

Abstract
Primary ciliary dyskinesia (PCD) is a genetically heterogeneous recessive disorder characterized by defective cilia and flagella motility. Chronic destructive-airway disease is caused by abnormal respiratory-tract mucociliary clearance. Abnormal propulsion of sperm flagella contributes to male infertility. Genetic defects in most individuals affected by PCD cause randomization of left-right body asymmetry; approximately half show situs inversus or situs ambiguous. Almost 70 years after the hy3 mouse possessing Hydin mutations was described as a recessive hydrocephalus model, we report HYDIN mutations in PCD-affected persons without hydrocephalus. By homozygosity mapping, we identified a PCD-associated locus, chromosomal region 16q21-q23, which contains HYDIN. However, a nearly identical 360 kb paralogous segment (HYDIN2) in chromosomal region 1q21.1 complicated mutational analysis. In three affected German siblings linked to HYDIN, we identified homozygous c.3985G>T mutations that affect an evolutionary conserved splice acceptor site and that subsequently cause aberrantly spliced transcripts predicting premature protein termination in respiratory cells. Parallel whole-exome sequencing identified a homozygous nonsense HYDIN mutation, c.922A>T (p.Lys307∗), in six individuals from three Faroe Island PCD-affected families that all carried an 8.8 Mb shared haplotype across HYDIN, indicating an ancestral founder mutation in this isolated population. We demonstrate by electron microscopy tomography that, consistent with the effects of loss-of-function mutations, HYDIN mutant respiratory cilia lack the C2b projection of the central pair (CP) apparatus; similar findings were reported in Hydin-deficient Chlamydomonas and mice. High-speed videomicroscopy demonstrated markedly reduced beating amplitudes of respiratory cilia and stiff sperm flagella. Like the hy3 mouse model, all nine PCD-affected persons had normal body composition because nodal cilia function is apparently not dependent on the function of the CP apparatus.
Introduction
Early in the evolution of the eukaryotic cell, cilia and flagella appeared as cell-surface appendages that enable protozoa and sperm to propel through fluid.1 They are highly conserved organelles with both motile and sensory functions; almost all motile cilia have the same 200 nm diameter and ultrastructural plan.2 The core axonemal structure of most motile cilia is based on microtubules—a central pair (CP) of microtubules is surrounded by nine peripheral microtubule doublets that, together with connecting radial spokes, act as a stabilizing framework. This framework runs along the cilium length and is attached at regular intervals to a number of other macromolecules, including the dynein arms and nexin links, also influencing ciliary motility and waveform.2,3
Primary ciliary dyskinesia (PCD [MIM 242650]) is a rare genetic disorder that is usually inherited as an autosomal-recessive trait and affects approximately 1 in 20,000 individuals. In PCD, defective mucociliary clearance, secondary to immotile or dyskinetic respiratory cilia, results in chronic infection and inflammation of the upper and lower airways. This often progresses to bronchiectasis.3 Dysfunctional sperm tails (flagella) frequently cause male infertility. Other congenital disorders, such as hydrocephalus, retinal degeneration, sensory hearing deficits, polycystic kidney disease, and intellectual disability, have also been observed in distinct PCD variants.3
Associated with most known PCD gene mutations, randomization of left-right asymmetry is a phenotype that was shown in mouse models to result from defective function of motile monocilia at the embryonic node.4 Thus, ∼50% of affected PCD individuals exhibit situs inversus, or more rarely, situs ambiguous associated with complex congenital heart disease.5 In contrast, a distinct minority of PCD cases are caused by observable defects of CP structures that are considered to be irrelevant for embryonic nodal cilia movement. These PCD variants are not associated with situs anomalies. In these cases, the cilia beat in a circular manner akin to that of nodal cilia.6,7 A proportion of CP defects are caused by mutations in the radial spoke head genes RSPH4A (MIM 612649) and RSPH9 (MIM 612650); these mutations cause an intermittent loss of the CP microtubules.7
Genetic defects in 12 additional genes have been identified in persons with nonsyndromic PCD. Genetic studies have identified autosomal-recessive mutations in DNAH5 (MIM 603335), DNAI1 (MIM 604366), DNAI2 (MIM 605483), DNAL1 (MIM 610062), TXNDC3 (MIM 607421), DNAH11 (MIM 603339), DNAAF1 (KTU [MIM 612517]), DNAAF2 (LRRC50 [MIM 613190]), DNAAF3 (MIM 614566), CCDC39 (MIM 613798), CCDC40 (MIM 613799), and CCDC103.8–21 The first six of these genes encode subunits of axonemal outer-dynein-arm (ODA) components. DNAAF1, DNAAF2, and DNAAF3 encode proteins displaying functions in the cytoplasmic preassembly of dynein-arm components. CCDC39 and CCDC40 mutations cause defects of the nexin links and inner-dynein-arm (IDA) components. CCDC103 encodes a protein that uniquely has dual localization both in the cytoplasm and in the axoneme, where it functions as a dynein-arm component involved in docking of the arms to ciliary microtubules.21 Two X-linked PCD variants associated with extra features suggestive of deficient sensory cilia function are accounted for by mutations in OFD1 (MIM 311200) and RPGR (MIM 312610).22,23
Recent molecular findings suggest that routine transmission electron microscopy (TEM), the conventional diagnostic gold standard, can miss certain PCD variants.15,24 Therefore, a combined approach of high-speed videomicroscopy (HVM) for analyzing ciliary beat patterns and subsequent confirmatory diagnostics such as TEM, immunofluorescence microscopy, or genetics should be the current standard diagnostic protocol according to the European Respiratory Society (ERS) recommendation.25
Here, we report on a PCD variant characterized by the absence of situs inversus in affected individuals and a subtle respiratory cilia beating defect of reduced amplitude creating a stiff beat pattern, which is caused by a deficiency of the CP-apparatus-associated protein HYDIN (MIM 610812).
Material and Methods
Affected Persons and Families
We studied DNA from a German consanguineous kindred and eight Faroe Island PCD individuals. Signed and informed consent was obtained from PCD-affected individuals and their family members according to protocols approved by the institutional ethics review boards.
Haplotype Analysis and Genome-wide SNP Mapping
We genotyped DNA samples from three affected persons and their parents (third-degree consanguineous parents) from the multiplex German family, OP-305, by using the Affymetrix GeneChip Human Mapping 10K Array v.2.0. Relationship errors were evaluated with the help of the program Graphical Relationship Representation.26 The program PedCheck was applied for detecting Mendelian errors.27 Non-Mendelian errors were identified with the program MERLIN.28 Linkage analysis was performed under the assumption of autosomal-recessive inheritance, full penetrance, and a disease gene frequency of 0.0001. Multipoint LOD scores were calculated with the program ALLEGRO29 and presented graphically with HaploPainter.30 All data handling was performed with the graphical user interface ALOHOMORA.31 SNP genotyping for the Faroe Island families was performed with the Illumina Linkage IV Panel of 6,008 SNPs with haplotypes constructed with GeneHunter v.2.1r5 from the easyLINKAGE Plus v.5.08 package as previously described.32
Whole-Exome Sequence Analysis
Exome sequencing of genomic DNA from two affected siblings from one Faroe Island family, UCL109, was performed at the Wellcome Trust Sanger Institute (Cambridge, UK) as part of the UK10K project. DNA (1–3 μg) was sheared to 100–400 bp with a Covaris E210 or LE220 (Covaris, Woburn, MA, USA). Sheared DNA was subjected to Illumina paired-end DNA library preparation and enriched for target sequences (Human All Exon 50 Mb, ELID S02972011, Agilent Technologies). Enriched libraries were sequenced with the HiSeq platform (Illumina) as paired-end 76 bp reads according to the manufacturer’s protocol. Sequencing reads that passed quality filtering were mapped to the reference genome sequence (hg19) with the Burrows-Wheeler Aligner 0.5.8c.33 GATK 1.1.534 was used for recalibrating base quality scores, realigning around indels, and marking duplicate reads. Variants were called independently on the resulting reads with both SAMtools mpileup (SAMtools 0.1.1335) and GATK UnifiedGenotyper,36 and the call sets were merged.
Filtering of the variants for novelty was performed by comparison to 181 UK10K exomes and by the exclusion of those with a frequency > 0.005 in the 1000 Genomes database.37 Variants not shared by both affected members of the pedigree and not consistent with recessive inheritance were excluded from the downstream analysis, which is detailed in Table S1, available online.
Mutational Analysis by Sanger Sequencing
Genomic DNA was isolated by standard methods. For analysis of family OP-305, amplification of 86 genomic fragments comprising all 86 exons of HYDIN (RefSeq accession number NM_001270974.1) was performed. Initially, for the detection of the Faroe Island sequence variant c.922A>T, DNA spanning HYDIN exon 8 from PCD-affected individuals and their family members was amplified with primers HYDINFor and HYDINRev. Subsequent allele-specific PCRs were performed with chromosome 1 HYDIN2-specific primers Ch1HYD2F and Ch1HYD2R and chromosome 16 HYDIN-specific primers Ch16HYDF and Ch16HYDR; we manually designed these primers by aligning the HYDIN and HYDIN2 genomic DNAs in BLAST to have them differ by only a single base pair at their 3′ ends. PCR conditions are available upon request. All amplified PCR products were sequenced bidirectionally. Primer sequences are presented in Figure S5 and Table S3.
cDNA Analysis of the Detected Splicing and Nonsense Mutation
RT-PCR was employed for determining the effect of the detected mutant variants on transcripts. We isolated RNA from respiratory epithelial cells obtained by nasal-brushing biopsies from individual OP-305 II1 and RNA from Epstein-Barr-virus-transformed lymphocytes from individual UCL109 II4 (OP-759) and controls. First-strand cDNA synthesis was performed with the RevertAid H minus First Strand cDNA Synthesis Kit (Fermentas) for OP-305 II1 and with the Transcriptor High Fidelity cDNA Synthesis Kit (Roche) for UCL109 II4. HYDIN cDNA was amplified with primers based on the Ensembl sequence (ENST00000393567) for HYDIN. Our cDNA experiments verified the predicted splice sites of the exon 25/exon 26 borders annotated by Ensembl (GenBank accession number JX501991). Further details and the locations of primers are presented in Figures S1A and S1B and Table S4.
Measurement of Nasal Nitric Oxide
Measurements of nasal nitric oxide (nNO) were obtained from family OP-305 with the use of an EcoMedics CLD88 chemiluminescence nitric oxide (NO) analyzer (Duernten, Switzerland); the aspiration sampling rate was 330 ml/min as recommended by the manufacturer. In Faroe Island individuals with PCD, nNO measurements were performed with Nitric Oxide Monitoring System (Aerocrine, Sweden) equipment as described with a flow sampling rate of 300 ml/min.38 An exhalation-against-resistance maneuver was performed while nNO was measured as per American Thoracic Society and European Respiratory Society guidelines.39 Measurements were recorded in parts per billion (ppb) and converted to nNO production rate in nl/min with the following equation: nNO (nl/min) = NO (ppb) × sampling rate (ml/min).39
TEM
The samples of nasal mucosa were fixed in 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer at 4°C, washed overnight, and postfixed in 1% osmium tetroxide. After dehydration, the samples were embedded in a mixture of propylene oxide and epoxy resin. After polymerization, several resin sections were cut. The sections were stained with aqueous 1% uranyl acetate and Reynold’s lead citrate. TEM was performed with a Philips CM10, Zeiss LEO912AB (zero-loss mode) EFTEM, or MORGAGNI 268 (Philips).
Image averages were designed as follows: contrast and brightness were adjusted with Adobe Photoshop CS4 and assembled with Adobe Illustrator CS4. In each case, six images of control and affected OP-305 II1 were set to 16% opacity and aligned manually.
Electron Microscopy Tomography
Sections 150 nm thick were cut with a Reichert Ultracut-E ultramicrotome and mounted onto copper grids. A 10 nm gold-particle solution was applied before staining with 2% methanolic uranyl acetate and Reynolds lead citrate. Tilt-series data were obtained with a Philips CM200 electron microscope with a tilt range of ± 70° and two increments. After the first tilt series was collected, the grid was subsequently rotated 90°, and a perpendicular tilt series was collected.
High-Speed Video Microscopy Analysis for Ciliary Beat Assessment
In the German samples, ciliary beat and sperm-tail function were assessed with the SAVA system.40 Respiratory epithelial cells and sperm cells were viewed with an Olympus IMT-2 microscope equipped with a Redlake ES-310Turbo monochrome high-speed video camera (Redlake, San Diego, CA) set at 125 or 250 frames per second. The nasal epithelial cells from the Faroe individuals were viewed with a Leica DMLB microscope equipped with a Mikrotron MC1302 camera set at 200 frames per second.
Immunofluorescence Analysis
Respiratory epithelial cells were obtained by nasal-brush biopsy and suspended in cell-culture medium. Samples were spread onto glass slides, air dried, and stored at –80°C until use. For immunofluorescence analyses, monoclonal DNAH5 and GAS11 antibodies, as well as polyclonal DNALI1 and CCDC39 antibodies, were used as described previously.16,19,41
Results
Clinical Findings in Affected Siblings of German and Faroe Island Families
Different approaches were taken in this study for the identification of causative mutations in PCD-affected families from Germany and the Faroe Islands. All affected individuals have normal situs composition (situs solitus). None of the affected individuals exhibit congenital heart disease or hydrocephalus.
The parents (OP-305 I1 and OP-305 I2) of the German consanguineous PCD-affected family, OP-305, are third-degree cousins (Figure 1A). The index person, OP-305 II1, is a 41-year-old affected female. After birth, she was hospitalized for several days because of a neonatal respiratory-distress syndrome. Since early infancy, she has suffered from chronic wet cough, recurrent bronchitis, and pneumonia. Upper-airway disease included recurrent otitis media and chronic rhinosinusitis with nasal polyps requiring repeated sinonasal surgery. Computed tomography of the lungs demonstrated marked bronchiectatic lung disease (Figure 1B). OP-305 II2 and OP-305 II3 are 38 and 34 years old, respectively, and are male affected brothers. Similarly to their sister OP-305 II1, both have suffered from neonatal respiratory distress syndrome, recurrent productive bronchitis, and pneumonia since early childhood, as well as chronic rhinosinusitis. Sinonasal surgery was performed because of marked nasal polyposis. In both persons, computed tomography showed bronchiectasis of the middle lobe. Neither brother has children.
Figure 1.
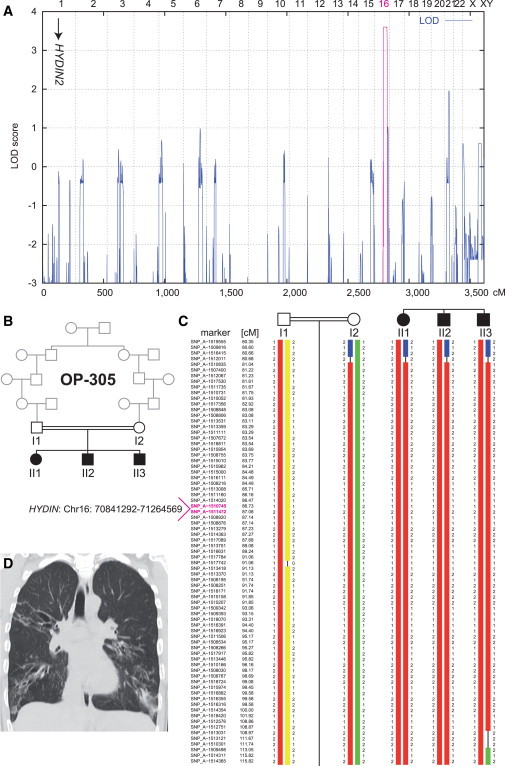
Linkage and Haplotype Analysis of Family OP-305
(A) Genome-wide linkage analysis of the consanguineous family OP-305 identifies a PCD gene locus on chromosome 16, which contains HYDIN. The position of the pseudogene HYDIN2 on chromosome 1 is marked with an arrow. A schematic representation of genome-wide LOD scores is shown. LOD scores are given along the y axis relative to genomic position in cM on the x axis. Note the significant peak (LOD = 3.6) in the region on chromosome 16.
(B) Pedigree of the OP-305 family.
(C) Haplotype reconstruction for the linkage region on chromosome 16. Disease-associated haplotypes are in red. The flanking markers SNP_A-1516415 (rs2221744) and SNP_A-1509498 (rs433325) span a region of 32.4 cM (corresponding to a genomic region of 21.8 Mb).
(D) High-resolution computed-tomography scans of individual OP-305 II1 show bronchiectasis of the right upper lobe, middle lobe, and lower lobes, peribronchial thickening, and airway collapse.
The three Faroe Island families (UCL96, UCL109, and UCL139; Figure 3B) are not immediately consanguineous; however, they originate from a historically isolated population, and an ancestral relationship between the paternal grandparent of UCL109 and the maternal grandparent of UCL96 was verbally reported by the families. In parallel, UCL109 family members were studied in Germany. Individuals were referred to as OP-921 (UCL109 II1), OP-836 (UCL109 II3), and OP-759 (UCL109 II4). Family UCL96 has two affected siblings (aged 30 and 24 years) who were first diagnosed at the age of 5 years (UCL96 II1) and 0.5 years (UCL96 II2). Both siblings had respiratory-distress syndrome as neonates and developed chronic wet cough, chronic bronchitis, and chronic rhinosinusitis and underwent tympanostomy-tube placement because of recurrent otitis media. Computed tomography showed bronchiectasis most prominent in the right middle lobe and the lingula. Family UCL109 has three affected siblings aged 22 (diagnosed at 6 years old), 19 (diagnosed at 3 years old) and 10 (diagnosed at 9 months old). In affected individual UCL109 II1, computed tomography demonstrated bronchiectatic lung disease of the right middle lobe. In affected siblings UCL109 II3 and UCL109 II4, chest X-rays showed chronic abnormalities of the right middle lobes. Individual UCL109 II4 is the only affected person who did not present with neonatal respiratory-distress syndrome. However, subsequent symptoms are typical for PCD and include chronic nasal discharge since early infancy, chronic bronchitis, chronic rhinosinusitis, and chronic otitis leading to grommets insertion. UCL139 II1, the only affected member of family UCL139, is a 25-year-old person diagnosed at the age of 8 years. Measurement of nNO in OP-305 II1 revealed a mean nNO concentration of 130 ppb, corresponding to a nNO production rate of 36.6 nl/min. Measurements were also taken in all six affected individuals from the Faroe Islands and showed a similar reduction; they ranged from 13 to 72 ppb, corresponding to a nNO production rate of 3.9–21.6 nl/min. These values are all significantly lower than current nNO cutoff values (58 nl/min).42
Figure 3.
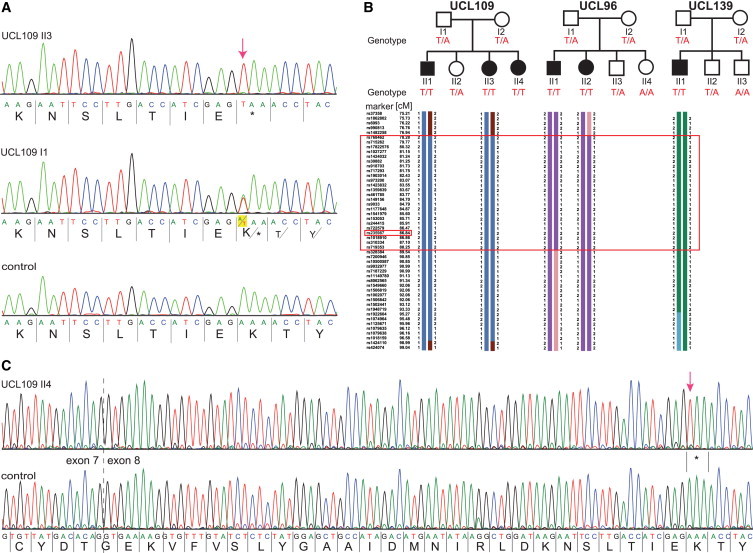
Identification of a Common HYDIN Mutation, c. 922A>T, in Faroe Island Families
(A) Sequence chromatographs of HYDIN exon 8 amplicons were generated with chromosome 16 HYDIN-specific primers (amplification-refractory mutation system [ARMS] method) and demonstrate homozygous A>T mutations (c.922A>T) predicted to create a homozygous AAA to TAA codon change, p.Lys307∗ (red arrow; amino acids are indicated below nucleotide sequence). Affected child UCL109 II3 (upper panel), homozygous for p.Lys307∗, and his father I1, a heterozygous carrier (middle), are compared to a control (bottom panel).
(B) Segregation-analysis genotypes confirmed that this nonsense variant cosegregates with disease status, which is inherited homozygously in all six affected children (T/T, black symbols), whereas all the parents and five unaffected siblings (white symbols) are carriers (T/A) or normal (A/A). Disease-associated haplotype reconstruction for the linkage region on chromosome 16 shows the flanking markers rs973200 and rs328384 spanning a 6.5 cM (8.8 Mb) region presumed to be a shared ancestral Faroe Island founder haplotype (large box). The location of HYDIN is indicated in the small box because marker rs235987 is intragenic.
(C) RT-PCR analysis of individual UCL109 II4 and control samples verified that the c.922A>T nonsense variant is contained within HYDIN on chromosome 16. All transcripts from the affected person (upper panel) showed the homozygous nonsense mutation (c.919A>T [p.Lys307∗]; red arrow); the HYDIN transcripts from a healthy control are shown in the lower sequence.
Identification of a PCD Locus on Chromosome 16
A total genome linkage scan using SNPs in the consanguineous German kindred OP-305 detected only a single chromosome 16q region with a significant LOD score >3.5 (Figures 1A and 1C). Simulation studies showed a maximum LOD score of 3.6, which is the maximum LOD score that can be expected in this family. A 21.8 Mb region of homozygosity shared between the three affected offspring was identified in chromosomal region 16q21-q23 between flanking markers SNP_A-1516415 (rs2221744) and SNP_A-1509498 (rs433325), consistent with recessive inheritance and homozygosity by descent (Figure 1C). The region contains 251 identified genes, two of which were of interest for PCD. These were HYDIN and DYNLRB2 (MIM 607168). DYNLRB2 encodes a member of the LC7/Roadblock family of light dynein chains (LCs). In LC7 mutant Chlamydomonas flagella, axonemal ODA and IDA multiprotein complexes are defective.43 Because our TEM and immunofluorescence analyses demonstrate the presence of ODAs and IDAs in mutant axonemes, we considered HYDIN the most promising candidate gene.
In hy3 mice possessing homozygous-recessive Hydin mutations, there is lethality in the first weeks of life because of hydrocephalus caused by abnormal ependymal ciliary motility.44–47 Unlike other PCD mouse models, such as Mdnah548- and Ccdc4020-deficient mice that also develop hydrocephalus, hy3 mice do not exhibit randomization of left-right body asymmetry. On the basis of the disease phenotype (primary ciliary dyskinesia without situs inversus) in all three affected members of family OP-305, we considered HYDIN a promising candidate gene for this PCD variant.
Mutational Analysis Identifies Recessive HYDIN Mutations
Mutational analysis of HYDIN on chromosome 16 was complicated by the fact that an interchromosomal duplication during human evolution created HYDIN2 (MIM 610813), located on chromosome 1, which contains a highly conserved gene within a conserved genomic interval containing an identical intron-exon structure spanning exons 6–83 of the total 86-exon-containing HYDIN open reading frame (Figure 2A).49 Therefore, we had to accept that when we amplified exons 6–83 of HYDIN, we also amplified and sequenced near-identical HYDIN2 alleles. Careful analysis of sequence reads identified the DNA sequence variant c.3985G>T in exon 27 (Figure 2). This mutation exchanges the first base of exon 27, affecting the evolutionary conserved acceptor splice site (Figure 2B and 2C). Because we were unable to distinguish by PCR amplification and sequencing whether this DNA exchange affects HYDIN on chromosome 16 or HYDIN2 on chromosome 1, we carefully studied the segregation of the mutant allele within the OP-305 family. Genotype analyses for this mutation indeed showed cosegregation with the disease status in the OP-305 family. As a result of interchromosomal duplication of the HYDIN locus, the sequences represent two mutant and two wild-type alleles for the affected children and three wild-type and one mutant allele for the parents (Figure 2B). Because we did not identify any linkage to the HYDIN2 locus on chromosome 1 by whole-genome SNP analysis (Figures 1A and 1C), results of the segregation analyses indicate that the mutation is located within HYDIN in chromosomal region 16q22.
Figure 2.
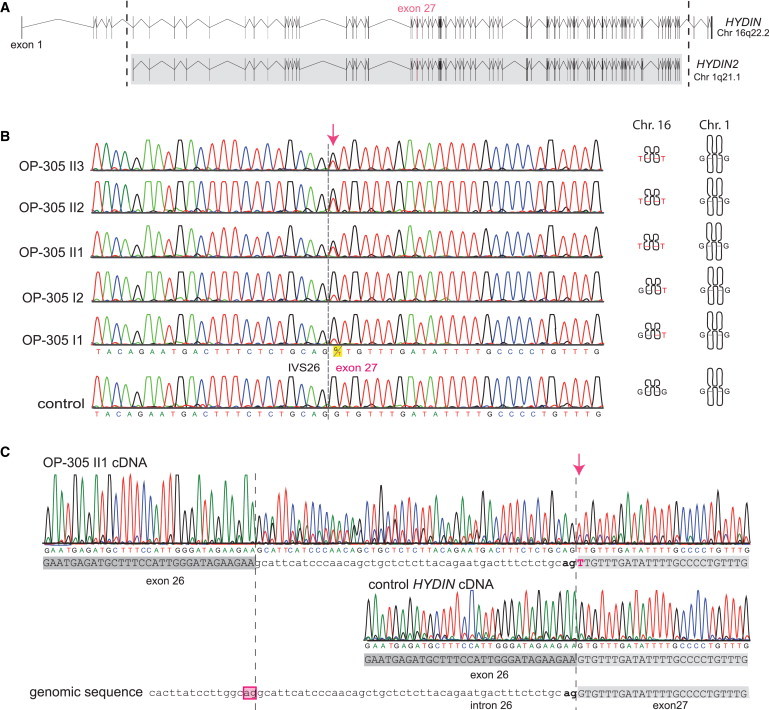
Identification of Recessive HYDIN Mutations in OP-305 and Verification that the c.3985G>T Splicing Mutation Causes Aberrant Splicing
(A) The ancestral duplication of the HYDIN locus from 16q22.2 to HYDIN2 (1q21.1) includes major parts of HYDIN (exons 6–83). The borders of the duplicated region are indicated by the dotted lines, and the gray box defines the paralogous genomic regions between the two loci HYDIN and HYDIN2. Because the duplication occurred very recently during human evolution, even intronic sequences are still conserved between HYDIN and HYDIN2. Thus, by PCR amplification of exon 27, four alleles are amplified, two alleles originate from HYDIN (chromosome 16), and two alleles originate from HYDIN2 (chromosome 1).
(B) Sequence chromatographs of exon 27 amplicons depict relative levels of the G>T DNA variant (c.3985G>T) in individuals from OP-305 at the first position of the conserved splice acceptor site of exon 27. Segregation analysis confirmed that this DNA variant, like SNP markers of the HYDIN locus (Figure 1), cosegregates with disease status. In contrast, SNP markers of the HYDIN2 locus do not show linkage (Figure 1). Thus, the segregation analysis indicates that the detected splicing mutation is located in HYDIN and not in the paralogous HYDIN2.
(C) To provide further evidence that the splicing variant is located within HYDIN, we performed HYDIN-specific RT-PCR analysis on individual OP-305 II1 and control respiratory epithelial cells (Figure S1 and Table S4). Identified in the sample from OP-305 II1 were exclusively abnormal transcripts resulting from malfunction of the HYDIN exon 27 splice acceptor site. The transcripts (upper panel) show a T nucleotide at c.3985 (arrow) at the first base of exon 27, and contained between the exon 26 and exon 27 boundary is an aberrant 47 bp insertion comprising the 3′ most end of IVS26. A control cDNA sequence (middle panel) is shown for comparison with the homozygous reference G nucleotide at c.3985 and normal exon 26/exon 27 splicing. The reference genomic sequence is shown in the bottom panel. The aberrant insertion can be explained by utilization of an upstream cryptic IVS26 splice acceptor (boxed) compared to the normal exon 27 splice acceptor (bold).
The HYDIN c.3985G>T Splicing Mutation Causes Aberrant Splicing
We performed RT-PCR analysis on individual OP-305 II1 and control respiratory epithelial cells. A cDNA fragment comprising exons 25–28 was amplified in two rounds of amplifications with a first-strand primer spanning the junction between exons 29 and 30 (see Figure S1A). The first round of PCR amplification was carried out with a HYDIN-specific forward primer located in the nonduplicated area (exon 5). We identified in the control samples only normal transcripts. We found in the affected person predominantly abnormal transcripts, which is consistent with malfunction of the HYDIN exon 27 splice acceptor site (c.3985G>T). The abnormal HYDIN transcript contained the c.3985G>T substitution and had a 47 bp insertion between exon 26 and exon 27 apparently as a result of the use of a cryptic splice acceptor site located within IVS26 (Figure 2C). The insertion leads to a frameshift and premature stop codon after the insertion of 62 novel amino acids (r.[3985-47_3985-1ins; 3985G>U]). Consistent with a homozygous loss-of function mutation present in OP-305 II1, we did not identify any normal HYDIN transcripts in this material. The background sequence notable in OP-305 II1 (Figure 2C) results from amplification of HYDIN2 transcripts with normal splicing and of the wild-type c.3985G sequence.
Whole-Exome Sequencing Identifies Recessive HYDIN Mutations
In parallel, next-generation whole-exome sequence analysis identified recessive HYDIN mutations in six Faroe PCD-affected persons. We performed whole-exome analysis on two affected siblings from PCD-affected family UCL109, which is a pedigree from a previously studied cohort of eight families originating from the Faroe Islands (Figure 3B).32 Sequence results were equivalent between the two samples (6.6 Gb of total bases generated per sample). More than 98% of the sequence reads were mapped with high mapping accuracy in both individuals (Table S2). A filtering process for assessing variants for novelty identified 17 homozygous and 328 heterozygous variations shared between the affected individuals (Table S1). Analysis of the 328 heterozygous variants showed that only one gene, ZNF295, had biallelic nonsynonymous variants, indicating inheritance of compound-heterozygous changes consistent with recessive inheritance. However, ZNF295 is not present in the cilia proteome and has no known ciliary functions.50 Of the 17 homozygous shared variants, we prioritized those in exonic regions and splice sites. Only two genes had stop-gain mutations, splice-site mutations, or nonsynonymous changes; these were DPYSL2 (MIM 602463), which is not present in the cilia proteome and has no known ciliary functions, and HYDIN.
Thus, whole-exome filtering identified HYDIN variants shared between both affected individuals: a homozygous A-to-T substitution at hg19 genomic position 71,171,175 corresponding to c.922A>T in exon 8 of HYDIN. This is predicted to create nonsense alteration p.Lys307∗ (Figures 3A and 3C and Figure S5). There was high sequence coverage with a read depth of 129 across this variant; however, a low sequence quality score was recorded. From the total of 129 reads, only nine reads could be mapped unambiguously to HYDIN on chromosome 16. The nine mapped reads all carried the aberrant T nucleotide resulting in a homozygous annotation of this change because of the 120 reads that could not be unambiguously mapped, nearly 50% carried the T variant and the rest carried an A. This suggests that only reads from chromosome 16 HYDIN were fully 100% aligned, and none of the reads were generated from the chromosome 1 HYDIN2 homolog. This recording of the nucleotide as a homozygous T in both persons is a useful finding in the whole-exome data in view of the difficulty of distinguishing the duplicated HYDIN from HYDIN2 by Sanger sequencing. The low depth of 9 resulted in a very low quality score, and this variant could have been missed if more stringent quality filtering had been applied.
The c.922A>T variant is not found in dbSNP or in exomes sequenced in the 1000 Genomes Project and National Heart, Lung, and Blood Institute (NHLBI) Grand Opportunity (GO) Exome Sequencing Project. Segregation analysis of c.922A>T within the entire cohort of eight Faroe Island families was performed with Sanger sequencing. Primers designed to amplify the exon 8 (chromosome 16) HYDIN sequence showed a similar result in chromatograms to that of the German OP-305 sample testing (Figure 2B)—the A:T dose ratio needed to be carefully assessed for determining genotypes due to background amplification of the equivalent section of HYDIN2. This analysis confirmed inheritance of a homozygous mutation in affected persons (Figure S5).
We therefore designed chromosome-specific primers in order to create a unique amplicon corresponding specifically to HYDIN and HYDIN2 (see Material and Methods). The primers were designed according to the principles of amplification refractory mutation system (ARMS) testing, whereby oligonucleotide primers with a single mismatched 3′ residue will not function as primers in PCR under sufficiently stringent conditions.51 Representative sequence data from family UCL109 for the chromosome-16-specific HYDIN amplicon show that affected children have an unambiguous homozygous c.922A>T change, whereas unaffected family members have heterozygous changes (Figure 3A). The mutation segregated in all three pedigrees in accordance with recessive disease (Figure 3B).
Analysis of the available SNP haplotypes across the HYDIN locus was entirely consistent with this mutation inheritance given that an 8.8 Mb ancestral haplotype was found to be shared between the affected individuals from these three families, consistent with the reported ancestral relationship between UCL109 and UCL96 (Figure 3B).
To confirm that the detected mutation is indeed located within HYDIN and not within HYDIN2 on chromosome 1, we performed nested cDNA amplification with HYDIN-specific forward primers in exons 4 and 5, which are not duplicated (Figure S1B). The control samples showed normal sequences, whereas the sequence of affected individual UCL109 II4 demonstrated a homozygous A>T exchange creating a stop codon (Figure 3C).
TEM and Electron Microscopy Tomography Show that HYDIN-Mutant Cilia Have a Subtle CP Defect
To characterize the sequelae of HYDIN mutations in the three affected siblings (OP-305 II1, OP-305 II2, and OP-305 II3), we examined TEM of respiratory cilia. Visualization of cilia cross-sections showed identical findings in all three affected individuals. Most cilia exhibited a normal 9+2 axonemal composition, characterized by nine outer microtubule doublets surrounding two single central microtubules (Figures 4A and 4B). ODAs and IDAs showed no alterations. Visualization of the CP-apparatus projections did not reveal any obvious abnormalities. 9+0 cilia (not shown) and 8+1 cilia (ciliary transposition defect where the CP is absent and replaced by one of the peripheral microtubules) were only rarely observed (Figure 4C). Thus, most cross-sections appeared entirely normal. In addition, TEM was performed in four (UCL109 II1, UCL109 II3, UCL109 II4, and UCL139 II1) of the six Faroe PCD-affected persons. Here, we found similar findings (Figure S4). Interestingly, in the three affected members of the UCL109 family, no ciliary transposition defects (8+1 cilia) were detected. Notably, a few 9+3 cilia (additional central microtubules) were only identified in UCL109 II1 (Figure S4). However, overall, routine TEM findings were nonspecific and not sufficient for diagnosis of PCD.
Figure 4.
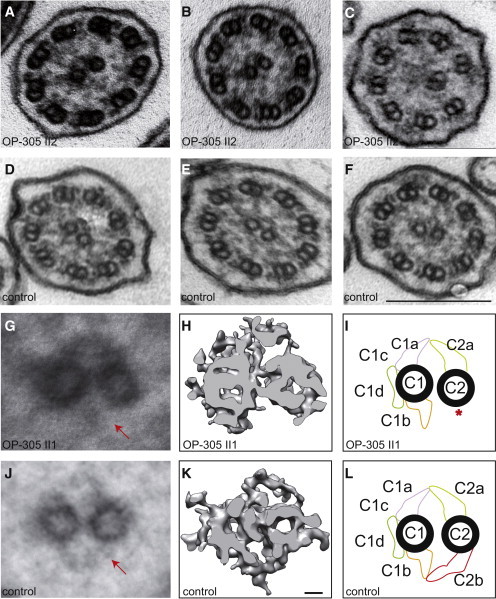
HYDIN Mutant Respiratory Ciliary Axonemes Show Defects of the CP Apparatus
Routine TEM cross-sections of HYDIN-mutant individual OP-305 II2 (A and B) exhibit in most cross-sections a normal 9+2 axonemal composition. When compared with control samples (D–F), the cross-sections of affected individuals rarely show 9+0 (not shown) or 8+1 (ciliary transposition defect in C) compositions. Please note that routine TEM cannot discern CP-apparatus projections. Image averages based on six affected (G) and six control (J) images aid visualization of the CP appendages. Note that the C2b projection is missing (marked with arrow) in the affected individuals. A detailed analysis of the CP apparatus by electron microscopic tomography (H and I) identified the absence of the C2b projection in HYDIN-mutant cilia from individual OP-305 II1 compared to control samples (K and L). Each rendered image (H and K) represents six cross-sections averaged from a dual-axis tomogram made up of 140 images at 2° tilts. The scale bars represent 10 nm (rendered picture) and 200 nm (TEM pictures).
These findings are consistent with our results by immunofluorescence microscopy. Using antibodies directed against ODA DNAH5 heavy chain, IDA DNALI1 light chain (Figure S2), and dynein regulatory complex (DRC) components GAS11 and CCDC39 (Figure S3), we found no evidence for alterations in the structure or composition of the ODA, IDA, and DRC complexes.
Because the projections of the CP apparatus cannot be visualized accurately by TEM, we performed image averages that aid visualizing the CP appendages (Figure 4G). To affirm these findings, we utilized electron microscopy tomography, which has a much higher resolution (Figure 4H). Electron microscopy tomography clearly showed that in HYDIN mutant respiratory cilia of OP-305 II1, the projection C2b is absent at the CP apparatus (Figure 4H). Movies S1 and S2 contain the original electron-microscopy images used for calculating the rendered images of the CP apparatus (Figures 4H and 4K).
HYDIN Mutant Respiratory Cilia and Sperm Flagella Exhibit Abnormal Axonemal Bending
To understand the effect of HYDIN mutations at the functional level, we performed high-speed videomicroscopic evaluation of respiratory cilia beating and sperm-tail motility. We were able to perform nasal-brushing biopsy in all affected siblings of family OP-305 and in individuals UCL139 II1 and UCL109 II1 to visualize respiratory-cilia function. In all affected individuals, we recognized identical abnormalities of the beating pattern (Movies S3, S4, S5, S6, S7, S8, S9, S10, S11, and S12 and Figure 5A). Respiratory cilia lacked coordinated beating activity, and the fact that individual cilia exhibited a reduced bending capacity along the ciliary axoneme resulted in reduced beating amplitudes. In comparison to control cilia, they appeared to be more rigid. As a consequence, mutant cilia did not generate distinct effective or recovery strokes and had a reduced capacity to transport mucus or particles (Movies S3, S4, S5, S6, S7, S8, S9, S10, S11, and S12). The ciliary beat frequency was normal (4–7 Hz at room temperature) in most of the areas analyzed. However, immotile cilia were also found in a few regions (Movies S3 and S4). Consistent with the occasional defects of ciliary transposition (indicated by 9+0 and 8+1 axonemes) present in some cross-sections documented by electron microscopy, some ciliary bundles also showed rotatory movement (Movies S5 and S6).
Figure 5.
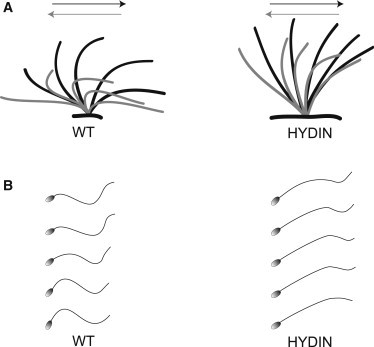
Functional Analysis of Respiratory Ciliary Beating and Sperm-Tail Movement
(A) Schematic of the effective stroke (black) and recovery stroke (gray) from wild-type respiratory cilia and an affected person of family OP-305 with HYDIN mutations.
(B) Schematic of sperm-tail movement of control and HYDIN-mutant sperm cells. Both HYDIN-mutant respiratory cilia and sperm tails exhibit a markedly reduced bending capacity. Note that most of the HYDIN-mutant sperm cells are immotile, and only a few show some residual motility. The bending of the proximal sperm flagellum and respiratory cilium appears to be more affected than distal bending.
In adult male OP-305 II2, ejaculate was available for high-speed videomicroscopic sperm analysis (Movies S11 and S12). Sperm motility was markedly decreased—69% of sperms were completely immotile, and 23% of sperms exhibited only some residual motility. Only 8% of sperms showed some degree of progressive motility. Analysis of these sperm tails revealed an aberrant flagellar beating pattern with a markedly reduced bending capacity, especially of the proximal part of the flagellum (Figure 5B). Like respiratory cilia, the HYDIN mutant sperm tails appeared rigid.
Discussion
We were very fortunate to study a large consanguineous PCD-affected family, enabling the identification of a single gene locus for PCD on chromosome 16q, which includes HYDIN (Figure 1), and we found homozygous HYDIN mutations in all those affected. For our mutation-detection strategy, we took into account that PCR amplification of HYDIN genomic fragments comprising exons 6–83 results in amplification of four alleles (two of HYDIN and two of HYDIN2). Thus, we actively searched for DNA sequence reads with equal strong peaks for wild-type and variant alleles in all three affected individuals. With this rather unusual strategy, we indeed identified recessive homozygous HYDIN mutations in the OP-305 family (Figure 2) and consistent data in Faroe Island families (Figure S5). However, using this approach for the identification of individuals with compound-heterozygous mutations in HYDIN will be very challenging because detection of one mutant allele on the background of three wild-type alleles could easily be overlooked (see also Figure 2 and Figure S5 sequence reads of the parents). The newer high-throughput DNA sequencing technologies applied in the Faroe Island cohort proved here to be more suitable for identifying HYDIN mutations because they produce allele-specific sequence reads. Despite the successful use of whole-exome sequencing in this case, determining whether the identified sequence variants represent HYDIN or HYDIN2 variants will still remain a major challenge in genetic testing, and cDNA expression analyses will be very important as a confirmatory test. Here, we could clearly demonstrate that the c.3985G>T HYDIN mutations affecting the evolutionarily conserved splice acceptor site of exon 27 cause aberrant cDNA processing and HYDIN transcripts with predicted premature stop of translation (Figure 2). A historical PCD locus (CILD5) on chromosome 16 was previously described in a cohort of eight Faroe Island families comprising all of the known PCD cases on these isolated islands located in the North sea and governed by Denmark.32 Whole-exome sequencing was used in this cohort for identifying a second variant, c.922A>T in HYDIN, predicted to lead to premature termination of the protein product (p.Lys307∗). This mutation accounted for all six individuals in three out of the total eight families, confirming at the molecular level the genetic heterogeneity within this population.
To understand the molecular HYDIN defect in more detail, we performed TEM in all the affected families, and this revealed only subtle abnormalities comprising a few 9+0, 9+3, or 8+1 axonemes (ciliary transposition defects; Figure 4 and Table S5). Most cross-sections appeared unremarkable. These TEM findings also parallel findings obtained in Hydin-deficient cilia of Trypanosoma, where abnormalities of the CP apparatus were reported in only a small proportion of cross-sections.45 To characterize the defect of the CP apparatus in detail, we performed high-resolution electron microscopy tomography and were able to clearly demonstrate that HYDIN mutant respiratory cilia lack the C2b projection of the CP apparatus (Figure 4 and Movies S1 and S2). Identical observations were reported in Hydin-deficient mice47 and Hydin-deficient unicellular green Chlamydomonas alga.46 On the basis of identical findings in three different organisms (Chlamydomonas, mouse, and man), it is very likely that the C2b projection represents the site of HYDIN/Hydin localization at the CP apparatus.
To understand the functional consequences of HYDIN dysfunction, we performed high-speed videomicroscopic analyses of the ciliary beat of respiratory cilia obtained by nasal-brushing biopsy in all three affected siblings. A small proportion of the mutant cilia were immotile. For the mutant cilia with preserved motile ciliary beating, the beat frequencies were apparently within normal ranges. However, detailed analyses of the ciliary beat patterns demonstrated a difference in the ciliary beat amplitude; stiff uncoordinated cilia had a reduced bending capacity. These were unable to move particles and mucus, explaining the defect in airway clearance observed in the affected persons. Interestingly, similar functional defects of the ciliary beat were observed in respiratory and ependymal cilia in Hydin-mutant hy3 mice.47
The HYDIN-related motility defect is distinct from that of cilia largely or completely lacking the entire CP apparatus as a result of RSPH4A or RSPH9 mutations.7 These gene mutations cause a much higher proportion of cilia to display transposition defects (9+0 or 8+1) and to have a predominantly circular motion reminiscent of the CP-less embryonic-node cilia. The HYDIN-mutant cilia were only occasionally observed to show rotatory movement, reflecting the occasional ciliary-transposition defects observed in some cross-sections.
In one of the affected adult male persons (OP-305 II3), ejaculate was available for assessment of sperm-tail (flagella) function. Most sperm tails were immotile, and the majority of remaining sperm flagella only showed twitchy stiff movements or markedly reduced bending amplitude, especially in the proximal part of the flagellum. This provokes an asymmetric bending pattern (Figure 5B and Movies S13 and S14). These findings resemble observations obtained in flagella of Hydin-deficient Chlamydomonas alga.46 Thus, the function of the CP-apparatus-associated HYDIN/Hydin appears to be evolutionarily conserved from the green Chlamydomonas alga to Trypanosoma, mouse, and man.
Two additional mouse models with CP-apparatus defects have been reported. Mice lacking the CP-apparatus protein Pcdp1 also develop hydrocephalus with normal situs composition and display abnormal respiratory ciliary beating. Like those of Hydin-mutant mice, TEM analyses appeared normal.52 Only after careful TEM analyses in Chlamydomonas alga deficient for the orthologous protein was the lack of the CP-associated appendage CP1d noted.53 Therefore, PCDP1 mutations might also cause PCD in humans. In the flagella of PF20-mutant Chlamydomonas alga, absence of the whole CP apparatus is apparent. PF20 is associated with the C2 tubule of the CP apparatus and is probably responsible for CP integrity.54 Mice homozygous for a knockout of the C-terminal transcript of the human PF20 ortholog (SPAG16L) are infertile as a result of sperm-motility defects but show apparently no other defects, such as hydrocephalus. This is probably explained by the fact that SPAG16L is exclusively expressed in sperm flagella, making this gene an interesting candidate for male infertility.55
We previously demonstrated in mice that integrity of the cilia motility of ependymal cells is mandatory for maintaining patency of the aqueduct of Sylvii, which connects the third and fourth brain ventricles.48 Thus, disruption of ependymal flow regularly causes hydrocephalus in mice. In contrast, human PCD-affected individuals carry only an increased risk of developing hydrocephalus. This difference is probably best explained by the much larger brain, brain ventricle, and aqueduct size in humans compared to mice.48 Therefore, it is not unexpected that all nine PCD-affected individuals possessing recessive HYDIN mutations do not suffer from hydrocephalus. They also do not have any left-right asymmetry defects like situs inversus or situs ambiguous, and this is also consistent with findings observed in hy3 mice. This is best explained by the fact that the motile node monocilia, which are responsible for determination of left-right asymmetry, contain a 9+0 axonemal composition and therefore naturally lack the CP apparatus.2 Thus, our findings are consistent with the interpretation that the motility of nodal cilia in humans and mice probably does not depend on the function of the CP apparatus. In contrast, ependymal and respiratory cilia, as well as sperm flagella, all contain 9+2 axonemes.
In a recent survey conducted in more than 1,000 individuals affected by PCD in 26 countries throughout Europe, the age at diagnosis and the risk factors for late diagnosis were determined.24 It was demonstrated that the diagnosis of PCD is often delayed, especially in children without situs inversus. It is currently considered that early diagnosis and appropriate respiratory management have a significantly positive effect on the long-term outcomes and life expectancy of persons with PCD. Delayed diagnosis is readily explained by the lack of availability of specific diagnostic tools for PCD. Thus, the molecular understanding of PCD variants without situs inversus is of great importance for improving PCD diagnostics.
Diagnosing PCD in persons possessing mutations of HYDIN will be a major challenge because these individuals do not have left-right asymmetry abnormalities and because routine TEM reveals predominantly normal findings in most cilia cross-sections. Our findings highlight the important role of nNO measurement as a screening tool and of high-speed videomicroscopy for the diagnostic workup in persons suspected to suffer from PCD.
Acknowledgments
We are grateful to the affected persons and their families for their participation in this study. We thank the German patient support group “Kartagener Syndrom und Primaere Ciliaere Dyskinesie e.V.” We thank Angelina Heer, Martina Herting, Denise Nergenau, Carmen Kopp, and Dinu Antony for excellent technical assistance. The UK10K project (http://www.uk10k.org/) is funded by the Wellcome Trust. Contributing investigators of the UK10K project are listed in the Supplemental Data. H.M.M. thanks R. Mark Gardiner and Maggie Meeks for originating the Faroe Island project and Peter J. Scambler and Philip L. Beales for their work on the UK10K project. This work was supported by grants from the Newlife Foundation for Disabled Children and Action Medical Research (to H.M.M.), the Kindness for Kids Foundation, Deutsche Forschungsgemeinschaft (DFG Om 6/4), GRK 1104, SFB 592, and the Cell Dynamics and Disease graduate program, as well as the European Community’s SYSCILIA project (to H.O.). The manuscript has been seen and approved by all authors.
Contributor Information
Hannah M. Mitchison, Email: h.mitchison@ucl.ac.uk.
Heymut Omran, Email: heymut.omran@ukmuenster.de.
Supplemental Data
The video contains the original images used for calculating the rendered image of the CP apparatus (Figure 4E). The cross-sections of the respiratory cilia show the absence of the CP-apparatus C2b projection in some sectional planes.
These pictures were used for calculating the rendered image of the normal CP apparatus (Figure 4I).
Individual cilia exhibit a reduced bending capacity along the ciliary axoneme, resulting in a reduced beating amplitude. Ciliary beat frequency was normal (4–7 Hz at room temperature) in most of the areas analyzed. However, immotile cilia were observed in a few regions.
A clear whirling phenotype that can be found in 9+0 cilia lacking a CP apparatus is not present. Again, the reduced intercilia coordination can be seen.
Note the persistence of mucus and debris as a result of lacking cilia-generated fluid flow.
In addition, a rigid beating pattern with reduced beating amplitude is observed.
The respiratory ciliary beating patterns appear rigid and have a reduced beating amplitude.
Here, representative examples of sperm tails with residual motility are shown. These sperm show an aberrant flagella beating pattern with a markedly reduced bending capacity.
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Project, http://www.1000genomes.org
Exome Variants Analysis and Reporting (EVAR), http://www.exome.info
NHLBI GO Exome Sequencing Project Exome Variant Server, http://snp.gs.washington.edu/EVS
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Satir P., Mitchell D.R., Jékely G. How did the cilium evolve? Curr. Top. Dev. Biol. 2008;85:63–82. doi: 10.1016/S0070-2153(08)00803-X. [DOI] [PubMed] [Google Scholar]
- 2.Fliegauf M., Benzing T., Omran H. When cilia go bad: Cilia defects and ciliopathies. Nat. Rev. Mol. Cell Biol. 2007;8:880–893. doi: 10.1038/nrm2278. [DOI] [PubMed] [Google Scholar]
- 3.Zariwala M.A., Knowles M.R., Omran H. Genetic defects in ciliary structure and function. Annu. Rev. Physiol. 2007;69:423–450. doi: 10.1146/annurev.physiol.69.040705.141301. [DOI] [PubMed] [Google Scholar]
- 4.Nonaka S., Tanaka Y., Okada Y., Takeda S., Harada A., Kanai Y., Kido M., Hirokawa N. Randomization of left-right asymmetry due to loss of nodal cilia generating leftward flow of extraembryonic fluid in mice lacking KIF3B motor protein. Cell. 1998;95:829–837. doi: 10.1016/s0092-8674(00)81705-5. [DOI] [PubMed] [Google Scholar]
- 5.Kennedy M.P., Omran H., Leigh M.W., Dell S., Morgan L., Molina P.L., Robinson B.V., Minnix S.L., Olbrich H., Severin T. Congenital heart disease and other heterotaxic defects in a large cohort of patients with primary ciliary dyskinesia. Circulation. 2007;115:2814–2821. doi: 10.1161/CIRCULATIONAHA.106.649038. [DOI] [PubMed] [Google Scholar]
- 6.Chilvers M.A., Rutman A., O’Callaghan C. Ciliary beat pattern is associated with specific ultrastructural defects in primary ciliary dyskinesia. J. Allergy Clin. Immunol. 2003;112:518–524. doi: 10.1016/S0091-6749(03)01799-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Castleman V.H., Romio L., Chodhari R., Hirst R.A., de Castro S.C., Parker K.A., Ybot-Gonzalez P., Emes R.D., Wilson S.W., Wallis C. Mutations in radial spoke head protein genes RSPH9 and RSPH4A cause primary ciliary dyskinesia with central-microtubular-pair abnormalities. Am. J. Hum. Genet. 2009;84:197–209. doi: 10.1016/j.ajhg.2009.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Olbrich H., Häffner K., Kispert A., Völkel A., Volz A., Sasmaz G., Reinhardt R., Hennig S., Lehrach H., Konietzko N. Mutations in DNAH5 cause primary ciliary dyskinesia and randomization of left-right asymmetry. Nat. Genet. 2002;30:143–144. doi: 10.1038/ng817. [DOI] [PubMed] [Google Scholar]
- 9.Pennarun G., Escudier E., Chapelin C., Bridoux A.M., Cacheux V., Roger G., Clément A., Goossens M., Amselem S., Duriez B. Loss-of-function mutations in a human gene related to Chlamydomonas reinhardtii dynein IC78 result in primary ciliary dyskinesia. Am. J. Hum. Genet. 1999;65:1508–1519. doi: 10.1086/302683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Loges N.T., Olbrich H., Fenske L., Mussaffi H., Horvath J., Fliegauf M., Kuhl H., Baktai G., Peterffy E., Chodhari R. DNAI2 mutations cause primary ciliary dyskinesia with defects in the outer dynein arm. Am. J. Hum. Genet. 2008;83:547–558. doi: 10.1016/j.ajhg.2008.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mazor M., Alkrinawi S., Chalifa-Caspi V., Manor E., Sheffield V.C., Aviram M., Parvari R. Primary ciliary dyskinesia caused by homozygous mutation in DNAL1, encoding dynein light chain 1. Am. J. Hum. Genet. 2011;88:599–607. doi: 10.1016/j.ajhg.2011.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duriez B., Duquesnoy P., Escudier E., Bridoux A.M., Escalier D., Rayet I., Marcos E., Vojtek A.M., Bercher J.F., Amselem S. A common variant in combination with a nonsense mutation in a member of the thioredoxin family causes primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2007;104:3336–3341. doi: 10.1073/pnas.0611405104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bartoloni L., Blouin J.L., Pan Y., Gehrig C., Maiti A.K., Scamuffa N., Rossier C., Jorissen M., Armengot M., Meeks M. Mutations in the DNAH11 (axonemal heavy chain dynein type 11) gene cause one form of situs inversus totalis and most likely primary ciliary dyskinesia. Proc. Natl. Acad. Sci. USA. 2002;99:10282–10286. doi: 10.1073/pnas.152337699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schwabe G.C., Hoffmann K., Loges N.T., Birker D., Rossier C., de Santi M.M., Olbrich H., Fliegauf M., Failly M., Liebers U. Primary ciliary dyskinesia associated with normal axoneme ultrastructure is caused by DNAH11 mutations. Hum. Mutat. 2008;29:289–298. doi: 10.1002/humu.20656. [DOI] [PubMed] [Google Scholar]
- 15.Omran H., Kobayashi D., Olbrich H., Tsukahara T., Loges N.T., Hagiwara H., Zhang Q., Leblond G., O’Toole E., Hara C. Ktu/PF13 is required for cytoplasmic pre-assembly of axonemal dyneins. Nature. 2008;456:611–616. doi: 10.1038/nature07471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Loges N.T., Olbrich H., Becker-Heck A., Häffner K., Heer A., Reinhard C., Schmidts M., Kispert A., Zariwala M.A., Leigh M.W. Deletions and point mutations of LRRC50 cause primary ciliary dyskinesia due to dynein arm defects. Am. J. Hum. Genet. 2009;85:883–889. doi: 10.1016/j.ajhg.2009.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Duquesnoy P., Escudier E., Vincensini L., Freshour J., Bridoux A.M., Coste A., Deschildre A., de Blic J., Legendre M., Montantin G. Loss-of-function mutations in the human ortholog of Chlamydomonas reinhardtii ODA7 disrupt dynein arm assembly and cause primary ciliary dyskinesia. Am. J. Hum. Genet. 2009;85:890–896. doi: 10.1016/j.ajhg.2009.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mitchison H.M., Schmidts M., Loges N.T., Freshour J., Dritsoula A., Hirst R.A., O’Callaghan C., Blau H., Al Dabbagh M., Olbrich H. Mutations in axonemal dynein assembly factor DNAAF3 cause primary ciliary dyskinesia. Nat. Genet. 2012;44:381–389, S1–S2. doi: 10.1038/ng.1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Merveille A.C., Davis E.E., Becker-Heck A., Legendre M., Amirav I., Bataille G., Belmont J., Beydon N., Billen F., Clément A. CCDC39 is required for assembly of inner dynein arms and the dynein regulatory complex and for normal ciliary motility in humans and dogs. Nat. Genet. 2011;43:72–78. doi: 10.1038/ng.726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Becker-Heck A., Zohn I.E., Okabe N., Pollock A., Lenhart K.B., Sullivan-Brown J., McSheene J., Loges N.T., Olbrich H., Haeffner K. The coiled-coil domain containing protein CCDC40 is essential for motile cilia function and left-right axis formation. Nat. Genet. 2011;43:79–84. doi: 10.1038/ng.727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Panizzi J.R., Becker-Heck A., Castleman V.H., Al-Mutairi D.A., Liu Y., Loges N.T., Pathak N., Austin-Tse C., Sheridan E., Schmidts M. CCDC103 mutations cause primary ciliary dyskinesia by disrupting assembly of ciliary dynein arms. Nat. Genet. 2012;44:714–719. doi: 10.1038/ng.2277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Budny B., Chen W., Omran H., Fliegauf M., Tzschach A., Wisniewska M., Jensen L.R., Raynaud M., Shoichet S.A., Badura M. A novel X-linked recessive mental retardation syndrome comprising macrocephaly and ciliary dysfunction is allelic to oral-facial-digital type I syndrome. Hum. Genet. 2006;120:171–178. doi: 10.1007/s00439-006-0210-5. [DOI] [PubMed] [Google Scholar]
- 23.Moore A., Escudier E., Roger G., Tamalet A., Pelosse B., Marlin S., Clément A., Geremek M., Delaisi B., Bridoux A.M. RPGR is mutated in patients with a complex X linked phenotype combining primary ciliary dyskinesia and retinitis pigmentosa. J. Med. Genet. 2006;43:326–333. doi: 10.1136/jmg.2005.034868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kuehni C.E., Frischer T., Strippoli M.P., Maurer E., Bush A., Nielsen K.G., Escribano A., Lucas J.S., Yiallouros P., Omran H., ERS Task Force on Primary Ciliary Dyskinesia in Children Factors influencing age at diagnosis of primary ciliary dyskinesia in European children. Eur. Respir. J. 2010;36:1248–1258. doi: 10.1183/09031936.00001010. [DOI] [PubMed] [Google Scholar]
- 25.Barbato A., Frischer T., Kuehni C.E., Snijders D., Azevedo I., Baktai G., Bartoloni L., Eber E., Escribano A., Haarman E. Primary ciliary dyskinesia: A consensus statement on diagnostic and treatment approaches in children. Eur. Respir. J. 2009;34:1264–1276. doi: 10.1183/09031936.00176608. [DOI] [PubMed] [Google Scholar]
- 26.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. GRR: Graphical representation of relationship errors. Bioinformatics. 2001;17:742–743. doi: 10.1093/bioinformatics/17.8.742. [DOI] [PubMed] [Google Scholar]
- 27.O’Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Abecasis G.R., Cherny S.S., Cookson W.O., Cardon L.R. Merlin—Rapid analysis of dense genetic maps using sparse gene flow trees. Nat. Genet. 2002;30:97–101. doi: 10.1038/ng786. [DOI] [PubMed] [Google Scholar]
- 29.Gudbjartsson D.F., Jonasson K., Frigge M.L., Kong A. Allegro, a new computer program for multipoint linkage analysis. Nat. Genet. 2000;25:12–13. doi: 10.1038/75514. [DOI] [PubMed] [Google Scholar]
- 30.Thiele H., Nürnberg P. HaploPainter: A tool for drawing pedigrees with complex haplotypes. Bioinformatics. 2005;21:1730–1732. doi: 10.1093/bioinformatics/bth488. [DOI] [PubMed] [Google Scholar]
- 31.Rüschendorf F., Nürnberg P. ALOHOMORA: A tool for linkage analysis using 10K SNP array data. Bioinformatics. 2005;21:2123–2125. doi: 10.1093/bioinformatics/bti264. [DOI] [PubMed] [Google Scholar]
- 32.Jeganathan D., Chodhari R., Meeks M., Faeroe O., Smyth D., Nielsen K., Amirav I., Luder A.S., Bisgaard H., Gardiner R.M. Loci for primary ciliary dyskinesia map to chromosome 16p12.1-12.2 and 15q13.1-15.1 in Faroe Islands and Israeli Druze genetic isolates. J. Med. Genet. 2004;41:233–240. doi: 10.1136/jmg.2003.014084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.McKenna A., Hanna M., Banks E., Sivachenko A., Cibulskis K., Kernytsky A., Garimella K., Altshuler D., Gabriel S., Daly M., DePristo M.A. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010;20:1297–1303. doi: 10.1101/gr.107524.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.1000 Genomes Project Consortium A map of human genome variation from population-scale sequencing. Nature. 2010;467:1061–1073. doi: 10.1038/nature09534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Marthin J.K., Nielsen K.G. Choice of nasal nitric oxide technique as first-line test for primary ciliary dyskinesia. Eur. Respir. J. 2011;37:559–565. doi: 10.1183/09031936.00032610. [DOI] [PubMed] [Google Scholar]
- 39.American Thoracic Society. European Respiratory Society ATS/ERS recommendations for standardized procedures for the online and offline measurement of exhaled lower respiratory nitric oxide and nasal nitric oxide, 2005. Am. J. Respir. Crit. Care Med. 2005;171:912–930. doi: 10.1164/rccm.200406-710ST. [DOI] [PubMed] [Google Scholar]
- 40.Sisson J.H., Stoner J.A., Ammons B.A., Wyatt T.A. All-digital image capture and whole-field analysis of ciliary beat frequency. J. Microsc. 2003;211:103–111. doi: 10.1046/j.1365-2818.2003.01209.x. [DOI] [PubMed] [Google Scholar]
- 41.Rashid S., Breckle R., Hupe M., Geisler S., Doerwald N., Neesen J. The murine Dnali1 gene encodes a flagellar protein that interacts with the cytoplasmic dynein heavy chain 1. Mol. Reprod. Dev. 2006;73:784–794. doi: 10.1002/mrd.20475. [DOI] [PubMed] [Google Scholar]
- 42.Mateos-Corral D., Coombs R., Grasemann H., Ratjen F., Dell S.D. Diagnostic value of nasal nitric oxide measured with non-velum closure techniques for children with primary ciliary dyskinesia. J. Pediatr. 2011;159:420–424. doi: 10.1016/j.jpeds.2011.03.007. [DOI] [PubMed] [Google Scholar]
- 43.DiBella L.M., Sakato M., Patel-King R.S., Pazour G.J., King S.M. The LC7 light chains of Chlamydomonas flagellar dyneins interact with components required for both motor assembly and regulation. Mol. Biol. Cell. 2004;15:4633–4646. doi: 10.1091/mbc.E04-06-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Davy B.E., Robinson M.L. Congenital hydrocephalus in hy3 mice is caused by a frameshift mutation in Hydin, a large novel gene. Hum. Mol. Genet. 2003;12:1163–1170. doi: 10.1093/hmg/ddg122. [DOI] [PubMed] [Google Scholar]
- 45.Dawe H.R., Shaw M.K., Farr H., Gull K. The hydrocephalus inducing gene product, Hydin, positions axonemal central pair microtubules. BMC Biol. 2007;5:33. doi: 10.1186/1741-7007-5-33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lechtreck K.F., Witman G.B. Chlamydomonas reinhardtii hydin is a central pair protein required for flagellar motility. J. Cell Biol. 2007;176:473–482. doi: 10.1083/jcb.200611115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lechtreck K.F., Delmotte P., Robinson M.L., Sanderson M.J., Witman G.B. Mutations in Hydin impair ciliary motility in mice. J. Cell Biol. 2008;180:633–643. doi: 10.1083/jcb.200710162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ibañez-Tallon I., Pagenstecher A., Fliegauf M., Olbrich H., Kispert A., Ketelsen U.P., North A., Heintz N., Omran H. Dysfunction of axonemal dynein heavy chain Mdnah5 inhibits ependymal flow and reveals a novel mechanism for hydrocephalus formation. Hum. Mol. Genet. 2004;13:2133–2141. doi: 10.1093/hmg/ddh219. [DOI] [PubMed] [Google Scholar]
- 49.Doggett N.A., Xie G., Meincke L.J., Sutherland R.D., Mundt M.O., Berbari N.S., Davy B.E., Robinson M.L., Rudd M.K., Weber J.L. A 360-kb interchromosomal duplication of the human HYDIN locus. Genomics. 2006;88:762–771. doi: 10.1016/j.ygeno.2006.07.012. [DOI] [PubMed] [Google Scholar]
- 50.Gherman A., Davis E.E., Katsanis N. The ciliary proteome database: An integrated community resource for the genetic and functional dissection of cilia. Nat. Genet. 2006;38:961–962. doi: 10.1038/ng0906-961. [DOI] [PubMed] [Google Scholar]
- 51.Newton C.R., Graham A., Heptinstall L.E., Powell S.J., Summers C., Kalsheker N., Smith J.C., Markham A.F. Analysis of any point mutation in DNA. The amplification refractory mutation system (ARMS) Nucleic Acids Res. 1989;17:2503–2516. doi: 10.1093/nar/17.7.2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lee L., Campagna D.R., Pinkus J.L., Mulhern H., Wyatt T.A., Sisson J.H., Pavlik J.A., Pinkus G.S., Fleming M.D. Primary ciliary dyskinesia in mice lacking the novel ciliary protein Pcdp1. Mol. Cell. Biol. 2008;28:949–957. doi: 10.1128/MCB.00354-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.DiPetrillo C.G., Smith E.F. Pcdp1 is a central apparatus protein that binds Ca(2+)-calmodulin and regulates ciliary motility. J. Cell Biol. 2010;189:601–612. doi: 10.1083/jcb.200912009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Smith E.F., Lefebvre P.A. PF20 gene product contains WD repeats and localizes to the intermicrotubule bridges in Chlamydomonas flagella. Mol. Biol. Cell. 1997;8:455–467. doi: 10.1091/mbc.8.3.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Nagarkatti-Gude D.R., Jaimez R., Henderson S.C., Teves M.E., Zhang Z., Strauss J.F., 3rd Spag16, an axonemal central apparatus gene, encodes a male germ cell nuclear speckle protein that regulates SPAG16 mRNA expression. PLoS ONE. 2011;6:e20625. doi: 10.1371/journal.pone.0020625. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The video contains the original images used for calculating the rendered image of the CP apparatus (Figure 4E). The cross-sections of the respiratory cilia show the absence of the CP-apparatus C2b projection in some sectional planes.
These pictures were used for calculating the rendered image of the normal CP apparatus (Figure 4I).
Individual cilia exhibit a reduced bending capacity along the ciliary axoneme, resulting in a reduced beating amplitude. Ciliary beat frequency was normal (4–7 Hz at room temperature) in most of the areas analyzed. However, immotile cilia were observed in a few regions.
A clear whirling phenotype that can be found in 9+0 cilia lacking a CP apparatus is not present. Again, the reduced intercilia coordination can be seen.
Note the persistence of mucus and debris as a result of lacking cilia-generated fluid flow.
In addition, a rigid beating pattern with reduced beating amplitude is observed.
The respiratory ciliary beating patterns appear rigid and have a reduced beating amplitude.
Here, representative examples of sperm tails with residual motility are shown. These sperm show an aberrant flagella beating pattern with a markedly reduced bending capacity.