

Abstract
Sequencing of gene-coding regions (the exome) is increasingly used for studying human disease, for which copy-number variants (CNVs) are a critical genetic component. However, detecting copy number from exome sequencing is challenging because of the noncontiguous nature of the captured exons. This is compounded by the complex relationship between read depth and copy number; this results from biases in targeted genomic hybridization, sequence factors such as GC content, and batching of samples during collection and sequencing. We present a statistical tool (exome hidden Markov model [XHMM]) that uses principal-component analysis (PCA) to normalize exome read depth and a hidden Markov model (HMM) to discover exon-resolution CNV and genotype variation across samples. We evaluate performance on 90 schizophrenia trios and 1,017 case-control samples. XHMM detects a median of two rare (<1%) CNVs per individual (one deletion and one duplication) and has 79% sensitivity to similarly rare CNVs overlapping three or more exons discovered with microarrays. With sensitivity similar to state-of-the-art methods, XHMM achieves higher specificity by assigning quality metrics to the CNV calls to filter out bad ones, as well as to statistically genotype the discovered CNV in all individuals, yielding a trio call set with Mendelian-inheritance properties highly consistent with expectation. We also show that XHMM breakpoint quality scores enable researchers to explicitly search for novel classes of structural variation. For example, we apply XHMM to extract those CNVs that are highly likely to disrupt (delete or duplicate) only a portion of a gene.
Introduction
Copy-number variants (CNVs) have emerged in the last decade as a category of structural genetic diversity that plays a key role in human health and common disease.1 A number of studies have implicated deletion and duplication CNVs in cancer susceptibility, metastasis, gene expression, and treatment.2 Similarly, rare CNVs are enriched in individuals with severe neuropsychiatric conditions, such as autism (MIM 209850), schizophrenia (MIM 181500), intellectual disability, and epilepsy.3–6 In fact, copy-number changes are the variants that have the largest known effect on the risk of schizophrenia possibly as a result of the constant introduction of de novo germline mutations.7 However, knowledge regarding the scope of CNV effects on disease is still incomplete. This results from the need for additional samples, as well as from a lack of fine-grained genomic resolution in existing technologies, such as microarray-based approaches (SNP arrays or array comparative genomic hybridization [aCGH]), with which most work on CNV detection has been performed. Although aCGH has gained resolution over the years, its widespread use might be limited because of its focus on CNV discovery at the expense of other variation.
The introduction of next-generation-sequencing (NGS) technology has provided a window into the genome at base-pair resolution and has the advantage of allowing simultaneous discovery of single-nucleotide, indel, and structural (translocation, inversion, and copy-number) variation. In practice, many recent disease studies have chosen high-depth targeted exome sequencing, i.e., focusing on known coding regions of the genome.8,9 The key reasons for this are the lower cost as compared to that of whole-genome sequencing, the expectation from Mendelian disorders that the exome will be enriched for disease mutations, and the interpretability of a variant’s effect on gene product. Such studies are likely to continue into the foreseeable future because exome sequencing of many individuals will have more statistical power to detect disease association than will whole-genome sequencing of fewer individuals. However, to maximize the impact on disease, these studies need to integrate the full spectrum of genetic variation ascertainable by using sequencing even though this is fraught with difficulty.10
Numerous tools exist for discovering CNVs from array intensities, and recent work has placed CNV calling on solid ground for whole-genome sequencing data by utilizing diverse information, including unusual mapping of read mate pairs to the reference genome, “split” reads that span breakpoints, and sequencing depth of coverage, i.e., “read depth.”11 In contrast, because exome sequencing takes aim at a sparse (∼1%) set of noncontiguous genomic targets (the exons), most CNV breakpoints will not be sequenced, leaving read depth as the predominant indicator of CNVs. However, the quantitative relationship between true copy number and depth is distorted by target- and sample-specific biases in exome hybridization (“capture”), PCR amplification, sequencing efficiency, and in silico read mapping, all of which are in turn affected by GC content of the targets, target size and sequence complexity, proximity to segmental duplications, nucleotide-level variation (SNPs), DNA concentration, hybridization temperature, experimental sample batching, and the complex interplay among these and various indeterminate factors (Figure S1, available online). The resulting differences are dramatic in that the number of reads varies by an order of magnitude or more (Figure S2), even for diploid regions (copy number = 2). Hence, whole-genome read-depth methods are not applicable to targeted sequencing if the extra biases are not accounted for.
Previously, researchers tailored CNV methods to targeted sequencing and used read-depth normalization to account for a small set of predefined factors, including background depth, GC content, and analysis window size.12 When “split” read evidence exists, it has been used for augmenting detection of CNVs,13 as well as other structural variants, indels, and copy-number-polymorphic-processed pseudogenes.14 Cancer studies have afforded themselves the use of per-sample case-control matching (tumor versus normal) to simplify depth normalization.15
To augment the repertoire of tools for “variation hunting,” we developed XHMM (exome hidden Markov model, Figure 1), a statistical toolset for detecting exon-resolution CNVs from exome sequence data with a disease-motivated focus on rare (<5%) events (see Material and Methods). XHMM extracts copy-number signal from noisy read depth by leveraging the large-scale nature of sequencing projects to discern patterns of read-depth biases. Specifically, we ran a principal-component analysis (PCA) on the sample-by-target-depth matrix by “rotating” the high-dimensional data to find the main modes in which depth varies across multiple samples and targets, and we removed the largest of such effects. This resulted in rigorous data-driven normalization (Figure S2) without the requirement of detailed knowledge of the particular confounders, although we did observe correlation with expected ones such as GC content (Figure S3). After this, we used a hidden Markov model (HMM) to discover CNVs spanning adjacent targets, where depletion or enrichment in normalized read depth implies a deletion or duplication, respectively. Our model takes into account genome-wide CNV rates, length, and distance between exome targets. Next, we derived HMM-based quality scores that measure the certainty we have regarding a CNV, its breakpoints, not having a CNV, and other metrics (see Material and Methods), which we implemented as a multisample quantitative genotyping module that estimates which samples show some (or no) evidence of a CNV discovered in another sample. Thus, XHMM is uniquely suited to detect de novo CNVs and other events requiring high-confidence accuracy, e.g., CNVs whose breakpoints fall within a gene and leave only a partial segment16 where such “gene disruptions” have been implicated in disease.6,17,18,19
Figure 1.
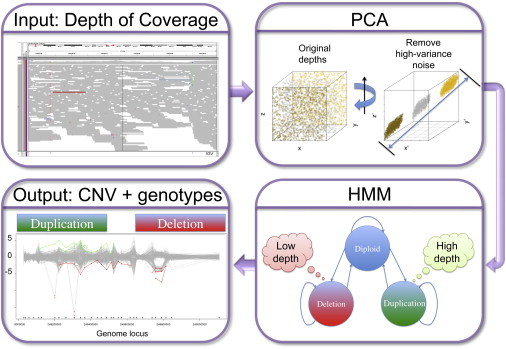
XHMM Pipeline for Discovery and Genotyping of CNVs from Exome Read-Depth Information
The XHMM framework starts with aligned exome read BAM files to: (1) calculate depth of coverage (top left panel), (2) normalize read depth by using principal-component analysis (PCA) (top right panel), (3) train and run a hidden Markov model (HMM) (bottom right panel), and (4) output CNV calls and genotype qualities for all samples (bottom left panel).
Material and Methods
Primary CNV-Calling Pipeline
We now detail the six steps in the XHMM framework for CNV detection from exome sequencing data.
Coverage: Per-Sample, Per-Target Depth
To start, XHMM requires sequencing reads aligned to the reference genome (in a BAM file), for which we use the Picard/Genome Analysis ToolKit (GATK) NGS data-processing pipeline implemented at the Broad Institute, as previously described.20 In brief, the Burrows-Wheeler Aligner (BWA)21 was used for read mapping and was followed by local realignment around known indels, marking of PCR duplicates, and base-quality-score recalibration. Next, XHMM uses GATK to calculate raw depth-of-coverage values across the exome. Specifically, at a given genomic position, the depth of coverage is defined as the number of sequenced reads aligning to that base (Figure 1, top left), and a minimum read mapping quality of 20 is required by default. This quality threshold is intended for the removal of reads that are spuriously mapped to a target or that have the potential to equally map to multiple genomic loci (MQ0 reads), but this value can be changed without grossly affecting results. For each exon target, the depth-of-coverage values are averaged over its extent, yielding a raw read-depth matrix of samples by targets (Figure S1); the value of a matrix entry is the mean number of reads covering each base in the corresponding target for a particular sequenced sample.
Filter I: Extreme Targets and Samples
The purpose of this prenormalization step is to ensure relative homogeneity in the samples and targets and prevent deviant values from adversely affecting the subsequent PCA. To do this, XHMM performs an outlier removal step, in which it filters out targets with extreme GC content (<0.1 or >0.9), targets with a significant stretch of low-complexity sequence (>10% of target bases soft masked by RepeatMasker22 in the hg19 human reference sequence), targets less than 10 bp or larger than 10 kb, or targets with very low coverage (<10× averaged over all samples) or very high coverage (>500×) in our experiment. Because the samples here were sequenced to an average coverage of 100–150×, we next removed samples with coverage values that were empirical outliers with respect to the full set of samples—those with unexpectedly low coverage (<50× averaged over all targets), high coverage (>200×), or extreme variance (standard deviation > 120 over all targets). As an example, the read-depth distributions for individual samples and targets are plotted in Figure S4. For other studies, we recommend examining the read-depth distributions and removing appropriate outliers.
PCA Normalization of Read Depth
The read depth of exome sequencing for an exon target is a function of a number of biochemical properties of the genome, in addition to experimental and bioinformatic steps including genomic fragmentation, array hybridization (“capture”), PCR,23 sequencing,24 and in silico alignment to the reference genome (“mapping”).25,26 Direct readout of copy number from depth of coverage is not possible because of local genomic-context effects (e.g., GC content,27 repeats, or low-complexity sequence), the inherent biases in each experimental step, and the dynamic range of ambient conditions during the handling of different sample batches.
We thus hypothesized that most of the read-depth variation within a sample and between targets is in fact dominated by effects unrelated to and independent of copy number. To normalize out what is effectively noise when looking for CNV signal, we applied PCA to find the main sources of confounding, i.e., orthogonal high-dimensional axes in which the read depth varies (Figure 1, top right). In detail, we first centered the target read depths about their means and used the singular value decomposition (SVD) implementation of PCA on the individual-by-target read-depth matrix. To better understand the nature of the systematic biases on target depth, we calculated the correlation between the top 100 principal components and various signals that we expected to possibly be involved in determining the read depth of exome sequencing, and we observed the strongest correlations with sample batch, exome-wide mean sample and target depths, target GC content, population, and combinations of these factors (Figure S5). Importantly, some of the highest components were not apparently correlated with any of these predefined phenomena, yet they clearly were not indicative of CNV levels. In practice, a scree plot (Figure S6) is often statistically informative for visually aiding the selection of components with highest variance if one looks for an “elbow”, i.e., a sharp drop in the read-depth variance contributed by later principal components, in the plot.
More formally, to find the high-variance components that are presumed to explain most of the read-depth variation (due to systematic biases), XHMM follows the empirical rule of thumb by calculating the relative variance of each component and removing the K components with a value of 0.7 / n or higher,28 where n is the number of components (in this case, number of samples) and 0.7 is a user-tunable XHMM parameter. To remove these K components, we subtract them out from the matrix of all samples’ read depths R to obtain the normalized read-depth matrix R∗:
| (Equation 1) |
where ci is the ith principal component of R to be normalized out of the depth signal.
As an example, the left panel of Figure S2 demonstrates the large positional and sample effects of the raw read depths for 500 samples in a region of 26 targets across almost 200 kb; the most striking observation is that each target has a characteristic mean shift of sequencing coverage. Furthermore, particular samples show consistently higher (or lower) coverage possibly as a result of batching effects, global extent of sequencing for that sample, or a real signal of CNVs. The read depths for a number of samples suspected of having duplication events are highlighted in green, but these CNVs can be directly read off only after normalization (right panel).
Filter II: Extremely Variable Targets
After the PCA normalization, there were still a number of targets with extreme variability in normalized depth. Thus, by default, XHMM filters out targets with a standard deviation of normalized read depth > 30 in an effort to ensure homogeneity in the input for the next stage and remove any outliers not normalized in previous steps. The filtering performed at this stage might need to be adapted to the features of a particular experiment.
Discovery: Per-Sample CNV Detection with a HMM
In the next step, XHMM discovers CNVs in each sample by using a HMM algorithm for segmentation of the exome into “diploid,” “deletion,” or “duplication” regions, which correspond to average, below-average, and above-average read depth, respectively. To do this, XHMM first transforms the PCA-normalized read depths by using a Z score calculation for each sample separately so that target-depth values are on a similar scale. These Z scores are used as input to a 3-state HMM (Figure 1, bottom right) that is conceptually similar to that used for whole-genome methods,29 but it takes into account exome-wide CNV rates and length distributions, as well as the distance between exome targets (this makes it more likely to continue a CNV within a single gene than across distant genes). The underlying homogeneous HMM state-transition matrix is given in Table 1, where 0 < p < 1 is the exome-wide CNV rate, q = 1 / T, and T > 0 is the mean number of targets in a CNV (geometrically distributed with parameter q). Note that, for simplicity, this model is symmetric with respect to deletions (copy number < 2) and duplications (copy number > 2), although it need not be so. In order to take into account the distance between targets in the exome (denoted by d), we overlay onto the matrix in Table 1 a distance-dependent exponential attenuation factor, f = e−d/D, where D is the expected distance between targets in a CNV (in bases). For longer distances between targets (weighted by 1 − f), we want the probability of being in a CNV to approach that of starting a CNV from a previously diploid state (middle row). This results in the final nonhomogeneous state-transition probability matrix for two targets at a distance of d base pairs (Table 2).
Table 1.
Basic HMM Transition Matrix
| From ↓ To → | Deletion | Diploid | Duplication |
|---|---|---|---|
| Deletion | 1 − q | q | 0 |
| Diploid | p | 1 − 2p | p |
| Duplication | 0 | q | 1 − q |
This matrix takes into account the exome-wide CNV rate (p) and the mean number of targets in a CNV (1 / q).
Table 2.
Distance-Dependent HMM Transition Matrix
| From ↓ To → | Deletion | Diploid | Duplication |
|---|---|---|---|
| Deletion | f(1 − q) + (1 − f)p | fq + (1 − f)(1 − 2p) | (1 − f)p |
| Diploid | p | 1 − 2p | p |
| Duplication | (1 − f)p | fq + (1 − f)(1 − 2p) | f(1 − q) + (1 − f)p |
For two exons located d base pairs apart, this matrix takes into account exome-wide CNV rate, the mean number of CNV targets (see Table 1), and the attenuation of CNV rates at distance d (f = e−d/D, where D is the mean distance between targets in a CNV).
Because we normalize the read-depth values into Z scores, the read-depth emission probability function is symmetrically taken to be a normal distribution of variance 1 centered at −M, 0, and +M, for deletion, diploid, and duplication, respectively.
To perform CNV discovery, XHMM makes copy-number calls by using the standard HMM Viterbi algorithm, which provides the most likely copy-number state given all of the sample’s read-depth data (Figure 1, bottom left) and fixed HMM parameters. To choose HMM parameters, we used the trio samples to perform a grid search to find a combination of all parameters minimizing both the number of putative “de novo” CNVs and the deviation from a 50% transmission rate. We considered p to be between 10−4 and 10−8, T to be between 1 and 6, D to be between 103 and 5 × 105, and M to be between 1 and 6. On the basis of the trio data, we chose CNV rate p = 10−8, mean targets per CNV T = 6, mean within-CNV target distance D = 70,000 (70 kb), and depth Z score threshold M = 3. These parameters, although by no means the “optimal” ones, are reasonable in nature, and we expect them to be broadly applicable for similar experimental exome data sets without significant fine tuning. Importantly, these parameters still give a liberally large CNV call set that should maximize sensitivity toward finding almost all CNV calls for which there is sufficient read-depth signal, whereas we use the quality metrics defined below to achieve higher specificity.
Genotype: Assign HMM-Based Quality Metrics to All Samples for Discovered CNVs
After running the per-sample HMM Viterbi algorithm to discover CNV in each sample, we leveraged the HMM framework to “genotype” each such event across all samples in the data set. Specifically, we derived metrics from the rich field of HMMs to calculate posterior probabilities of, for example, having the whole delineated region as deleted, having some deleted target in the region, not having any deleted targets at all, or having the breakpoint occurring exactly as called in the discovery step. These quantities can be calculated efficiently with the HMM chain structure and can be used for defining quality scores regarding the event occurring (or not occurring) in a particular sample. The important point is that these can be applied to any sample with read-depth data and not just the sample in which the CNV was originally discovered.
As a concrete example, let us assume that a deletion was discovered in some sample ranging between targets t1 and t2, and we would like to genotype this event in a different sample, whose normalized read-depth Z score and underlying copy-number vectors we denote by y1:E and x1:E, respectively (E is the number of exome-wide targets). For simplicity, we denote deletion, diploid, and duplication states as 1, 2, and 3, respectively.
Now, running the standard HMM forward-backward algorithm on this sample gives the quantities:
Forward probability of copy number xt at target t:
Backward probability of copy number xt at target t:
Data likelihood:
With HMM theory, it can be shown that the probability of copy-number sequence given the sample’s read depths is
where Pr(xt | xt−1) and Pr(yt | xt) are the transition and emission probabilities, respectively, defined above. We thus obtain , , and as the probabilities of the sample’s copy-number state being deletion, diploid, or duplication, respectively, for all targets between t1 and t2.
Another quantity of interest is the probability of the copy number being restricted to certain categories (in this case, no duplications):
where the denominator is the standard likelihood defined above and the numerator is a modified likelihood that we calculate by locally rerunning the forward-backward algorithm with the added constraint that (no duplications allowed).
Finally, we define the relevant CNV genotyping qualities as:
where given the probability of a particular CNV genotype quantity c, the Phred-scaled quality of c not being an error is
and a higher quality score implies greater probability that the quantity is supported by the data. Note that it always holds that SQ ≥ EQ because SQ is the CNV deletion quality based on the probability that at least one of the targets is deleted, whereas EQ requires that all targets in the range be most likely deleted. However, only LQ and RQ explicitly require that the particular breakpoints at t1 and t2 be highly likely. Note that similar metrics are of course defined for duplication events.
Applications of Genotype Quality Metrics
When we wish to know whether a particular individual carries the event in question or a similar event (e.g., any deletion in a region), we apply the above-defined genotype qualities depending on the context. For example, we used the trios to calibrate the XHMM parameters and genotype quality thresholds to enable us to converge to accurate, yet sensitive, CNV calls by considering the number of calls, the implied de novo CNV rates, and Mendelian transmission rates from parent to child (Figure 2). We will denote by Q a global quality threshold (in this paper, a value of 60 was ultimately derived from the trio data). We now step through some typical use cases in which these scores can be applied for making statistically informed conclusions about the CNV in question despite the presence of noise. Note that these metrics can also be applied in the case of CNV regions defined externally, where XHMM will use the overlapping targets in a region to calculate the full list of metrics above.
Figure 2.
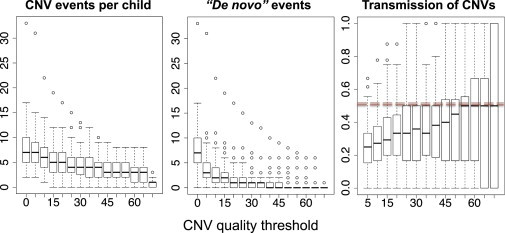
Calibration of XHMM CNV Quality Parameters with 90 Schizophrenia Trio Samples
We calibrate the XHMM parameters by considering how the number of rare CNVs per child (left panel), putative de novo events (middle panel), and parent-to-child transmission rates (right panel) vary as a function of increasingly stringent quality filtering. Boxes denote the interquartile range over all 90 trios. Horizontal solid lines indicate the median, and whiskers extend to the most extreme data points at most 1.5× the interquartile range from the box.
De Novo CNV
For a deletion CNV event discovered in a child, we would like to ask whether the parents have strong evidence for being diploid over the entire span of exon targets (thus implying a de novo deletion). To answer this in the affirmative, we require that SQ ≥ Q in the child and that NQ ≥ Q in each of the parents so that we are confident that the read depths support at least some deletion event existing in the child’s exome and not a hint of a deletion in either parent.
CNV Transmission from Parents
When a CNV is discovered in a parent sample with the HMM Viterbi step, we want to test whether this was, or was not, transmitted to the child. For this, we require that SQ ≥ Q in this parent (to be confident in, at least a portion of, the parental CNV) and require that the child’s genotype qualities satisfy SQ ≥ Q (transmitted) or NQ ≥ Q (not transmitted); if the child’s call does not satisfy either of these criteria, then it is effectively marked as “missing” and we do not include it in this analysis because of the uncertainty. The “missing” genotype permits us to make the important distinction between the absence of a Viterbi call and actually being confident that a sample is diploid. Note that we also ensure that NQ ≥ Q in the other parent so that we know that only one parent could have transmitted this CNV.
Disruptive CNV
To detect high-quality gene-disrupting CNVs, we use the standard quality threshold Q to ensure that there is a significant signal (SQ ≥ Q) of the called CNV. Also, to be certain that at least one of the breakpoint locations (5′ or 3′) is of high quality, we require that LQ ≥ Q′ or RQ ≥ Q′, where Q′ ≤ Q is some more relaxed threshold (we chose Q′ = Q / 2 = 30 as a reasonable value in this study). Lastly, we require that the breakpoint with high certainty actually falls within a gene transcript and not at the edge of the gene.
Comparison of Overlapping CNV
Note that a similar approach to that for finding disruptive CNV could be used for inferring whether overlapping CNVs in two different individuals are in fact the same event or not. Specifically, we would require high-quality CNV breakpoints and would compare their respective locations for the two samples. This test can be used for investigating the possibilities of recurrent events that have different mechanisms, have the same mechanism, or are identical by descent.
Genotyping an Entire Genomic Interval
Finally, for the case of genotyping a sample as being either diploid, deleted, duplicated, or “no call” over an entire particular region, XHMM applies one of the four rules below.
-
1.
Call as diploid if DQ ≥ Q (and SQdel < Q and SQdup < Q).
-
2.
Call as deletion if EQdel ≥ Q (and NQdel < Q).
-
3.
Call as duplication if EQdup ≥ Q (and NQdup < Q).
-
4.
Otherwise, no call is made (“missing” genotype).
Note that these determine the actual hard genotype calls present in the VCF file output by XHMM; in order to correct for the fact that DQ and EQ will be strongly correlated with the number of targets in the region, XHMM chooses Q here on a call-by-call basis as the minimum EQ in the samples in which this call was discovered.
Focus on Rare Variation
We have optimized XHMM for rare variation (frequency < 0.05) because of the typical application of exome sequencing for complex diseases and the fact that common CNVs (copy-number polymorphisms [CNPs]) do not explain much risk for these diseases.30 Specifically, the PCA normalization and HMM parameters have been tuned under the assumption that most read-depth variation at a given locus is due to noise, whereas a CNP would not fit into this mold. More generally, the user might need to adjust some of the parameters in the description above in order to maximize the trade-off between false positives and false negatives. However, under reasonable experimental settings, we expect the default values noted to give a liberally called (but not too large) set of CNVs, which can then be easily and effectively filtered by frequency and with the use of the CNV quality scores output by XHMM.
Exome Data Sets Used
In this work, we adopted the following two neuropsychiatric data sets (in the context of large schizophrenia studies currently underway) as a focus for our methods:
-
•
90 trios (a child with schizophrenia and his or her parents), which are part of a larger ongoing sequencing effort of over 600 trios from Bulgaria.7
-
•
1,017 individuals from a Swedish schizophrenia case-control sample (50% cases and 50% controls).31
All samples were whole-exome sequenced at the Broad Institute with the use of whole-blood DNA as previously described.9 One of the driving forces in deriving a rigorous data-driven normalization technique, which does not require explicit knowledge of how systematic effects cause read depth to vary, was the fact that these data had somewhat varying sequencing coverage and experimental batches; this scenario is typical as workflows are frequently updated. Indeed, sample batch was correlated with a few of the highest variance principal components removed during normalization (Figure S5). The use of human subjects for this research was approved by an institutional review board.
Comparison with Affymetrix SNP Microarray CNV Calls
For the case-control data set, all samples were previously genotyped on Affymetrix 5.0 or 6.0 arrays and CNVs were called with Birdsuite32 on the intensity data as previously described.31 The trio samples were also run on the Affymetrix 6.0 platform, and array-detectable de novo CNVs were called and validated as recently described.7
Results
To calibrate XHMM parameters, we used the family-based data set of 90 schizophrenia trios to estimate HMM transition and emission parameters on a grid search (see Material and Methods). We then examined a range of call quality-score thresholds (Figure 2) and considered only those CNV calls of two targets or more. As expected, the number of rare (<5%) CNV events decreases as more stringent (SQ) quality filtering is applied (Figure 2, left panel). For each CNV discovered in a child, we used the genotyping quality scores to detect de novo CNVs in a quantitative fashion, i.e., by requiring high certainty that the child has a deletion and the parents do not. That is, applying score filters results in only a handful of trio offspring that have any de novo CNVs, as expected given Mendelian inheritance and a low mutation rate (Figure 2, middle panel). Moreover, when parental CNVs are genotyped in the child, the median transmission rate from parent to child converges to 50%, as expected from random Mendelian segregation (Figure 2, right panel). We repeated these analyses by limiting to shorter CNVs (<100 kb) and had almost identical results, indicating our confidence in XHMM CNV calls of various lengths. In addition to globally ensuring CNV call quality, we note that of the three independently validated array-based de novo CNV calls in these 90 samples (overlapping exome targets),7 XHMM detected two of these with high quality (Figures S7 and S8). The remaining one overlaps only two exome targets, for which we observe lower overall concordance rates with Affymetrix (see below), but manual inspection indicates a subthreshold de novo deletion that is observable only as a result of the PCA normalization (Figure S9).
Next, we took the trio-calibrated parameters and applied them to the set of 1,017 schizophrenia case-control samples. Running XHMM yielded a total of 2,315 rare (frequency < 1%, calculated by PLINK33) CNVs and a median of two rare CNVs per individual (one deletion and one duplication); over 80% of all CNV were <100 kb (Figure S10). To corroborate our calls, we utilized similarly rare, reliable (>100 kb) Birdsuite calls that were made on these same samples with Affymetrix 6.0 arrays and that overlap one or more exome targets (544 in total; Table S1). XHMM detected 67% of these, and this rate increased to 85% for Affymetrix calls overlapping five or more exome targets (Table 3); see Table S2 for separate sensitivities to deletions and duplications. We conclude from the case-control and trio sets that XHMM performs well in detecting high-quality rare CNVs that span three or more exons because of its sensitivity of 79% and its high specificity corresponding to a 50% transmission rate in trios.
Table 3.
Sensitivity of XHMM to Affymetrix-Based Calls
| t Exome Targets | Affymetrix-Based Calls Overlapping ≥ t Targets | XHMM Sensitivity to Affymetrix | Median Affymetrix CNV Length (kb) |
|---|---|---|---|
| 1 | 544 | 367 (67%) | 214 |
| 2 | 483 | 365 (76%) | 218 |
| 3 | 452 | 357 (79%) | 219 |
| 4 | 409 | 330 (81%) | 202 |
| 5 | 362 | 309 (85%) | 205 |
| 6 | 321 | 282 (88%) | 232 |
| 8 | 288 | 260 (90%) | 240 |
| 10 | 247 | 227 (92%) | 259 |
Using the trio-calibrated XHMM quality scores (Figure 2), we applied XHMM to a sample of 1,017 schizophrenia case-control samples and measured what fraction of high-quality rare Affymetrix-based CNV calls (that overlap at least t exome targets) are captured by the XHMM calls.
We leveraged the resolution of exome sequencing to detect CNV events that fall within a gene and disrupt said gene by requiring high confidence of a copy-number change within a single transcript; the XHMM CNV in Figures S7 and S8 are examples of partial gene deletions. We implement this search by thresholding on the XHMM-estimated breakpoint quality metrics, LQ and RQ. When searching the case-control sets, we detected 182 rare, high-quality disruptive deletions and significantly greater burden in cases (rate of 0.22 versus 0.14 in controls, p = 0.007 according to PLINK33), suggesting that gene-disruptive CNVs might play a role in the etiology of schizophrenia. In contrast, we observed no overall enrichment of CNVs in general (either from exome sequencing or the Affymetrix calls) in this n = 1,017 subsample. As we continue our sequencing of this cohort, we will systematically follow up on this result, which is consistent with the hypothesis that disruptive events are more likely to be pathogenic.16
While this manuscript was under review, the CoNIFER method for detection of copy-number changes was published.34 The depth-normalization approach in XHMM and CoNIFER is similar; both effectively use the SVD implementation of PCA to detect and remove large read-depth variations due to non-CNV signals. However, the methods significantly diverge thereafter in their use of these normalized data. CoNIFER makes calls on the basis of consecutive runs of at least three targets with values above or below a hard threshold, whereas XHMM takes advantage of the full power of HMMs to make and assess the quality of the CNV calls. Moreover, we note the important distinction between how the CoNIFER paper and the present paper use the term “genotype.” In the former, “genotype” is used in the strict sense of determining absolute copy number at common CNP loci, whereas the “statistical genotyping” performed by XHMM is actually a probabilistic assessment (for a genomic interval) of the copy-number states (here, diploid, deletion, or duplication) we consider given the observed read-depth data across the exome. As in CoNIFER, hard copy-number calls can be made from these genotyping scores, although we do not discard these scores because they play an important role in the analysis of the data. Our approach differentiates between not making a call and actually declaring with high certainty that the individual has diploid copy number.
The high sensitivity (79%–85% for CNVs overlapping three to five targets; see Table 3) that we observed in detecting rare chip-based CNVs by using XHMM is similar to the estimate (between 76% and 84%) reported in the CoNIFER paper. With this in mind, we sought to characterize the differences between XHMM and CoNIFER and particularly focused on the specificity of the call sets. We started by comparing the overlap of the rare CNV calls made by the two algorithms for the 90 schizophrenia trios (Figure 3 and Table 4). For the sake of comparison, we used the same BWA-mapped reads as input to the two algorithms, and we ran CoNIFER by using default parameters. A single low-depth outlier sample that was removed by XHMM was also noted by the CoNIFER protocol. For the autosomes, we used CoNIFER to remove six principal components, and visual inspection of the singular values confirmed that this was near the inflection point of the scree plots. Here, we considered only those XHMM CNVs consisting of three targets or more for an equal comparison with CoNIFER’s behavior.
Figure 3.
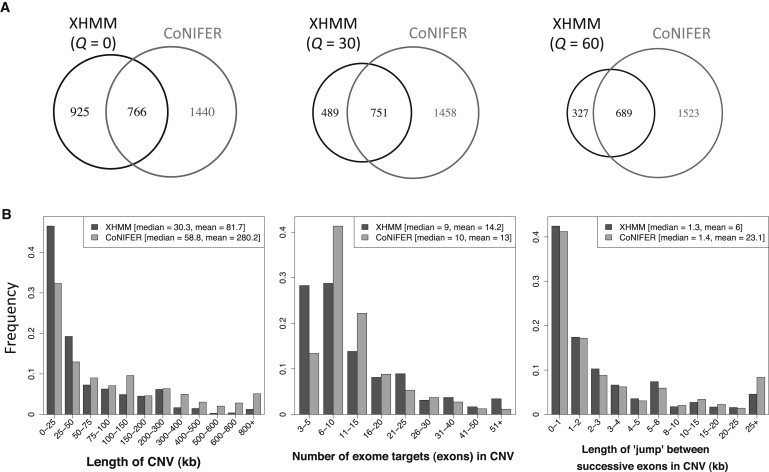
Comparison of XHMM and CoNIFER CNV Calls
(A) Overlap between XHMM and CoNIFER rare (<5%) CNV calls made on the 90 schizophrenia trios, for which XHMM calls are filtered at progressively higher quality filters (Q). Note that overlapping calls are counted as one event.
(B) Comparison of the properties of the XHMM Q = 60 and CoNIFER CNV calls: genomic length of CNV (left panel), number of exome targets (exons) in a CNV (middle panel), and the distance between consecutive exons in a CNV (right panel).
Table 4.
Mendelian Inheritance Analysis for XHMM and CoNIFER
| Median Number of Child CNVs | Median Number of De Novo CNVs | Proportion of Children with One or More De Novo CNVs | Median Transmission Rate | |
|---|---|---|---|---|
| Hard Calls | ||||
| XHMM (Q = 60) | 3 | 1 | 62% | 33% |
| CoNIFER | 8 | 3 | 91% | 29% |
| CoNIFER (unique) | 5 | 3 | 88% | 23% |
| CoNIFER (XHMM overlap) | 2 | 0 | 37% | 43% |
| Statistical Genotyping | ||||
| XHMM (Q = 60) | 3 | 0 | 13% | 50% |
From top to bottom, the high-quality XHMM call set, the CoNIFER set, the calls unique to CoNIFER, the overlap set between XHMM and CoNIFER, and the statistical genotyping in XHMM were analyzed for Mendelian violations and inheritance patterns. See main text for details.
The main findings, which we detail below, are:
-
•
CoNIFER calls possess a high rate of Mendelian violations, whereas XHMM statistical genotyping leads to very few (Table 4).
-
•
CoNIFER makes more calls than XHMM, whereas XHMM provides quality scores to obtain calls with higher confidence (Figure 3A).
-
•
CoNIFER calls are longer than corresponding XHMM calls (Figure 3B) and extend into additional genes.
Considering rare (<5% frequency) CNVs, CoNIFER makes 30% more calls (n = 2,206) than does XHMM (n = 1,691 raw [Q = 0] calls) (Figure 3A). Going to higher-quality XHMM calls (Q = 60), 689 (68%) of the XHMM calls have evidence from a CoNIFER call, whereas these overlapping calls make up only 31% of the CoNIFER calls. Thus, the quality scores we have developed serve as an intrinsic mechanism for selectively obtaining these reliable calls (with secondary “confirmation”) and as a complement to successful filtering based on segmental duplications or similar genomic features.34
We observed striking differences in the properties of the XHMM (Q = 60) and CoNIFER CNV sets in terms of size, number of exons called, and distance between exons in CNV calls (Figure 3B). We found that, overall, the CoNIFER calls are longer than the XHMM calls (Figure 3B, left histogram, mean of 82 kb versus 280 kb, t test p = 4 × 10−11). However, although XHMM does have a higher proportion of calls consisting of between three and five targets (Figure 3B, center, first pair of bars), it is not the case that the CoNIFER calls always include more exons—XHMM makes relatively more calls of 21 targets or more. This most likely results from the ability of the HMM caller to effectively smooth out the normalized read-depth signal and call some large CNVs spanning noisier genomic regions. Notwithstanding, CoNIFER has a significant tendency to include more distant targets within the same CNV call, as measured by the distance between consecutive exons called in a particular CNV (Figure 3B, right, mean of 6 kb versus 23 kb, p = 6 × 10−13). These results are consistent with the implementation of CoNIFER as making calls across extreme-depth exome targets irrespective of their relative genomic distance. On the other hand, XHMM conservatively requires additional evidence (i.e., more extreme normalized read depth) to extend a CNV call across larger genomic intervals (because parameter D attenuates the transition probabilities as a function of distance) while still smoothing out the signal by allowing calls to sometimes extend across noisier regions.
Restricting the above analysis to the 689 high-quality CNVs overlapping in the two call sets, we observed similar significant differences between these intrinsic CNV-call features. Moreover, whereas we noted above that CoNIFER often seems to overextend CNV calls by not regarding intertarget distance, it also inversely tends to break up XHMM calls at a higher rate than XHMM does with respect to CoNIFER calls (17 XHMM calls broken up into two or more separate CoNIFER calls, but only one CoNIFER call is split in two by XHMM). This presumably results from the lack of CoNIFER smoothing that is available to XHMM in the form of an HMM calling procedure. For these CNVs overlapping between XHMM and CoNIFER, we found the CoNIFER calls to have a marked mean increase in CNV length (180 kb longer, paired t test p = 9 × 10−15) and a mean of 3.9 more targets (p = 4 × 10−15).
We then assessed how the above differences in CNV sizes between XHMM and CoNIFER affect which genes are implicated by a particular call. As an example, if one considers the validated de novo CNVs overlapping DLGAP1 (MIM 605445) (Figure S7) and EHMT1 (MIM 607001) (Figure S8), the CoNIFER calls seemingly overextend the XHMM and validated calls and overlap an “additional” gene in one case (for the DLGAP1 deletion). Overall, comparing only calls with some overlap between XHMM and CoNIFER, we found that the genes implicated by both algorithms are copy-number variable in 7.2 exons on average, whereas the genes implicated by the CoNIFER-unique part of the same CNV call include only 4.1 exons (p < 10−16). This is consistent with our assessment that it is critical to account for genomic distance in calling CNVs so that a small number of targets with a similar read-depth trend in a neighboring gene do not artificially extend a particular CNV call. We thus conclude that compared with XHMM, CoNIFER might often add extra targets (as well as extra genes with fewer supporting targets in the CNV call) and that, overall, XHMM might give more accurate CNV breakpoints for its calls. We expect the resolution of exome-based CNV breakpoints to be particularly critical in: (1) gene-set enrichment analysis of genes hit by CNVs, where the inclusion of false-positive genes will decrease testing power by adding noise to the tests; and (2) gene-disruption analysis, where it is especially important to resolve the correct breakpoint in the CNV call, or at least provide a measure of breakpoint quality (output by XHMM as LQ and RQ), so that we can know when a CNV is more likely to affect only part of a gene transcript.
Finally, Table 4 presents the results of Mendelian-inheritance analysis (counting putative de novo CNVs and the fraction of CNVs transmitted from parent to child) for various subsets of the calls. We found that CoNIFER makes more de novo calls (91% of children with at least one de novo call) and fewer transmitted parent calls (29%) than the quality-filtered XHMM calls with hard genotyping (33%). When the CoNIFER calls are split into those with XHMM overlap and those without, the overlapping ones have much better Mendelian metrics (only 37% of children with a de novo and 43% transmission). It is important to emphasize that both the CoNIFER and Q = 60 XHMM calls have Mendelian violations that result from the use of hard genotyping without the assessment of the actual confidence of being diploid. On the other hand, statistically genotyping the XHMM calls results in a large majority of trios that have no de novo CNV calls, but the trios still maintain a median of 50% of parental CNV that is transmitted to the children (these data are identical to those in Figure 2 for Q = 60). Thus, we conclude that both quality filtering and statistical genotyping will play an important role in constructing a prioritized list of a smaller number of (de novo) CNV calls that we expect to be experimentally validated.
Discussion
In this paper, we present a tool (XHMM) to comprehensively normalize sequencing coverage in large-scale exome sequencing and use this rich information to discover CNVs while providing quality metrics that indicate how strongly the data support a particular CNV. We demonstrate that XHMM has high specificity (few Mendelian violations in trios) along with high sensitivity to reliable Affymetrix calls. Using CNV-breakpoint quality metrics, the XHMM framework also permits high-resolution discovery of partial gene disruptions, a form of structural variation potentially involved in disease pathology,16 and we observed a possible burden of gene-disrupting deletions in schizophrenia.
To use the XHMM suite for smaller-scale targeted sequencing, the main limitations in decreasing the number of targets or number of samples are a function of the PCA normalization step, which will degrade in performance because the read-depth aberrations (due to true copy-number changes) of any single target or sample might be filtered out as “batch” effects. In addition, in order to detect and remove underlying experimental artifact (e.g., GC bias), the PCA will need to see a nontrivial subset of samples or targets with that particular trend. Thus, in practice, we would recommend using XHMM with on the order of at least 1,000 targeted regions and 50 (unrelated) samples. For the case of related samples, we propose using principal components derived from larger sets of samples sequenced contemporaneously under similar conditions so that familial CNVs do not dominate the read-depth signal and are not picked up by PCA. Thus, we explicitly chose a parameterization of read-depth normalization (Equation 1) so that the principal components (ci) need not be derived from the matrix R to be normalized, as long as they are defined for the same target set.
In terms of coverage, the important point is that the dynamic range of the read-depth signal be large enough that decreases can be assigned to underlying deletions and increases to duplications. Thus, although the sequencing coverage for the samples here was at least 100× on average, XHMM could be applied with lower mean sequencing coverage (say, 50×), as long as the observed coverages do not essentially degenerate into a discrete all-or-none value. XHMM could also potentially be adapted in the context of whole-genome sequencing, e.g., by the division of the genome into “pseudotargets” with the use of a sliding-window approach.13 For simplicity, XHMM does not explicitly consider homozygous deletions or duplications (or copy number above 3), although we expect such events to be correctly called as copy-number variable. These can be modeled by the augmentation of the HMM with states corresponding to these copy numbers. Also, although XHMM can work with sex chromosomes, we performed all analyses herein on the autosomes to limit cryptic sex-specific effects, for which additional normalization might be required.
In summary, the distinguishing features of XHMM are: (1) efficient data-driven whole-exome read-depth normalization with the use of PCA for thousands of individuals (it does not rely on a single reference sample or any predefined notions of read-depth confounders), (2) incorporation of genomic distance into the calling procedure for well-calibrated CNV lengths, (3) use of a HMM for smoothing over noisy regions and taking into account exome-wide CNV rates, (4) HMM-based quality scores for filtering good calls from bad calls, and (5) using these quality scores and breakpoint metrics for statistically genotyping discovered CNVs in other individuals and detecting de novo and gene-disrupting events. We have demonstrated that these features endow XHMM with superior performance in the analysis of real data sets, and we expect exome-based CNV analysis to be a useful complement to array-based approaches because of their differential strengths and biases.
Acknowledgments
We would like to thank Mark DePristo, Joeseph Buxbaum, and Edward Scolnick for their helpful discussions. This work was supported by National Institute of Health grants RC2MH089905 (to principal investigators S.M.P. and P.S.) and R01HG005827 (to S.M.P.) and by the Sylvan Herman Foundation.
Contributor Information
Menachem Fromer, Email: menachem.fromer@mssm.edu.
Shaun M. Purcell, Email: shaun.purcell@mssm.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
XHMM Software, http://atgu.mgh.harvard.edu/xhmm/
References
- 1.Sharp A.J., Locke D.P., McGrath S.D., Cheng Z., Bailey J.A., Vallente R.U., Pertz L.M., Clark R.A., Schwartz S., Segraves R. Segmental duplications and copy-number variation in the human genome. Am. J. Hum. Genet. 2005;77:78–88. doi: 10.1086/431652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Shlien A., Malkin D. Copy number variations and cancer susceptibility. Curr. Opin. Oncol. 2010;22:55–63. doi: 10.1097/CCO.0b013e328333dca4. [DOI] [PubMed] [Google Scholar]
- 3.Stone J.L., O’Donovan M.C., Gurling H., Kirov G.K., Blackwood D.H., Corvin A., Craddock N.J., Gill M., Hultman C.M., Lichtenstein P., International Schizophrenia Consortium Rare chromosomal deletions and duplications increase risk of schizophrenia. Nature. 2008;455:237–241. doi: 10.1038/nature07239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Stefansson H., Rujescu D., Cichon S., Pietiläinen O.P., Ingason A., Steinberg S., Fossdal R., Sigurdsson E., Sigmundsson T., Buizer-Voskamp J.E., GROUP Large recurrent microdeletions associated with schizophrenia. Nature. 2008;455:232–236. doi: 10.1038/nature07229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Walsh T., McClellan J.M., McCarthy S.E., Addington A.M., Pierce S.B., Cooper G.M., Nord A.S., Kusenda M., Malhotra D., Bhandari A. Rare structural variants disrupt multiple genes in neurodevelopmental pathways in schizophrenia. Science. 2008;320:539–543. doi: 10.1126/science.1155174. [DOI] [PubMed] [Google Scholar]
- 6.Cooper G.M., Coe B.P., Girirajan S., Rosenfeld J.A., Vu T.H., Baker C., Williams C., Stalker H., Hamid R., Hannig V. A copy number variation morbidity map of developmental delay. Nat. Genet. 2011;43:838–846. doi: 10.1038/ng.909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Kirov G., Pocklington A.J., Holmans P., Ivanov D., Ikeda M., Ruderfer D., Moran J., Chambert K., Toncheva D., Georgieva L. De novo CNV analysis implicates specific abnormalities of postsynaptic signalling complexes in the pathogenesis of schizophrenia. Mol. Psychiatry. 2012;17:142–153. doi: 10.1038/mp.2011.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Teer J.K., Mullikin J.C. Exome sequencing: the sweet spot before whole genomes. Hum Mol Genet. 2010;19(R2):R145–R151. doi: 10.1093/hmg/ddq333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kiezun A., Garimella K., Do R., Stitziel N.O., Neale B.M., McLaren P.J., Gupta N., Sklar P., Sullivan P.F., Moran J.L. Exome sequencing and the genetic basis of complex traits. Nat. Genet. 2012;44:623–630. doi: 10.1038/ng.2303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Koboldt D.C., Ding L., Mardis E.R., Wilson R.K. Challenges of sequencing human genomes. Brief. Bioinform. 2010;11:484–498. doi: 10.1093/bib/bbq016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Handsaker R.E., Korn J.M., Nemesh J., McCarroll S.A. Discovery and genotyping of genome structural polymorphism by sequencing on a population scale. Nat. Genet. 2011;43:269–276. doi: 10.1038/ng.768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Love M., Myšičková A., Sun R., Kalscheuer V., Vingron M., Haas S. Modeling read counts for CNV detection in exome sequencing data. Stat Appl in Genet. Mol. Bio. 2011;10 doi: 10.2202/1544-6115.1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nord A., Lee M., King M.-C., Walsh T. Accurate and exact CNV identification from targeted high-throughput sequence data. BMC Genomics. 2011;12:184. doi: 10.1186/1471-2164-12-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karakoc E., Alkan C., O’Roak B.J., Dennis M.Y., Vives L., Mark K., Rieder M.J., Nickerson D.A., Eichler E.E. Detection of structural variants and indels within exome data. Nat. Methods. 2012;9:176–178. doi: 10.1038/nmeth.1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sathirapongsasuti J.F., Lee H., Horst B.A.J., Brunner G., Cochran A.J., Binder S., Quackenbush J., Nelson S.F. Exome sequencing-based copy-number variation and loss of heterozygosity detection: ExomeCNV. Bioinformatics. 2011;27:2648–2654. doi: 10.1093/bioinformatics/btr462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mills R.E., Walter K., Stewart C., Handsaker R.E., Chen K., Alkan C., Abyzov A., Yoon S.C., Ye K., Cheetham R.K., 1000 Genomes Project Mapping copy number variation by population-scale genome sequencing. Nature. 2011;470:59–65. doi: 10.1038/nature09708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee C., Scherer S.W. The clinical context of copy number variation in the human genome. Expert Rev. Mol. Med. 2010;12:e8. doi: 10.1017/S1462399410001390. [DOI] [PubMed] [Google Scholar]
- 18.Durand C.M., Betancur C., Boeckers T.M., Bockmann J., Chaste P., Fauchereau F., Nygren G., Rastam M., Gillberg I.C., Anckarsäter H. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat. Genet. 2007;39:25–27. doi: 10.1038/ng1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vacic V., McCarthy S., Malhotra D., Murray F., Chou H.H., Peoples A., Makarov V., Yoon S., Bhandari A., Corominas R. Duplications of the neuropeptide receptor gene VIPR2 confer significant risk for schizophrenia. Nature. 2011;471:499–503. doi: 10.1038/nature09884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DePristo M.A., Banks E., Poplin R., Garimella K.V., Maguire J.R., Hartl C., Philippakis A.A., del Angel G., Rivas M.A., Hanna M. A framework for variation discovery and genotyping using next-generation DNA sequencing data. Nat. Genet. 2011;43:491–498. doi: 10.1038/ng.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Smit, A., Hubley, R., and Green, P. (2010). RepeatMasker Open-3.0. Institute for Systems Biology. http://www.repeatmasker.org.
- 23.Aird D., Ross M.G., Chen W.S., Danielsson M., Fennell T., Russ C., Jaffe D.B., Nusbaum C., Gnirke A. Analyzing and minimizing PCR amplification bias in Illumina sequencing libraries. Genome Biol. 2011;12:R18. doi: 10.1186/gb-2011-12-2-r18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bentley D.R., Balasubramanian S., Swerdlow H.P., Smith G.P., Milton J., Brown C.G., Hall K.P., Evers D.J., Barnes C.L., Bignell H.R. Accurate whole human genome sequencing using reversible terminator chemistry. Nature. 2008;456:53–59. doi: 10.1038/nature07517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Campbell P.J., Stephens P.J., Pleasance E.D., O’Meara S., Li H., Santarius T., Stebbings L.A., Leroy C., Edkins S., Hardy C. Identification of somatically acquired rearrangements in cancer using genome-wide massively parallel paired-end sequencing. Nat. Genet. 2008;40:722–729. doi: 10.1038/ng.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Alkan C., Kidd J.M., Marques-Bonet T., Aksay G., Antonacci F., Hormozdiari F., Kitzman J.O., Baker C., Malig M., Mutlu O. Personalized copy number and segmental duplication maps using next-generation sequencing. Nat. Genet. 2009;41:1061–1067. doi: 10.1038/ng.437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Clark M.J., Chen R., Lam H.Y., Karczewski K.J., Chen R., Euskirchen G., Butte A.J., Snyder M. Performance comparison of exome DNA sequencing technologies. Nat. Biotechnol. 2011;29:908–914. doi: 10.1038/nbt.1975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Everitt B., Dunn G. Second Edition. Great Britain; Arnold: 2001. Applied Multivariate Data Analysis. [Google Scholar]
- 29.Yoon S., Xuan Z., Makarov V., Ye K., Sebat J. Sensitive and accurate detection of copy number variants using read depth of coverage. Genome Res. 2009;19:1586–1592. doi: 10.1101/gr.092981.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Craddock N., Hurles M.E., Cardin N., Pearson R.D., Plagnol V., Robson S., Vukcevic D., Barnes C., Conrad D.F., Giannoulatou E., Wellcome Trust Case Control Consortium Genome-wide association study of CNVs in 16,000 cases of eight common diseases and 3,000 shared controls. Nature. 2010;464:713–720. doi: 10.1038/nature08979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bergen S.E., O’Dushlaine C.T., Ripke S., Lee P.H., Ruderfer D.M., Akterin S., Moran J.L., Chambert K.D., Handsaker R.E., Backlund L. Genome-wide association study in a Swedish population yields support for greater CNV and MHC involvement in schizophrenia compared with bipolar disorder. Mol. Psychiatry. 2012;17:880–886. doi: 10.1038/mp.2012.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Korn J.M., Kuruvilla F.G., McCarroll S.A., Wysoker A., Nemesh J., Cawley S., Hubbell E., Veitch J., Collins P.J., Darvishi K. Integrated genotype calling and association analysis of SNPs, common copy number polymorphisms and rare CNVs. Nat. Genet. 2008;40:1253–1260. doi: 10.1038/ng.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Purcell S., Neale B., Todd-Brown K., Thomas L., Ferreira M.A., Bender D., Maller J., Sklar P., de Bakker P.I., Daly M.J., Sham P.C. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007;81:559–575. doi: 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Krumm N., Sudmant P.H., Ko A., O’Roak B.J., Malig M., Coe B.P., Quinlan A.R., Nickerson D.A., Eichler E.E., NHLBI Exome Sequencing Project Copy number variation detection and genotyping from exome sequence data. Genome Res. 2012;22:1525–1532. doi: 10.1101/gr.138115.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.