

Abstract
The discovery of mutations causing human disease has so far been biased toward protein-coding regions. Having excluded all annotated coding regions, we performed targeted massively parallel resequencing of the nonrepetitive genomic linkage interval at Xq28 of family MRX3. We identified in the binding site of transcription factor YY1 a regulatory mutation that leads to overexpression of the chromatin-associated transcriptional regulator HCFC1. When tested on embryonic murine neural stem cells and embryonic hippocampal neurons, HCFC1 overexpression led to a significant increase of the production of astrocytes and a considerable reduction in neurite growth. Two other nonsynonymous, potentially deleterious changes have been identified by X-exome sequencing in individuals with intellectual disability, implicating HCFC1 in normal brain function.
Main Text
Intellectual disability is one of the most genetically heterogeneous human disorders.1 Among these, X-chromosome-linked intellectual disability (XLID) has been the best characterized at >92 genes involved.2 Recent large-scale X-exome resequencing3 complemented by high-resolution copy-number profiling4 failed to provide imminent answers for >50% of 208 families studied. Both studies used exome-centric and thus coding-sequence-biased mutation searches. Currently, about 86% of ∼100,000 known mutations causing human disease reside in the coding sequences.5 Noncoding variants are frequently overlooked or deemed too hard to be investigated. However, the advent of more systematic whole-genome sequencing,6 together with constantly improving annotation of the human genome (ENCODE), is facilitating mutation inquiry also into the noncoding and regulatory sequences.
MRX3 is a family of northern European descent and is affected by nonsyndromic intellectual disability (MIM 309541), which was mapped to Xq28-qter in 1991.7 The affected males in the MRX3 family are generally facially nondysmorphic; a few of the males have been described as having subtle dysmorphism, such as a small chin, high arched palate, or tapered fingers, but no feature is consistent throughout the family. Body habitus is normal and consists of below-average height (<3rd to 25th percentile) and a head shape that is often brachycephalic and of variable size (5th to 97th percentile). Affected males function within a variable range, from mild to moderate intellectual disability. Some males live semi-independently with supervised employment, whereas others need full-time supervision. Many of the males show behavioral problems, including aggression, and are not easily managed. In addition, others are described as reclusive and display a number of autistic symptoms, including ritualized and obsessive behaviors, narrow interests, and little socialization. Only one of the affected males (V-14 in Figure S1, available online; now deceased) has been described as having psychotic symptoms. One male (VII-7 in Figure S1), who had absence seizures during childhood, has normal cerebral imaging. No other affected males have epilepsy or have had brain imaging. Obligate carrier females appear to be normal and show high (>96%) skewing of their X inactivation, as measured on white blood cells (data not shown). This study was approved by the Women’s and Children’s Health Network Human Research Ethics Committee, and informed consent was obtained from all participants.
The 5.6 Mb linkage interval of the MRX3 family is known to be rich in genes8 and harbors 108 protein-coding genes (Figure 1A), accounting for ∼2.42% of the whole sequence in the interval. For over 20 years, extensive studies, including the most recent large-scale approaches,3,4 have failed to identify the causative mutation. In the presence of solid genetic evidence for X chromosome involvement, we hypothesized that the mutation might reside outside the coding region. To identify potential noncoding variants, we designed a custom capture array targeted to all coding regions and putative functional noncoding regions in the linkage interval (Figure 1A). Genomic DNA from individual V-4 in the MRX3 family7 was enriched to an estimated mean of 1,416× (Figure S1 and data not shown). The captured DNA fragments were sequenced with the Illumina GAIIx massively parallel sequencing platform, and 4.78 × 106 65 bp reads (51.64% of the total number of reads) mapped to a repeat-masked, human genome reference sequence from UCSC (hg18) as described previously9 and covered 1.29 × 106 bp of unique sequences in the linkage interval (Figures S2 and S3). Combined with our previous X-exome Sanger sequencing data,3 only 356 bp (<0.25% of the coding region from the linkage interval) distributed over four genes showed zero coverage (Tables S1 and S2). These zero-coverage coding regions (which included the first exon of SLC6A8 [MIM 300036], a gene which has been shown to be mutated in XLID) were sequenced manually and were excluded from having DNA sequence variation in the MRX3 family.
Figure 1.
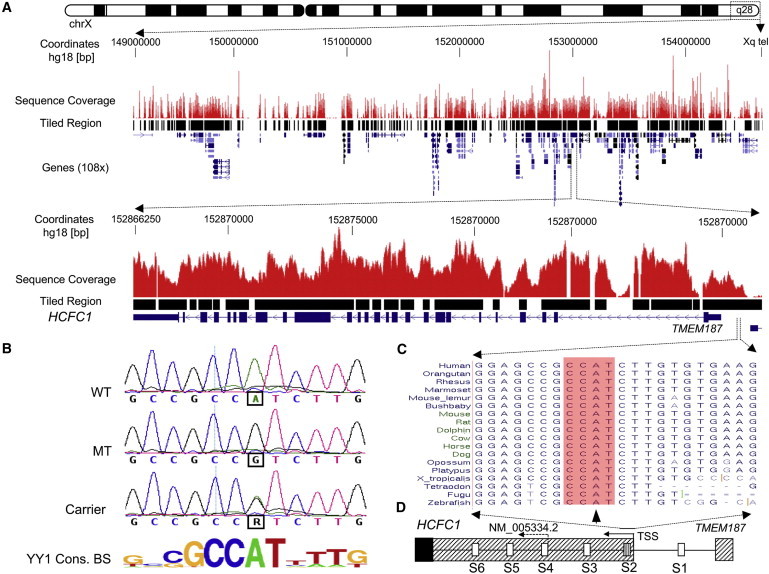
Sequencing, Mapping, and Identification of the chrX:152,890,455A>G Variant in a Core YY1 Binding Site of the HCFC1 Regulatory Region
(A) The upper panel shows targeted resequencing of the linkage interval in Xq28, an overview of the X chromosome, and custom tracks representing sequence-coverage histograms and tiled regions. The 108 genes in the region are displayed in the UCSC genome browser (hg18). The tiled region (∼5.5 Mb) on the capture array includes all exons from GenBank, mRNA and all ESTs, putative minimal promoter regions 2,000 bp upstream of the first exon in GenBank mRNAs, all snoRNAs, and miRNAs. The lower panel shows an overview of the genomic structure of HCFC1 and the sequence-coverage histograms.
(B) Partial sequence chromatograms showing the chrX:152,890,455A>G variant in the YY1 consensus DNA-binding site (BS; shown below) in a control male (WT), an affected male from the MRX3 family (MT), and a carrier female (carrier).
(C) Comparison of the sequences harboring the chrX:152,890,455A>G variant across 18 vertebrate species. The chrX:152,890,455A sequence is localized in the intergenic region between HCFC1 and TMEM187. The core motif (CCAT) of the YY1 binding site is highlighted with an arrowhead pointing at the chrX:152,890,455A nucleotide.
(D) Schematic of the partial genomic structure around the 5′ ends of HCFC1 and TMEM187. There are six predicted YY1 binding sites (marked as S1–S6) in the region. The arrows show the direction of HCFC1 transcription and the transcription start sites (TSSs) associated with reference sequence NM_005334.2. The new TSS was extended beyond the S2 YY1 binding site with the use of ESTs and other publicly available resources (ENCODE and DBTSS). The sequence of the chrX:152,890,455A>G variant is located within the S2 YY1 binding site. The hatched region indicates the 5′ UTR, whereas the solid box indicates the HCFC1 open reading frame.
In total, 383 genetic variants were called. We prioritized variants not present in dbSNP130 for further analysis (Figure S3). This prioritization identified 15 unique noncoding variants. We found no unique sequence variants (synonymous or nonsynonymous) in the protein-coding regions. Among the 15 unique noncoding variants, only the chrX:152,890,455A>G variant (UCSC hg18) is highly conserved (Table S3). This variant lies within the intergenic region between host cell factor C1 (HCFC1 or HCF-1 [MIM 300019]) and TMEM187 (or CXorf12 [MIM 300059]). Subsequently, we found that the chrX:152,890,455A>G variant localizes in a putative binding site for the transcription factor Yin-Yang-1 (YY1) (Figures 1B–1D). A recent study reported a de novo YY1 (MIM 600013) mutation as a plausible cause of intellectual disability.10 With TFSEARCH,11 the variant was predicted to abolish YY1 binding. Other variants were weakly conserved or poorly annotated and were not investigated for functional effect(s). There are six YY1 binding sites (S1–S6) in the same orientation in the region between HCFC1 and TMEM18712 (Figure 1D). We postulated that HCFC1 is the most likely affected gene. The S2 chrX:152,890,455A>G variant completely segregated with intellectual disability in the original and extended MRX3 pedigrees (Figure S1).
According to the HCFC1 reference sequence, NM_005334.2, the S2 chrX:152,890,455A>G variant would be in the intergenic region. However, our analysis of human expressed sequence tags (ESTs) and other publicly available resources (e.g., ENCODE and DBTSS) strongly suggests that the predominant HCFC1 transcription start site (TSS) is 450 bp upstream of NM_005334.2 at chrX: 152,890,463 (see Figure 2D) This makes the chrX:152,890,455A>G variant (and the S2 YY1 binding site) part of the HCFC1 5′ UTR, only ∼8 bp from the TSS of HCFC1 (Figures 1D and 2D).
Figure 2.
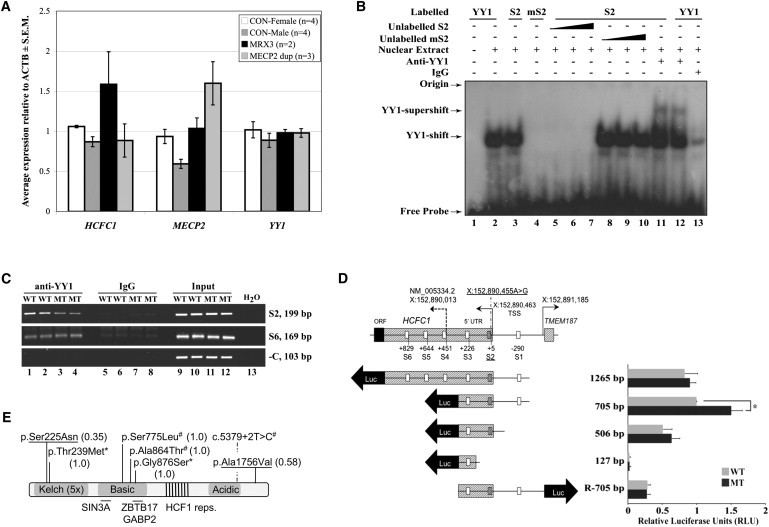
The chrX:152890455A>G Variant Leads to Upregulation of HCFC1 mRNA Expression through Loss of the YY1 Binding Site within Region S2
(A) qRT-PCR results show that affected MRX3 family members (n = 2) have significantly more upregulation of HCFC1 expression in LCLs than do controls (n = 4 for females and n = 4 for males) (p < 0.006 for MRX3 males against all controls) and individuals with MECP2 duplications (n = 3). Relative expression normalized against the ACTB mRNA is shown for HCFC1, MECP2, and YY1. qRT-PCR was performed as previously described13 with primers listed in Table S4. Error bars represent ± standard error of the mean (SEM).
(B) An electrophoretic mobility shift assay (EMSA) shows that the chrX:152,890,455A>G variant of S2 (mS2) abolishes YY1 binding. IgG is the unrelated rabbit IgG. Unlabelled probes are 25×, 100×, and 500× higher than labeled probes. The positions of the YY1 shift and YY1 supershift (antibody from Santa Cruz; sc-7341X) are indicated by arrows. Isolation of nuclear extracts and EMSA was performed as previously described14,15 with oligonucleotide probes listed in Table S5.
(C) The chrX:152,890,455A>G variant abolishes YY1 binding. A ChIP assay was performed on formaldehyde crosslinked LCL lysates from normal (WT; n = 2) and MRX3 (MT; n = 2) males with the use of rabbit anti-YY1 antibody or control IgG as previously described.16 The level of YY1 binding to the S2 region of HCFC1 was determined by semiquantitative PCR. The S6 YY1 binding site and an unrelated region of DNA around HUWE1 were used as negative controls. All primers are listed in Table S6. Whereas the region of S2 YY1 binding site shows a clear decrease of PCR product in the MRX3 males with respect to controls, this is not seen for a more distant S6 YY1 binding site PCR. A decrease rather than a complete loss of S2 PCR products in the MRX3 male LCL lysates is most likely due to immunoprecipitation of closely located YY1 binding sites (e.g., S1 and S3) rather than through the S2 site itself. Inputs (lanes 9–12) are equivalent to 1% of the immunoprecipitated samples (lanes 1–4 and 5–8).
(D) The left panel shows a schematic representation of the genomic structure of the 5′ ends of HCFC1 and TMEM187 and the pGL4 constructs for luciferase reporter assays. The positions of the HCFC1 TSS (including the one associated with NM_005334.2, as well as the new TSS site) and the TMEM187 TSS are indicated with UCSC hg18 coordinates and arrows. The positions of the YY1 S1–S6 sites are indicated with respect to the new HCFC1 TSS at chrX:152,890,463. The S2 site is crosshatched. Each different-length reporter plasmid has been made with wild-type (WT; A) and mutant (MT; G) nucleotides as follows: the 1,265 bp (chrX: 152,889,593−152,890,857), 705 bp (chrX: 152,890,153–152,890,857), and R-705 bp (chrX: 152,890,153–152,890,857) fragments were amplified from genomic DNAs of normal or affected individuals in MRX3 family with the use of primers listed in Table S7 and were cloned into pGEMT (Promega). The 506 bp (chrX: 152,890,153–152,890,658) and 127 bp (chrX: 152,890,153–152,890,279) fragments were obtained with restriction-enzyme digests with XhoI and HindIII (for the 506 bp fragment) and AvrII and HindIII (for the 127 bp fragment) from pGEMT containing 705 bp inserts. All fragments were subsequently cloned into the multiple cloning site of the pGL4.1 luciferase reporter (Promega). The arrows show the direction of transcription. The right panel shows the results of the corresponding luciferase reporter assays conducted in HEK 293T cells as previously described.14 Values are normalized against the 705 bp wild-type construct. Error bars represent standard deviation of at least three independent experiments. ∗p < 0.0007 by Student’s t test.
(E) Schematic of HCFC1 domain structure with the 5× Kelch domain region, the basic region (containing SIN3A17 and ZBTB1718 protein interacting regions), HCF1 repeats, and the acidic region. The two unique variants identified as part of this study are underlined. The two variants identified by Piton et al.19 are indicated by an asterisk (∗), and those identified by Tarpey et al.3 are indicated by a number sign (#). The values in brackets are PolyPhen 2 scores. The diagram is not drawn to scale.
To assess the impact of the S2 chrX:152,890,455A>G variant on HCFC1 expression, we tested the relative levels of total HCFC1 transcripts by using quantitative RT-PCR (qRT-PCR). The HCFC1 mRNA expression in affected individuals’ (n = 2) lymphoblastoid cell lines (LCLs) was >1.6× higher (p < 0.006, Student’s one-tailed t test) than that in male and female controls (Figure 2A and Figures S4–S6). Given that HCFC1 is duplicated in the majority of individuals with MECP2 (MIM 300005) duplications, we also tested HCFC1 expression in cell lines of these affected individuals (Figure 2A and data not shown). No significant difference in HCFC1 expression was identified, suggesting that HCFC1 is not upregulated when duplicated. This also suggested to us that the YY1 binding site at position S2 is a repressive cis-regulator rather than an enhancer.12 We also tested the possibility that S2 could function as a remote cis-regulatory sequence for neighboring genes, namely TMEM187 and, more distantly, RENBP (MIM 312420)20 and MECP2.21 The expression of TMEM187 is highly variable and not responsive to S2 variation in the affected individuals from the MRX3 family. Although RENBP expression is significantly (2-fold) upregulated in individuals with MECP2 duplications, it is only slightly upregulated in the affected individuals from the MRX3 family. MECP2 expression is higher in the affected individuals from the MRX3 family, but this was not statistically significant (Figure 2A and Figure S4). We therefore concluded that HCFC1 is likely to be the only gene significantly affected by the S2 chrX:152,890,455A>G variant in the YY1 binding site.
To further investigate whether the chrX:152,890,455A>G variant has a functional effect, we performed an electrophoretic mobility shift assay (EMSA) by using the nuclear extracts from human embryonic kidney (HEK) 293T cells. The presence of the variant completely abolished YY1 binding (Figure 2B). This was supported by chromatin immunoprecipitation with a YY1 antibody in LCLs from affected individuals from the MRX3 family (Figure 2C). To study the effect of the S2 YY1 binding site on HCFC1 expression and explore the activity of the other neighboring YY1 binding sites, we generated wild-type and mutant S2 luciferase reporter constructs with different lengths of the HCFC1 and TMEM187 UTRs and intergenic regions (Figure 2D). The construct containing S1, S2, and S3 was the only one significantly impacted by the S2 chrX:152,890,455A>G variant (Figure 2D).
That HCFC1 is highly expressed during embryonic brain development20 (Figure S7) led us to investigate the effect that HCFC1 overexpression has on the behavior of embryonic murine neural cells, namely neural stem cells (NSCs) and embryonic hippocampal neurons. We employed nucleofection to deliver expression plasmids encoding enhanced green fluorescent protein (EGFP) and either an empty-vector control or a HCFC1 expression plasmid into ex-vivo cultures of these neural populations, resulting in cotransfection of 30%–40% of cells (data not shown). In NSCs grown nonadherently (i.e., as neurospheres), overexpression of HCFC1 slightly reduced the proliferation of the entire culture (Figure 3A). To resolve further, we used the 5-ethynyl-2′-deoxyuridine (EdU) pulse label assay to identify cells in S phase (i.e., proliferative cells). To restrict analysis to only transfected cells (i.e., those expressing EGFP), we employed either fluorescent-activated cell sorting (FACS) or immunofluorescent microscopy. Overexpression of HCFC1 resulted in a significant reduction in the percentage of EdU-labeled cells (∼11% lower than controls) grown nonadherently (Figure 3B), whereas under adherent culture conditions more permissive to differentiation, the percentage of EdU-labeled cells was even further reduced (∼28% lower than controls) (Figure 3C and Figure S8). Because analysis of the cell-cycle length revealed no significant difference (Figure 3D), we explored the possibility that HCFC1 overexpression might instead promote cell-cycle exit. We used immunofluorescent microscopy to quantify the differentiation status of transfected cells. The overexpression of HCFC1 significantly increased the production of astrocytes (∼30% higher than controls), whereas the percentages of neurons and oligodendrocytes were unaffected (Figures 3E and 3F and Figure S8). Next, we addressed the effect of HCFC1 overexpression on the outgrowth of freshly isolated embryonic hippocampal neurons. HCFC1 overexpression resulted in a significant reduction in neurite growth as evidenced by a greater than 50% reduction in primary axon growth at day 4 of culture and a 33% reduction at day 8 (Figures 3G and 3H). This result was coupled with reductions in the degree of neurite arborization, as reported by the number of both axonal and dendritic termini, and high amounts of neuronal death (Figure S9). Together, these data strongly suggest that HCFC1 is a potent regulator of embryonic neural development. Overexpression in NSCs promoted cell-cycle exit coupled with astrocyte production, whereas in neurons, overexpression hindered neurite growth and viability.
Figure 3.
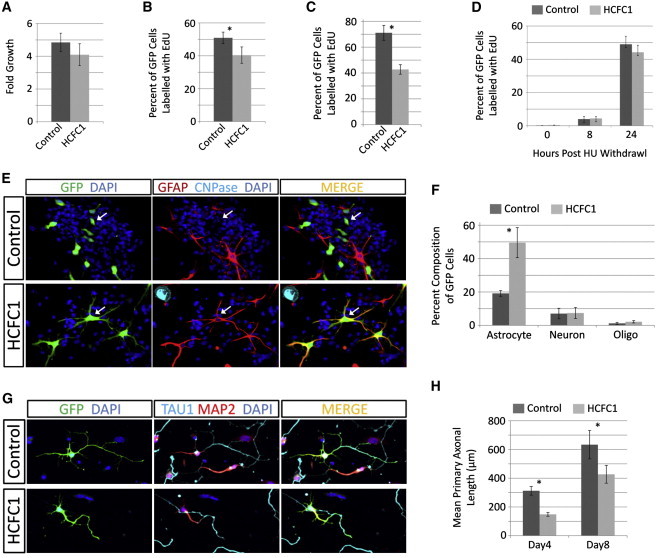
HCFC1 Overexpression Alters the Behavior of Embryonic Neural Cells
Ex vivo cultures of embryonic NSCs or embryonic hippocampal neurons were isolated and manipulated as previously described.22,23 Cells were transfected with plasmids expressing EGFP (pMAX-EGFP; Lonza) together with either an empty vector control (pcDNA3.1; Invitrogen) or HCFC1 expression plasmid (pCGN-HCFC1). All experiments were done in biological triplicates. All graphs are the average of triplicate experiments ± the standard deviation (error bars). ∗p < 0.05 by Student’s two-tailed t test.
(A) Proliferation of transfected NSC cultures grown in nonadherent conditions over 4 days was assessed with the Cell Titer96 AQueous Cell Proliferation Assay as per the manufacturer’s (Promega) instructions. Note that only ∼30% of the cells are transfected.
(B–C) HCFC1 overexpression reduces the number of NSCs in S phase. 5-ethynyl-2′-deoxyuridine (EdU) labeling and detection were done with the Click-it EdU AlexaFluor647-Azide kits (Invitrogen) as per the manufacturer’s instructions. In (B), EdU pulse labeling (6 hr) of transfected NSCs in nonadherent culture was analyzed with FACS (FACS AriaII flow cytometer, BD Bioscience), and in (C), 12 hr pulse labeling of transfected NSCs after 3 days of adherent culture was analyzed by immunofluorescent microscopy.
(D) HCFC1 overexpression does not alter the length of the NSC cycle. NSCs were synchronized in S phase with hydroxyurea (HU) and were allowed to re-enter the cell cycle at the G2 phase in the presence of EdU, as previously described.24 Cell-cycle length is inferred by the number of transfected cells that have transitioned through G2, M, G1, and back into S, which is reported by the number of cells labeled by EdU at 0, 8, and 24 hr as previously described.24
(E–F) HCFC1 overexpression promotes differentiation of NSCs. In (E) are representative immunofluorescent images after 3 days of adherent culture. Transfected cells were identified via EGFP expression (green), and their differentiation status was obtained with antibodies against cell-type-specific marker proteins: GFAP was used for astrocytes (red), and CNPase was used for oligodendrocytes (cyan). Nuclei were stained with DAPI (blue). The upper-panel arrowheads indicate unidentified transfected cells, and the lower-panel arrowheads indicate transfected cells of astrocyte identity. The quantification of cellular identities is shown in (F). At least 300 cells were counted for each experiment (conducted in triplicate).
(G–H) HCFC1 overexpression reduces primary axonal outgrowth in hippocampal neurons. In (G) is a representative immunofluorescent image of 4-day-old cultures. Transfected neurons were identified with GFP expression (green) and stained with antibodies specific for axons (Tau1; cyan) and dendrites (MAP2; red). Nuclei were stained with DAPI (blue). In (H) is the quantification of primary axonal length at days 4 and 8 of culture, conducted as previously described.23 All staining and microscopy were conducted as previously described.23,25
The expression of the evolutionarily conserved transcription regulator HCFC1 is tightly regulated by YY1, which suggested to us that this is necessary to ensure that HCFC1 target genes are also strictly regulated. To investigate this further, we performed microarray expression profiling by using MRX3 (n = 2) and control male (n = 4) and female (n = 5) LCLs on HT-12v4 (WGDASL) arrays (Illumina) (the NCBI Gene Expression Omnibus accession number for array data is GSE39326).
Background-subtracted expression values were quantile normalized with the lumi package from bioconductor.26 Significant differential expression on probes with a detection threshold > 0 was determined with the limma package from bioconductor.27 We identified 218 significantly deregulated genes (p < 0.01; false discovery rate [FDR] < 0.8), at least five of which have been identified by chromatin immunoprecipitation (ChIP) sequencing as targets of HCFC128 (these five are MRPL34 [MIM 611840], LSG1 [MIM 610780], NDUFB5 [MIM 603841], MRPL47 [MIM 611852], and NDUFS7 [MIM 601825]). With the exception of LSG1, all these genes are involved in mitochondrial function or biogenesis, a process in which HCFC1 has been previously implicated.29 Further analysis of the 218 genes identified a cluster of 24 genes whose products localize to mitochondria and show significant enrichment in this data set (p = 0.0012 with medium classification stringency on DAVID30). Significant enrichment was also noted for genes and their acetylation-involved proteins, which included those that bind to or directly acetylate histones, consistent with the known function of HCFC1 as a chromatin-associated transcriptional regulator. Overall, the array results support HCFC1 upregulation due to a mutation in the YY1 binding site as having an impact on downstream regulation of gene expression. The biological processes associated with known functions of HCFC1, together with the neural functions reported in this study, rank highly on functional-annotation tables of differentially expressed genes (Tables S8–S10).
Finally, we identified two additional unique HCFC1 variants by using systematic X chromosome exome sequencing of probands from unresolved families affected by X-chromosome-linked or X-chromosome-suggestive intellectual disability (Figures S10–S12 and Table S11; unpublished data). These include c.674G>A (p.Ser225Asn) (RefSeq accession number NM_005334.2) within one of the Kelch domains and c.5267C>T (p.Ala1756Val) (RefSeq NM_005334.2). We have also found a c.2626G>A (p.Gly876Ser) variant, which lies within the ZBTB17/GABP2 binding domain of HCFC1 and has been identified in a frequency of 1/1,207 chromosomes (or 7/8,454; National Heart, Lung, and Blood Institute [NHLBI] Exome Variant Server). Variant p.Ser225Asn segregated in family D144 (Figure S11 and Table S11), and variant p.Ala1756Val, as well as missense change c.356A>G (p.Gln119Arg) (RefSeq NM_005096.3) in another gene, ZMYM3 (MIM 300061), was found in the index affected individual of a family with four affected males in two generations (family D82, Table S11) but could not be tested for cosegregation with XLID because DNA from other family members was unavailable. The c.2626G>A (p.Gly876Ser) variant was found in a simplex case who had intellectual disability and whose mother had 100% skewing of X inactivation (family D147; Table S11). The X-exome sequencing of the affected individual from family D147 also identified a potentially deleterious change (c.3101T>G [p.Phe1034Cys] [RefSeq NM_005120.2]) in MED12 (MIM 300188), mutations in which are known to cause XLID31 (MIM 305450 and 309520). No other obviously deleterious X-exome variants were identified in these families, making involvement of these HCFC1 variants plausible in at least families D144 and D82. Interestingly, Piton et al.19 identified several unique HCFC1 variants, including p.Gly876Ser (annotated as p.Gly779Ser19) in a male individual with autism spectrum disorder and p.Thr239Met (annotated as p.Thr142Met; not present in the Exome Variant Server data) in a female individual with schizophrenia (Figure 2E). Tarpey et al.3 found two unique nonsynonymous changes and a splice-site change (c.5379+2T>C [RefSeq NM_005334.2]) affecting the canonical 5′ donor splice site of intron 21. Inspection of dbSNP identified additional unique nonsynonymous changes and a frameshift of HCFC1 of unknown frequency. Although many of these variants are predicted to be damaging (Piton et al.19 and data not shown), their functional effect has not been tested. All six amino acid positions are highly conserved across multiple species (Figure S10). All together, the combined data strongly suggest that mutations of HCFC1 lead to different neurological phenotypes, which include but are not restricted to intellectual disability.
There are several lines of evidence that implicate HCFC1 in biological processes important for normal neuronal function and development. HCFC1 was first identified as a crucial VP-16/Oct-1-interacting cell-proliferation factor involved in herpes simplex virus infection (for review, see Wysocka and Herr32). Since then, HCFC1 has been shown to be involved with many other proteins in regulating gene transcription. Among others, HCFC1 recruits mixed-lineage leukemia (MLL, encoded by MLL [MIM 159555]) and Set-1 histone H3 lysine 4 methyltransferases to E2F responsive promoters and induces histone methylation and transcriptional activation.17 HCFC1 has also been implicated with other known chromatin-modifying proteins that are known to be involved in the pathology of intellectual disability. For example, a protein complex containing the histone lysine demethylase PHF8 (encoded by PHF8 [MIM 300560]) and another protein, ZNF71133 (encoded by ZNF711 [MIM 314990]), known to be implicated in XLID when mutated, remove the repressive H4K20me1 mark from the promoters of a subset of HCFC1-E2F1-regulated genes.34 Some of the known targets of this complex are also other genes with known mutations associated with XLID; one such gene is KDM5C (MIM 314690), which encodes another histone H3 lysine 4 demethylase (also known as JARID1C).33,35 HCFC1 directly associates with YY1,36 and through the interaction of YY1 with MECP2, it regulates mitochondrial adenine nucleotide translocase ANT137 (MIM 103220) and thus might contribute to the pathology of Rett syndrome (MIM 312750). Our microarray data on cells from affected MRX3 family members overexpressing HCFC1 further support the role of HCFC1 in mitochondrial function or biogenesis. The crucial role of HCFC1 in cell-cycle regulation and particularly in cell growth is supported by its interaction with the deubiquitinating enzyme BAP136 (encoded by BAP1 [MIM 603089]), recently implicated in various human cancers.38,39
The role of HCFC1 in neuronal cells is yet to be understood despite its high expression during embryonic brain development.20,40 We investigated HCFC1 overexpression, as observed in affected individuals from the MRX3 family, in embryonic neural cell types. In NSCs, overexpression resulted in cell-cycle exit coupled with the production of astrocytes. As implicated by others, our finding identifies HCFC1 as a regulator of stem cell behavior.28,41,42 Studies of HCFC1 in cell-cycle regulation have revealed requirements during M-phase43 and G0/G1-S phase progression.17,44 In static cell-culture models, HCFC1 is known to promote S-phase initiation;17 however, evidence has also suggested that mutated HCFC1 can promote G0/G1 arrest. For example, during G0/G1, HCFC1 is known to occupy and repress promoters of S-phase genes by bridging interactions between the E2F4 (encoded by E2F4 [MIM 600659]) transcription factor and the Sin3 histone deacetylase, thus inhibiting S-phase initiation.17 HCFC1 is also a key transcriptional coactivator of the retinoblastoma protein (Rb, encoded by Rb1 [MIM 614041]), a potent inhibitor of S-phase genes.45 During muscle cell differentiation, this regulation is required for the cell-cycle exit of myoblasts and terminal differentiation into myotubes.45 Finally, loss-of-function studies of the HCFC1 antagonist PDCD246 (encoded by PDCD2 [MIM 600866]) implicate HCFC1 activity as a negative regulator of embryonic stem cell maintenance.42 The above evidence suggests that the effect that HCFC1 has on the cell cycle is likely to be dependent on the cellular context; this is consistent with its ability to interact with multiple transcription factors and opposing histone modifiers in a pleiotropic manner.
We extended our studies beyond the roles of HCFC1 in NSCs to include postmitotic hippocampal neurons. Neurons overexpressing HCFC1 displayed significant reductions in neurite growth and viability. The reduction in viability might be a result of increased apoptosis. Deregulation of the complex containing HCFC1, the MLL histone methyltransferase, and E2F1 (encoded by E2F1 [MIM 189971]), which normally governs the transition from the G1-S phase of the cell cycle, is known to cause both DNA damage and orchestrate an ensuing apoptotic process.47 Alternatively, other cellular insults mediated by global deregulation of gene expression might affect neuronal cell viability. For example, consistent with ChIP studies,28 our microarray studies using cell lines from affected MRX3 family members identify mitochondrial function, which is known to impact neuronal viability, as a prominent process regulated by HCFC1.
Massively parallel sequencing is revolutionizing the annotation of human DNA variation and thus leads to a dramatic shift in the understanding of the genetic architecture of human traits and heritable disorders in particular. Although the recent successes are heavily biased toward the discovery of disease-associated variation in coding exonic regions of the human genome,5 our work demonstrates that mutations outside these regions can and should also be tackled.
Acknowledgments
We would like to express our gratitude to all participating individuals, L. Hobson for assistance with Sanger sequencing, M. Bienek for assistance with X-exome sequencing, K. Friend for linkage analysis, C. Schwartz for additional information about the c.5379+2T>C mutation, and F. Lammers and W. Herr for kindly providing HCFC1 full-length cDNA and rabbit anti-HCFC1 antibody. L.H. was supported by a scholarship under the State Scholarship Fund from China Scholarship Council. This work was supported by the German Federal Ministry of Education and Research through the German Mental Retardation Network (grant 01GS08167 to D.W. and grant 01GS08161 to H.H.R.), the European Union’s Seventh Framework Program under grant agreement number 241995, project GENCODYS, and grants from the National Health and Medical Research Council of Australia (project grant 1008077 and Principal Research Fellowship 508043 to J.G.).
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Database of Transcriptional Start Sites, http://dbtss.hgc.jp/
NCBI Gene Expression Omnibus, http://www.ncbi.nlm.nih.gov/geo/
NHLBI Exome Variant Server, http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen 2, http://genetics.bwh.harvard.edu/pph2/index.shtml
UCSC Genome Browser, http://genome.ucsc.edu
Accession Numbers
The NCBI Gene Expression Omnibus accession number for the microarray data reported in this paper is GSE39326.
Note Added in Proof
During the publication of this work, a most likely pathogenic mutation in ARID1B was identified in the simplex case D147 and might explain his phenotype.
References
- 1.Ropers H.H. Genetics of intellectual disability. Curr. Opin. Genet. Dev. 2008;18:241–250. doi: 10.1016/j.gde.2008.07.008. [DOI] [PubMed] [Google Scholar]
- 2.Gécz J., Shoubridge C., Corbett M. The genetic landscape of intellectual disability arising from chromosome X. Trends Genet. 2009;25:308–316. doi: 10.1016/j.tig.2009.05.002. [DOI] [PubMed] [Google Scholar]
- 3.Tarpey P.S., Smith R., Pleasance E., Whibley A., Edkins S., Hardy C., O’Meara S., Latimer C., Dicks E., Menzies A. A systematic, large-scale resequencing screen of X-chromosome coding exons in mental retardation. Nat. Genet. 2009;41:535–543. doi: 10.1038/ng.367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Whibley A.C., Plagnol V., Tarpey P.S., Abidi F., Fullston T., Choma M.K., Boucher C.A., Shepherd L., Willatt L., Parkin G. Fine-scale survey of X chromosome copy number variants and indels underlying intellectual disability. Am. J. Hum. Genet. 2010;87:173–188. doi: 10.1016/j.ajhg.2010.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cooper D.N., Chen J.M., Ball E.V., Howells K., Mort M., Phillips A.D., Chuzhanova N., Krawczak M., Kehrer-Sawatzki H., Stenson P.D. Genes, mutations, and human inherited disease at the dawn of the age of personalized genomics. Hum. Mutat. 2010;31:631–655. doi: 10.1002/humu.21260. [DOI] [PubMed] [Google Scholar]
- 6.Cirulli E.T., Goldstein D.B. Uncovering the roles of rare variants in common disease through whole-genome sequencing. Nat. Rev. Genet. 2010;11:415–425. doi: 10.1038/nrg2779. [DOI] [PubMed] [Google Scholar]
- 7.Gedeon A., Kerr B., Mulley J., Turner G. Localisation of the MRX3 gene for non-specific X linked mental retardation. J. Med. Genet. 1991;28:372–377. doi: 10.1136/jmg.28.6.372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kolb-Kokocinski A., Mehrle A., Bechtel S., Simpson J.C., Kioschis P., Wiemann S., Wellenreuther R., Poustka A. The systematic functional characterisation of Xq28 genes prioritises candidate disease genes. BMC Genomics. 2006;7:29. doi: 10.1186/1471-2164-7-29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Corbett M.A., Schwake M., Bahlo M., Dibbens L.M., Lin M., Gandolfo L.C., Vears D.F., O’Sullivan J.D., Robertson T., Bayly M.A. A mutation in the Golgi Qb-SNARE gene GOSR2 causes progressive myoclonus epilepsy with early ataxia. Am. J. Hum. Genet. 2011;88:657–663. doi: 10.1016/j.ajhg.2011.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vissers L.E., de Ligt J., Gilissen C., Janssen I., Steehouwer M., de Vries P., van Lier B., Arts P., Wieskamp N., del Rosario M. A de novo paradigm for mental retardation. Nat. Genet. 2010;42:1109–1112. doi: 10.1038/ng.712. [DOI] [PubMed] [Google Scholar]
- 11.Heinemeyer T., Wingender E., Reuter I., Hermjakob H., Kel A.E., Kel O.V., Ignatieva E.V., Ananko E.A., Podkolodnaya O.A., Kolpakov F.A. Databases on transcriptional regulation: TRANSFAC, TRRD and COMPEL. Nucleic Acids Res. 1998;26:362–367. doi: 10.1093/nar/26.1.362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim J.D., Hinz A.K., Bergmann A., Huang J.M., Ovcharenko I., Stubbs L., Kim J. Identification of clustered YY1 binding sites in imprinting control regions. Genome Res. 2006;16:901–911. doi: 10.1101/gr.5091406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tarpey P.S., Raymond F.L., Nguyen L.S., Rodriguez J., Hackett A., Vandeleur L., Smith R., Shoubridge C., Edkins S., Stevens C. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat. Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shoubridge C., Tan M.H., Seiboth G., Gécz J. ARX homeodomain mutations abolish DNA binding and lead to a loss of transcriptional repression. Hum. Mol. Genet. 2012;21:1639–1647. doi: 10.1093/hmg/ddr601. [DOI] [PubMed] [Google Scholar]
- 15.Yu J., Hai Y., Liu G., Fang T., Kung S.K., Xie J. The heterogeneous nuclear ribonucleoprotein L is an essential component in the Ca2+/calmodulin-dependent protein kinase IV-regulated alternative splicing through cytidine-adenosine repeats. J. Biol. Chem. 2009;284:1505–1513. doi: 10.1074/jbc.M805113200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kumar R., Selth L.A., Schulz R.B., Tay B.S., Neilsen P.M., Callen D.F. Genome-wide mapping of ZNF652 promoter binding sites in breast cancer cells. J. Cell. Biochem. 2011;112:2742–2747. doi: 10.1002/jcb.23214. [DOI] [PubMed] [Google Scholar]
- 17.Tyagi S., Chabes A.L., Wysocka J., Herr W. E2F activation of S phase promoters via association with HCF-1 and the MLL family of histone H3K4 methyltransferases. Mol. Cell. 2007;27:107–119. doi: 10.1016/j.molcel.2007.05.030. [DOI] [PubMed] [Google Scholar]
- 18.Piluso D., Bilan P., Capone J.P. Host cell factor-1 interacts with and antagonizes transactivation by the cell cycle regulatory factor Miz-1. J. Biol. Chem. 2002;277:46799–46808. doi: 10.1074/jbc.M206226200. [DOI] [PubMed] [Google Scholar]
- 19.Piton A., Gauthier J., Hamdan F.F., Lafrenière R.G., Yang Y., Henrion E., Laurent S., Noreau A., Thibodeau P., Karemera L. Systematic resequencing of X-chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol. Psychiatry. 2011;16:867–880. doi: 10.1038/mp.2010.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wilson A.C., Parrish J.E., Massa H.F., Nelson D.L., Trask B.J., Herr W. The gene encoding the VP16-accessory protein HCF (HCFC1) resides in human Xq28 and is highly expressed in fetal tissues and the adult kidney. Genomics. 1995;25:462–468. doi: 10.1016/0888-7543(95)80046-o. [DOI] [PubMed] [Google Scholar]
- 21.Liu J., Francke U. Identification of cis-regulatory elements for MECP2 expression. Hum. Mol. Genet. 2006;15:1769–1782. doi: 10.1093/hmg/ddl099. [DOI] [PubMed] [Google Scholar]
- 22.Giachino C., Basak O., Taylor V. Isolation and manipulation of mammalian neural stem cells in vitro. Methods Mol. Biol. 2009;482:143–158. doi: 10.1007/978-1-59745-060-7_9. [DOI] [PubMed] [Google Scholar]
- 23.Corbett M.A., Bahlo M., Jolly L., Afawi Z., Gardner A.E., Oliver K.L., Tan S., Coffey A., Mulley J.C., Dibbens L.M. A focal epilepsy and intellectual disability syndrome is due to a mutation in TBC1D24. Am. J. Hum. Genet. 2010;87:371–375. doi: 10.1016/j.ajhg.2010.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kouroupi G., Lavdas A.A., Gaitanou M., Thomaidou D., Stylianopoulou F., Matsas R. Lentivirus-mediated expression of insulin-like growth factor-I promotes neural stem/precursor cell proliferation and enhances their potential to generate neurons. J. Neurochem. 2010;115:460–474. doi: 10.1111/j.1471-4159.2010.06939.x. [DOI] [PubMed] [Google Scholar]
- 25.Jolly L.A., Taylor V., Wood S.A. USP9X enhances the polarity and self-renewal of embryonic stem cell-derived neural progenitors. Mol. Biol. Cell. 2009;20:2015–2029. doi: 10.1091/mbc.E08-06-0596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du P., Kibbe W.A., Lin S.M. lumi: a pipeline for processing Illumina microarray. Bioinformatics. 2008;24:1547–1548. doi: 10.1093/bioinformatics/btn224. [DOI] [PubMed] [Google Scholar]
- 27.Smyth G.K. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 2004;3:e3. doi: 10.2202/1544-6115.1027. [DOI] [PubMed] [Google Scholar]
- 28.Dejosez M., Levine S.S., Frampton G.M., Whyte W.A., Stratton S.A., Barton M.C., Gunaratne P.H., Young R.A., Zwaka T.P. Ronin/Hcf-1 binds to a hyperconserved enhancer element and regulates genes involved in the growth of embryonic stem cells. Genes Dev. 2010;24:1479–1484. doi: 10.1101/gad.1935210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Vercauteren K., Gleyzer N., Scarpulla R.C. PGC-1-related coactivator complexes with HCF-1 and NRF-2beta in mediating NRF-2(GABP)-dependent respiratory gene expression. J. Biol. Chem. 2008;283:12102–12111. doi: 10.1074/jbc.M710150200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dennis G., Jr., Sherman B.T., Hosack D.A., Yang J., Gao W., Lane H.C., Lempicki R.A. DAVID: Database for Annotation, Visualization, and Integrated Discovery. Genome Biol. 2003;4:3. [PubMed] [Google Scholar]
- 31.Risheg H., Graham J.M., Jr., Clark R.D., Rogers R.C., Opitz J.M., Moeschler J.B., Peiffer A.P., May M., Joseph S.M., Jones J.R. A recurrent mutation in MED12 leading to R961W causes Opitz-Kaveggia syndrome. Nat. Genet. 2007;39:451–453. doi: 10.1038/ng1992. [DOI] [PubMed] [Google Scholar]
- 32.Wysocka J., Herr W. The herpes simplex virus VP16-induced complex: The makings of a regulatory switch. Trends Biochem. Sci. 2003;28:294–304. doi: 10.1016/S0968-0004(03)00088-4. [DOI] [PubMed] [Google Scholar]
- 33.Kleine-Kohlbrecher D., Christensen J., Vandamme J., Abarrategui I., Bak M., Tommerup N., Shi X., Gozani O., Rappsilber J., Salcini A.E., Helin K. A functional link between the histone demethylase PHF8 and the transcription factor ZNF711 in X-linked mental retardation. Mol. Cell. 2010;38:165–178. doi: 10.1016/j.molcel.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu W., Tanasa B., Tyurina O.V., Zhou T.Y., Gassmann R., Liu W.T., Ohgi K.A., Benner C., Garcia-Bassets I., Aggarwal A.K. PHF8 mediates histone H4 lysine 20 demethylation events involved in cell cycle progression. Nature. 2010;466:508–512. doi: 10.1038/nature09272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Jensen L.R., Amende M., Gurok U., Moser B., Gimmel V., Tzschach A., Janecke A.R., Tariverdian G., Chelly J., Fryns J.P. Mutations in the JARID1C gene, which is involved in transcriptional regulation and chromatin remodeling, cause X-linked mental retardation. Am. J. Hum. Genet. 2005;76:227–236. doi: 10.1086/427563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yu H., Mashtalir N., Daou S., Hammond-Martel I., Ross J., Sui G., Hart G.W., Rauscher F.J., 3rd, Drobetsky E., Milot E. The ubiquitin carboxyl hydrolase BAP1 forms a ternary complex with YY1 and HCF-1 and is a critical regulator of gene expression. Mol. Cell. Biol. 2010;30:5071–5085. doi: 10.1128/MCB.00396-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Forlani G., Giarda E., Ala U., Di Cunto F., Salani M., Tupler R., Kilstrup-Nielsen C., Landsberger N. The MeCP2/YY1 interaction regulates ANT1 expression at 4q35: Novel hints for Rett syndrome pathogenesis. Hum. Mol. Genet. 2010;19:3114–3123. doi: 10.1093/hmg/ddq214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wiesner T., Obenauf A.C., Murali R., Fried I., Griewank K.G., Ulz P., Windpassinger C., Wackernagel W., Loy S., Wolf I. Germline mutations in BAP1 predispose to melanocytic tumors. Nat. Genet. 2011;43:1018–1021. doi: 10.1038/ng.910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Testa J.R., Cheung M., Pei J., Below J.E., Tan Y., Sementino E., Cox N.J., Dogan A.U., Pass H.I., Trusa S. Germline BAP1 mutations predispose to malignant mesothelioma. Nat. Genet. 2011;43:1022–1025. doi: 10.1038/ng.912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kristie T.M. The mouse homologue of the human transcription factor C1 (host cell factor). Conservation of forms and function. J. Biol. Chem. 1997;272:26749–26755. doi: 10.1074/jbc.272.42.26749. [DOI] [PubMed] [Google Scholar]
- 41.Dejosez M., Krumenacker J.S., Zitur L.J., Passeri M., Chu L.F., Songyang Z., Thomson J.A., Zwaka T.P. Ronin is essential for embryogenesis and the pluripotency of mouse embryonic stem cells. Cell. 2008;133:1162–1174. doi: 10.1016/j.cell.2008.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mu W., Munroe R.J., Barker A.K., Schimenti J.C. PDCD2 is essential for inner cell mass development and embryonic stem cell maintenance. Dev. Biol. 2010;347:279–288. doi: 10.1016/j.ydbio.2010.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Julien E., Herr W. A switch in mitotic histone H4 lysine 20 methylation status is linked to M phase defects upon loss of HCF-1. Mol. Cell. 2004;14:713–725. doi: 10.1016/j.molcel.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 44.Goto H., Motomura S., Wilson A.C., Freiman R.N., Nakabeppu Y., Fukushima K., Fujishima M., Herr W., Nishimoto T. A single-point mutation in HCF causes temperature-sensitive cell-cycle arrest and disrupts VP16 function. Genes Dev. 1997;11:726–737. doi: 10.1101/gad.11.6.726. [DOI] [PubMed] [Google Scholar]
- 45.Deléhouzée S., Yoshikawa T., Sawa C., Sawada J., Ito T., Omori M., Wada T., Yamaguchi Y., Kabe Y., Handa H. GABP, HCF-1 and YY1 are involved in Rb gene expression during myogenesis. Genes Cells. 2005;10:717–731. doi: 10.1111/j.1365-2443.2005.00873.x. [DOI] [PubMed] [Google Scholar]
- 46.Scarr R.B., Sharp P.A. PDCD2 is a negative regulator of HCF-1 (C1) Oncogene. 2002;21:5245–5254. doi: 10.1038/sj.onc.1205647. [DOI] [PubMed] [Google Scholar]
- 47.Tyagi S., Herr W. E2F1 mediates DNA damage and apoptosis through HCF-1 and the MLL family of histone methyltransferases. EMBO J. 2009;28:3185–3195. doi: 10.1038/emboj.2009.258. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.