splicing occurs, a secondary 177 bp PCR product is seen with primers against the minigene exon sequences. If CABP2 exon 6 splice sites are used, a 325 bp product is amplified; this was the case with WT cDNA (Figure 2B). Analysis of cDNA prepared from COS-7 cells transfected with a MT c.637+1G>T 5′ donor site only revealed a PCR product of 177 bp, indicating that the mutation at c.637+1G>T leads to a complete skipping of exon 6 (Figure 2B). The correct sequence of all secondary PCR products was confirmed by Sanger DNA sequencing. Skipping of exon 6 is expected to lead to a shifted reading frame and a premature truncation of the protein (p.Phe164Serfs∗4 [RefSeq NP_057450.2]).
Figure 2.
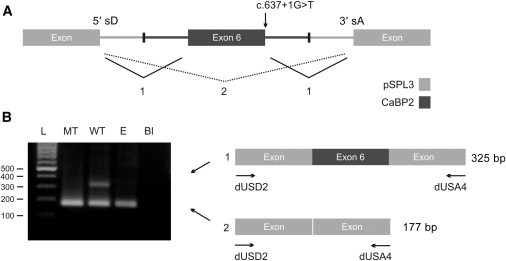
CABP2 c.637+1G>T Leads to Exon Skipping
(A) A schematic illustration of the CABP2-pSPL3 exon trap. CABP2 exon 6 and flanking introns (dark gray)— with a WT or MT (c.637+1G>T) 5′ donor site—were cloned into the pSPL3 vector (light gray). The pSPL3 vector contains a portion of the HIV-1 tat gene, an intron containing the multiple cloning site, and functional 5′ splice donor (5′ sD) and 3′ splice acceptor (3′ sA) sites.
(B) COS-7 cells were transfected with WT or MT CABP2 or an empty pSPL3 vector (“E”). After RNA isolation, splicing products were analyzed by RT-PCR and visualized by agarose-gel electrophoresis. In case of an empty vector (“E”), only vector-vector splicing (2) will occur, leading to a 177 bp secondary product. If exon 6 contains functional 5′ sD and 3′ sA sites (WT), both vector-exon splicing (1) and vector-vector splicing (2) will occur and will lead to 177 bp and 325 bp secondary PCR products, respectively. Mutant plasmids containing the c.637+1G>T (MT) 5′ donor splice-site mutation only showed vector-vector splicing (2), confirming that this mutation results in exon skipping of exon 6. Secondary PCR products were confirmed by sequencing. The following abbreviation is used: Bl, blank.
CABP2 c.637+1G>T Is a Founder Mutation
To identify additional individuals with mutations in CABP2, we screened 39 consanguineous Iranian families affected by variable degrees of prelingual hearing loss by genotyping three microsatellite markers (D11S913, D11S1889, and D11S1337) near CABP2. We identified two families (Sh11 and He) with affected individuals who were homozygous in this region (Figure 1). Affected family members presented with an audiogram similar to the affected individuals in the Sh10 family (Figure 3). Sanger DNA sequencing of all seven exons and intron-exon boundaries of CABP2 revealed the same c.637+1G>T mutation in these families. To investigate whether the c.637+1G>T splice-site mutation in Iranian families Sh10, Sh11, and He is a founder mutation, we analyzed an additional set of microsatellite markers (n = 5) and SNPs (n = 13) in the region (Figures S2–S4). This showed that affected individuals from the three families shared the same haplotype in a region of 0.52 Mb (Figure S5), which suggests that the mutation found in these three families arose on the same ancestral haplotype.
Figure 3.
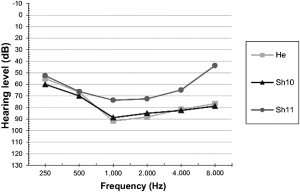
Audiogram
Mean air conduction (AC) thresholds of the left ear for each family. Hearing loss is symmetric.
Mutations in CABP2 Cause Moderate-to-Severe Hearing Loss
All affected individuals with the c.637+1G>T mutation in CABP2 present with a prelingual, stable, and bilateral form of hearing loss with a similar audiometric profile. The hearing loss is moderate to severe and is more pronounced in the mid frequencies, resulting in a typical U-shaped audiogram (Figure 3). Average speech-reception thresholds were 64 db (right) and 64 db (left) for Sh10, 82 db (right) and 72 db (left) for Sh11, and 75 db (right) and 80 db (left) for He. ABR recordings showed comparable hearing thresholds to those of pure-tone audiograms. TEOAEs were negative in all affected individuals. Routine clinical examinations did not reveal any special ophthalmological findings. Upon cardiovascular investigation in the Iranian families, no sign of bradycardia was noted, although this had been previously reported for alterations in Cav1.3 (encoded by CACNA1D [MIM 114206]).20 However, physical characteristics of the affected individuals showed ectomorphic and Marfanoid features. Because mutations in CABP4 (MIM 608965) cause night blindness21 and CABP2 is also expressed in the retina, ophthalmologic investigations were performed in affected individuals from the Iranian families, but they did not reveal any eye disorder except in one patient from family He; this patient was diagnosed with both myopia and astigmatism.
Effects of Exon Skipping Due to CABP2 c.637+1G>T on Ca2+ Binding of CaBP2
The frame shift and premature stop are expected to truncate CaBP2 such that it lacks the Ca2+ binding EF hands 3 and 4. Because the remaining portion of the MT protein would possess only one functional EF hand, we hypothesized that the truncation should alter Ca2+ binding. To test this hypothesis, we performed ITC by using His6-tagged WT and p.Phe164∗ (a premature stop predicted to result from exon skipping) CaBP2 purified from E. coli. Both proteins were readily purified and remained soluble in the absence and presence of Ca2+ (Figure S6). Ca2+ favored the monomeric form and, hence, was only removed prior to the ITC in the experiments shown. By ITC, we found an endothermic Ca2+-binding reaction (probably mediated by EF hands 1, 3, and 4; Figure 4A) for WT CaBP2 in three independent experiments. In contrast, the truncated protein displayed a weaker exothermic reaction (probably mediated by EF hand 1, Figure 4B) in three independent experiments. These results confirm that the truncation predicted to arise from the c.637+1G>T mutation via exon skipping alters Ca2+ binding and thereby might interfere with its role as a Ca2+ sensor.
Figure 4.
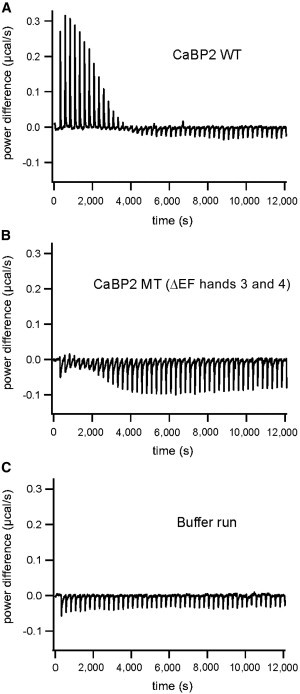
Isothermal Titration Calorimetry of WT and Truncated CaBP2
(A) Trace of the calorimetric titration of 5 μl aliquots (after one initial injection of 2 μl) of 5 mM CaCl2 into 1.3 ml of 110 μM recombinant WT apo-CaBP2. A robust endothermic binding reaction was observed.
(B) Trace of the calorimetric titration of 5 μl aliquots (after one initial injection of 2 μl) of 5 mM CaCl2 into 1.3 ml of 114 μM recombinant truncated apo-CaBP2 (MT CaBP2 [ΔEF hands 3 and 4]). In contrast to the WT apo-CaBP2, the truncated protein leads to a weaker and exothermic reaction.
(C) Trace of the blank titration curve for both experiments A and B with the same parameters but without protein in the sample cell.
Effects of Exon Skipping Due to CABP2 c.637+1G>T on Cav1.3 Ca2+ Currents
To gain further insights into the functional consequences of the c.637+1G>T mutation, we compared the abilities of the WT and truncated CaBP2 to modulate Cav1.3 Ca2+ channels. These channels mediate presynaptic Ca2+ influx and subsequent exocytosis of glutamate in auditory inner hair cells (IHCs).22,23 CaBP2 is also localized in IHCs11 and so might play a role in regulating Cav1.3 and IHC synaptic transmission. In transfected HEK 293T cells, CaBP family members, including CaBP1 (encoded by CABP1 [MIM 605563]) and CaBP4, strongly suppress CDI of Cav1.3.11,12 CDI is a negative feedback regulation of Cav channels and is mediated by incoming Ca2+ ions and calmodulin binding to the pore-forming Cav α1 subunit.8 CaBPs inhibit Cav1.3 CDI in part by competing with calmodulin for binding to the channel.11 Thus, we compared the effects of WT and MT CaBP2 on the inactivation of Cav1.3 Ca2+ currents in transfected HEK 293T cells.
In initial immunoblot experiments to confirm the protein levels of WT and MT CaBP2, we noticed that the MT CaBP2 exhibited ∼10-fold higher protein levels than did the WT CaBP2 (Figure 5A). To account for this difference, we compared CDI in cells expressing WT or MT CaBP2 after transfection with 10-fold more cDNA for WT CaBP2 than for MT CaBP2. Ca2+ currents were evoked by 500 ms step depolarizations, and CDI strength was measured as the fraction of residual current at the end of the pulse relative to the peak current amplitude (r500). CDI was significantly weaker in cells transfected with maximal levels of WT CaBP2 (r500 = 0.92 ± 0.01, n = 6; p < 0.001) than in cells transfected with Cav1.3 alone (r500 = 0.19 ± 0.02, n = 7) (Figure 5B). At protein levels comparable to those of the WT CaBP2 at maximal transfection, the MT CaBP2 showed weaker CDI suppression than did the WT (r500 = 0.62 ± 0.06, n = 9; p < 0.02). At this protein level, the MT CaBP2 was still effective in suppressing CDI because r500 was significantly greater than for Cav1.3 alone (p = 0.001). This difference in WT and MT CaBP2 was negated at lower protein levels of both (r500 = 0.32 ± 0.11 and n = 6 for WT versus r500 = 0.36 ± 0.03 and n = 6 for MT CaBP2; p = 0.09).
Figure 5.
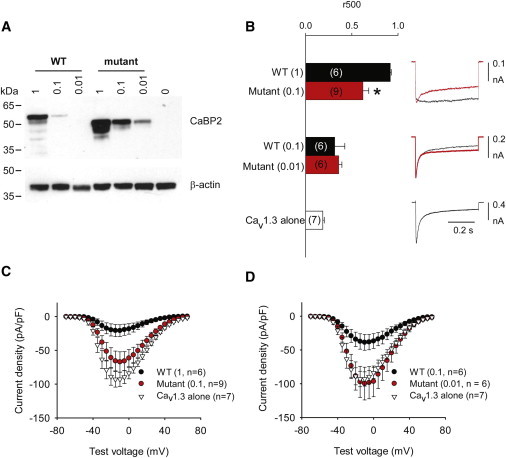
c.637+1G>T Mutation Impairs CaBP2 Regulation of Cav1.3 Ca2+ Channels
(A) The MT CaBP2 shows greater protein levels in transfected HEK 293T cells. Lysates of cells cotransfected with Cav1.3 subunits and WT or MT CaBP2 were subject to immunoblotting with CaBP2 antibodies (top panel). WT and MT CaBP2 were cotransfected at different molar ratios as indicated. The lower panel was probed with β-actin antibodies for confirming equal loading between groups.
(B) The MT CaBP2 shows weaker regulation of CDI. HEK 293T cells were cotransfected as in (A). Ca2+ currents were evoked by 0.5 s pulses from −90 mV to −10 mV. Inactivation was measured as r500, which was the current amplitude at the end of the pulse normalized to the peak current amplitude. So that different protein levels (also in C) could be accounted for, comparisons to WT were made with cells transfected with cDNA encoding MT CaBP2 at levels ten times lower than those of WT CaBP2. Parentheses indicate numbers of cells. ∗p < 0.05. Error bars represent the SEM.
(C and D) The MT CaBP2 shows a weaker ability to suppress Cav1.3 current density than does the WT CaBP2. Ca2+ currents were evoked by 50 ms pulses to variable voltages from −90 mV. Current amplitude was normalized to cell capacitance and plotted against test voltage. Error bars represent the SEM.
In HEK 293T cells, CaBPs also have a second effect of inhibiting Cav1.3 current density. WT CaBP2 also significantly inhibited Cav1.3 current density (maximally ∼78%; p < 0.001; Figure 5C). By contrast, the MT CaBP2 had no effect on Cav1.3 current density (p = 0.35 compared to Cav1.3 alone). Taken together, these results show that c.637+1G>T impairs the function of CaBP2 regulating Cav1.3 channels and potentially other targets important for hearing.
Evaluation of CABP2 Mutations in Affected Individuals with Other Ethnicities and Individuals with Usher Syndrome
We screened 24 unrelated affected Belgian and Dutch individuals with a moderate-to-severe hearing loss for mutations in CABP2 by sequence analysis of the exons and exon-intron boundaries, but we could not identify a clear damaging mutation in any of them. In addition, given that CaBP2 is localized in both the retina and inner ear, we considered it as a candidate gene in which mutations could cause Usher syndrome. We performed genotype analysis for three flanking microsatellite markers—D11S1883, D11S1889, and D11S4113—in 14 small Usher-syndrome-affected families with one to three affected individuals per family. For one USH1-affected family and one USH2-affected family, genotypes were compatible with linkage. However, sequence analysis of exons and exon-intron boundaries did not reveal any putatively pathogenic sequence variant.
Discussion
Like other CaBP family members, CABP2 is closely related to calmodulin. Different from calmodulin, CaBP2 has only three functional Ca2+-binding EF-hand domains (EF hands 1, 3, and 4) because EF hand 2 contains a 3 amino acid deletion that would inhibit Ca2+ binding.6 Although calmodulin is localized in nearly all eukaryotic cells, CaBP2 has thus far been detected in the retina6 and the inner ear.11 In this study, we identified CABP2 mutations in affected individuals who are from the DFNB93-affected family and who present with moderate-to-severe hearing loss.
We identified three Iranian families (Sh10, Sh11, and He) affected by a splice-site CABP2 mutation, c.637+1G>T, that segregates with hearing loss (Figure 1 and Figure S1). To evaluate the effect of the splice-site mutation on splicing, we developed an exon-trapping system (Figure 2). This indicated that the c.637+1G>T mutation most likely leads to skipping of exon 6. Skipping of exon 6, if RNA is not degraded by nonsense-mediated mRNA decay, is predicted to lead to a shifted reading frame, a premature truncation of the protein (p.Phe164Serfs∗4), and a complete loss of EF hands 3 and 4. To check whether the c.637+1G>T mutation in three Iranian families arose on the same ancestral haplotype, we genotyped several microsatellite markers and SNPs near CABP2 (Figure S5). We found that affected individuals from the three families shared a haplotype of 0.52 Mb, suggesting that the c.637+1G>T mutation is a founder mutation inherited from an ancient common ancestor. The small size of the shared region suggests that this mutation might be hundreds or even thousands of years old.
Structural analyses of Ca2+ binding to CaBP1, which, like CaBP2, has a nonfunctional EF hand 2, indicate that high-affinity Ca2+ binding to EF hands 3 and 4 induces conformational protein changes that might be required for Ca2+-dependent regulation of effectors. In contrast, Ca2+ binds with relatively low affinity to EF hand 1.24 Indeed, the CaBP2 truncation predicted by c.637+1G>T altered Ca2+ binding in our ITC experiments (Figure 4), which is consistent with the expectation that EF hands 3 and 4 are required for high-affinity Ca2+ binding in WT CaBP2.
We showed previously that although CaBP1, CaBP2, CaBP4, and CaBP5 are present in mouse IHCs, CaBP1 most strongly suppressed CDI of Cav1.3 channels.11 Using higher levels of CaBP2 cDNA than in our previous study, we now report that CaBP2, like CaBP1, can nearly abolish CDI. It is important to note that factors governing the protein levels and localization of CaBPs in IHCs might differ from those in transfected HEK 293T cells. For example, IHCs might possess sorting and trafficking mechanisms that favor the abundance of CaBP2 at presynaptic Cav1.3 channel clusters, such that CaBP2, indeed, could play an important role in modulating IHC Ca2+ influx.
Our electrophysiological results show that the truncation due to the c.637+1G>T mutation impairs but does not abolish the ability of CaBP2 to modulate Cav1.3. By analogy with CaBP1/Cav1.2 interactions,25 the CaBP2 C-terminal, which is absent in the truncated mutant, might be necessary for anchoring CaBP2 to Cav1.3 and permitting efficient competition with calmodulin. However, the major determinants of CaBP1 regulation of Cav1 channels are the N-terminal lobe and linker region.25 The corresponding regions are spared in the truncated MT CaBP2, which likely permits residual modulation of Cav1.3 CDI.
The observed reduction of Cav1.3 current density by CaBP2 is consistent with the effects of CaBP1 and CaBP4 on Cav1.3 channels in transfected HEK 293T cells.11,12 CaBPs can inhibit Cav1 channel current density by antagonizing the acceleration of forward trafficking of Cav1 channels by calmodulin.26,27 The CaBP2 truncation was more potent in preventing this effect on current density than in impairing the inhibition of CDI (Figure 5C). This suggests that the CaBP2 C-terminal lobe, which is absent in the c.637+1G>T mutant, might be more important in regulating plasma membrane levels of Cav1.3 than in regulating CDI.
On the basis of our present findings and the colocalization of CaBP2 and Cav1.3 in IHCs,11 dysregulation of IHC synaptic transmission could be one pathogenic mechanism underlying hearing impairment in DFNB93. Cav1.3 channels are the primary voltage-gated Ca2+ channels in IHCs and outer hair cells and are required for hearing in mice and humans.20,22,23 In IHCs, the c.637+1G>T mutation in CABP2 would most likely enhance inactivation of synaptic Ca2+ influx. This, in turn, could reduce rates of transmitter release and consequently diminish spiral ganglion neuron firing and ascending auditory-pathway activation. If, like in HEK 293T cells, CaBP2 normally reduces Cav1.3 current density in IHCs, the c.637+1G>T mutation in CABP2 could also have deleterious effects via comparatively higher Cav1.3 current density. Excessive Ca2+ entry and the ensuing increased glutamate release could ultimately be toxic and lead to degeneration of hair cells, afferent synapses, and spiral ganglion neurons. The presence of CaBPs other than CaBP2 in IHCs could partially compensate for the effects of the c.637+1G>T mutation in CABP2, which might explain the moderate hearing loss associated with DFNB93.
It is noteworthy that the structural CaBP2 defect caused by the c.637+1G>T mutation in DFNB93 is analogous to that in CaBP4 of individuals who have congenital cone-rod synaptic disorder and who carry a CABP4 mutation (c.646C>T [p.Arg216∗]).28 Individuals with a c.646C>T (p.Arg216∗) mutation exhibit reduced visual acuity and abnormal color vision, which could result from disruption of CaBP4 interactions with Cav1.4 Ca2+ channels.28 Like Cav1.3 in IHCs and hearing, Cav1.4 in photoreceptors regulates glutamate release and is required for vision.29,30 Moreover, CaBP4 is localized in photoreceptor presynaptic terminals and enhances Cav1.4 function, although via a different mechanism than CaBP2 regulation of Cav1.3.16 In affected individuals with the c.646C>T (p.Arg216∗) mutation, mRNA encoding the truncated CaBP4 is not subject to nonsense-mediated decay and is still detected at levels comparable to those of the mRNA encoding WT CaBP4.28 If the same is true for the c.637+1G>T mutation in CABP2, our electrophysiological results most likely pinpoint one disease mechanism in DFNB93-affected individuals. The fact that similar alterations in the Cav1 regulators, CaBP2 and CaBP4, lead to analogous defects in hearing and vision, respectively, supports the physiological significance of CaBP-Cav1 interactions for sensory transmission.
However, CaBP2 might be required for normal hearing through roles in other cell types besides IHCs. CaBP1 and CaBP5 are localized in multiple neuronal cell types in the retina and likewise,6 CaBP2 could be localized in spiral ganglion neurons, as well as in hair cells. Moreover, CaBP2 could also interact with targets in addition to Cav1.3.31–33
In conclusion, we identified mutations in CABP2 in individuals with moderate-to-severe hearing loss. Mutations in CABP2 cause an audiometric phenotype that is similar to those related to TECTA mutations but differs from the typical profound deafness phenotype that is seen in most families segregating arNSHL. Our results suggest the importance of screening for mutations in CABP2, as well as in TECTA, in families with this milder audiometric phenotype.
Acknowledgments
This work was supported by grants from the University of Antwerp to G.V.C.; grants from the State of Lower Saxony through the “Audiologie Initiative Niedersachsen” program, from the Federal Ministry for Research and Technology through the Bernstein Center for Computational Neuroscience Göttingen (01GQ1005A), and from the German Research Foundation (through the Priority Program 1608, MO896/3-1) to T.M.; a Shahrekord University of Medical Sciences grant to M.H.C.; National Institutes of Health grants (R01 DC009433, R01 HL087120, and R01 EY020850) and a Carver Research Program of Excellence to A.L.; and grants from the Oticon Foundation (09-3742) and ZonMW (40-00812-98-09047 to H.K. I.S. is a postdoctoral fellow of the Fonds Wetenschappelijk Onderzoek-Vlaanderen. We would like to thank Maarten Vanwesemael for assistance with the exon-trapping experiments; the NXTGNT sequencing centre at Ghent University for support with next-generation sequencing; Kai Tittmann for advice on the isothermal titration calorimetry; and Mercedes Suske, Christiane Senger-Freitag, and Sandra Gerke for excellent technical assistance.
Contributor Information
Tobias Moser, Email: tmoser@gwdg.de.
Guy Van Camp, Email: guy.vancamp@ua.ac.be.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Ensembl, http://ensembl.org/
Hereditary Hearing Loss Homepage, http://hereditaryhearingloss.org/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Primer3 Software, http://frodo.wi.mit.edu/
References
- 1.Morton C.C., Nance W.E. Newborn hearing screening—a silent revolution. N. Engl. J. Med. 2006;354:2151–2164. doi: 10.1056/NEJMra050700. [DOI] [PubMed] [Google Scholar]
- 2.Meyer N.C., Alasti F., Nishimura C.J., Imanirad P., Kahrizi K., Riazalhosseini Y., Malekpour M., Kochakian N., Jamali P., Van Camp G. Identification of three novel TECTA mutations in Iranian families with autosomal recessive nonsyndromic hearing impairment at the DFNB21 locus. Am. J. Med. Genet. A. 2007;143A:1623–1629. doi: 10.1002/ajmg.a.31718. [DOI] [PubMed] [Google Scholar]
- 3.Alasti F., Sanati M.H., Behrouzifard A.H., Sadeghi A., de Brouwer A.P., Kremer H., Smith R.J., Van Camp G. A novel TECTA mutation confirms the recognizable phenotype among autosomal recessive hearing impairment families. Int. J. Pediatr. Otorhinolaryngol. 2008;72:249–255. doi: 10.1016/j.ijporl.2007.09.023. [DOI] [PubMed] [Google Scholar]
- 4.Francey L.J., Conlin L.K., Kadesch H.E., Clark D., Berrodin D., Sun Y., Glessner J., Hakonarson H., Jalas C., Landau C. Genome-wide SNP genotyping identifies the Stereocilin (STRC) gene as a major contributor to pediatric bilateral sensorineural hearing impairment. Am. J. Med. Genet. A. 2012;158A:298–308. doi: 10.1002/ajmg.a.34391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tabatabaiefar M.A., Alasti F., Shariati L., Farrokhi E., Fransen E., Nooridaloii M.R., Chaleshtori M.H., Van Camp G. DFNB93, a novel locus for autosomal recessive moderate-to-severe hearing impairment. Clin. Genet. 2011;79:594–598. doi: 10.1111/j.1399-0004.2010.01593.x. [DOI] [PubMed] [Google Scholar]
- 6.Haeseleer F., Sokal I., Verlinde C.L., Erdjument-Bromage H., Tempst P., Pronin A.N., Benovic J.L., Fariss R.N., Palczewski K. Five members of a novel Ca(2+)-binding protein (CABP) subfamily with similarity to calmodulin. J. Biol. Chem. 2000;275:1247–1260. doi: 10.1074/jbc.275.2.1247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Haeseleer F., Imanishi Y., Sokal I., Filipek S., Palczewski K. Calcium-binding proteins: Intracellular sensors from the calmodulin superfamily. Biochem. Biophys. Res. Commun. 2002;290:615–623. doi: 10.1006/bbrc.2001.6228. [DOI] [PubMed] [Google Scholar]
- 8.Christel C., Lee A. Ca(2+)-dependent modulation of voltage-gated Ca(2+) channels. Biochim. Biophys. Acta. 2012;1820:1243–1252. doi: 10.1016/j.bbagen.2011.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Moser T., Neef A., Khimich D. Mechanisms underlying the temporal precision of sound coding at the inner hair cell ribbon synapse. J. Physiol. 2006;576:55–62. doi: 10.1113/jphysiol.2006.114835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Matthews G., Fuchs P. The diverse roles of ribbon synapses in sensory neurotransmission. Nat. Rev. Neurosci. 2010;11:812–822. doi: 10.1038/nrn2924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cui G., Meyer A.C., Calin-Jageman I., Neef J., Haeseleer F., Moser T., Lee A. Ca2+-binding proteins tune Ca2+-feedback to Cav1.3 channels in mouse auditory hair cells. J. Physiol. 2007;585:791–803. doi: 10.1113/jphysiol.2007.142307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yang P.S., Alseikhan B.A., Hiel H., Grant L., Mori M.X., Yang W., Fuchs P.A., Yue D.T. Switching of Ca2+-dependent inactivation of Ca(v)1.3 channels by calcium binding proteins of auditory hair cells. J. Neurosci. 2006;26:10677–10689. doi: 10.1523/JNEUROSCI.3236-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rozen S., Skaletsky H.J. Primer3 on the WWW for general users and for biologist programmers. In: Misener S., Krawetz S.A., editors. Methods in Molecular Biology vol. 132: Bioinformatics Methods and Protocols. Humana Press; Totawa, NJ: 2000. pp. 365–386. [DOI] [PubMed] [Google Scholar]
- 15.Helfmann S., Neumann P., Tittmann K., Moser T., Ficner R., Reisinger E. The crystal structure of the C2A domain of otoferlin reveals an unconventional top loop region. J. Mol. Biol. 2011;406:479–490. doi: 10.1016/j.jmb.2010.12.031. [DOI] [PubMed] [Google Scholar]
- 16.Haeseleer F., Imanishi Y., Maeda T., Possin D.E., Maeda A., Lee A., Rieke F., Palczewski K. Essential role of Ca2+-binding protein 4, a Cav1.4 channel regulator, in photoreceptor synaptic function. Nat. Neurosci. 2004;7:1079–1087. doi: 10.1038/nn1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Desmet F.O., Hamroun D., Lalande M., Collod-Béroud G., Claustres M., Béroud C. Human Splicing Finder: An online bioinformatics tool to predict splicing signals. Nucleic Acids Res. 2009;37:e67. doi: 10.1093/nar/gkp215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reese M.G., Eeckman F.H., Kulp D., Haussler D. Improved splice site detection in Genie. J. Comput. Biol. 1997;4:311–323. doi: 10.1089/cmb.1997.4.311. [DOI] [PubMed] [Google Scholar]
- 19.Brunak S., Engelbrecht J., Knudsen S. Prediction of human mRNA donor and acceptor sites from the DNA sequence. J. Mol. Biol. 1991;220:49–65. doi: 10.1016/0022-2836(91)90380-o. [DOI] [PubMed] [Google Scholar]
- 20.Baig S.M., Koschak A., Lieb A., Gebhart M., Dafinger C., Nürnberg G., Ali A., Ahmad I., Sinnegger-Brauns M.J., Brandt N. Loss of Ca(v)1.3 (CACNA1D) function in a human channelopathy with bradycardia and congenital deafness. Nat. Neurosci. 2011;14:77–84. doi: 10.1038/nn.2694. [DOI] [PubMed] [Google Scholar]
- 21.Zeitz C., Kloeckener-Gruissem B., Forster U., Kohl S., Magyar I., Wissinger B., Mátyás G., Borruat F.X., Schorderet D.F., Zrenner E. Mutations in CABP4, the gene encoding the Ca2+-binding protein 4, cause autosomal recessive night blindness. Am. J. Hum. Genet. 2006;79:657–667. doi: 10.1086/508067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Platzer J., Engel J., Schrott-Fischer A., Stephan K., Bova S., Chen H., Zheng H., Striessnig J. Congenital deafness and sinoatrial node dysfunction in mice lacking class D L-type Ca2+ channels. Cell. 2000;102:89–97. doi: 10.1016/s0092-8674(00)00013-1. [DOI] [PubMed] [Google Scholar]
- 23.Brandt A., Striessnig J., Moser T. CaV1.3 channels are essential for development and presynaptic activity of cochlear inner hair cells. J. Neurosci. 2003;23:10832–10840. doi: 10.1523/JNEUROSCI.23-34-10832.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wingard J.N., Chan J., Bosanac I., Haeseleer F., Palczewski K., Ikura M., Ames J.B. Structural analysis of Mg2+ and Ca2+ binding to CaBP1, a neuron-specific regulator of calcium channels. J. Biol. Chem. 2005;280:37461–37470. doi: 10.1074/jbc.M508541200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Findeisen F., Minor D.L., Jr. Structural basis for the differential effects of CaBP1 and calmodulin on Ca(V)1.2 calcium-dependent inactivation. Structure. 2010;18:1617–1631. doi: 10.1016/j.str.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao T., Bunemann M., Gerhardstein B.L., Ma H., Hosey M.M. Role of the C terminus of the alpha 1C (CaV1.2) subunit in membrane targeting of cardiac L-type calcium channels. J. Biol. Chem. 2000;275:25436–25444. doi: 10.1074/jbc.M003465200. [DOI] [PubMed] [Google Scholar]
- 27.Wang H.G., George M.S., Kim J., Wang C., Pitt G.S. Ca2+/calmodulin regulates trafficking of Ca(V)1.2 Ca2+ channels in cultured hippocampal neurons. J. Neurosci. 2007;27:9086–9093. doi: 10.1523/JNEUROSCI.1720-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Littink K.W., van Genderen M.M., Collin R.W., Roosing S., de Brouwer A.P., Riemslag F.C., Venselaar H., Thiadens A.A., Hoyng C.B., Rohrschneider K. A novel homozygous nonsense mutation in CABP4 causes congenital cone-rod synaptic disorder. Invest. Ophthalmol. Vis. Sci. 2009;50:2344–2350. doi: 10.1167/iovs.08-2553. [DOI] [PubMed] [Google Scholar]
- 29.Mansergh F., Orton N.C., Vessey J.P., Lalonde M.R., Stell W.K., Tremblay F., Barnes S., Rancourt D.E., Bech-Hansen N.T. Mutation of the calcium channel gene Cacna1f disrupts calcium signaling, synaptic transmission and cellular organization in mouse retina. Hum. Mol. Genet. 2005;14:3035–3046. doi: 10.1093/hmg/ddi336. [DOI] [PubMed] [Google Scholar]
- 30.Bech-Hansen N.T., Naylor M.J., Maybaum T.A., Pearce W.G., Koop B., Fishman G.A., Mets M., Musarella M.A., Boycott K.M. Loss-of-function mutations in a calcium-channel alpha1-subunit gene in Xp11.23 cause incomplete X-linked congenital stationary night blindness. Nat. Genet. 1998;19:264–267. doi: 10.1038/947. [DOI] [PubMed] [Google Scholar]
- 31.Zhu M.X. Multiple roles of calmodulin and other Ca(2+)-binding proteins in the functional regulation of TRP channels. Pflugers Arch. 2005;451:105–115. doi: 10.1007/s00424-005-1427-1. [DOI] [PubMed] [Google Scholar]
- 32.Kasri N.N., Holmes A.M., Bultynck G., Parys J.B., Bootman M.D., Rietdorf K., Missiaen L., McDonald F., De Smedt H., Conway S.J. Regulation of InsP3 receptor activity by neuronal Ca2+-binding proteins. EMBO J. 2004;23:312–321. doi: 10.1038/sj.emboj.7600037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yang J., McBride S., Mak D.O., Vardi N., Palczewski K., Haeseleer F., Foskett J.K. Identification of a family of calcium sensors as protein ligands of inositol trisphosphate receptor Ca(2+) release channels. Proc. Natl. Acad. Sci. USA. 2002;99:7711–7716. doi: 10.1073/pnas.102006299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.