

Abstract
Mutations in the genes composing the mitochondrial translation apparatus are an important cause of a heterogeneous group of oxidative phosphorylation (OXPHOS) disorders. We studied the index case in a consanguineous family in which two children presented with severe encephalopathy, lactic acidosis, and intractable seizures leading to an early fatal outcome. Blue native polyacrylamide gel electrophoretic (BN-PAGE) analysis showed assembly defects in all of the OXPHOS complexes with mtDNA-encoded structural subunits, and these defects were associated with a severe deficiency in mitochondrial translation. Immunoblot analysis showed reductions in the steady-state levels of several structural subunits of the mitochondrial ribosome. Whole-exome sequencing identified a homozygous missense mutation (c.1250G>A) in an uncharacterized gene, RMND1 (required for meiotic nuclear division 1). RMND1 localizes to mitochondria and behaves as an integral membrane protein. Retroviral expression of the wild-type RMND1 cDNA rescued the biochemical phenotype in subject cells, and siRNA-mediated knockdown of the protein recapitulated the defect. BN-PAGE, gel filtration, and mass spectrometry analyses showed that RMND1 forms a high-molecular-weight and most likely homopolymeric complex (∼240 kDa) that does not assemble in subject fibroblasts but that is rescued by expression of RMND1 cDNA. The p.Arg417Gln substitution, predicted to be in a coiled-coil domain, which is juxtaposed to a transmembrane domain at the extreme C terminus of the protein, does not alter the steady-state level of RMND1 but might prevent protein-protein interactions in this complex. Our results demonstrate that the RMND1 complex is necessary for mitochondrial translation, possibly by coordinating the assembly or maintenance of the mitochondrial ribosome.
Main Text
Mitochondria contain their own translation machinery for the synthesis of the 13 mtDNA-encoded polypeptides that are essential structural components of the oxidative phosphorylation (OXPHOS) complexes. Whereas the mitochondrial genome (mtDNA) itself codes for the two rRNAs and 22 tRNAs necessary for translation, all of the other components of the translation apparatus are encoded in the nucleus and must be targeted to the mitochondrion. Mutations in genes affecting mitochondrial translation are common causes of OXPHOS disorders in adults and children.1,2 For instance, mutations in all of the mitochondrial tRNAs have been associated with disease, and although they make up less than 10% of mtDNA, they account for a large proportion of mtDNA mutations associated with human mitochondrial disease.3 Most of the diseases that have been identified in the nuclear-encoded components of the translation machinery are early-onset, fatal disorders that are inherited in an autosomal-recessive fashion.2 Although not universally true, many of the translation defects cause deficiencies in more than one OXPHOS complex. There is, however, a great deal of heterogeneity (often gene specific, but not always) in phenotypic presentation, which is an enduring mystery. In addition, it is likely that many translation factors remain unknown as new factors continue to be identified by the investigation of individuals with translation defects.4
In this study, we investigated a subject who presented with an encephalopathy and lactic acidosis. Informed consent was obtained, and the institutional review board at the Montreal Neurological Institute approved the research studies. The subject, a girl, was the fourth child of consanguineous parents. There are three older female siblings, the first two of whom are normal. The third daughter developed intractable seizures at 2 months of age, and a computed tomography (CT) scan showed cerebral atrophy and microcephaly. She died at the age of 13 months. The proband was normal at birth but developed seizures on day 6. She was hypotonic and required tube feeding. At 4 months of age, she had unremitting seizures and her head circumference was not increasing. The liver was slightly enlarged, but there was no splenomegaly. The blood lactate concentration was 3.56 mmol/l (the normal range is 0.63–2.44 mmol/l), and the cerebrospinal-fluid lactate concentration was 3.43 mmol/l (the normal range is 0.90–2.80 mmol/l). The lactate to pyruvate ratio in the blood was 21.2 (the normal ratio is 10–20). Blood ammonia, urine amino acids, and organic acids and lysosomal-enzyme studies were all normal. Cytochrome c oxidase (COX) activity in cultured fibroblasts was 4 nmol/mg protein/min (the normal range is 30–90), and the COX to citrate synthase ratio was 0.11 (normal is >1). The subject died at 5 months of age. At autopsy, the brain showed marked atrophy of the cortex but a relatively normal cerebellum and brain stem. The cord was also atrophic. There was symmetrical ventricular dilatation, and the corpus callosum was thin. Microscopically, there was widespread vacuolation of cortical gray matter and status spongiosus and extensive loss of myelin in the brain stem.
To further investigate the nature of the biochemical defect, we first immortalized the subject fibroblasts as described previously.5 The COX activity in these cells was 26 ± 11% of control levels (n = 34). BN-PAGE analysis6 revealed marked assembly defects in complexes I, III, IV, and V (Figure 1A), suggesting a mitochondrial-translation defect. Confirming this prediction, the synthesis of the mtDNA-encoded polypeptides, investigated by pulse labeling the mitochondrial translation products with a mixture of [35S] methionine and [35S] cysteine,7 was severely and uniformly decreased to <20% of control levels (Figure 1B). Although this pattern of mitochondrial-translation deficiency is similar to that found in other subjects with genetically defined mitochondrial-translation defects,8–10 it represents the most severe generalized mitochondrial-translation decrease that we have observed.
Figure 1.
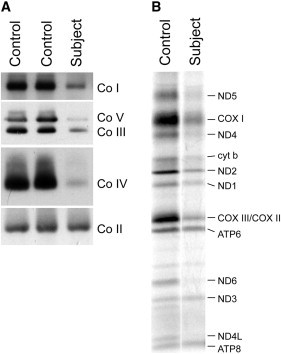
Characterization of Biochemical and Molecular Defects in Subject Fibroblasts
Control and subject fibroblasts were analyzed by BN-PAGE (A) and by pulse labeling of the mtDNA-encoded polypeptides (B).
(A) Each of the five OXPHOS complexes (I–V) was visualized with a subunit-specific antibody that recognizes the native complex as follows: CoI (NDUFA9), CoII (SDHA), CoIII (UQCRC1), CoIV (COX4), CoV (ATP5A1). Complex II is the loading control.
(B) The seven subunits of complex I (ND), one subunit of complex III (cyt b), three subunits of complex IV (COX), and two subunits of complex V (ATP) are indicated to the right of the figure.
Southern blot analysis of subject fibroblasts showed no decrease in the level of mtDNA when normalized to the cytoplasmic 18S rRNA gene (Figure 2A), excluding a mtDNA depletion syndrome. Northern blot analysis of several mitochondrial mRNAs (normalized to actin mRNA) showed an increase of 2–5 fold in all of those examined (Figure 2B). The 12S mitochondrial rRNA level was also 1.5× higher in subject fibroblasts than in controls; however, the 16S rRNA was relatively decreased (0.8× the control) (Figure 2B). Immunoblot analysis, carried out on fibroblasts solubilized in 1.5% lauryl maltoside, showed near control levels of the mitochondrial-translation elongation factors but severely decreased levels of several mitochondrial ribosomal subunits (MRPL13, MRPL32, and MRPS2) (Figure 2C), suggesting that the mitochondrial-translation defect could be due to impaired assembly or maintenance of the mitochondrial ribosome.
Figure 2.
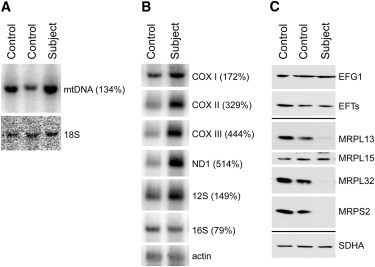
Steady-State Levels of Mitochondrial DNA, mRNAs, rRNAs, and Mitochondrial-Translation Proteins
(A) Southern blot analysis of genomic DNA extracted from control and subject fibroblasts. Hybridization was performed with probes directed against a 16 kb fragment of the mitochondrial genome, and the nuclear 18S rRNA gene was used as a loading control.
(B) Northern blot analysis carried out with total RNA extracted from control and subject fibroblasts. Hybridization was performed with probes specific to mitochondrial mRNAs encoding the three COX subunits, one of the complex I subunits (ND1), and the 12S and 16S mitochondrial ribosomal RNAs. Beta-actin was used as the loading control.
(C) Immunoblot analysis of control and subject fibroblasts with antibodies against the mitochondrial-translation elongation factors (EFG1and EFTs) and the mitochondrial ribosomal proteins MRPL32 (a kind gift of T. Langer, Cologne), MRPL13, MRPL15, and MRPS2 (kind gifts of L. Spremulli, UNC Chapel Hill). The 70 kDa subunit of complex II (SDHA) was used as a loading control.
Microcell-mediated chromosome-transfer studies11 and sequencing of several candidate genes failed to reveal a causative mutation, so we used whole-exome sequencing to search for a homozygous mutation. DNA from exonic regions was captured with the Agilent SureSelect Human All Exon Kit v.1 and sequenced on one lane of an Illumina GAIIx with single-end sequencing of 76 bp reads. Read alignment and variant identification were performed as previously described.12 The average coverage in the National Center for Biotechnology Information Consensus Coding Sequence was 23.5×, resulting in approximately 65,000 variants passing the SAMtools varFilter13 (default parameters, as well as a minimum SNP quality of 20 and a minimum indel quality of 50, were used). This number was significantly reduced to approximately 1,000 candidates after we filtered any previously identified variants in dbSNP (v.131), the 1000 Genomes Project (July 2010 release), and ten local exomes. In addition, because of the subject’s consanguinity, homozygous nonsynonymous alleles were prioritized, leading to only 22 candidates. Finally, we searched for known and predicted mitochondrial proteins (by using the Mitocarta database14), and RMND1 (required for meiotic nuclear division 1), an uncharacterized gene, emerged as the only candidate. Sanger sequencing of RMND1 in the subject cDNA and genomic DNA confirmed the homozygous missense mutation (c.1250G>A) identified by whole-exome sequencing (Figure 3A); this mutation was predicted to cause amino acid substitution p.Arg417Gln (RefSeq accession number NM_017909.2). DNA was not available for analysis from any of the other family members.
Figure 3.
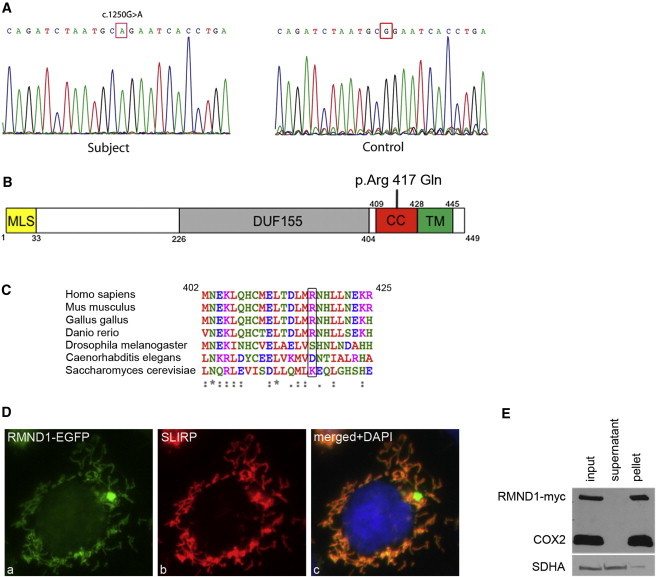
Mutational Analysis of RMND1 and Mitochondrial Localization of the Protein
(A) DNA sequence analysis of RMND1 cDNA indicates the position of the homozygous c.1250G>A mutation in the subject compared to the control.
(B) A schematic representation of RMND1 (not to scale) shows the predicted domains and the position of the p.Arg417Gln substitution. The following abbreviations are used: MLS, mitochondrial localization signal; DUF155, domain of unknown function 155; CC, coiled-coil domain; and TM, transmembrane domain.
(C) The alignment of the amino acid sequences of RMND1 homologs in different species shows that the mutated arginine (black rectangle) is conserved only in the vertebrates.
(D) Control fibroblasts transiently expressing RMND1-EGFP (left panel, green) were incubated with an antibody against the mitochondrial protein SLIRP (middle panel, red). The far right panel showing the overlay is counterstained with DAPI for visualization of the nucleus.
(E) Alkaline carbonate extraction of mitochondria from HEK cells stably expressing a C-terminal Myc-tagged RMND1. Immunoblot analysis with an Myc antibody shows that RMND1 is an integral membrane protein. SDHA (soluble, membrane-associated protein) and COX subunit 2 (integral inner membrane protein) were used as controls.
Bioinformatic analysis of the protein sequence revealed the presence of a DUF155 domain (Figure 3B) that is shared by members of the RMD1/Sif2 family comprising RMD1 (the yeast homolog of RMND1) and four other yeast genes, viz., RMD8, SIF2, SIF3, and YDR282C. RMND1 is also predicted to contain a mitochondrial localization signal at the N terminus and a coiled-coil domain (where the mutation is localized) juxtaposed to a transmembrane domain at the extreme C terminus (Figure 3B).
RMND1 is evolutionarily conserved down to yeast (from which the name is derived), but the S. cerevisiae gene product is reported to be cytosolic in a high-throughput study of the cellular localization of the gene products expressed from genes chromosomally tagged with green fluorescent protein.15 All the other members of the SIF2 family are thought to have either nuclear or cytosolic localizations. Alignment of the amino acid sequences of RMND1 homologs (Figure 3C) shows that the mutated residue is only conserved in vertebrates (40 of 40 vertebrates for which the complete RMND1 sequence is available in Ensembl). For confirming the mitochondrial localization of RMND1, control fibroblasts transiently expressing RMND1-EGFP were grown on coverslips, fixed with 4% paraformaldehyde, and analyzed by immunofluorescence. This analysis showed a nearly perfect colocalization of the EGFP signal with mitochondria detected with a SLIRP antibody (Figure 3D). Alkaline carbonate extraction of isolated mitochondria5 from a stable human embryonic kidney (HEK) cell line expressing RMND1-Myc demonstrated that it behaves as an integral membrane protein (Figure 3E). Similar results were obtained for the native protein in both control and subject fibroblasts (data not shown).
To confirm the pathogenicity of the RMND1 mutation, we used a retroviral vector to express the wild-type RMND1 cDNA in subject fibroblasts. The RMND1 cDNA was amplified by OneStep RT-PCR (QIAGEN) with specific primers modified for Gateway-compatible vectors and was cloned into pBABE, modified to be Gateway-compatible.4 The Phoenix packaging cell line was transiently transfected with the retroviral construct according to the HBS/Ca3(PO4)2 method (see Web Resources). Fibroblasts were infected 48 hr later by exposure to virus-containing medium in the presence of 4 μg/ml of polybrene.4 Expression of the wild-type RMND1 cDNA rescued the assembly defects in the OXPHOS complexes (Figure 4A). (RMND1 C-terminally tagged with either a flag or Myc epitope also rescued the assembly defects; data not shown). Immunoblot analysis with a polyclonal antibody (Sigma) raised against a peptide antigen (amino acids 336–412) of RMND1 showed a specific 35 kDa band that increased in intensity when the cDNA was expressed from the retroviral vector (Figure 4B), suggesting that RMND1 is processed beyond the mitochondrial localization sequence (amino acid residue 32, predicted by MITOPROT) because the molecular weight of that species would be 48 kDa. Consistent with this interpretation, analysis of immunoprecipitated RMND1 by mass spectrometry (n = 7) failed to identify a peptide N terminal of amino acid 127, whereas the coverage of the protein C terminal to this residue was 73%. The steady-state level of RMND1 in subject fibroblasts was similar to that in controls (Figure 4B), indicating that the mutation does not destabilize the protein and that the phenotype is most likely due to a loss of function of the mutant RMND1. The steady-state levels of all analyzed OXPHOS-complex components, as well as mitochondrial ribosomal proteins in subject fibroblasts expressing the wild-type RMND1 cDNA, were also comparable to levels in controls (Figure 4B). Finally, we tested whether expression of the wild-type RMND1 rescued the mitochondrial-translation defect in subject fibroblasts. Mitochondrial protein synthesis was completely restored by retroviral expression of either RMND1-Myc or RMND1-flag in subject fibroblasts (Figure 4C).
Figure 4.
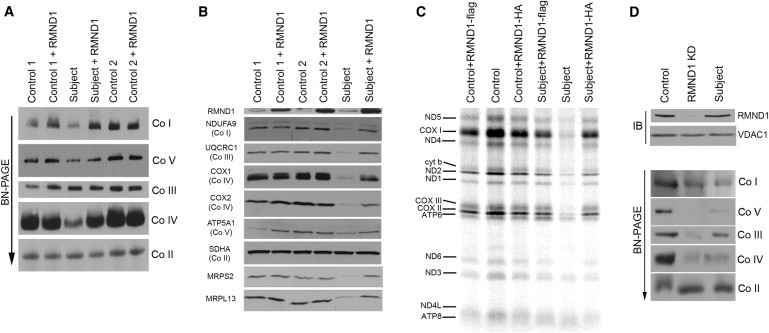
Rescue of the Biochemical Phenotype by RMND1 Expression and Recapitulation of the Defect by siRNA-Mediated Knockdown of the Protein
(A) BN-PAGE analysis of controls and subject fibroblasts expressing RMND1 from a retroviral vector (pBABE). Each of the five OXPHOS complexes (I–V) was visualized with a subunit-specific antibody.
(B) Immunoblot analysis of the same samples as in (A) for expression of RMND1, individual structural subunits of the five OXPHOS complexes, and two mitochondrial ribosomal subunits. The 70 kDa subunit of complex II (SDHA) was used as a loading control.
(C) Analysis of mitochondrial translation products in control and subject fibroblasts transduced with retroviral vectors expressing RMND1-HA or RMND1-flag.
(D) Stealth RNAi-mediated knockdown of RMND1. The upper panel shows the level of knockdown of RMND1 on an immunoblot with VDAC1 (porin) as a loading control. The bottom panel shows the BN-PAGE analysis in the control, RMND1 knockdown, and subject cells.
To further investigate the function of RMND1, we used RNAi-mediated knockdown of the protein. Three different Stealth RNAi duplexes were designed with Block-iT RNAi Designer (Invitrogen). The different RNAi constructs were transiently transfected into control fibroblasts with Lipofectamine RNAimax (Invitrogen) at a final concentration of 12 nM. The transfections were repeated on days 3 and 6, and the cells were harvested and analyzed on day 9. The Stealth KD2 duplex (5′−CAAACCAAAUCUGUUGGGUUCUAAA-3′) produced the most robust RMND1 knockdown (Figure 4D, top panel), so we continued the BN-PAGE analysis with this construct. The OXPHOS-assembly defects produced by RNAi-mediated knockdown of RMND1 were very similar to those found in the RMND1 subject (Figure 4D), confirming that RMND1 is required for normal assembly of the OXPHOS complexes.
We next asked whether RMND1 was part of a protein complex. We isolated mitochondria from HEK cells stably expressing RMND1-Myc, solubilized them, and analyzed them by gel filtration as previously described.16 RMND1-Myc and the endogenous RMND1 eluted in the fractions between the peak fractions from COX (230 kDa, Figure 5A) and LRPPRC10 (250 kDa, Figure 5A). To confirm that RMND1 formed a high-molecular-weight complex, we carried out a similar analysis by using BN-PAGE. The complex containing RMND1 ran slightly slower than that of COX on the first-dimension BN-PAGE, confirming that it is approximately 240 kDa in size. Strikingly, this complex almost completely failed to assemble in the subject cells but was restored by retroviral expression of wild-type RMND1-Myc (Figure 5B). These data indicate that the mutation in the coiled-coil domain of RMND1 prevents the formation of a higher-order complex and that the complex is essential for RMND1 function. To establish whether the complex contained other proteins, we immunoprecipitated RMND1-Myc with an Myc antibody and ran the immunoprecipitate on a BN-PAGE gel. Mass-spectrometry analysis of the purified complex identified only RMND1 (data not shown). Although we cannot rule out the presence of other proteins at the moment, this analysis suggests that RMND1 very likely assembles into a homopolymeric complex.
Figure 5.
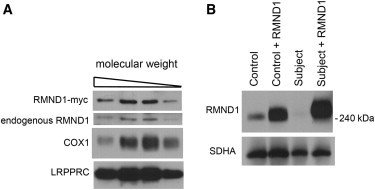
RMND1 Is Part of a High-Molecular-Weight Protein Complex that Does Not Assemble in the Subject
(A) Size-exclusion-chromatography analysis was carried out with mitochondria from HEK cells expressing RMND1-Myc. Immunoblotting with antibodies against either the native protein or the Myc epitope demonstrated that both the endogenous and overexpressed proteins are part of a complex of about 240 kDa. The COX subunit 1 of COX (230 kDa) and LRPPRC (250 kDa) were used as molecular-weight references.
(B) BN-PAGE analysis of control and subject fibroblasts expressing RMND1-Myc from a retroviral vector. An antibody directed against the native protein shows that RMND1 forms a 250 kDa complex that does not assemble in the subject. The complex is restored in subject cells expressing RMND1-Myc. SDHA was used as a loading control.
Although we do not know the exact molecular function of RMND1, our data show that it is part of a large mitochondrial-membrane complex that is required for the translation of the mitochondrially encoded polypeptides and is thus essential for the biogenesis of the OXPHOS complexes. The gene products of all the other members of the RMD/SIF2 family localize to the cytoplasm or nucleus and have a function in some aspect of meiosis or sporulation in yeast species. Sif2p, for example, is part of the Set3 histone deacetylase complex that represses a set of meiotic genes.17 Although the function of Rmd1p, the S. cerevisae homolog of RMND1, remains unknown, there is no evidence that it has a role in mitochondrial translation or OXPHOS function. RMND1 has apparently evolved a new function related to mitochondrial translation in mammalian cells. The substantial decreases in the steady-state levels in some of the mitochondrial ribosomal subunits and the relative decrease in the 16S rRNA in the RMND1 subject cells both point to a role for RMND1 in some aspect of the assembly or maintenance of the mitochondrial ribosome.
Ribosome assembly is a complex, highly coordinated process in which protein binding and posttranscriptional nucleotide modifications drive and stabilize rRNA structures until the native state is reached. How this is regulated in mammalian mitochondria is largely unknown, although a few ribosome-biogenesis factors have been identified. ERAL118,19 and the PPR protein PTCD320 bind and stabilize the 12S rRNA, and the mitochondrial transcription factor TFB1M is responsible for dimethylating two highly conserved adenines in a stem loop near the 3′ end of the 12S rRNA.21,22 A GTPase, C4orf14 (NOA1), associates with the mt-SSU, but its precise molecular role has not been defined.23 Knockdown of any of these factors compromises mt-SSU assembly and mitochondrial translation.
The only known protein that binds the 16S rRNA is mTERF4, which forms a complex with the RNA dimethyl transferase NSUN4 presumably to methylate a cytosine residue (not yet identified) on the 16S rRNA.24 Interestingly, a tissue-specific knockout of Mterf4 in the heart results in a large increase in both mt-SSU and mt-LSU without a corresponding increase in monosome formation, suggesting that it is not required for the assembly or stability of the individual subunits per se. The GTPase MTG1 is thought to play a role in mt-LSU assembly on the basis of its ability to partially rescue a deletion of the yeast ortholog, but its precise role has not been defined.25 Assembly of the mature ribosome requires proteolytic processing of MRPL32, a task performed by the m-AAA protease.26 C7orf30, a protein of the widely expressed DUF143 family, was shown to associate with the mt-LSU,27,28 specifically with MRPL14,29 but its role in the assembly of mt-LSU and the downstream effects on translation remain controversial. Aside from the m-AAA protease in the list above, RMND1 stands out because of all the proteins involved in ribosome biogenesis, it is the only one that forms a membrane-anchored complex. We speculate that this could act to stabilize or position the mitochondrial ribosome perhaps by linking it through to the outer membrane. Further detailed studies will be necessary for testing this hypothesis.
Acknowledgments
This work was supported in part by Canadian Institutes of Health Research (CIHR) grants to E.A.S. and J.M. E.L. was supported by a CIHR fellowship. A.J. is supported by a postdoctoral fellowship from the Réseau de Médecine Génétique Appliquée. E.A.S. is an International Scholar of the Howard Hughes Medical Institute. J.M. is a recipient of a Canada Research Chair. The authors wish to acknowledge the contribution of the high-throughput sequencing platform of the McGill University and Genome Québec Innovation Centre (Montréal, Canada). We thank T. Johns and S. Salomon for technical assistance.
Web Resources
The URLs for data presented herein are as follows:
BLOCK-iT RNAi Designer, http://rnaidesigner.invitrogen.com/rnaiexpress
Transfection protocol for Phoenix retroviral packaging cell line, http://www.stanford.edu/group/nolan/protocols/pro_helper_dep.html
References
- 1.Smits P., Smeitink J., van den Heuvel L. Mitochondrial translation and beyond: Processes implicated in combined oxidative phosphorylation deficiencies. J. Biomed. Biotechnol. 2010;2010:737385. doi: 10.1155/2010/737385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Koopman W.J., Willems P.H., Smeitink J.A. Monogenic mitochondrial disorders. N. Engl. J. Med. 2012;366:1132–1141. doi: 10.1056/NEJMra1012478. [DOI] [PubMed] [Google Scholar]
- 3.Greaves L.C., Reeve A.K., Taylor R.W., Turnbull D.M. Mitochondrial DNA and disease. J. Pathol. 2012;226:274–286. doi: 10.1002/path.3028. [DOI] [PubMed] [Google Scholar]
- 4.Weraarpachai W., Sasarman F., Nishimura T., Antonicka H., Auré K., Rötig A., Lombès A., Shoubridge E.A. Mutations in C12orf62, a factor that couples COX I synthesis with cytochrome c oxidase assembly, cause fatal neonatal lactic acidosis. Am. J. Hum. Genet. 2012;90:142–151. doi: 10.1016/j.ajhg.2011.11.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Yao J., Shoubridge E.A. Expression and functional analysis of SURF1 in Leigh syndrome patients with cytochrome c oxidase deficiency. Hum. Mol. Genet. 1999;8:2541–2549. doi: 10.1093/hmg/8.13.2541. [DOI] [PubMed] [Google Scholar]
- 6.Leary S.C., Sasarman F. Oxidative phosphorylation: Synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol. Biol. 2009;554:143–162. doi: 10.1007/978-1-59745-521-3_10. [DOI] [PubMed] [Google Scholar]
- 7.Sasarman F., Shoubridge E.A. Radioactive labeling of mitochondrial translation products in cultured cells. Methods Mol. Biol. 2012;837:207–217. doi: 10.1007/978-1-61779-504-6_14. [DOI] [PubMed] [Google Scholar]
- 8.Antonicka H., Sasarman F., Kennaway N.G., Shoubridge E.A. The molecular basis for tissue specificity of the oxidative phosphorylation deficiencies in patients with mutations in the mitochondrial translation factor EFG1. Hum. Mol. Genet. 2006;15:1835–1846. doi: 10.1093/hmg/ddl106. [DOI] [PubMed] [Google Scholar]
- 9.Antonicka H., Ostergaard E., Sasarman F., Weraarpachai W., Wibrand F., Pedersen A.M., Rodenburg R.J., van der Knaap M.S., Smeitink J.A., Chrzanowska-Lightowlers Z.M., Shoubridge E.A. Mutations in C12orf65 in patients with encephalomyopathy and a mitochondrial translation defect. Am. J. Hum. Genet. 2010;87:115–122. doi: 10.1016/j.ajhg.2010.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sasarman F., Brunel-Guitton C., Antonicka H., Wai T., Shoubridge E.A., LSFC Consortium LRPPRC and SLIRP interact in a ribonucleoprotein complex that regulates posttranscriptional gene expression in mitochondria. Mol. Biol. Cell. 2010;21:1315–1323. doi: 10.1091/mbc.E10-01-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu Z., Yao J., Johns T., Fu K., De Bie I., Macmillan C., Cuthbert A.P., Newbold R.F., Wang J., Chevrette M. SURF1, encoding a factor involved in the biogenesis of cytochrome c oxidase, is mutated in Leigh syndrome. Nat. Genet. 1998;20:337–343. doi: 10.1038/3804. [DOI] [PubMed] [Google Scholar]
- 12.Lalonde E., Albrecht S., Ha K.C., Jacob K., Bolduc N., Polychronakos C., Dechelotte P., Majewski J., Jabado N. Unexpected allelic heterogeneity and spectrum of mutations in Fowler syndrome revealed by next-generation exome sequencing. Hum. Mutat. 2010;31:918–923. doi: 10.1002/humu.21293. [DOI] [PubMed] [Google Scholar]
- 13.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pagliarini D.J., Calvo S.E., Chang B., Sheth S.A., Vafai S.B., Ong S.E., Walford G.A., Sugiana C., Boneh A., Chen W.K. A mitochondrial protein compendium elucidates complex I disease biology. Cell. 2008;134:112–123. doi: 10.1016/j.cell.2008.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Huh W.K., Falvo J.V., Gerke L.C., Carroll A.S., Howson R.W., Weissman J.S., O’Shea E.K. Global analysis of protein localization in budding yeast. Nature. 2003;425:686–691. doi: 10.1038/nature02026. [DOI] [PubMed] [Google Scholar]
- 16.Kaufman B.A., Durisic N., Mativetsky J.M., Costantino S., Hancock M.A., Grutter P., Shoubridge E.A. The mitochondrial transcription factor TFAM coordinates the assembly of multiple DNA molecules into nucleoid-like structures. Mol. Biol. Cell. 2007;18:3225–3236. doi: 10.1091/mbc.E07-05-0404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cerna D., Wilson D.K. The structure of Sif2p, a WD repeat protein functioning in the SET3 corepressor complex. J. Mol. Biol. 2005;351:923–935. doi: 10.1016/j.jmb.2005.06.025. [DOI] [PubMed] [Google Scholar]
- 18.Dennerlein S., Rozanska A., Wydro M., Chrzanowska-Lightowlers Z.M., Lightowlers R.N. Human ERAL1 is a mitochondrial RNA chaperone involved in the assembly of the 28S small mitochondrial ribosomal subunit. Biochem. J. 2010;430:551–558. doi: 10.1042/BJ20100757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uchiumi T., Ohgaki K., Yagi M., Aoki Y., Sakai A., Matsumoto S., Kang D. ERAL1 is associated with mitochondrial ribosome and elimination of ERAL1 leads to mitochondrial dysfunction and growth retardation. Nucleic Acids Res. 2010;38:5554–5568. doi: 10.1093/nar/gkq305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies S.M., Rackham O., Shearwood A.M., Hamilton K.L., Narsai R., Whelan J., Filipovska A. Pentatricopeptide repeat domain protein 3 associates with the mitochondrial small ribosomal subunit and regulates translation. FEBS Lett. 2009;583:1853–1858. doi: 10.1016/j.febslet.2009.04.048. [DOI] [PubMed] [Google Scholar]
- 21.Seidel-Rogol B.L., McCulloch V., Shadel G.S. Human mitochondrial transcription factor B1 methylates ribosomal RNA at a conserved stem-loop. Nat. Genet. 2003;33:23–24. doi: 10.1038/ng1064. [DOI] [PubMed] [Google Scholar]
- 22.Metodiev M.D., Lesko N., Park C.B., Cámara Y., Shi Y., Wibom R., Hultenby K., Gustafsson C.M., Larsson N.G. Methylation of 12S rRNA is necessary for in vivo stability of the small subunit of the mammalian mitochondrial ribosome. Cell Metab. 2009;9:386–397. doi: 10.1016/j.cmet.2009.03.001. [DOI] [PubMed] [Google Scholar]
- 23.He J., Cooper H.M., Reyes A., Di Re M., Kazak L., Wood S.R., Mao C.C., Fearnley I.M., Walker J.E., Holt I.J. Human C4orf14 interacts with the mitochondrial nucleoid and is involved in the biogenesis of the small mitochondrial ribosomal subunit. Nucleic Acids Res. 2012;40:6097–6108. doi: 10.1093/nar/gks257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cámara Y., Asin-Cayuela J., Park C.B., Metodiev M.D., Shi Y., Ruzzenente B., Kukat C., Habermann B., Wibom R., Hultenby K. MTERF4 regulates translation by targeting the methyltransferase NSUN4 to the mammalian mitochondrial ribosome. Cell Metab. 2011;13:527–539. doi: 10.1016/j.cmet.2011.04.002. [DOI] [PubMed] [Google Scholar]
- 25.Barrientos A., Korr D., Barwell K.J., Sjulsen C., Gajewski C.D., Manfredi G., Ackerman S., Tzagoloff A. MTG1 codes for a conserved protein required for mitochondrial translation. Mol. Biol. Cell. 2003;14:2292–2302. doi: 10.1091/mbc.E02-10-0636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nolden M., Ehses S., Koppen M., Bernacchia A., Rugarli E.I., Langer T. The m-AAA protease defective in hereditary spastic paraplegia controls ribosome assembly in mitochondria. Cell. 2005;123:277–289. doi: 10.1016/j.cell.2005.08.003. [DOI] [PubMed] [Google Scholar]
- 27.Wanschers B.F., Szklarczyk R., Pajak A., van den Brand M.A., Gloerich J., Rodenburg R.J., Lightowlers R.N., Nijtmans L.G., Huynen M.A. C7orf30 specifically associates with the large subunit of the mitochondrial ribosome and is involved in translation. Nucleic Acids Res. 2012;40:4040–4051. doi: 10.1093/nar/gkr1271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rorbach J., Gammage P.A., Minczuk M. C7orf30 is necessary for biogenesis of the large subunit of the mitochondrial ribosome. Nucleic Acids Res. 2012;40:4097–4109. doi: 10.1093/nar/gkr1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Häuser R., Pech M., Kijek J., Yamamoto H., Titz B., Naeve F., Tovchigrechko A., Yamamoto K., Szaflarski W., Takeuchi N. RsfA (YbeB) Proteins Are Conserved Ribosomal Silencing Factors. PLoS Genet. 2012;8:e1002815. doi: 10.1371/journal.pgen.1002815. [DOI] [PMC free article] [PubMed] [Google Scholar]