

Abstract
Catecholaminergic polymorphic ventricular tachycardia (CPVT) is a devastating inherited disorder characterized by episodic syncope and/or sudden cardiac arrest during exercise or acute emotion in individuals without structural cardiac abnormalities. Although rare, CPVT is suspected to cause a substantial part of sudden cardiac deaths in young individuals. Mutations in RYR2, encoding the cardiac sarcoplasmic calcium channel, have been identified as causative in approximately half of all dominantly inherited CPVT cases. Applying a genome-wide linkage analysis in a large Swedish family with a severe dominantly inherited form of CPVT-like arrhythmias, we mapped the disease locus to chromosome 14q31-32. Sequencing CALM1 encoding calmodulin revealed a heterozygous missense mutation (c.161A>T [p.Asn53Ile]) segregating with the disease. A second, de novo, missense mutation (c.293A>G [p.Asn97Ser]) was subsequently identified in an individual of Iraqi origin; this individual was diagnosed with CPVT from a screening of 61 arrhythmia samples with no identified RYR2 mutations. Both CALM1 substitutions demonstrated compromised calcium binding, and p.Asn97Ser displayed an aberrant interaction with the RYR2 calmodulin-binding-domain peptide at low calcium concentrations. We conclude that calmodulin mutations can cause severe cardiac arrhythmia and that the calmodulin genes are candidates for genetic screening of individual cases and families with idiopathic ventricular tachycardia and unexplained sudden cardiac death.
Main Text
Idiopathic ventricular tachycardia (VT) is a cardiac arrhythmia that is seen in individuals without structural heart disease. Depending on the ECG characteristics, it can be classified as monomorphic VT or polymorphic VT, the latter comprising a number of uncommon, often malignant familial disorders, including catecholaminergic polymorphic VT (CPVT [MIM 604772]).1 CPVT is characterized by episodic syncope and/or sudden cardiac arrest induced by exercise or acute emotion.2,3 The ECG is usually within normal limits at rest and often displays prominent U waves, but ventricular arrhythmias might arise at times of adrenergic activation.4,5 The arrhythmias, typically bidirectional and/or polymorphic VT, can develop into ventricular fibrillation and sudden death, leading to a high mortality rate (30%–50% by the age of 30) for this disorder.6 CPVT often manifests in childhood, and a family history of juvenile sudden death and stress-induced syncope is present in approximately one-third of the cases. It can present as sudden death in children without any prior signs or warning,6 and it has been estimated to cause up to 15% of unexplained sudden cardiac deaths in young people.7 Previously, familial assessment revealed little evidence of the condition in many victims of unexplained sudden death.8,9 Mutations in the ryanodine receptor 2 gene (RYR2 [MIM 180902]) are known to cause dominantly inherited CPVT10 (CPVT1 [MIM 604772], and more than 70 different mutations are currently known. A less common autosomal-recessive form of the disorder (CPVT2 [MIM 611938]) is caused by mutations in the calsequestrin-2 gene11 (CASQ2 [MIM 114251]). Mutations in these genes together explain a little more than half of all familial CPVT cases. Mutations in KCNJ2 [MIM 600681] and ANK2 [MIM 106410], normally causing Andersen-Tawil Syndrome (ATS [MIM 170390]) and type 4 long-QT syndrome (LQT4 [MIM 600919]), respectively, have in addition been found in a low number of individuals diagnosed with CPVT.12,13 Finally, a locus for a severe form of CPVT (CPVT3 [MIM 614021]) was in 2007 localized to chromosome 7 without the identification of the disease gene.14
Tightly controlled cycling of the intracellular calcium concentration is the basis for cardiac muscle contraction and determination of the heart rhythm. RYR2 is the sarcoplasmic reticulum (SR) calcium release channel. Upon a small increase in local intracellular Ca2+ concentration (from approximately 100 nM to a few micromolar), this channel switches from a closed to an open conformation, resulting in a large influx of Ca2+ from the SR storage and ultimately causing muscle contraction. The current molecular understanding of RYR2-associated VT is that RYR2 mutations render the tetrameric RYR2 complex “leaky,” thereby leading to increased local Ca2+ concentrations (Ca2+ sparks), untimely activation of nearby RYR2 clusters through calcium-induced calcium release (CICR), and eventual arrhythmia.15,16 The precise molecular mechanism is, however, still unclear, and it has been put forward that an abnormal interaction with one or more of the associated proteins or ions, such as FKBP12.6 or CASQ2, or abnormal activation by extraluminal or intraluminal Ca2+ ions also might play a role, as reviewed by Kontula et al.16 and Mohler et al.17
Here we report a Swedish multiplex family presenting with a history of ventricular arrhythmias, syncopes and sudden death, predominantly in association with physical exercise or stress (Figure 1A). The index case (II:6), a now 42-year old man of Swedish ethnic origin, presented with syncope while playing football at the age of 12. The ECG at admission (Figure 1B, upper panel) showed sinus bradycardia (HR = 45/min) and prominent U-wave in V2 and V3, but no evidence of QT prolongation (QTc = 0.42 s). He had a history of loss of consciousness on at least four occasions during physical activity and once in connection with a fire alarm. A 24 hr ECG registration revealed ventricular extrasystoles (VES), bigemini, and paired VES (Figure 1B, lower panel) during football training, but no symptoms were reported. He was started on β1-adrenergic-receptor blocker treatment. At follow up, a 24 hr ECG and exercise test still showed some VES and bigemini under conditions of increasing load and heart rate. A genomic DNA sample from the index case was later screened for mutations in a panel of arrhythmia-associated genes, including RYR2 and CASQ2, where no mutations were identified. An older brother (II:4), aged 23, had a history of repeated syncope during exercise. During an exercise test, he displayed polymorphic VT (an ECG from that time is not available). After treatment with β1-adrenergic-receptor blocker, a follow-up exercise test showed VES at high loads. The family history also included a brother (II:3) who drowned during a swimming competition at age 15 after having had prior episodes of syncope and an older sister (II:2) who suffered from fits or syncope. After a later episode of ventricular fibrillation (VF), she was stabilized by treatment with β1-adrenergic-receptor blocker and became asymptomatic. A younger sister (II:8) presented at the age of 7 with a history of repeated syncope during intense physical activity. One younger family member (III:5), with reported syncope from age 2.5, died suddenly at age 13, having been treated with β1-adrenergic-receptor blocker for several years. Another family member (III:4) started having syncope at age 4, was asymptomatic under treatment with β1-adrenergic-receptor blocker, and suffered cardiac arrest at age 16. After rapid defibrillation and resuscitation, she recovered and had an ICD implanted. An older sister (III:2) presented with syncope from age 6–7 and became asymptomatic under treatment with β1-adrenergic-receptor blocker. A younger sister (III:6) presented with syncope at age 3–4. Family members III:9, III:12, and IV:1 were put on β1-adrenergic-receptor blocker after having suffered syncope and attacks of dizziness. According to the family, subject I:1 suffered multiple cases of syncope in her youth and was on medication. She died at 60 years old. Thus, the phenotypic picture of the family is characterized by CPVT-like features with symptoms including frequent syncope and three cases of sudden death or cardiac arrest. The affected family members display no other apparent clinical manifestations.
Figure 1.
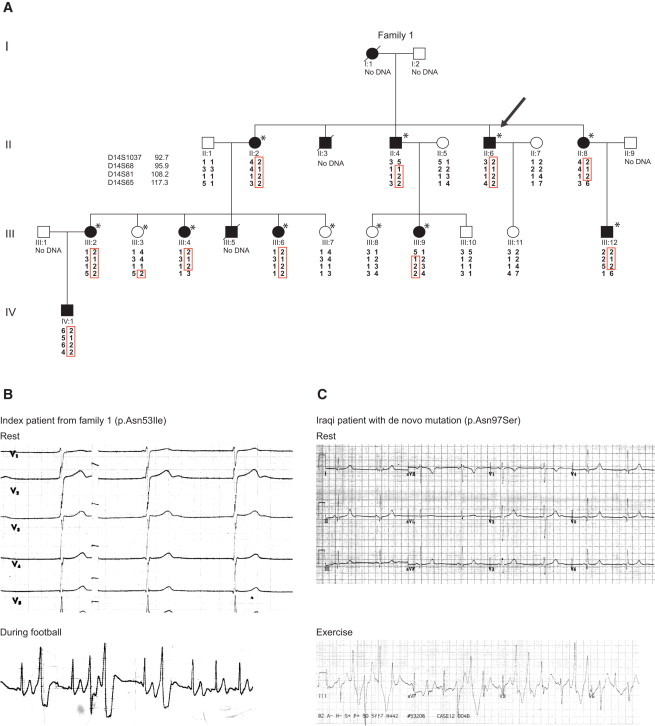
Pedigree and Electrocardiography
(A) Pedigree of Swedish family with the chromosome 14 haplotypes presented graphically with HaploPainter.18 The index case is indicated with an arrow. The individuals included in the initial SNP genome-wide linkage analysis are marked by an asterisk.
(B) The ECG during rest and an arrhythmia event (while the individual was playing football) occurring during a 24 hr ECG registration of the index case II:6 of family 1.
(C) Resting and exercise ECGs from a second, unrelated individual of Iraqi origin. Both resting ECGs demonstrate prominent U waves in the anterior leads, and bidirectional ventricular ectopy is seen during physical activity or exercise.
All procedures were in accordance with the ethical standards of the responsible committees (institutional and national) on human experimentation. After obtaining informed consent from all the subjects, we extracted genomic DNA from peripheral blood samples from all the subjects to perform linkage analysis. Initially, 12 subjects (nine affected and three unaffected, indicated by an asterisk in Figure 1A) were genotyped with the Human Mapping 50K SNP Xba240 Array (Affymetrix, High Wycombe, UK). The unaffected subjects were all older than 16 years at the time of inclusion. Genotypes were called by the Genotyping Console (Affymetrix) and uploaded to the BCSNP data management platform (BC Platforms, Espoo, Finland) for quality-control filtering (58,958 markers). Markers with Mendelian errors were detected with MERLIN19 and removed from the data set. Removal of monomorphic markers and LD pruning (for which a sliding window of 50 SNPs and an r2 threshold of 0.5 was used) was performed with PLINK20 and resulted in a filtered data set of 7,199 markers in approximate linkage equilibrium with each other. MERLIN was used for the identification of unlike genotypes, resulting in the removal of 292 genotypes from the data set. A parametric linkage analysis assuming autosomal-dominant inheritance with complete penetrance and a disease gene frequency of 0.0001 was carried out with MERLIN with allele frequencies from the Affymetrix 100K frequency files (CEU population) and genetic distances from the Affymetrix 100K Marshfield cM map. Two possible linked loci were identified (on chromosomes 14q31-32 and 6q27-qter), each with a maximum LOD score of 3.01 (Figure S1 in the Supplemental Data available online). A follow-up analysis with microsatellite markers (D14S1037, D14S68, D14S81, D14S65, D6S305, D6S386, and D6S1590) in these two regions was performed and included an additional six subjects from the family (one affected, IV:1; two unaffected, III:10 and III:11; and three healthy married-in individuals, II:1, II:5, and II:7). In total, 18 individuals with the microsatellite markers were genotyped. Primer sequences were retrieved from the NCBI UniSTS database. The microsatellite analysis excluded the locus on chromosome 6 and mapped the disease locus to chromosome 14 (LOD score 3.9) (Figure 2A).
Figure 2.
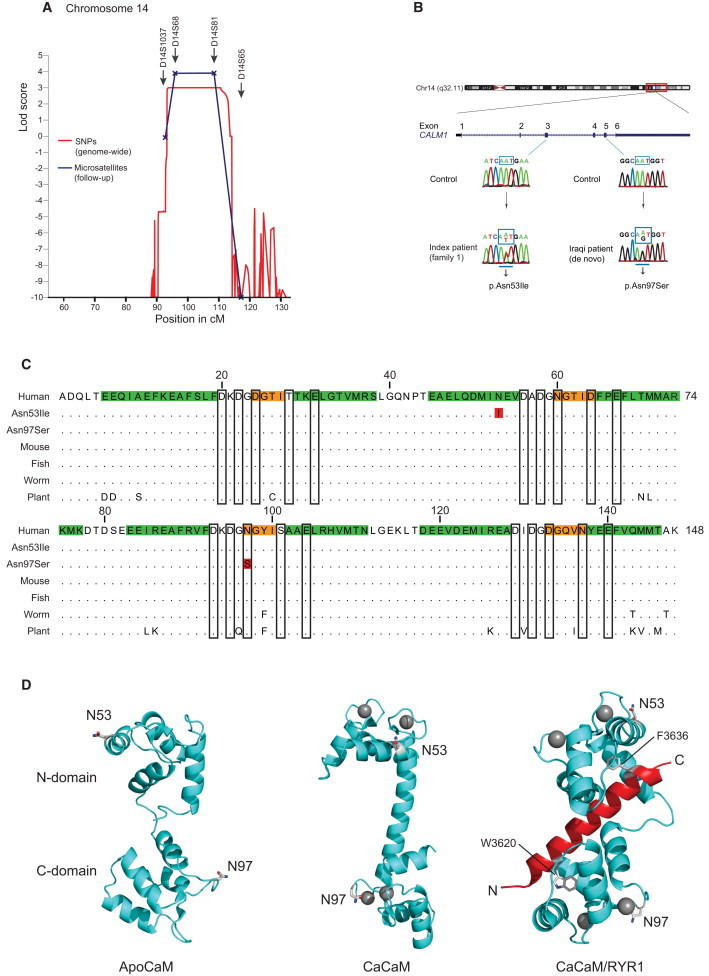
Genome-wide Linkage Analysis and Position of the Calmodulin Mutations
(A) A locus for a severe dominantly inherited form of CPVT-like arrhythmia is shown at position 14q31-32 (LOD score 3.9) and harboring approximately 70 known genes.
(B) CALM1 structure and position of the c.161A>T and c.293A>G mutations identified in two different individuals with ventricular tachycardia and the sequencing electropherograms of exon 3 and 5 in the case and a control subject.
(C) Alignment of calmodulin amino acid sequences from different species; the p.Asn53Ile and p.Asn97Ser substitutions are indicated (red residues). Secondary-structure elements are shown in green and orange (α-helix and β strand, respectively), and the Ca2+-binding residues are framed. Dots indicate conserved residues, showing that calmodulin is 100% conserved in all vertebrates and only displays a few changes in round worms and plants.
(D) The position of the substituted residues in the three-dimensional structural model of native calmodulin prepared with Pymol (Schrödinger, LLC). Left panel: apo-calmodulin (Protein Data Bank [PDB] ID: 1DMO21), Middle panel: Ca2+-saturated calmodulin (pdb ID: 1CLL22), Right panel: Ca2+ bound calmodulin-RYR1 peptide complex (PDB ID: 2BCX23). Calcium ions are shown as gray spheres; the RYR1 peptide is in red. The two aromatic hydrophobic binding anchors in RYR1 are shown in stick representation. The residue Asn53 is positioned on the solvent-exposed side of α-helix C, not in contact with Ca2+, peptide ligand, or other calmodulin domains. Asn97 is one of the Ca2+-coordinating residues of Ca2+-binding site III. The calmodulin N domain is positioned on top, and the C domain is at the bottom. The Ca2+- and RYR1-complexed calmodulin structures have been rotated roughly 90 and 180 degrees counterclockwise, respectively, around a vertical axis after structural alignment of the N domains with the apo-calmodulin structure.
A haplotype analysis confirmed the presence of the chromosome 14 disease haplotype in all affected and none of the unaffected individuals (Figure 1A), suggesting 100% penetrance of the mutation in this family. The peak on chromosome 14 extended approximately 21 cM, from rs9323782 (86.6Mb) to rs721403 (95.7Mb) (hg19) (Table S2), a region that harbors around 70 genes (Figure S2 and Table S3). Because of the pivotal role of calmodulin in calcium signaling and heart contraction, CALM1 was selected for sequencing. Primers amplifying the coding regions, adjacent splice sites, and the 5′- and 3′- untranslated regions of CALM1 were designed with Primer324 (Table S1). The amplified products were purified and sequenced on both strands at MWG (Eurofins MWG Operon, Ebersberg, Germany). Sequencing one affected family member (the index case) and one unaffected family member revealed a heterozygous missense mutation c.161A>T [GenBank NM_006888.4] in exon 3. This mutation results in a change from asparagine (Asn) to isoleucine (Ile), p.Asn53Ile (UniProtKB P62158, notated without the initiator methionine residue to reflect the mature processed calmodulin) in the index case (Figure 2B). A genotyping assay for p.Asn53Ile for the LightCycler 480 instrument (Roche, Hvidovre, Denmark) was developed by TIB MOLBIOL (Berlin, Germany) (Table S1). Genotyping with LightCycler 480 Genotyping Master Mix (Roche) confirmed that the p.Asn53Ile substitution was present in all affected individuals and absent in all of the unaffected individuals in the pedigree. Genotyping of 1,200 control individuals (500 Danish medical student volunteers from Aarhus University and 700 anonymous Danish blood donors) showed that the mutation was absent among these 2,400 control chromosomes. This sample size provided an 80% power to detect a variant with a minor-allele frequency of 0.001,25 verifying that the mutation is highly unlikely to be a normal but rare sequence variation.
To systematically screen for other mutations in CALM1, we developed a mutation-detection assay for each of the five coding exons on the basis of high-resolution melting (HRM) of amplified PCR products. All assays were carried out with 480 High Resolution Melting Master (Roche) and HPLC purified primers (MWG). Sixty-three individuals, referred for RYR2 mutation analysis at the Statens Serum Institute in Denmark (where genetic testing failed to identify any RYR2 mutation in 61 of these individuals), were analyzed in duplicate. All samples with aberrant melting curves, as well as ten control samples with normal melting curves, were sequenced (primer sequences are listed in Table S1). From this screening a second heterozygous CALM1 missense mutation was identified in an individual of Iraqi origin. The mutation (c.293A>G) was located in exon 5 and resulted in an asparagine-to-serine substitution (p.Asn97Ser) (Figure 2B). DNA sequencing revealed that this mutation was absent in the mother and the father, neither of whom showed signs of heart arrhythmias, demonstrating the presence of a de novo mutation in this individual. A microsatellite marker analysis confirmed the paternity relationship. The individual with the de novo CALM1 mutation was a 23-year-old female who presented at age 4 with a successfully resuscitated, out-of-hospital cardiac arrest due to VF while she was running. She made a full neurological recovery and was stabilized by treatment with β1-adrenergic-receptor blocker. An initial ECG and echocardiogram were within normal limits, and there was no evidence of QT prolongation (not shown). Evaluation of her immediate family was unremarkable. An exercise ECG and electrophysiological study undertaken on full betablockade and right ventricular and coronary angiography were within normal limits. An initial diagnosis of idiopathic VF was made at the time. Follow-up ECGs demonstrated prominent U waves in the anterior leads but no evidence of the long-QT or Brugada syndromes (Figure 1C upper panel). When she was 12 years old, further investigation including signal-averaged ECG, echocardiography, cardiac MRI, and cold pressor and procainamide tests were unremarkable. An exercise ECG while she was off β1-adrenergic-receptor blocker demonstrated ventricular ectopy with couplets and triplets of varying morphology into recovery (Figure 1C, lower panel). These appeared to be bidirectional at times. On this basis, a diagnosis of CPVT was made, but genetic testing of RYR2, CASQ2, and KCNJ2 failed to identify any mutations in these genes. In a follow-up genetic test of other arrhythmia-associated genes, including KCNQ1 (MIM 607542), KCNH2 (MIM 152427), KCNE1 (MIM 176261), KCNE2 (MIM 603796), and SCN5A (MIM 600163), no mutations were found. She fainted twice more in her teens, and at age 15 she suffered a further cardiac arrest. After recovery, an ICD was implanted, and she remained well but was diagnosed with systemic lupus erythematosus (SLE [MIM 152700]). Her mother, aged 62, was asymptomatic until developing heart failure secondary to adriamycin-induced cardiomyopathy from breast cancer treatment. She did not experience syncope or arrhythmia. An ECG demonstrated left-bundle-branch block and leftward axis deviation. Her father, aged 66, had suffered nonexertional syncope in stressful situations, consistent with vasovagal syncope. His resting ECG was normal, and exercise testing for atypical chest pain had induced ischemic changes without arrhythmia. An angiogram was reported as showing unobstructed coronaries and vasospasm.
Both CALM1 mutations thus appear to induce an early-onset and highly penetrant phenotype that belongs to the severe end of the spectrum of CPVT-like arrhythmias. In accordance with this, an initial response to therapy with β1-adrenergic-receptor blocker was observed with subsequent recurrent symptoms both in the Iraqi case and in individuals III:4 and III:5 in family 1. The fact that p.Asn97Ser is a de novo substitution is fits well with the presentation of the associated disease as a highly malignant form with very early presentation and is consistent with the RYR2-associated disease, which is commonly caused by de novo mutations.26
To assess whether CALM1 missense mutations exist in the general population, a systematic HRM screen of all five CALM1 coding exons were performed in 500 Danish control individuals. No missense mutations were identified among these 1,000 control chromosomes. In contrast, three rare silent polymorphisms were identified (these were present among five control individuals) (Table S4), stressing the selection pressure against missense mutations in this gene. A specific Iraqi control population was not investigated because the mutation was de novo, and the mutation rate is likely to be the same in all populations. No missense mutations in CALM1 (ENST00000356978) were found from the 1000 Genomes Project (October 2011 release) interrogated through the Project Browser #ICHG2011 (Ensembl release 63 representing an integrated set of variant calls and INDELS from low-coverage and exome sequencing data of 1,092 individuals). A literature search also failed to present evidence for any previously identified calmodulin mutations. The lack of identified missense mutations is not surprising given that calmodulin is known as one of the most conserved molecules throughout evolution, displays an identifical protein sequence in all vertebrate species, and has evolved only slightly since the divergence from plants (Figure 2C). The presence of three independent genes in the human genome (CALM1-3 [MIM 114180, 114182 and 114183]), all encoding ubiquitously expressed identical calmodulin protein molecules, further underscores the selection pressure against any amino acid changes in this classic calcium-binding protein.
Calmodulin is a 148 amino acid α-helical protein containing four classical Ca2+-binding EF hands that bind one calcium ion each. It is shaped as a dumbbell and is capable of mediating a signal consisting of small intracellular Ca2+-concentration changes to a multitude of intracellular targets via fine-tuned differences in the N- and C-domain affinity for Ca2+.27 The N domain contains the lower-affinity Ca2+-binding sites I and II, and the C domain contains the high-affinity binding sites III and IV. The two identified substituted residues are located in opposite domains; the Asn53 residue is positioned on the solvent-exposed surface of the first α-helix of Ca2+-binding site II in the N domain, and the Asn97 residue is one of the Ca2+-binding residues of binding site III, located in the calmodulin C domain (Figure 2D). Calmodulin binds to and regulates the activity of a large number of intracellular proteins, but in none of the published high-resolution calmodulin structures available are the two substituted residues in direct contact with any bound protein or peptide, exemplified by the calmodulin- RYR1 peptide complex in Figure 2D.
To functionally characterize the substitutions, we used a nested priming approach to amplify the CALM1 cDNA (primers are listed in Table S1) from a cDNA preparation from liver mRNA,28 and we ligated this cDNA into a modified pMAL vector (New England Biolabs, Ipswich, USA) containing a Tobacco Etch Virus (TEV) protease cleavage site. Missense mutations were introduced with QuickChange Lightning (QIAGEN) (primer sequences are listed in Table S1). The calmodulin variants were expressed in Rosetta B cells (EMD Chemicals). All buffers and reagents were prepared with purified and deionized (>18.2 Ω resistance) water and plastic vessels, and all laboratory equipment was washed in 1 M HCl and purified water so that Ca2+ contamination would be minimized. The total Ca2+ concentration of the utilized buffers was determined to be between 1 and 3 μM with the Quin-2 fluorogenic calcium indicator and Calcium Calibration Buffer Kit #1 (Invitrogen) and an Infinite M1000 fluorescence microplate reader (Tecan, Switzerland). Calmodulin proteins were purified by affinity chromatography (Amylose Resin [New England Biolabs]), TEV cleavage, anion-exchange chromatography (Source 15Q 10/10 [GE Healthcare]), and size-exclusion chromatography (Superdex 75 16/60 [GE Healthcare]). Protein concentrations were determined by absorption at 280 nm. The identity and integrity of each protein preparation was confirmed by MALDI-TOF mass spectometry (Bruker Reflex III [Bruker-Daltronics]).
First, the Ca2+-binding property of native and variant calmodulin was investigated. Because calcium binding leads to a large conformational change in calmodulin and the C domain contains two tyrosine (Tyr) residues, it is possible to monitor Ca2+ binding to the C domain by measuring the changes in intrinsic Tyr fluorescence intensity.29 Calmodulin variants (15 μM) were titrated with CaCl2 while the intrinsic Tyr fluorescence intensity at 320 nm (277 nm excitation) was recorded on a FluoroMax 4 spectrofluorometer (HORIBA Jobin Yvon) equipped with a peltier temperature controller set at 37°C. The fractional C domain Ca2+ binding demonstrated a significant reduction in the Ca2+ affinity for the p.Asn97Ser variant (Figure 3), as evidenced by an apparent half-saturation concentration of ∼40 μM (equivalent to 2.7 Ca2+ ions per calmodulin molecule), compared to ∼25 μM total Ca2+ for native calmodulin (1.7 Ca2+ ions/calmodulin molecule). The p.Asn53Ile variant, on the other hand, demonstrated a small, but significantly increased, C domain Ca2+ saturation and an earlier half saturation of ∼21 μM (1.4 Ca2+ ions/calmodulin molecule). Because the p.Asn53Ile substitution is located in the N domain of calmodulin, we suggest that this “earlier” C domain Ca2+ saturation reflects a decreased N domain Ca2+ affinity and leads to increased availability of free Ca2+ for the C domain to bind at lower total Ca2+ concentration. A less likely possibility is that it reflects an increased positive inter-domain cooperativity between the N and C domains. Hence, we show that both variants lead to altered calcium binding property of calmodulin and that the substituted residues impose different effects on the distribution of bound Ca2+ between the N and C domains of calmodulin.
Figure 3.
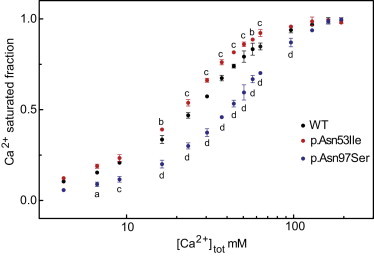
Calcium Binding
The relative Ca2+ binding of the C domain of calmodulin as a function of total Ca2+ concentration, determined as the change in the tyrosine fluorescence intensity. The data presents the average of three independent experiments (+/− SD); in most cases the error bars are smaller than the symbols. In comparison to native calmodulin, p values are as follows: a, p < 0.05; b, p < 0.01; c, p < 0.001; and d, p < 0.0001 (two-way repeated-measures ANOVA with the Bonferroni multiple comparisons post-hoc test).
Next, the interaction between the calmodulin variants and a peptide from RYR2 encompassing the calmodulin binding domain (CBD), RYR2-CBD (R3581-SKKAVWHKLLSKQRKRAVVACFRMAPLYN-L3611 [Genscript, USA]), was investigated. In the high-resolution 3D structure of calmodulin bound to the equivalent peptide from RYR1, the Ca2+-bound C domain of calmodulin fully encloses the single tryptophan (Trp) residue of the RYR peptide (RYR1 Trp3620, corresponding to RYR2 Trp3586, Figure 2D). Calcium binding to calmodulin leads to an N-terminal shift in its binding site on the RYR2-CBD peptide. This shift is evidenced by a large increase in fluorescence intensity of the RYR2 Trp3586 residue; calmodulin itself does not contain any Trp residues. The intrinsic fluorescence emission (290–450 nm, at 280 nm excitation) of RYR2 Trp3586 was monitored with and without addition of saturating amounts of calmodulin variants under four different free-Ca2+ concentrations: no free Ca2+ ([Ca2+]free < 1 nM, buffer including 5 mM EGTA), resting cardiomyocyte conditions ([Ca2+]free ∼100 nM), excited conditions ([Ca2+]free ∼1 μM),30 and saturating Ca2+ conditions (200 μM total Ca2+), all in 20 mM HEPES; 100 mM KCl (pH 7.2). The free-Ca2+ concentrations for excited and resting conditions were controlled with an EGTA-Ca2+ buffer system, as previously described.31 A concentration of 0.5 μM RYR2 peptide was used with no calmodulin variant added (RYR2 alone), 3 μM calmodulin variant added (zero and 100 nM [Ca2+]free), or 1 μM calmodulin variant added (1 μM [Ca2+]free and 200 μM total Ca2+). When saturating amounts of calmodulin were added at low calcium concentrations (100 nM free Ca2+), a remarkable difference in the RYR2-CBD binding mode was demonstrated for p.Asn97Ser as opposed to p.Asn53Ile and native calmodulin. The p.Asn97Ser variant only induced a slight blueshift and no increase in fluorescence intensity (Figure 4A), similar to the effect seen when calmodulin binds to the skeletal myocyte ryanodine receptor (RYR1)32 binding-domain peptide in the absence of Ca2+. In marked contrast, little or no difference in the RYR2-CBD binding mode was observed between p.Asn97Ser, p.Asn53Ile, and native calmodulin at moderate (1 μM free) or saturating (200 μM total) calcium concentrations (Figures 4B and 4C). Thus, our data demonstrate that for the p.Asn97Ser substitution, the calmodulin-RYR2-CBD interaction is defective at low intracellular Ca2+ concentrations and is restored at moderate to high Ca2+ concentrations.
Figure 4.

Interaction with RYR2 Peptide
Calmodulin binding to the RYR2-CBD peptide (Arg3581–Leu3611). The RYR2 Trp3586 fluorescence-emission spectrum, normalized to the maximal fluorescence intensity of the peptide alone, is shown without added calmodulin (RYR) and with a saturating amount of the indicated calmodulin variants added at (A) low intracellular (100 nM free Ca2+), (B) moderate (1 μM free Ca2+), and (C) saturating (200 μM Ca2+) concentrations.
It is well established that calmodulin binds directly to RYR2, and it has been demonstrated that calmodulin binding decreases the probability that RYR2 has an open conformation at free-Ca2+ concentrations down to 100 nM,33 corresponding to a low, diastolic Ca2+ concentration, at which we demonstrated a compromised calmodulin-RYR2-CBD interaction for the p.Asn97Ser substitution. Hence, the lack of a calmodulin-RYR2 interaction at low Ca2+ concentrations would most likely lead to an increased probability that RYR2 has an open conformation. On this basis, we suggest that p.Asn97Ser, being a loss-of-function mutation with regard to the RYR2 interaction, exerts a dominant-negative effect on the RYR2 channel complex and leads to “inappropriate RYR2 leakiness” similar to what is proposed for RYR2 mutations. This would also explain how one mutated calmodulin allele out of six encoding identical proteins is sufficient to cause a dominantly inherited phenotype.
Interestingly, several animal studies have recently suggested that a disturbed calmodulin RYR2 interaction plays a key role in arrhythmia and heart failure.33–35 For example, in a study using knock-in mice harboring the human RYR2 CPVT-type substitution p.Arg2474Ser, previously found to be a model for CPVT36,37 Xu and co-workers investigated the RYR2-calmodulin interaction and found a markedly reduced affinity for calmodulin in response to PKA phosphorylation, which led the authors to conclude that a defective interaction between RYR2 and calmodulin is part of the pathogenic mechanism for CPVT.35 Our results are the first human data to strongly support the molecular disease model in which an aberrant calmodulin-RYR2 interaction is causative of CPVT-like arrhythmias.
The p.Asn53Ile substitution did not demonstrate a significantly compromised RYR2 interaction in the experimental setup used here. One limitation of our work is that we studied the calmodulin-RYR2 interaction by using a small peptide fragment (RYR2-CBD) instead of the large RYR2 molecular complex. Thus, it is at present unclear whether the p.Asn53Ile substitution affects the function of RYR2 in vivo. Intriguingly, the Ca2+-binding property of p.Asn53Ile was markedly different from the native and p.Asn97Ser calmodulin, and it is possible that a different molecular pathogenic mechanism is in play in family 1. This could be reminiscent of the mechanism suggested for ANKB mutations in CPVT given that calmodulin regulates, and AnkB is required for correct assembly of, a number of ion channels and pumps involved in cardiomyocyte contraction. For example, such ion channels and pumps include the L-type calcium channel, Na+/K+ ATPase, Na+/Ca2+ exchanger, and the InsP(3) receptor.12
In the screened clinical sample, which included 63 individuals who had unspecified arrhythmias or sudden cardiac death and had been referred for RYR2 screening, only two CALM1 mutations were detected (both the family 1 index case and the Iraqi case were included in this sample). Thus, the frequency of CALM1 mutations is seemingly low from this finding. However, because only two individuals were found to be positive for RYR2 mutations, the frequencies of CALM1 and RYR2 mutations were identical in this sparsely characterized and heterogeneous sample collection. More accurately establishing the frequency and phenotypic variation of CALM1 mutations in CPVT and related disorders will require screening of large well-characterized clinical samples. Before this is done, we cannot exclude that CALM1 is a rare contributor to CPVT.
In conclusion, we have identified two functionally distinct CALM1 missense mutations that lead to development of early-onset CPVT or CPVT-like arrhythmia. Our finding will directly allow presymptomatic genetic diagnosis in families in which members with this severe disorder harbor CALM1 mutations, enabling the initiation of potentially life-saving treatment for children and young individuals with disease mutations. As a consequence of our findings, the three calmodulin genes are candidates for genetic screening of individuals with CPVT-like arrhythmia and unexplained sudden cardiac death.
Acknowledgments
We thank all participants and their families for their cooperation. We thank Anne Hedemand and Hanne K. Nielsen for expert technical assistance. This study was supported by the Obelske Family Foundation, the Fraenkel Foundation, the Edith Agnete Stern Foundation, the Carlsberg Foundation, and the Novo Nordisk Foundation.
Contributor Information
Mette Nyegaard, Email: nyegaard@hum-gen.au.dk.
Anders D. Børglum, Email: anders@hum-gen.au.dk.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes, http://www.1000genomes.org/home
Haplopainter, http://haplopainter.sourceforge.net/
NHLBI Exome Sequencing Project, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org/
Primer3 program, http://frodo.wi.mit.edu/primer3/
UCSC Genome Browser (assembly hg19), http://genome.ucsc.edu/
UniSTS database, http://www.ncbi.nlm.nih.gov/unists
Accession Numbers
The dbSNP database accession numbers for the variations reported in this study are rs267607276 for c.161A>T, rs267607277 for c.293A>G, rs267607278 for c.72C>T, and rs267607279 for c.421+16C>G (Table S4).
References
- 1.Badhwar N., Scheinman M.M. Idiopathic ventricular tachycardia: Diagnosis and management. Curr. Probl. Cardiol. 2007;32:7–43. doi: 10.1016/j.cpcardiol.2006.10.002. [DOI] [PubMed] [Google Scholar]
- 2.Coumel P., Fidelle J., Lucet V., Attuel P., Bouvrain Y. Catecholaminergic-induced severe ventricular arrhythmias with Adams-Stokes syndrome in children: Report of four cases. Br. Heart J. 1978;40(suppl):28–37. [Google Scholar]
- 3.Napolitano C., Priori S.G. Diagnosis and treatment of catecholaminergic polymorphic ventricular tachycardia. Heart Rhythm. 2007;4:675–678. doi: 10.1016/j.hrthm.2006.12.048. [DOI] [PubMed] [Google Scholar]
- 4.Aizawa Y., Komura S., Okada S., Chinushi M., Aizawa Y., Morita H., Ohe T. Distinct U wave changes in patients with catecholaminergic polymorphic ventricular tachycardia (CPVT) Int. Heart J. 2006;47:381–389. doi: 10.1536/ihj.47.381. [DOI] [PubMed] [Google Scholar]
- 5.Viitasalo M., Oikarinen L., Väänänen H., Kontula K., Toivonen L., Swan H. U-waves and T-wave peak to T-wave end intervals in patients with catecholaminergic polymorphic ventricular tachycardia, effects of beta-blockers. Heart Rhythm. 2008;5:1382–1388. doi: 10.1016/j.hrthm.2008.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Fisher J.D., Krikler D., Hallidie-Smith K.A. Familial polymorphic ventricular arrhythmias: A quarter century of successful medical treatment based on serial exercise-pharmacologic testing. J. Am. Coll. Cardiol. 1999;34:2015–2022. doi: 10.1016/s0735-1097(99)00438-6. [DOI] [PubMed] [Google Scholar]
- 7.Liu N., Ruan Y., Priori S.G. Catecholaminergic polymorphic ventricular tachycardia. Prog. Cardiovasc. Dis. 2008;51:23–30. doi: 10.1016/j.pcad.2007.10.005. [DOI] [PubMed] [Google Scholar]
- 8.Behr E.R., Dalageorgou C., Christiansen M., Syrris P., Hughes S., Tome Esteban M.T., Rowland E., Jeffery S., McKenna W.J. Sudden arrhythmic death syndrome: Familial evaluation identifies inheritable heart disease in the majority of families. Eur. Heart J. 2008;29:1670–1680. doi: 10.1093/eurheartj/ehn219. [DOI] [PubMed] [Google Scholar]
- 9.Tester D.J., Spoon D.B., Valdivia H.H., Makielski J.C., Ackerman M.J. Targeted mutational analysis of the RyR2-encoded cardiac ryanodine receptor in sudden unexplained death: A molecular autopsy of 49 medical examiner/coroner’s cases. Mayo Clin. Proc. 2004;79:1380–1384. doi: 10.4065/79.11.1380. [DOI] [PubMed] [Google Scholar]
- 10.Priori S.G., Napolitano C., Tiso N., Memmi M., Vignati G., Bloise R., Sorrentino V., Danieli G.A. Mutations in the cardiac ryanodine receptor gene (hRyR2) underlie catecholaminergic polymorphic ventricular tachycardia. Circulation. 2001;103:196–200. doi: 10.1161/01.cir.103.2.196. [DOI] [PubMed] [Google Scholar]
- 11.Lahat H., Pras E., Olender T., Avidan N., Ben-Asher E., Man O., Levy-Nissenbaum E., Khoury A., Lorber A., Goldman B. A missense mutation in a highly conserved region of CASQ2 is associated with autosomal recessive catecholamine-induced polymorphic ventricular tachycardia in Bedouin families from Israel. Am. J. Hum. Genet. 2001;69:1378–1384. doi: 10.1086/324565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mohler P.J., Splawski I., Napolitano C., Bottelli G., Sharpe L., Timothy K., Priori S.G., Keating M.T., Bennett V. A cardiac arrhythmia syndrome caused by loss of ankyrin-B function. Proc. Natl. Acad. Sci. USA. 2004;101:9137–9142. doi: 10.1073/pnas.0402546101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tester D.J., Arya P., Will M., Haglund C.M., Farley A.L., Makielski J.C., Ackerman M.J. Genotypic heterogeneity and phenotypic mimicry among unrelated patients referred for catecholaminergic polymorphic ventricular tachycardia genetic testing. Heart Rhythm. 2006;3:800–805. doi: 10.1016/j.hrthm.2006.03.025. [DOI] [PubMed] [Google Scholar]
- 14.Bhuiyan Z.A., Hamdan M.A., Shamsi E.T., Postma A.V., Mannens M.M., Wilde A.A., Al-Gazali L. A novel early onset lethal form of catecholaminergic polymorphic ventricular tachycardia maps to chromosome 7p14-p22. J. Cardiovasc. Electrophysiol. 2007;18:1060–1066. doi: 10.1111/j.1540-8167.2007.00913.x. [DOI] [PubMed] [Google Scholar]
- 15.Cerrone M., Napolitano C., Priori S.G. Catecholaminergic polymorphic ventricular tachycardia: A paradigm to understand mechanisms of arrhythmias associated to impaired Ca(2+) regulation. Heart Rhythm. 2009;6:1652–1659. doi: 10.1016/j.hrthm.2009.06.033. [DOI] [PubMed] [Google Scholar]
- 16.Kontula K., Laitinen P.J., Lehtonen A., Toivonen L., Viitasalo M., Swan H. Catecholaminergic polymorphic ventricular tachycardia: Recent mechanistic insights. Cardiovasc. Res. 2005;67:379–387. doi: 10.1016/j.cardiores.2005.04.027. [DOI] [PubMed] [Google Scholar]
- 17.Mohler P.J., Wehrens X.H. Mechanisms of human arrhythmia syndromes: Abnormal cardiac macromolecular interactions. Physiology (Bethesda) 2007;22:342–350. doi: 10.1152/physiol.00018.2007. [DOI] [PubMed] [Google Scholar]
- 18.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 19.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 20.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zhang M., Tanaka T., Ikura M. Calcium-induced conformational transition revealed by the solution structure of apo calmodulin. Nat. Struct. Biol. 1995;2:758–767. doi: 10.1038/nsb0995-758. [DOI] [PubMed] [Google Scholar]
- 22.Maximciuc A.A., Putkey J.A., Shamoo Y., Mackenzie K.R. Complex of calmodulin with a ryanodine receptor target reveals a novel, flexible binding mode. Structure. 2006;14:1547–1556. doi: 10.1016/j.str.2006.08.011. [DOI] [PubMed] [Google Scholar]
- 23.Chattopadhyaya R., Meador W.E., Means A.R., Quiocho F.A. Calmodulin structure refined at 1.7 A resolution. J. Mol. Biol. 1992;228:1177–1192. doi: 10.1016/0022-2836(92)90324-d. [DOI] [PubMed] [Google Scholar]
- 24.Rozen S., Skaletsky H. Primer3 on the WWW for general users and for biologist programmers. Methods Mol. Biol. 2000;132:365–386. doi: 10.1385/1-59259-192-2:365. [DOI] [PubMed] [Google Scholar]
- 25.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Priori S.G., Napolitano C., Memmi M., Colombi B., Drago F., Gasparini M., DeSimone L., Coltorti F., Bloise R., Keegan R. Clinical and molecular characterization of patients with catecholaminergic polymorphic ventricular tachycardia. Circulation. 2002;106:69–74. doi: 10.1161/01.cir.0000020013.73106.d8. [DOI] [PubMed] [Google Scholar]
- 27.Tadross M.R., Dick I.E., Yue D.T. Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell. 2008;133:1228–1240. doi: 10.1016/j.cell.2008.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Overgaard M.T., Oxvig C., Christiansen M., Lawrence J.B., Conover C.A., Gleich G.J., Sottrup-Jensen L., Haaning J. Messenger ribonucleic acid levels of pregnancy-associated plasma protein-A and the proform of eosinophil major basic protein: expression in human reproductive and nonreproductive tissues. Biol. Reprod. 1999;61:1083–1089. doi: 10.1095/biolreprod61.4.1083. [DOI] [PubMed] [Google Scholar]
- 29.VanScyoc W.S., Newman R.A., Sorensen B.R., Shea M.A. Calcium binding to calmodulin mutants having domain-specific effects on the regulation of ion channels. Biochemistry. 2006;45:14311–14324. doi: 10.1021/bi061134d. [DOI] [PubMed] [Google Scholar]
- 30.Peersen O.B., Madsen T.S., Falke J.J. Intermolecular tuning of calmodulin by target peptides and proteins: differential effects on Ca2+ binding and implications for kinase activation. Protein Sci. 1997;6:794–807. doi: 10.1002/pro.5560060406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patton C., Thompson S., Epel D. Some precautions in using chelators to buffer metals in biological solutions. Cell Calcium. 2004;35:427–431. doi: 10.1016/j.ceca.2003.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Rodney G.G., Moore C.P., Williams B.Y., Zhang J.Z., Krol J., Pedersen S.E., Hamilton S.L. Calcium binding to calmodulin leads to an N-terminal shift in its binding site on the ryanodine Receptor. J. Biol. Chem. 2001;276:2069–2074. doi: 10.1074/jbc.M008891200. [DOI] [PubMed] [Google Scholar]
- 33.Yamaguchi N., Xu L., Pasek D.A., Evans K.E., Meissner G. Molecular basis of calmodulin binding to cardiac muscle Ca(2+) release channel (ryanodine receptor) J. Biol. Chem. 2003;278:23480–23486. doi: 10.1074/jbc.M301125200. [DOI] [PubMed] [Google Scholar]
- 34.Ono M., Yano M., Hino A., Suetomi T., Xu X., Susa T., Uchinoumi H., Tateishi H., Oda T., Okuda S. Dissociation of calmodulin from cardiac ryanodine receptor causes aberrant Ca2+ release in heart failure. Cardiovasc. Res. 2010;87:609–617. doi: 10.1093/cvr/cvq108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu X., Yano M., Uchinoumi H., Hino A., Suetomi T., Ono M., Tateishi H., Oda T., Okuda S., Doi M. Defective calmodulin binding to the cardiac ryanodine receptor plays a key role in CPVT-associated channel dysfunction. Biochem. Biophys. Res. Commun. 2010;394:660–666. doi: 10.1016/j.bbrc.2010.03.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Suetomi T., Yano M., Uchinoumi H., Fukuda M., Hino A., Ono M., Xu X., Tateishi H., Okuda S., Doi M. Mutation-linked defective interdomain interactions within ryanodine receptor cause aberrant Ca2+release leading to catecholaminergic polymorphic ventricular tachycardia. Circulation. 2011;124:682–694. doi: 10.1161/CIRCULATIONAHA.111.023259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Uchinoumi H., Yano M., Suetomi T., Ono M., Xu X., Tateishi H., Oda T., Okuda S., Doi M., Kobayashi S. Catecholaminergic polymorphic ventricular tachycardia is caused by mutation-linked defective conformational regulation of the ryanodine receptor. Circ. Res. 2010;106:1413–1424. doi: 10.1161/CIRCRESAHA.109.209312. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.