

Abstract
Whole-exome sequencing was performed in a family affected by dominantly inherited inflammatory disease characterized by recurrent blistering skin lesions, bronchiolitis, arthralgia, ocular inflammation, enterocolitis, absence of autoantibodies, and mild immunodeficiency. Exome data from three samples, including the affected father and daughter and unaffected mother, were filtered for the exclusion of reported variants, along with benign variants, as determined by PolyPhen-2. A total of eight transcripts were identified as possible candidate genes. We confirmed a variant, c.2120C>A (p.Ser707Tyr), within PLCG2 as the only de novo variant that was present in two affected family members and not present in four unaffected members. PLCG2 encodes phospholipase Cγ2 (PLCγ2), an enzyme with a critical regulatory role in various immune and inflammatory pathways. The p.Ser707Tyr substitution is located in an autoinhibitory SH2 domain that is crucial for PLCγ2 activation. Overexpression of the altered p.Ser707Tyr protein and ex vivo experiments using affected individuals’ leukocytes showed clearly enhanced PLCγ2 activity, suggesting increased intracellular signaling in the PLCγ2-mediated pathway. Recently, our laboratory identified in individuals with cold-induced urticaria and immune dysregulation PLCG2 exon-skipping mutations resulting in protein products with constitutive phospholipase activity but with reduced intracellular signaling at physiological temperatures. In contrast, the p.Ser707Tyr substitution in PLCγ2 causes a distinct inflammatory phenotype that is not provoked by cold temperatures and that has different end-organ involvement and increased intracellular signaling at physiological temperatures. Our results highlight the utility of exome-sequencing technology in finding causal mutations in nuclear families with dominantly inherited traits otherwise intractable by linkage analysis.
Main Text
Autoinflammatory diseases are inherited immunologic disorders caused by aberrant activation of innate immune cells, primarily neutrophils and macrophages. In a broad sense, autoinflammatory diseases could be defined as primary immunodeficiencies because they result from defects in components of the innate immune response. The term autoinflammation was coined with the intent of distinguishing this category of illnesses from the more commonly recognized autoimmune inflammatory diseases that are largely mediated by effectors of the adaptive immune system, namely autoreactive T cells and autoantibodies. Autoinflammatory syndromes include both monogenic and polygenic diseases that manifest as life-long recurrent episodes of systemic inflammation and are accompanied by organ-specific involvement typically affecting the serosa, skin, joints, bones, gastrointestinal tract, and occasionally the CNS. Genome-wide linkage analysis, positional cloning, and candidate-gene screening have led to the identification of mutations in 12 genes associated with various monogenic autoinflammatory diseases.1
Discovering the genetic causes of autoinflammatory diseases has transformed our understanding of human immunobiology and particularly the importance of pathways regulating the cytokine IL-1β. Without advanced techniques in genetic analysis, many of these disorders might have gone unrecognized as members of a phenotypically diverse group. However, the identification of causative mutations in autoinflammatory diseases is limited by their rarity within the general population and the lack of large multicase families. A significant number of genetically uncharacterized affected subjects are sporadic cases or come from small families underpowered for linkage analysis. Recent advances in high-throughput sequencing technologies have allowed a search for causal variants in families that are affected by recessively inherited diseases and that have as few as one or two affected individuals. Nevertheless, small families affected by dominantly inherited and sporadic phenotypes remain a challenge for analysis.
The individuals described here include affected father II-1 and daughter III-2 (Figures 1 and 2), who both suffer from early-onset recurrent blistering skin lesions, nonspecific interstitial pneumonitis with respiratory bronchiolitis (NSIP), arthralgia, eye inflammation, enterocolitis, cellulitis, and mild immunodeficiency manifested by recurrent sinopulmonary infections and by a paucity of circulating antibodies (IgM and IgA). Both individuals tested negative for autoantibodies on multiple occasions. Both individuals developed a full-body epidermolysis-bullosa-like eruption in infancy, but this later evolved to recurrent erythmatous plaques and vesiculopustular lesions that worsen with heat and sun exposure (Figures 1A–1C). Biopsy of open skin lesions revealed a dense infiltrate of neutrophils, eosinophils, histiocytes, and lymphocytes (Figure 1D). At the age of 6 months, individual III-2 developed recurrent abdominal pain with bloody diarrhea and was colonoscopically diagnosed with ulcerative colitis at the age of 2 years. Eye involvement became evident at the age of 8 months when small blisters were discovered around her cornea; these blisters ultimately progressed into corneal erosions, ulcerations, intraocular hypertension, and more recently, cataracts. Her father (II-1) suffered similar dermatological lesions sensitive to the same stimuli, in addition to a history of recurrent sinopulmonary infections. At the age of 18 years, he was diagnosed with “lazy leukocyte syndrome” (impaired leukocyte chemotaxis [MIM 150550]).
Figure 1.
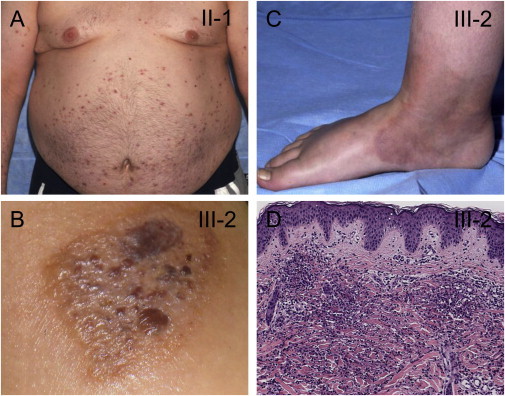
Clinical Manifestations in Two Individuals with the p.Ser707Tyr Substitution in PLCγ2
The inflammatory skin lesions observed in these affected individuals include recurrent eruptions of erythematous plaques (A) and vesiculopustular lesions (B) and the frequent evolution of skin lesions into cellulitis (C).
(D) A dense interstitial and perivascular inflammatory infiltrate in the dermis is composed of lymphocytes, histiocytes, eosinophils, and much karyorrhectic nuclear debris. Karyorrhectic nuclear debris is typically seen in neutrophilic dermatosis.
Figure 2.
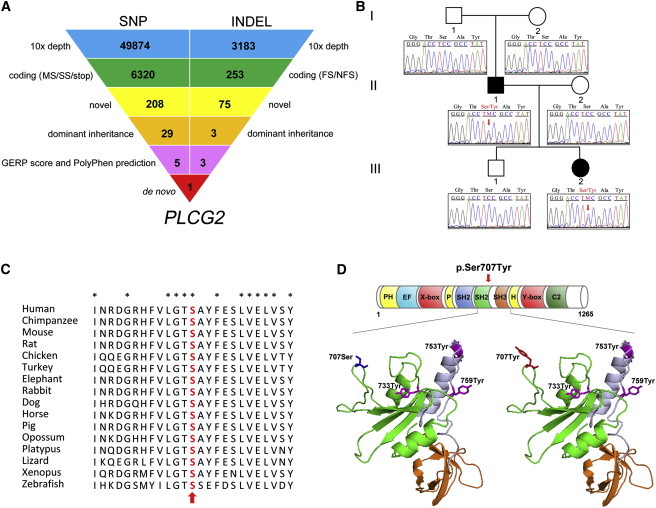
Exome Sequencing Identifies a De Novo PLCG2 c.2120C>A Mutation in a Family Affected by Systemic Inflammatory Disease
(A) Schematic representation of the exome-data-filtering approach under the assumption of dominant inheritance in the family. The following abbreviations are used: MS, missense variant; SS, splice-site variant; stop, stop-codon variant; FS, frameshift indel; and NFS, nonframeshift indel.
(B) A pedigree of the family shows the unaffected grandparents and the affected father and daughter and indicates that the disease-causative mutation (c.2120C>A) arose as a de novo event in the affected father (II-1) and was dominantly inherited in the third generation. Sanger sequencing confirmed complete cosegregation of p.Ser707Tyr in PLCγ2 within the family.
(C) The evolutionary conservation of the p.Ser707Tyr substitution of PLCγ2. Sequence alignment of PLCγ2 among various species shows that Ser707 (red indicated with an arrow) is a highly conserved residue.
(D) Structure of C-terminal SH2 (cSH2) and SH3 domains of PLCγ2. The top panel shows a schematic representation of PLCγ2 and the location of the p.Ser707Tyr substitution. The p.Ser707Tyr substitution is not located within the catalytic domain of PLCγ2 (X-box = 316–458; Y-box = 926–1,029) but is located in the cSH2 domain. The lower panel shows a three-dimensional structural model of the PLCγ2 cSH2 domain (green) and SH3 domain (orange) with the use of homology modeling by SWISS-Model. The substitution of Ser707 (blue) with Tyr (red) lies in the cSH2 domain. The three known phosphorylation sites are depicted in magenta; Tyr733 is located in the cSH2 domain, whereas Tyr753 and Tyr759 are located in the linker region (blue gray) between the cSH2 domain and the SH3 domain.
More recent lymphocyte phenotyping revealed a near complete absence of class-switched memory B cells (CD20+ CD27+ IgM- IgA+ or IgG+), potentially explaining the increased propensity for developing bacterial infections. The distribution and number of circulating naive and memory T cells were normal, as were those of NK cells; however, NK T cell numbers were lower in both individuals (Table S1, available online). Intracellular IFN-γ and IL-17 production of mitogen-activated T cells was similar to that of controls (data not shown), as was IFN-γ production in response to an array of TLR agonists; the only exception was a diminished response to a TLR8 agonist, ssRNA40 (Figures 3A and 3B). Both individuals were refractory to treatment with nonsteroidal anti-inflammatory drugs and TNF inhibitors, and they were partially responsive to an IL-1 inhibitor. Their inflammatory manifestations were ameliorated by high-dose corticosteroids, but the dosage and duration of treatment have been limited by side effects. The paternal grandparents, mother, and brother of individual III-2 were unaffected, and DNA samples from all family members were available for analysis. All individuals (or parents of minors) provided written informed consent as approved by the institutional review board at the National Institute of Arthritis and Musculoskeletal and Skin Diseases and National Institute of Diabetes and Digestive and Kidney Diseases.
Figure 3.
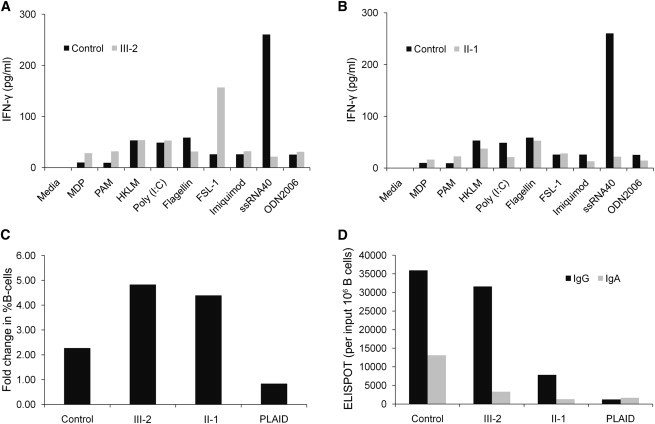
IFN-γ Production in Stimulated PBMCs and Immunophenotyping of Expanded Individual B Cells
(A and B) Peripheral-blood mononuclear cells (PBMCs) were isolated from heparinized blood and cultured in the presence of the indicated agonists for 48 hr. The harvested supernatant was analyzed for the IFN-γ production by multiplex cytokine analysis. The indicated control was representative of multiple different healthy controls.
(C) B cell expansion in response to CpG and SAC stimulation of the affected individual, control, and PLAID PBMCs for 4 days is shown here as the fold increase from baseline after expansion in the percentage of B cells. The percentage of B cells among input or cultured PBMC was measured by flow cytometry.
(D) IgG- or IgA-positive B cells from the cultures in (C) are measured by ELISPOT in the control, affected individual, and PLAID PBMCs. They are represented as Ig-positive cells per million B cells in the initial PBMC culture.
To identify the causative mutation in this family affected by uncharacterized systemic inflammatory disease and without similar cases reported in the literature, we sequenced the exomes of the affected father (II-1) and daughter (III-2) and the unaffected mother (II-2). Targeted exon enrichment was performed on 3 μg of DNA extracted from peripheral blood with the use of the SureSelect Human All Exon 50 Mb Kit (∼24,000 genes, Agilent Technologies). The exon-enriched DNA libraries were subjected to paired-end sequencing with the SOLiD 4.0 System platform (Applied Biosystems). Sequence data were mapped with SOLiD Bioscope software version 1.2 and hg18 human genome as a reference. Reads with mapping quality below 10 and potential duplicates were removed. We achieved an average of 15 Gb of mappable sequences with a mean coverage of 80×; 80% of targeted bases were covered sufficiently for variant calling (>10× coverage). Single-nucleotide substitutions and small indels were identified with SAMtools 0.1.11,2 bam2mpg,3 and Dindel.4 Calls with a read coverage less than 10× and a Phred-scaled variant quality less than 20 were filtered out.
For individuals III-2, II-1, and II-2, there remained 51,744, 50,855, and 47,023 SNPs, respectively, and 3,514, 2,645, and 3,389 indels, respectively, after quality screening. SeattleSeq SNP annotation and ANNOVAR programs5 were then used for categorizing all variants, of which 6,320 were identified as either missense, splice site, or nonsense variants and 253 were identified as coding indels (Figure 2A). Filtering for novel (not available in any database) variants was performed on the basis of comparisons to dbSNP132, the 1000 Genomes Pilot Project (May 2011), the National Heart, Lung, and Blood Institute (NHLBI) Exome Sequencing Project (ESP5400), and 65 additional exomes from internal data. Next, we applied filtering for dominant inheritance present in the affected father and daughter, but not in the unaffected mother, which reduced the number of variants to 32, including 29 SNPs and three indels. Five out of the 29 SNPs passed through the conservation analysis on the basis of the degree of nucleotide-level evolutionary conservation (with the use of Genomic Evolutionary Rate Profiling [GERP])6 and functional-impact prediction on protein by PolyPhen-2, resulting in a total of eight candidate variants (Figure 2A and Table S2).
Sanger sequencing of the eight variants in all family members (I-1, I-2, II-1, II-2, III-1, and III-2) validated a missense variant, a C to A change in exon 20 (c.2120C>A; RefSeq accession number NM_002661.3) of PLCG2 (MIM 600220); this mutation converts Ser707 to Tyr (p. Ser707Tyr) and is the only de novo mutation in the father and in complete segregation with the disease status in this family (Figure 2B). We evaluated the allele frequency of mutation c.2120C>A (p.Ser707Tyr) in a panel of 376 Ashkenazi Jewish healthy controls (paternal ancestry) and 368 control DNA samples of European descent (maternal ancestry) by using mass spectrometry (the Sequenom homogeneous MassExtend assay, Sequenom, San Diego, CA). With a total of 1,488 genotyped control chromosomes, we could sufficiently achieve greater than 95% power to detect a normal variant in the population with an allele frequency of 1%.7 The c.2120C>A (p.Ser707Tyr) mutation was not detected in any of the control DNA samples, suggesting that it is most likely a disease-causing variant in this family.
PLCG2 encodes phospholipase Cγ2 (PLCγ2), an enzyme responsible for ligand-mediated signaling in cells of the hematopoietic system, and plays a key role in the regulation of immune responses. The lipase activity of most phospholipases is basally repressed, and the X/Y linker inserted within the catalytic domain contributes to this autoinhibition. On the basis of studies of PLCγ1, it has been suggested that the autoinhibition can be relieved upon phosphorylation of the protein by receptor tyrosine kinases (RTKs) and subsequent conformational changes.8 The p.Ser707Tyr substitution in PLCγ2 affects a residue highly conserved throughout vertebrates (Figure 2C). The evolutionary conservation of p.Ser707Tyr was measured by the GERP score that was calculated on 34 mammalian species and by the PhastCons score9 based on 17 vertebrate species. A GERP score > 2 or a PhastCons score close to 1 is commonly used as an indication of evolutionary conservation. The GERP and PhastCons scores for p.Ser707Tyr are significantly high at 4.64 and 0.953, respectively. In addition, the substitution occurs in the highly conserved C-terminal copy of a tandem pair of PLCγ2 SH2 domains (cSH2), which, along with the nSH2 domain, SH3 domain, and a split PH domain, comprise the PLCγ-specific array, γSA. This regulatory region is placed within the X/Y linker and contains sites of interaction with regulatory proteins and also elements of autoinhibition. The cSH2 domain is crucial for activation of PLCγ2 as a result of its role in repression of the intrinsic enzyme’s activity because it incorporates the main autoinhibitory regions.8 In general, mutations affecting the SH2 domain in a variety of signaling proteins are directly involved in ligand binding, affect residues interacting with the catalytic domain, or cause SH2 domain destabilization.10 Our recent data suggest that Ser707 is located on the surface of the cSH2 domain that is directly involved in autoinhibition of the catalytic region of PLCγ2 (unpublished data).
p.Ser707Tyr is also in close proximity to the three known phosphorylation sites, of which Tyr733 is located in the same cSH2 domain, whereas Tyr753 and Tyr759 are both located within a linker region next to the cSH2 domain (Figure 2D), suggesting that the p.Ser707Tyr substitution might possibly create a new phosphorylation site in PLCγ2. In that case, the extra phosphorylation site might generate a more easily and efficiently inducible form of PLCγ2. On the basis of computational predictions with PolyPhen-2 (PSIC score difference = 2.21) and SIFT (tolerance index score = 0.02), it is likely that this variant might affect protein function, although determining the exact role that the p.Ser707Tyr substitution plays will require further molecular and structural analyses of PLCγ2. Given this information and the dominant inheritance of the trait, we predicted that the p.Ser707Tyr substitution might render an increased function in PLCγ2 signaling pathways.
Upon stimulation with various immune receptors, PLCγ2 cleaves the membrane-bound phospholipid phosphatidyl inositol-4, 5-biphosphate (PIP2) into the second messenger molecules inositol-1,4,5-trisphosphate (IP3) and diacylglycerol (DAG).11 IP3 increases intracellular calcium levels by inducing the release of endoplasmic reticulum (ER) calcium stores.12 The released intracellular Ca2+ then activates protein kinase C (PKC), which subsequently activates a number of pathways in the cell. DAG remains on the cell membrane and potentiates the signaling cascade by activating some PKC enzymes and other targets. In human T cells, DAG accumulation in the cell membrane causes RasGRP translocation and activation through its binding to DAG and thus leads to activation of the Raf/Mek/Erk pathway.13 Therefore, our working hypothesis was that if p.Ser707Tyr is a hypermorphic substitution, we should observe an increase in production of IP3, along with an increase in intracellular Ca2+ release and an increase in phosphorylation of a molecule downstream of DAG.
Transient transfection of the mutant p.Ser707Tyr construct in either human embryonic kidney (HEK) 293T cells or COS7 cells resulted in increased EGF-stimulated production of intracellular IP3 and/or IP1, which is a surrogate measure for IP3 production14 (Figures 4A and 4B). The binding of IP3, a product of PLCγ2 activity, to IP3 receptors in the membrane of the ER has been shown to cause a rapid and transient release of Ca2+ from the ER Ca2+ stores into the cytosol. Consistent with the observed increased IP3 production, cells transfected with the mutant p.Ser707Tyr construct exhibited increased intracellular Ca2+ release after stimulation (Figures 4C and 4D).
Figure 4.
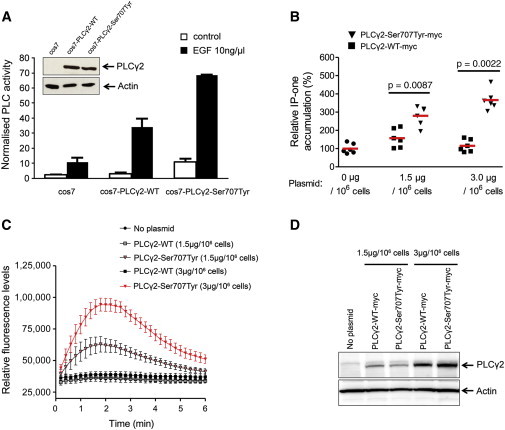
The p.Ser707Tyr Substitution Enhances PLCγ2 Activity and Intracellular Ca2+ Flux via the IP3 Signaling Pathway
(A) PLCγ2 activity was assayed in COS-7 cells transfected with wild-type (WT) or p.Ser707Tyr altered PLCγ2 constructs. [3H]inositol phosphate production was measured after stimulation with 10 ng/μl epidermal growth factor (EGF). PLCγ2 levels were analyzed by immunoblot assay (insets).
(B) The WT PLCγ2 construct (#RC200442, Origene, Rockville, MD) and the p.Ser707Tyr altered PLCγ2 construct (Site-Directed Mutagenesis Kit, QuikChange II, Stratagene-Agilent Technologies, Santa Clara, CA) were transiently introduced into 293T cells with Lipofectamine 2000 according to the manufacturers’ protocols. The transfected 293T cells were treated for 1 hr with EGF (150 ng/ml, #CN02, Cytoskeleton, Denver, CO) for Rac-mediated PLCγ2 activation and were subjected to the IP-one assay. The IP1 (surrogate for IP3) level in the cell lysate was measured by IP-One ELISA (#72IP1PEA, Cisbio, Bedford, MA) according to the manufacturer’s instructions.
(C) Increasing concentrations of PLCγ2 WT and p.Ser707Tyr altered constructs (0, 1.5, or 3.0 μg/106 cells) were transiently introduced into 293T cells for an assay of the intracellular Ca2+ level. The transfected 293T cells were plated onto 96-well plates (70,000 cells per well) and treated with EGF (150 ng/ml) and the calcium-indicating dye (Calcium Assay Kit 640176, BD Biosciences, San Diego, CA). The plates were cooled down to room temperature for 15 min, and the relative fluorescence was measured every 10 s for the duration of 6 min with the use of Victor (PerkinElmer, Waltham, MA) according the manufacturers’ manuals.
(D) The protein levels of PLCγ2 WT and p.Ser707Tyr altered constructs in 293T cells were confirmed by immunoblot assay.
All graphs represent three or four independent experiments, and error bard indicate the mean ± the standard error of the mean (SEM).
To further confirm the causal role of the p.Ser707Tyr substitution and to address the physiological significance of results from an overexpression cellular system, we measured IP1 production and intracellular Ca2+ release in affected individuals’ peripheral-blood mononuclear cells (PBMCs). Upon crosslinking stimulation with IgE, affected individuals’ cells produced significantly higher IP1 and released significantly higher amounts of Ca2+ into the cytosol than did control cells from an unaffected family member and another healthy control (Figures 5A and 5B). PLCγ2 is a crucial lipid-metabolizing effector enzyme that is known to activate IκB kinase (IKK) and extracellular signal-related kinase (ERK) in CD19 B cells. After stimulating CD19+ cells by crosslinking surface IgM, we observed that the levels of phosphorylated ERK in cells from both individuals carrying the p.Ser707Tyr substitution were well above those seen in healthy control samples (Figure 5C). All together, our data from exogenous expression and ex vivo experiments demonstrate evidence that p.Ser707Tyr in PLCγ2 is a hypermorphic substitution that enhances the activity of PLCγ2.
Figure 5.
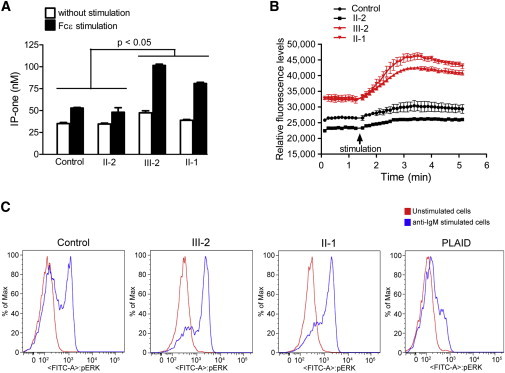
Affected Individuals’ PBMCs Exhibit Significantly Higher PLCγ2 Activity
PBMCs of two healthy controls—the unaffected mother (II-2) and another healthy control—and two affected individuals (III-2 and II-1) were isolated and subjected to IP-one (A) and intracellular Ca2+ (B) assays. PBMCs were coated with IgE anti-Dinitrophenyl (DNP) mAb (#D8406, 10 μg/ml, Sigma-Aldrich, St. Louis, MO) for 2 hr and then stimulated with DNP-albumin (#A6661, 100 ng/ml, Sigma-Aldrich). The graphs represent the mean ± SEM from three or four independent experiments. Error bars indicate the SEM.
(C) Affected individuals’ cells show increased ERK phosphorylation. CD19 B cells from individuals III-2 and II-1, a healthy control sample, and a PLAID-affected individual were stimulated by surface anti-IgM crosslinking and analyzed for expression of phosphorylated ERK. Lines indicate phosphostaining in unstimulated cells (red) and anti-IgM stimulated cells (blue). The data are representative of three independent experiments.
Recent work from our laboratory has identified PLCG2 genomic deletions in individuals presenting with a distinct inflammatory disease manifested by cold-induced urticaria and immune dysregulation including features of both immunodeficiency and autoimmunity. We have proposed the term PLAID (PLCγ2-associated antibody deficiency and immune dysregulation [MIM 614468]) to describe this disorder.15 The PLAID-associated genomic deletions disrupt the cSH2 domain of PLCγ2, causing failure of autoinhibition and resulting in constitutive phospholipase activity. Interestingly, the gain of constitutive enzymatic function results in the reduction of distal signaling and downstream function at physiologic temperatures (Figure 5C). Ca2+ release in unstimulated B cells from PLAID-affected individuals increased only upon exposure to low temperatures, and BCR-stimulated PLAID B cells exhibited increased ERK phosphorylation also with decreasing temperature. Thus, despite constitutive PLCγ2 enzymatic activity, PLAID-affected individuals have reduced PLCγ2-mediated signal transduction at physiologic temperatures most likely as a result of a negative feedback caused by constitutive activation. However, PLC signaling is enhanced at subphysiologic temperatures by a mechanism that underpins the pathophysiology of cold-induced skin rash and inflammation in PLAID-affected individuals.
The p.Ser707Tyr substitution in individuals reported here compromises autoinhibition, enhances activation, and exhibits exactly the opposite effects with increased cellular signaling in mutant cells at physiological temperatures. We speculate that the substitution of Tyr for Ser in the cSH2 domain might create an extra phosphorylation site and thus compromises autoinhibition and leads to a slight increase in basal enzymatic activity that is not sufficient for triggering negative feedback; this therefore causes enhanced PLCγ2 activity upon stimulation with a variety of upstream signals. Consequently, the individuals with the p.Ser707Tyr substitution have increased PLCγ2-dependent signaling after receptor crosslinking, as indicated by the elevated levels of IP3 production, intracellular Ca2+ release, and ERK phosphorylation.
Of interest, despite the divergent signaling effects seen in this family’s B cells when compared to PLAID B cells, both diseases lead to the near absence of circulating class-switched memory B cells. B cell expansion and IgG+ B cell accumulation were similar to those of the controls in stimulated PBMCs. Phenotypic and antibody-secreting cell frequencies of cultured cells also suggest a preferential expansion of IgG+ cells compared to IgA+ B cells; however, in PLAID individuals, all three measures were impaired (Figures 3C and 3D).15 This implies that the B cell memory defect in these individuals might be due to a different mechanism from that seen in PLAID. Potential mechanisms for this phenotype might include enhanced activation-induced deletion16 (although not detected in this in vitro assay), in vivo anergy due to enhanced cellular signaling,17 and enhanced negative selection of immature B cells.18 Alternatively, the different mechanism might be a reflection of the relative impact of exonic deletion versus a missense mutation (c.2120C>A [p.Ser707Tyr]).
Our finding that p.Ser707Tyr is a gain-of-function substitution in PLCγ2 is further corroborated by animal studies that have implicated the critical role for PLCγ2 in regulation of inflammation in mice.19 In the murine phospholipase Cg2 (Plcg2), two missense gain-of-function mutations introduced by ENU mutagenesis lead to severe spontaneous inflammation and autoimmunity. The Ali5 (abnormal limb 5) substitution, p.Asp993Gly, removes a negative charge from a critical region of the Plcγ2 molecule that ordinarily limits its interaction with the negatively-charged inner plasma membrane, thereby increasing enzymatic activity.20 The mice homozygous for the Ali5 substitution suffer from severe inflammation of the paws, severe skin and eye inflammation, and proliferative glomerulonephritis. In particular, eye findings include destruction of the different layers of the corneal epithelium with keratinization, resembling the signs observed in affected individual III-2. The inflammatory infiltrate in Ali5 mice is composed of neutrophils, eosinophils, and lymphocytes, consistent with histopathology findings in the affected individuals. Similar to our results, the production of IP3 is increased in B cells from Ali5 mice and in WEHI-231 cells expressing mutated Plcγ2. Retroviral transfection of altered Ali5 proteins, as compared to wild-type proteins, induces a stronger Ca2+ flux in primary LPS-activated wild-type B cells. Ali14, another mouse strain resulting from ENU mutagenesis, has the p.Tyr495Cys substitution, which is within the split pleckstrin homology spPH domain of γSA and enhances activation of Plcγ219 by different signals as a result of compromised autoinhibition. The Ali14/+ heterozygous mice show an abnormally high T cell to B cell ratio; upregulation of IgG1, IgG2b, and IgM; inflammatory arthritis; and metabolic defects.21
In contrast to mice with Ali5 and Ali14 gain-of-function mutations, Plcg2-deficient mice present with a different phenotype. Plcγ2-null mice exhibit increased perinatal lethality with gastrointestinal hemorrhages. Although viable mice can be obtained, they display major functional defects in B cells, platelets, mast cells, and NK cells and absent IgM-receptor-induced Ca2+ flux.22 Plcg2-deficient mice exhibit distinct phenotypes, and some aspects of these phenotypes are the opposite of those observed in mice with Plcg2 gain-of function mutations.
Although the precise changes that underlie enhanced signaling by hypermorphic alterations in PLCγ2 remain to be elucidated, our study offers an important advance in the field of PLCγ2 biology given that PLCG2 disease-associated missense mutations in humans have not been described in the past. Furthermore, in concert with the findings in individuals with PLCG2 deletions, the affected individuals show the wide phenotypic variability that can occur with mutations in the same region of the same gene. The wide phenotypic differences, along with the similarities between the two, namely impaired B cell memory and immunoglobulin production, suggest that a broad range of diseases could result from PLCG2 mutations, and the common theme is involvement of the humoral immune system and various forms of inflammation. We propose the term APLAID (autoinflammation and PLCγ2-associated antibody deficiency and immune dysregulation) to distinguish phenotypes caused by missense PLCG2 mutations from phenotypes caused by genomic deletions, such as in the case of PLAID-affected individuals. In conclusion, exome-sequencing technology in a family with only two affected members identified a mutation in the PLCG2 pathway as a cause of an autoinflammatory disease. These results highlight the importance of the whole-genome approach in elucidating the pathogenesis of inflammation that would be otherwise missed by a candidate-gene approach.
Acknowledgments
We would like to acknowledge Elaine Remmers and Jae Jin Chae for critical reading of the manuscript and Chyi-chia Lee for help with reviewing a histology slide. We would also like to acknowledge Nona Colburn, Maria Turner, Patrycja Hoffman, and Anne Jones for their help in caring for these affected individuals. This study has been supported by the National Human Genome Research Institute and the National Institute for Arthritis and Musculoskeletal and Skin Diseases, Bethesda, MD. Work in Matilda Katan’s laboratory has been supported by Cancer Research, UK.
Contributor Information
Daniel L. Kastner, Email: kastnerd@mail.nih.gov.
Ivona Aksentijevich, Email: aksentii@exchange.nih.gov.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
1000 Genomes Browser, http://browser.1000genomes.org/index.html
NHLBI Exome Sequencing Project (ESP), http://evs.gs.washington.edu/EVS/
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
PolyPhen-2, http://genetics.bwh.harvard.edu/pph2/
SeattleSeq Annotation, http://snp.gs.washington.edu/SeattleSeqAnnotation/
SIFT Genome, http://sift.jcvi.org/
SWISS-Model Server, http://swissmodel.expasy.org/
References
- 1.Aksentijevich I., Kastner D.L. Genetics of monogenic autoinflammatory diseases: Past successes, future challenges. Nat. Rev. Rheumatol. 2011;7:469–478. doi: 10.1038/nrrheum.2011.94. [DOI] [PubMed] [Google Scholar]
- 2.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Teer J.K., Bonnycastle L.L., Chines P.S., Hansen N.F., Aoyama N., Swift A.J., Abaan H.O., Albert T.J., Margulies E.H., Green E.D., NISC Comparative Sequencing Program Systematic comparison of three genomic enrichment methods for massively parallel DNA sequencing. Genome Res. 2010;20:1420–1431. doi: 10.1101/gr.106716.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Albers C.A., Lunter G., MacArthur D.G., McVean G., Ouwehand W.H., Durbin R. Dindel: Accurate indel calls from short-read data. Genome Res. 2011;21:961–973. doi: 10.1101/gr.112326.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wang K., Li M., Hakonarson H. ANNOVAR: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. doi: 10.1093/nar/gkq603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cooper G.M., Stone E.A., Asimenos G., Green E.D., Batzoglou S., Sidow A., NISC Comparative Sequencing Program Distribution and intensity of constraint in mammalian genomic sequence. Genome Res. 2005;15:901–913. doi: 10.1101/gr.3577405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collins J.S., Schwartz C.E. Detecting polymorphisms and mutations in candidate genes. Am. J. Hum. Genet. 2002;71:1251–1252. doi: 10.1086/344344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gresset A., Hicks S.N., Harden T.K., Sondek J. Mechanism of phosphorylation-induced activation of phospholipase C-gamma isozymes. J. Biol. Chem. 2010;285:35836–35847. doi: 10.1074/jbc.M110.166512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Siepel A., Bejerano G., Pedersen J.S., Hinrichs A.S., Hou M., Rosenbloom K., Clawson H., Spieth J., Hillier L.W., Richards S. Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 2005;15:1034–1050. doi: 10.1101/gr.3715005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Filippakopoulos P., Müller S., Knapp S. SH2 domains: modulators of nonreceptor tyrosine kinase activity. Curr. Opin. Struct. Biol. 2009;19:643–649. doi: 10.1016/j.sbi.2009.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bunney T.D., Katan M. PLC regulation: Emerging pictures for molecular mechanisms. Trends Biochem. Sci. 2011;36:88–96. doi: 10.1016/j.tibs.2010.08.003. [DOI] [PubMed] [Google Scholar]
- 12.Wilde J.I., Watson S.P. Regulation of phospholipase C gamma isoforms in haematopoietic cells: why one, not the other? Cell. Signal. 2001;13:691–701. doi: 10.1016/s0898-6568(01)00191-7. [DOI] [PubMed] [Google Scholar]
- 13.Jones D.R., Sanjuán M.A., Stone J.C., Mérida I. Expression of a catalytically inactive form of diacylglycerol kinase alpha induces sustained signaling through RasGRP. FASEB J. 2002;16:595–597. doi: 10.1096/fj.01-0762fje. [DOI] [PubMed] [Google Scholar]
- 14.Trinquet E., Fink M., Bazin H., Grillet F., Maurin F., Bourrier E., Ansanay H., Leroy C., Michaud A., Durroux T. D-myo-inositol 1-phosphate as a surrogate of D-myo-inositol 1,4,5-tris phosphate to monitor G protein-coupled receptor activation. Anal. Biochem. 2006;358:126–135. doi: 10.1016/j.ab.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 15.Ombrello M.J., Remmers E.F., Sun G., Freeman A.F., Datta S., Torabi-Parizi P., Subramanian N., Bunney T.D., Baxendale R.W., Martins M.S. Cold urticaria, immunodeficiency, and autoimmunity related to PLCG2 deletions. N. Engl. J. Med. 2012;366:330–338. doi: 10.1056/NEJMoa1102140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Donjerković D., Scott D.W. Activation-induced cell death in B lymphocytes. Cell Res. 2000;10:179–192. doi: 10.1038/sj.cr.7290047. [DOI] [PubMed] [Google Scholar]
- 17.Goodnow C.C., Cyster J.G., Hartley S.B., Bell S.E., Cooke M.P., Healy J.I., Akkaraju S., Rathmell J.C., Pogue S.L., Shokat K.P. Self-tolerance checkpoints in B lymphocyte development. Adv. Immunol. 1995;59:279–368. doi: 10.1016/s0065-2776(08)60633-1. [DOI] [PubMed] [Google Scholar]
- 18.Meffre E. The establishment of early B cell tolerance in humans: Lessons from primary immunodeficiency diseases. Ann. N Y Acad. Sci. 2011;1246:1–10. doi: 10.1111/j.1749-6632.2011.06347.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Everett K.L., Bunney T.D., Yoon Y., Rodrigues-Lima F., Harris R., Driscoll P.C., Abe K., Fuchs H., de Angelis M.H., Yu P. Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J. Biol. Chem. 2009;284:23083–23093. doi: 10.1074/jbc.M109.019265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yu P., Constien R., Dear N., Katan M., Hanke P., Bunney T.D., Kunder S., Quintanilla-Martinez L., Huffstadt U., Schröder A. Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity. 2005;22:451–465. doi: 10.1016/j.immuni.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 21.Abe K., Fuchs H., Boersma A., Hans W., Yu P., Kalaydjiev S., Klaften M., Adler T., Calzada-Wack J., Mossbrugger I. A novel N-ethyl-N-nitrosourea-induced mutation in phospholipase Cγ2 causes inflammatory arthritis, metabolic defects, and male infertility in vitro in a murine model. Arthritis Rheum. 2011;63:1301–1311. doi: 10.1002/art.30280. [DOI] [PubMed] [Google Scholar]
- 22.Wang D., Feng J., Wen R., Marine J.C., Sangster M.Y., Parganas E., Hoffmeyer A., Jackson C.W., Cleveland J.L., Murray P.J., Ihle J.N. Phospholipase Cgamma2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13:25–35. doi: 10.1016/s1074-7613(00)00005-4. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.