

Abstract
The decreasing cost of whole-genome and whole-exome sequencing has resulted in a renaissance for identifying Mendelian disease mutations, and for the first time it is possible to survey the distribution and characteristics of these mutations in large population samples. We conducted carrier screening for all autosomal-recessive (AR) mutations known to be present in members of a founder population and revealed surprisingly high carrier frequencies for many of these mutations. By utilizing the rich demographic, genetic, and phenotypic data available on these subjects and simulations in the exact pedigree that these individuals belong to, we show that the majority of mutations were most likely introduced into the population by a single founder and then drifted to the high carrier frequencies observed. We further show that although there is an increased incidence of AR diseases overall, the mean carrier burden is likely to be lower in the Hutterites than in the general population. Finally, on the basis of simulations, we predict the presence of 30 or more undiscovered recessive mutations among these subjects, and this would at least double the number of AR diseases that have been reported in this isolated population.
Introduction
Founder populations have contributed disproportionally to the discovery of autosomal-recessive (AR) disease-causing mutations because affected individuals from these populations are typically homozygous for founder mutations that reside on relatively long haplotypes, facilitating mutation discovery by identity by descent (IBD) and haplotype-sharing methods.1–4 Moreover, despite their negative impact on fitness, many disease-causing founder mutations occur at relatively high frequencies in these populations presumably as a result of the effects of random genetic drift following the founding bottleneck. Classic examples of high-frequency founder mutations are those causing Ellis-van Creveld syndrome (MIM 225500) in the Amish,5 congenital chloride diarrhea (MIM 214700) in the Finnish,6,7 Tay-Sachs disease (MIM 272800) in Ashkenazi Jews,8 Charlevoix-Saguenay spastic ataxia (MIM 270550) in the Charlevoix-Saguenay-Lac-Saint-Jean region of Quebec,9 and cystic fibrosis (MIM 219700) in the Hutterites.10 However, in nearly all cases, our understanding of the impact and fate of founder mutations is based on identifying homozygous individuals with the disease and studying these individuals and their close relatives. As a result, our understanding of the frequency spectrum, carrier burden, and penetrance of AR disease-causing founder mutations is limited to those that are observed in homozygous affected individuals. The recent explosion of next-generation-sequencing approaches to mutation discovery facilitates more comprehensive surveys of AR disease-causing mutations and, for the first time, unbiased estimates of genetic parameters of deleterious mutations segregating in founder populations.
The North American Hutterites are one of the best-characterized young founder populations.11–14 To date, >28 AR diseases have been observed in members of this population,13 and causal mutations have been identified in more than half. We initiated this study to determine the frequency spectrum of known AR disease-causing mutations in United States Schmeideleut (S-leut) Hutterites and to identify the AR disease-causing mutations that are present among the participants in our genetic studies of complex phenotypes and common diseases.14 Using a combination of exome sequencing, targeted Sanger sequencing, and direct genotyping, we identified the mutations causing three AR conditions in the Hutterites and determined carrier frequencies and carrier burdens for 14 mutations associated with 13 AR diseases in 1,644 United States S-leut Hutterites. We report here remarkably high carrier frequencies of all mutations and temporal trends that predict increasing incidences of some of these conditions.
Subjects and Methods
Subjects
The Hutterites are an Anabaptist religious group that originated during the 1500s in the Tyrolean Alps. To escape religious persecution, the Hutterites lived throughout central and eastern Europe for the next >300 years. In the 1870s they migrated from Russia to the United States and settled on three communal farms (called colonies) in what is now South Dakota. These three colonies gave rise to the three major Hutterite subdivisions, referred to as the Schmiedeleut (S-leut), Lehrerleut (L-leut), and Dariusleut (D-leut). The population has since undergone rapid expansion, and today >400 Hutterite colonies of all three “leut” are located in the north central plain states of the United States and western provinces of Canada; marriages between leut have been uncommon for at least the past 90 years. Detailed genealogical records that extend back to the early 1700s during their tenure in Russia trace the >40,000 extant members of this founder population to fewer than 90 ancestors.15
Our studies have focused on the United States S-leut Hutterites residing primarily in South Dakota; some participants are from colonies in North Dakota and Minnesota. Over the past nearly 30 years, we have conducted genetic studies of fertility,16,17 asthma and other common diseases,14,18 and quantitative phenotypes.19–21 All of our genetic studies of complex phenotypes have been population based and have included from ten colonies all members (six years of age and older) who were home during our field trips, as well as Hutterite visitors to those colonies on the days of our field work. Our fertility studies include from an additional 39 colonies couples in their childbearing years at the time of enrollment; some of these individuals were enrolled through the mail. DNA for all subjects was obtained from blood collected during field trips or from saliva samples collected through the mail. The final sample for this study includes 1,644 Hutterites, 6–92 years old at the time of our studies, from 56 colonies. These individuals are related to each other in a 13-generation pedigree that includes 3,671 individuals, all of whom can be traced to 64 founders.
Written informed consent was obtained from all participants 18 years of age or older and from parents of subjects under 18 years of age; written assent was obtained from subjects between 6 and 18 years of age. These studies were approved by the institutional review board at the University of Chicago.
Selection of Disease Mutations
Of the 30 AR diseases described in the Hutterites (Table S1, available online, adapted from Boycott et al., 2008), mutations were known for 20 prior to this study. We focused these studies on 14 AR disease-causing mutations that fell into one of the following three groups: (1) known mutations for diseases that were either observed or reported to have occurred in study subjects or their children (n = 5), (2) known disease mutations that were present in the exome sequences of 25 Hutterites in our sample (n = 6), or (3) disease mutations that were discovered in our laboratory during the course of this study (n = 3). These 14 disease mutations are described in Table 1. The eight remaining AR disease-causing mutations that have been reported in the Hutterites were either not present or could not be reliably assessed (i.e., insertions and deletions) in the 25 exomes, and, to our knowledge, the diseases attributed to those mutations are not present in the families in our studies.
Table 1.
Thirteen AR Diseases and the Corresponding 14 Mutations in the Hutterites
| Disease | MIM Number | Chromosome | Gene | Mutation (Common Name) | Mutation (HGVS Name) | Reference Cluster ID | Group | Mutation Detected in 25 Exomes |
|---|---|---|---|---|---|---|---|---|
| Restrictive dermopathy | 275210 | 1 | ZMPSTE24 | c.1085dupT | NM_005857.3: c.1078dupT | rs137854889 | 3 | no |
| NP_005848.2: p.Cys359_Phe360?fs | ||||||||
| Joubert syndrome | 614424 | 2 | TMEM237 | R18X | NM_001044385.1: c.52C>T | rs199469707 | 2 | yes |
| NP_001037850.1: p.Arg18Ter | ||||||||
| Sitosterolemia | 210250 | 2 | ABCG8 | S107X | NM_022437.2: c.320C>G | rs137854891 | 2 | yes |
| NP_071882.1: p.Ser107Ter | ||||||||
| Dilated cardiomyopathy with ataxia syndrome | 610198 | 3 | DNAJC19 | IVS3-1G>C | NM_145261.3: c.130-1G>C | rs137854888 | 2 | yes |
| Spinal muscular atrophy type III | 253400 | 5 | SMN1 | exon 7 del | N/A | N/A | 1 | no |
| Cystic fibrosis | 219700 | 7 | CFTR | F508del | NM_000492.3: c.1521_1523delCTT | rs113993960 | 1 | no |
| NP_000483.3: p.Ile507_Phe508? | ||||||||
| M1101K | NM_000492.3: c.3302T>A | rs36210737 | 1 | yes | ||||
| NP_000483.3: p.Met1101Lys | ||||||||
| Limb girdle muscular dystrophy 2H | 254110 | 9 | TRIM32 | D487N | NM_001099679.1: c.1459G>A | rs111033570 | 2 | yes |
| NP_001093149.1: p.Asp487Asn | ||||||||
| Usher syndrome type 1F | 602083 | 10 | PCDH15 | c.1471delT | NM_001142763.1: c.1101delT | rs199469706 | 2 | yes |
| NP_001136235.1: p.Leu368fs∗ | ||||||||
| Oculocutaneous albinism type 1A | 203100 | 11 | TYR | C91Y | NM_000372.4: c.272G>A | rs137854890 | 3 | yes |
| NP_000363.1: p.Cys91Tyr | ||||||||
| Nonsyndromic deafness | 220290 | 13 | GJB2 | c.35delG | NM_004004.5: c.35delG | rs80338939 | 3 | no |
| NP_003995.2: p.Gly12Valfs | ||||||||
| Bardet-Biedl syndrome | 209900 | 16 | BBS2 | IVS3-2A>G | NM_031885.3: c.472-2A>G | rs137854887 | 2 | yes |
| Limb girdle muscular dystrophy 2I | 607155 | 19 | FKRP | L276I | NM_024301.4: c.826C>A | rs28937900 | 1 | yes |
| NP_077277.1: p.Leu276Ile | ||||||||
| Nonsyndromic mental retardation | 614020 | 19 | TECR | P182L | NM_138501.5: c.545C>T | rs199469705 | 1 | yes |
| NP_612510.1: p.Pro182Leu |
This table is ordered by chromosome. Group 1 mutations correspond to diseases that were either previously observed or reported22–24 to have occurred in study subjects or their children. Group 2 mutations were previously reported in the Hutterites25–32 and were present in the exome sequences of 25 Hutterites. Group 3 mutations were discovered in our laboratory during the course of this study. Both the “common” mutation names, as well as HGVS names, are provided; in some cases, the coding sequence has been revised since the original mutation publication because the common name is outdated. We use the common names throughout this manuscript to retain consistency with existing literature on these mutations.
Exome Sequencing
We selected 25 Hutterites for exome sequencing with the goal of capturing as much of the genetic variation present in the Hutterites in our sample as possible.33 To achieve that goal, we selected individuals who were relatively unrelated to each other (mean pairwise kinship = 0.038 versus 0.041 in the entire sample) and who had the largest number of genotyped descendants in the pedigree (median number of genotyped descendants = 31). After sequencing, we searched among the called variants for previously reported AR mutations;13,22,25,34 all mutations identified in the exome sequences were confirmed by Sanger sequencing. Three (out of five) mutations from group 1 and one (out of three) mutation from group 3 were also present in the 25 exomes. Group 2 was composed of six additional mutations that were present in the exomes but for which no individuals affected by these diseases had been observed or reported in our study participants or their families.
Mutation Discovery
The third group included three mutations that were identified in our laboratory in DNA from affected individuals or from obligate carrier parents. Here, we studied individuals and families with oculocutaneous albinism (OCA1A [MIM 203100]), parents of children with nonsyndromic deafness (DFNB1A [MIM 220290]), and parents of a child who died of restrictive dermopathy (RD [MIM 275210]). Sanger sequencing of all five exons of TYR (MIM 606933) was performed in an adult with albinism; variants were identified by comparison to hg18, and a mutation (RefSeq accession number NM_000372.4: c.272G>A) resulting in a protein alteration, p.Cys91Tyr, was identified at a conserved site. This mutation was then confirmed in DNA from parents whose children had albinism and did not participate in our studies (Figure S1A). The GJB2 (MIM 121011) c.35delG mutation (RefSeq NM_004004.5) commonly causes nonsyndromic AR deafness in European populations35 but had not been identified as a cause of AR deafness in the Hutterites. This mutation was identified and confirmed by sequencing in both parents of multiple deaf children (Figure S1B). The ZMPSTE24 (MIM 606480) c.1085dupT (RefSeq NM_005857.3) mutation causing RD was discovered simultaneously in our and another laboratory.26 Sequences were analyzed with 4Peaks (Mekentosj, Amsterdam).
Mutation Screening
We determined the frequency of each of these 14 mutations in 1,644 subjects by direct genotyping (Table S2). In addition, we used a haplotype-based method to study the SMN1 (MIM 600354) deletion causing spinal muscular atrophy type III (SMA [MIM 253300]) in 1,415 subjects, as previously described.23 All genotypes were in Hardy-Weinberg equilibrium according to a test that adjusts for the Hutterite population structure and relatedness.36 Mendelian errors were identified by Pedcheck;37 these errors were corrected, but if they could not be unambiguously resolved, the implicated nuclear families were removed from further analyses. Four couples in our sample were ascertained because they had children with cystic fibrosis (CF [MIM 219700]);24 these couples and their children were excluded from population-based estimates of carrier and homozygote frequencies for CF.
Haplotype Analysis
To determine whether each mutation occurred on a single founder haplotype, we used genotypes for 1,415 subjects who were genotyped on the Affymetrix 500k, 5.0, or 6.0 SNP arrays. SNPs underwent quality control (QC) checks as previously described.20 We included in these studies the 271,486 SNPs that were present on all three arrays and passed all QC checks.
For the seven mutations that occurred in at least one homozygous individual in our sample, we identified the haplotype carrying the mutation directly in those individuals. However, to identify the haplotype(s) shared by carriers of the seven mutations that did not occur as homozygous in our sample, we used a phasing algorithm33 that uses both the known IBD structure of the population and the Affymetrix genotypes to generate a “reference haplotype” from a heterozygote that was selected at random. Then, we used a Perl script to identify the extent to which the reference haplotype was shared among all carriers. The algorithm starts at the mutation locus in each carrier and scans each consecutive SNP on one side of the mutation; it checks for identity by state (IBS) ≥ 1 with the reference haplotype until it reaches a SNP for which IBS = 0. The same procedure is used for scanning SNPs on the other side of the mutation, and the total length of IBS ≥ 1 sharing is determined by the addition of the segments on both sides. The algorithm was modified to allow for n mismatches by tracking the lengths of segments with 0 to n mismatches on both sides of the mutation and determining the total extent of sharing by identifying the longest combined length of segments that contain exactly n total mismatches. We manually chose the final value of n for the analysis of each mutation after trying increasing numbers of allowed mismatches and selecting the n that maximized the number of subjects whose haplotypes agreed on a particular start or endpoint.
Gene-Dropping Simulations
We performed gene-dropping simulations in the 3,671 person pedigree to assess the probability that the observed frequencies of each mutation were consistent with neutral expectations. In the simulations, a unique mutation was assigned to each founder and propagated downward through the pedigree—in each of 100,000 iterations—under two different models. In the first model, we assumed that the mutation was neutral (i.e., it had no effect on fitness) in both heterozygotes and homozygotes (50% likelihood of transmission regardless of genotype). In the second model, we assumed that homozygotes for the mutation had fitness equivalent to zero (homozygosity for the mutation prevents them from reproducing) and that the fitness of heterozygotes was not affected. Because the pedigree only includes individuals who are ancestral to a genotyped subject in our study, it follows that all pedigree members must have reproduced and could not have been homozygous for an AR mutation with fitness = 0. Therefore, when simulating the number of mutations inherited by an offspring of two heterozygous parents, we implemented zero fitness by giving the offspring a 0% chance of being homozygous for the mutation, a 66% chance of being heterozygous, and a 33% chance of being homozygous for the major allele. This is equivalent to resampling the offspring’s alleles from the parents until the offspring is not homozygous for the mutation; this is the method used by Heyer to simulate lethal mutations in the Saguenay pedigree.38 The first model (neutral) is more reflective of late-onset diseases (e.g., limb girdle muscular dystrophy 2H [LGMD2H (MIM 254110)]) or those that have an early onset and that have limited or no effect on reproductive fitness (e.g., oculocutaneous albinism or sitosterolemia [STSL (MIM 210250)]), whereas the latter model (lethal or zero fitness) is more reflective of early-onset diseases that are either lethal prior to reproduction (e.g., RD) or limit or preclude reproduction in surviving adults (e.g., CF [MIM 219700]).
To determine the probability that a mutation present in a single Hutterite founder would reach a given carrier frequency, we calculated the proportion of iterations in which the founder’s mutation reached the frequency of the mutation observed in the study sample. We used a regression-based test designed for large complex pedigrees (general two-allele model)39 to test for an association between carrier status and fertility in the Hutterites. The fertility phenotypes, family size, and birth rate have been described elsewhere.40 These analyses were performed with and without couples in which both individuals were carriers of the same mutation in case some parents altered their reproductive plans after the diagnosis of a child with the corresponding disease.
Carrier Burden
We defined carrier burden as the number of AR disease-causing mutations per genome. This was assessed in 686 individuals who had a nonmissing genotype at all 14 mutations studied. In addition, we assessed their expected mean carrier burden for simulated founder mutations. Using the same gene-dropping simulations described above, we assigned all of the founders one, two, three, or five unique unlinked mutations each and dropped those mutations through the pedigree. We then determined the total number of founder alleles carried by each individual. To increase simulation efficiency, we assigned the same number of mutations (x) to each founder, although this is mathematically equivalent to treating the number of mutations per founder as independent and identically distributed random variables with mean x (in other words, giving each founder exactly three mutations will produce equivalent results to randomly picking each founder’s number of mutations from a distribution with a mean of 3). Finally, we calculated the 95% confidence intervals for the mean carrier burden and expected number of surviving mutations by taking the middle 95% of values. In all cases, 100,000 trials were performed. Code for all gene-dropping and carrier-burden simulations is available online.
Results
Carrier Frequencies
Seven of the 14 mutations present in the Hutterites are also present in other populations, and four of these are the most common for each disease in Europeans—they are a SMN1 (MIM 600354) deletion causing SMA, p.Phe508del (RefSeq NM_000492.3: c.1521_1523delCTT) causing CF, c.35delG causing DFNB1A, and p.Leu276Ile (RefSeq NM_024301.4: c.826C>A) causing limb girdle muscular dystrophy 2I (LGMD2I [MIM 607155]). In contrast, TRIM32 p.Asp487Asn (RefSeq NM_001099679.1: c.1459G>A) and ZMPSTE24 c.1085dupT are rare outside the Hutterites—there are only a few case reports worldwide of each mutation in non-Hutterites—and CFTR p.Met1101Lys (RefSeq NM_000492.3: c.3302T>A) has only been reported in one individual with CF from the South Tyrol,41 the home of some of the Hutterite founders. The remaining seven mutations have not been reported outside of the Hutterites (these mutations are referred to as “private” mutations in Table 2).
Table 2.
Results of Carrier Screening for 14 AR Mutations in 1,644 Hutterites from South Dakota
| Disease | Gene Mutation | Number of Heterozygotes | Number of Homozygotes | Number Screened | Carrier Frequency in Hutterites | Carrier Frequency in Other Populations |
|---|---|---|---|---|---|---|
| Limb girdle muscular dystrophy 2H | TRIM32 p.Asp487Asn | 228 | 9 | 1,493 | 0.153 (1 in 6.5) | unknown (only two non-Hutterite cases reported42) |
| Oculocutaneous albinism type 1A | TYR p.Cys91Tyr | 180 | 3 | 1,281 | 0.141 (1 in 7) | private mutation |
| Spinal muscular atrophy type III | SMN1 exon 7 del | 179 | 2 | 1,415 | 0.127 (1 in 8) | 1 in 3543 |
| Limb girdle muscular dystrophy 2I | FKRP p.Leu276Ile | 121 | 3 | 1,127 | 0.107 (1 in 9.5) | 1 in 30044 |
| Sitosterolemia | ABCG8 p.Ser107Ter | 127 | 4 | 1,515 | 0.084 (1 in 12) | private mutation |
| Joubert syndrome | TMEM237 p.Arg18Ter | 122 | 0 | 1,520 | 0.080 (1 in 12.5) | private mutation |
| Cystic fibrosis | CFTR p.Met1101Lys | 108 | 6 | 1,473 | 0.073 (1 in 13.5) | unknown (only one non-Hutterite case reported41) |
| Nonsyndromic mental retardation | TECR p.Pro182Leu | 103 | 5 | 1,496 | 0.069 (1 in 14.5) | private mutation |
| Restrictive dermopathy | ZMPSTE24 c.1085dupT | 87 | 0 | 1,361 | 0.064 (1 in 15.5) | unknown (<60 cases worldwide45) |
| Nonsyndromic deafness | GJB2 c.35delG | 54 | 0 | 1,510 | 0.036 (1 in 28) | 1 in 4035 |
| Dilated cardiomyopathy with ataxia syndrome | DNAJC19 IVS3-1G>C | 42 | 0 | 1,504 | 0.028 (1 in 36) | private mutation |
| Bardet-Biedl syndrome | BBS2 IVS3-2A>G | 42 | 0 | 1,518 | 0.028 (1 in 36) | private mutation |
| Usher syndrome type 1F | PCDH15 c.1471delT | 38 | 0 | 1,524 | 0.025 (1 in 40) | private mutation |
| Cystic fibrosis | CFTR p.Phe508del | 32 | 0 | 1,482 | 0.022 (1 in 45.5) | 1 in 3046 |
This table is ordered by carrier frequency. The combined carrier frequency for cystic fibrosis (c.1521_1523delCTT + c.3302T>A) is 0.093 (1 in 11). We use common mutation names to retain consistency with the existing literature.
The frequencies of the 14 mutations varied greatly; carrier frequencies ranged from 1 in 6.5 for TRIM32 (MIM 602290) p.Asp487Asn, which causes LGMD2H (MIM 254110), to 1 in 45 for CFTR p.Phe508del (RefSeq NM_000492.3: c.1521_1523delCTT), which causes CF (Table 2). We were aware of individuals with oculocutaneous albinism type 1A (OCA1A) (n = 3), SMA (n = 1), LGMD2I (n = 3), nonsyndromic mental retardation (NSMR [MIM 614020]) (n = 5), and CF (n = 6) in our sample, and each was homozygous for the expected disease mutation. However, 14 individuals were homozygous for three AR mutations but were unaware of their status: nine individuals (ages 10–42 years) were homozygous for the LGMD2H-causing mutation, four individuals (one pair of teenage siblings and one pair of adult siblings) were homozygous for the STSL mutation, and one adult was homozygous for the SMA deletion mutation as previously reported.23 On a subsequent exam, all four of the subjects with STSL-causing mutations had elevated sitosterol levels (4.29–19.0 mg/100 ml), which is diagnostic for STSL (D.J. Waggoner, personal observation). The adult woman who was homozygous for the SMA-causing deletion was 41 years old and asymptomatic at the time of our study; she died of cancer at age 50 without any symptoms related to SMA according to her close relatives. No additional information is available for the other subjects.
Haplotype Analyses
A priori, we expected that all private mutations would have been introduced by a single founder and that each of those mutations would be present on a single haplotype background. Although the expectation is less clear for mutations that have been observed in non-Hutterite populations, it would not be surprising if some of these mutations (e.g., the SMN1 deletion or GJB2 c.35delG) were introduced by more than one founder and present on more than one haplotype background. To address this, we characterized the extent of haplotype sharing around the mutations in homozygotes, if available, and in heterozygotes if no homozygotes were present in our sample. The lengths of the shared haplotypes around each mutation and the number of SNPs defining each haplotype are described in Table 3.
Table 3.
Analysis of Shared Haplotypes Bearing AR Mutations
| Mutation (Gene) |
Heterozygotes |
Homozygotes |
|||||||
|---|---|---|---|---|---|---|---|---|---|
| n | Kinship Coefficient (Mean [Min, Max]) | Consensus Length (bp) | Consensus Length (SNPs) | n | Kinship Coefficient (Mean [Min, Max]) | Consensus Length (bp) | Consensus Length (SNPs) | Mismatches Allowed | |
| p.Asp487Asn (TRIM32) | 189 | 0.049 (0.01, 0.30) | 449,953 | 90 | 8 | 0.054 (0.02, 0.29) | 2,553,354 | 417 | 2 |
| p.Cys91Tyr (TYR) | 155 | 0.049 (0.01, 0.30) | 4,086,749 | 416 | 3 | 0.046 (0.03, 0.07) | 15,476,024 | 1812 | 1 |
| exon 7 del (SMN1) | 177 | 0.053 (0.01, 0.29) | 2,834,415 | 84 | 2 | 0.027 (0.03, 0.03) | 9,990,562 | 917 | 3 |
| p.Leu276Ile (FKRP) | 116 | 0.051 (0.01, 0.29) | 1,433,672 | 48 | 3 | 0.079 (0.04, 0.16) | 8,695,515 | 444 | 2 |
| p.Ser107Ter (ABCG8) | 117 | 0.057 (0.01, 0.29) | 2,120,092 | 290 | 4 | 0.134 (0.07, 0.28) | 9,041,651 | 500 | 2 |
| p.Arg18Ter (TMEM237) | 106 | 0.052 (0.01, 0.30) | 3,666,069 | 336 | 0 | − | − | − | 2 |
| p.Met1101Lys (CFTR)a | 98 | 0.055 (0.02, 0.29) | 3,011,533 | 229 | 5 | 0.131 (0.04, 0.28) | 8,221,623 | 659 | 2 |
| p.Pro182Leu (TECR) | 93 | 0.058 (0.02, 0.29) | 1,941,199 | 138 | 5 | 0.286 (0.29, 0.29) | 2,455,513 | 196 | 2 |
| c.1085dupT (ZMPSTE24) | 85 | 0.066 (0.02, 0.29) | 692,209 | 63 | 0 | − | − | − | 2 |
| c.35delG (GJB2) | 46 | 0.058 (0.02, 0.30) | 1,995,634b | 162 | 0 | − | − | − | 2 |
| IVS3-1G>C (DNAJC19) | 41 | 0.127 (0.02, 0.29) | 2,073,507 | 122 | 0 | − | − | − | 2 |
| IVS3-2A>G (BBS2) | 42 | 0.081 (0.01, 0.28) | 2,118,302 | 230 | 0 | − | − | − | 2 |
| c.1471delT (PCDH15) | 31 | 0.082 (0.03, 0.29) | 6,681,213 | 829 | 0 | − | − | − | 3 |
| p.Phe508del (CFTR) | 27 | 0.077 (0.02, 0.30) | 4,260,744 | 296 | 0 | − | − | − | 2 |
This table is ordered by carrier frequency. We use common mutation names to retain consistency with the existing literature. The following abbreviations are used: min, minimum; and max, maximum.
The c.3302T>A mutation is present on two Hutterite haplotypes,10 one of which is rare. Our sample included one c.3302T>A homozygote who was heterozygous for the two haplotypes. Because there were no other c.3302T>A carriers on this haplotype background, the consensus data are presented for the common haplotype only.
The GJB2 c.35delG haplotype might be up to ∼0.25 Mb longer than reported here because the proximal end of the haplotype is near the centromere, and no SNPs within that region were genotyped in our sample.
Zielenski et al.10 previously identified two CFTR (MIM 602421) p.Met1101Lys-bearing Hutterite haplotypes—a common haplotype (b) and a rare haplotype (c) that was only present in a single family—and hypothesized that haplotype c was derived from b by a single recombination proximal to p.Met1101Lys (RefSeq NM_000492.3: c.3302T>A). Five p.Met1101Lys homozygotes in our sample had two copies of the common b haplotype across ∼8 Mb (659 SNPs) surrounding CFTR, and examination of 98 p.Met1101Lys carriers revealed that all 98 also carried the b haplotype. We identified a sixth p.Met1101Lys homozygote who carried this alteration on haplotypes b and c. However, haplotype analysis revealed that the c haplotype differs from the b haplotype at multiple SNPs both proximal and distal to p.Met1101Lys (Figure S2). Therefore, two recombinations would be required for explaining the origin of p.Met1101Lys -bearing haplotypes c from b. Alternatively, two founders might have introduced p.Met1101Lys into the Hutterites on different haplotype backgrounds.
The remaining 13 mutations are present on a single extended haplotype, suggesting that one founder introduced each of these mutations into the population. This is consistent with previous studies of BBS2 (MIM 606151) IVS3-2A>G (RefSeq NM_031885.3: c.472-2A>G),27 TECR (MIM 610057) p.Pro182Leu (RefSeq NM_138501.5: c.545C>T),22 DNAJC19 (MIM 608977) IVS3-1G>C (RefSeq NM_145261.3: c.130-1G>C),28 ZMPSTE24 c.1085dupT,26 FKRP p.Leu276Ile,44 TRIM32 p.Asp487Asn,29 and the SMN1 deletion.23 The ZMPSTE24 c.1085dupT mutation was present on a relatively long (>1 Mb) haplotype in the majority of carriers, and four other carriers shared <1 Mb of the large haplotype. These carriers were members of a single family in which a mother and three of her children were heterozygous for ZMPSTE24 c.1085dupT but only had ∼0.6 Mb of the long shared haplotype; the mother’s brother carried the alteration on the long haplotype (Figure S3). In this family, it is likely that the mother inherited c.1085dupT on a recombinant haplotype and passed it on to her offspring.
Gene-Dropping Simulations
To characterize the expected frequencies of the founder mutations causing AR diseases, we performed gene-dropping simulations to generate null distributions of mutation frequencies given the exact structure of the Hutterite pedigree. This was performed under two models, one in which the homozygotes for the mutation have zero fitness (i.e., do not reproduce) and one in which the mutation is neutral. As expected, the founders with the most descendants in our study had the greatest variance in simulated carrier frequencies such that in one trial (1/100,000) the carrier frequency for the founder’s mutation was as high as 40%, whereas in nearly half of the trials (42,209/100,000) the same mutation went to extinction in early generations (Figure 1A). Similarly, we observed a clear and unsurprising correlation between the number of individuals descended from each founder in the current population and the expected carrier frequency for each founder’s mutation. For example, mutations introduced by any one of six founders (three founder couples) who were ancestral to all or nearly all members of the sample had between 50% and 74% probability of reaching carrier frequencies of 5% or greater and a ≥10% probability of reaching carrier frequencies of 10% or greater. In fact, if one assumes that the number of unique (rare) variants carried by each of the 64 Hutterite founders was drawn from the same distribution, those six founders are expected to collectively account for 15.4% of the variants currently segregating in the sample.
Figure 1.
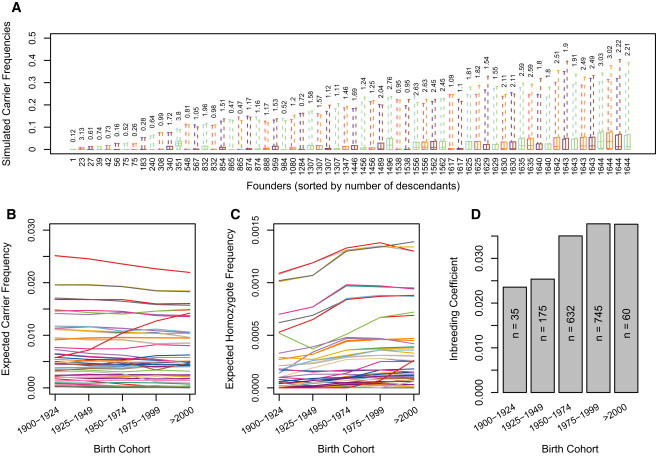
Results of Gene-Dropping Simulations
(A–C) Simulated carrier frequency in the current population for each unique founder mutation. Box plots show the distribution (first quartile, median, and third quartile; dashed lines show minimum and maximum) of frequencies that each founder mutation reached in 100,000 trials under two models. The box plots are ordered in pairs from left to right by the number of descendants (shown along x axis beneath box plots) in the study sample from each founder. The first box plot in each pair shows the simulated frequencies under a neutral model, and the second box plot shows the simulated frequencies under a zero fitness (lethal) model. In the current population, the expected percentage of variants that are attributable to a given founder is shown above the box plots. In (B) and (C), each line represents the simulated frequency of (B) heterozygous carriers and (C) homozygotes for a neutral founder mutation in 25 year birth cohorts. For many founder mutations, the frequency of homozygotes increases over time, whereas there is no such pattern observed for the frequency of carriers.
(D) Inbreeding coefficient by birth cohorts. The increase in consanguinity corresponds to the increase in the frequency of homozygotes in (C).
Given the evidence suggesting that 13 out of the 14 mutations were each introduced by a single founder on a single haplotype, we next asked how often this would result in mutations reaching frequencies as high as those observed in the Hutterites. For each mutation, we limited the potential contributing founders to those who are ancestral to all carriers of each mutation and determined the probability of reaching a target frequency under two models, one in which the homozygotes for the mutation have zero fitness and one in which the mutation is neutral. Diseases in which homozygotes do not reproduce are best modeled in the first scenario, whereas diseases with late ages of onset or reduced penetrance are modeled better in the second scenario. For each mutation, the probability of reaching the current carrier frequency under the more appropriate model is indicated in Table 4. Most of the mutations except the three most common (TRIM32 p.Asp487Asn, TYR p.Cys19Tyr, and the SMN1 deletion) had ≥5% probability of reaching the current carrier frequency by drift alone. To determine whether a heterozygote fertility advantage could have contributed to the high carrier frequencies of these three common mutations, we tested whether carrier status was associated with increased fertility, as measured by number of offspring, parents’ age at the birth of their last child, and birth rate.40 None of the mutations were associated with fertility in this sample (p ≥ 0.19 for all phenotypes), although the numbers of carriers were relatively small (fertility phenotypes were available for ≤45 carriers for any single mutation) and provided limited power for detecting small effects. This lack of association was robust to the inclusion or exclusion of couples in which both partners carried the same mutation.
Table 4.
Results of Gene-Dropping Simulations
| Disease | Gene Mutation | Carrier Frequency in Hutterites | Number of Founders Ancestral to All Carriers | Most Likely Model | Probability of Reaching Frequency under Neutral Model (95% CI) | Probability of Reaching Frequency under 0 Fitness Model (95% CI) |
|---|---|---|---|---|---|---|
| Limb girdle muscular dystrophy 2H | TRIM32 p.Asp487Asn | 0.153 | 12 | 1 | 0.0318 (3.1 × 10−4) | 0.0188 (2.4 × 10−4) |
| Oculocutaneous albinism type 1A | TYR p.Cys91Tyr | 0.141 | 15 | 1 | 0.0382 (3.1 × 10−4) | 0.0256 (2.5 × 10−4) |
| Spinal muscular atrophy Type III | SMN1 exon 7 del | 0.127 | 13 | 0 | 0.0574 (4.0 × 10−4) | 0.0427 (3.5 × 10−4) |
| Limb girdle muscular dystrophy 2I | FKRP p.Leu276Ile | 0.107 | 17 | 1 | 0.0711 (3.9 × 10−4) | 0.0575 (3.5 × 10−4) |
| Sitosterolemia | ABCG8 p.Ser107Ter | 0.084 | 16 | 1 | 0.1256 (5.1 × 10−4) | 0.1110 (4.9 × 10−4) |
| Joubert syndrome | TMEM237 p.Arg18Ter | 0.080 | 17 | 0 | 0.1405 (5.2 × 10−4) | 0.1261 (5.0 × 10−4) |
| Cystic fibrosis | CFTR p.Met1101Lys | 0.073 | 18 | 0 | 0.1198 (4.7 × 10−4) | 0.1089 (4.6 × 10−4) |
| Nonsyndromic mental retardation | TECR p.Pro182Leu | 0.069 | 18 | 0 | 0.1631 (5.4 × 10−4) | 0.1513 (5.2 × 10−4) |
| Restrictive dermopathy | ZMPSTE24 c.1085dupT | 0.064 | 26 | 0 | 0.1518 (4.4 × 10−4) | 0.1432 (4.3 × 10−4) |
| Nonsyndromic deafness | GJB2 c.35delG | 0.036 | 27 | 1 | 0.2607 (5.2 × 10−4) | 0.2546 (5.2 × 10−4) |
| Dilated cardiomyopathy with ataxia syndrome | DNAJC19 IVS3-1G>C | 0.029 | 28 | 0 | 0.2909 (5.3 × 10−4) | 0.2862 (5.3 × 10−4) |
| Bardet-Biedl syndrome | BBS2 IVS3-2A>G | 0.028 | 22 | 0 | 0.3214 (6.2 × 10−4) | 0.3164 (6.2 × 10−4) |
| Usher syndrome type 1F | PCDH15 c.1471delT | 0.025 | 20 | 0 | 0.3221 (6.5 × 10−4) | 0.3175 (6.5 × 10−4) |
| Cystic fibrosis | CFTR p.Phe508del | 0.022 | 30 | 0 | 0.3437 (5.4 × 10−4) | 0.3399 (5.4 × 10−4) |
This table is ordered by carrier frequency. The probability that each of the 14 AR mutations reached the carrier frequency observed in our study sample is shown in the last two columns. Probabilities are given for two extreme models: a neutral model and a zero fitness (lethal) model. The more appropriate model based on clinical phenotypes is indicated in column 4 (1 = neutral model; 0 = lethal model). We use common mutation names to retain consistency with the existing literature. The following abbreviation is used: CI, confidence interval.
Temporal Changes in Carrier Frequency
From the gene-dropping simulations, we also estimated genotype frequencies for each unique founder mutation by birth cohort. Although the carrier frequencies changed little over successive generations (Figure 1B), the frequency of homozygotes for most founder mutations increased in each generation (Figure 1C) most likely as a result of the increasing consanguinity in the population (Figure 1D). Consistent with our simulations, there was no obvious pattern of change in carrier frequencies for the 14 disease mutations in successive 25 year birth cohorts in this sample (Figure 2A), but the frequency of couples in which both parents are carriers (and at risk for having affected children) (Figure 2B) and the frequency of homozygotes (Figure 2C) have been increasing over time.
Figure 2.
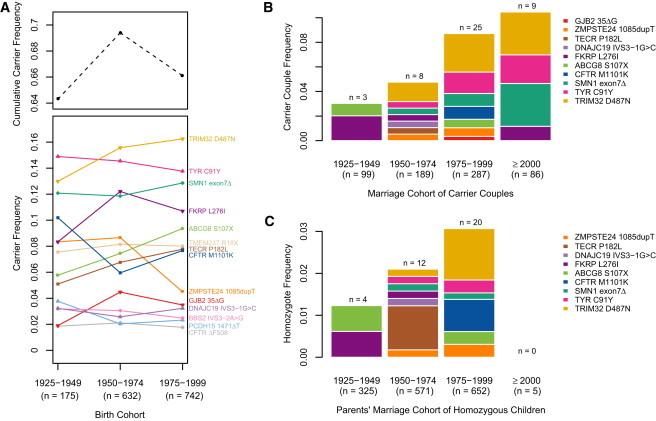
Observed Frequencies of Carriers, Carrier Couples, and Homozygotes over Time for 14 Disease Mutations
Four CF carrier couples were excluded because they were previously ascertained for having children with CF.24
(A) Heterozygote frequencies in 25 year birth cohorts. The cohorts born before 1925 and after 2000 were excluded because of small sample sizes (n < 60). The black dashed line shows the cumulative carrier frequency for all 14 mutations.
(B) Frequency of carrier couples. Both partners of one couple were carriers of DNAJC19 IVS3-1G>C and TECR p.Pro182Leu and are included twice in this figure.
(C) Frequency of homozygotes born to each couple in (B). There are no homozygotes shown among the children of the ≥2000 marriage cohort because our study included children who were 6 years of age or older at the time of our last study in 2006–2009. Although we are aware that these families include children homozygous for SMA (n = 4) and CF (n = 2), we did not include them because we do not know the status of children born to all carrier couples. In addition, because infants homozygous for ZMPSTE24 c.1085dupT, causing RD, die within a few days of birth, homozygotes for this mutation would not have been included in our studies. However, family-history interviews with carrier couples and acquisition of medical records allowed us to identify homozygous children, who are included in this figure.
Carrier Burden
The 686 individuals with nonmissing genotypes at all 14 disease mutations had a mean carrier burden of 1.096 mutations—67% of them carried at least one mutation, and some individuals carried up to five of mutations (Figure 3). These data are consistent with the expectation that carrier burden follows a Poisson distribution (p = 0.28 that the data is not Poisson distributed). In addition, we decomposed the total mean carrier burden into the mean carrier burden of neutral and nearly neutral versus lethal (zero fitness) mutations and found that the mean carrier burden of lethal and neutral mutations (not shown) is approximately equal (0.55 for lethal versus 0.54 for neutral mutations per individual). Finally, we “seeded” each founder with a set number of mutations and performed gene-dropping simulations to generate expectations for the distribution of carrier burden. Mean carrier burden for neutral mutations remained constant from the founders to the current generation, as expected. In contrast, in simulations assuming that mutations are homozygous lethal, the mean carrier burden in the study subjects decreased to 92% of the mean carrier burden in the founders (see Table 5, lines correspond to one mutation per founder).
Figure 3.
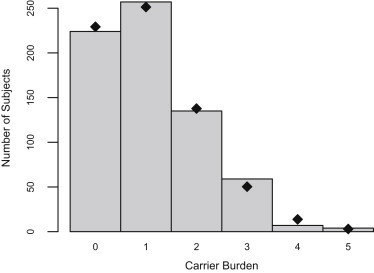
Distribution of Carrier Burden in 686 Subjects Who Were Genotyped for All 14 Mutations in This Study
The mean carrier burden (defined as the number of mutations per individual) is 1.096 mutations. Black dots represent the theoretical expected number of subjects in each bin of mutations with a Poisson distribution with λ = 1.096.
Table 5.
Results of Simulations of Carrier Burden
| Mean Mutations per Founder | Total Founder Mutations | Expected Mean Carrier Burden in Study Sample [95% CI] | Expected Number of Mutations Surviving in Study Sample [95% CI] |
|---|---|---|---|
| Neutral Fitness of Homozygotes | |||
| 1 | 64 | 1.00 [0.63, 1.42] | 24.4 [19, 30] |
| 2 | 128 | 2.00 [1.47, 2.58] | 48.8 [41, 57] |
| 3 | 192 | 3.00 [2.34, 3.71] | 73.1 [63, 83] |
| 5 | 320 | 5.00 [4.14, 5.91] | 121.9 [109, 135] |
| Lethal Fitness of Homozygotes | |||
| 1 | 64 | 0.92 [0.60, 1.28] | 24.4 [19, 30] |
| 2 | 128 | 1.84 [1.37, 2.35] | 48.7 [41, 57] |
| 3 | 192 | 2.77 [2.18, 3.38] | 73.0 [63, 83] |
| 5 | 320 | 4.61 [3.86, 5.40] | 121.7 [109, 135] |
The expected mean carrier burden in the study sample is shown and assumes that each of the 64 founders introduced a specified number of unique mutations (column 1) into the population. Expected mean carrier burden for independently segregating neutral mutations will not change, but expected mean carrier burden for lethal mutations will be reduced from the mean carrier burden in the founders. Expected mean carrier burden is reported to two decimal places of accuracy and might appear as integers because of extremely small errors in our simulations (e.g., the expected mean burden given one mutation per founder is 1.000059). The following abbreviation is used: CI, confidence interval.
Because we did not genotype all mutations causing AR diseases (Table S1) in the Hutterites, the true mean carrier burden must certainly be higher than 1.096 mutations per individual. We calculated expectations for the true mean carrier burden (Table 5) by assigning different numbers of mutations to each founder and dropping those mutations through the pedigree. If we assume that, on average, each of the 64 Hutterite founders carried three unique AR mutations,47 our simulations show that 73 mutations (38% of founder mutations) are expected to still be present in our study subjects; this corresponds to an expected mean carrier burden between 2.76 and 3 mutations per Hutterite (depending on the relative proportions of lethal and neutral mutations).
Because some of the 64 Hutterite founders were most likely related,48 we also considered the possibility that some mutations might have been introduced into the population by more than one founder and might thereby have resulted in a higher initial carrier frequency. However, the simulations indicate that under the neutral model, the expected mean burden in our study subjects will always be equivalent to the mean burden in the founders, regardless of the initial carrier frequency. However, under the lethal mutation model, as compared to the model shown in Table 5, the expected mean burden in the current population will be even lower than the initial mean burden in the founders because mutations occurring at a higher frequency in the founders will be eliminated at a faster rate.49
Discussion
Influence of Drift on Mutations in an Isolated Population
This study provides empiric illustration of the powerful effects of drift on rare recessive mutations in a founder population. On one hand, the founder effect reduces the amount of variation present among the founders, and drift results in the further loss of the majority of rare variants from those founders during the first few generations of the pedigree. These phenomena are illustrated by our simulations and the observation that many “common” Mendelian disease mutations, such as those causing phenylketonuria, hemophilias, Fragile X Syndrome, and Duchenne muscular dystrophy, have not been observed in the Hutterites. On the other hand, the surviving variants can drift to high frequencies over subsequent generations. High-frequency disease-causing mutations in founder populations have been the subject of many investigations into whether the high carrier frequencies might be evidence of balancing (overdominant) selection.38,50–53 For example, allele-dropping simulations in the Saguenay population also show that after the introduction of an AR mutation by just a single founder, the stochastic effects of drift can generate high carrier frequencies and that the lethality of a mutation has relatively little effect on the frequencies it reaches in later generations.38 Similarly, although we cannot exclude the possibility of selective advantage in carriers of the mutations studied here, we show that the high carrier frequencies for 11 out of 14 mutations in this study are consistent with expectations from a founder effect followed by population expansion. Furthermore, we failed to find a fertility advantage for carriers of the three remaining mutations. In fact, the probabilities that the mutations could have reached the current carrier frequencies (Table 4) are most likely underestimates because our simulations assumed that each mutation was present in only one founder. Instead, the mutations might have been segregating in the Hutterites’ ancestral populations, and some of the founders might have been related;48 hence, multiple founders could have introduced the same mutation on a single haplotype. This possibility is supported by the fact that four of the mutations (TRIM32 p.Asp487Asn, FKRP p.Leu276Ile, CFTR p.Met1101Lys, and ZMPSTE24 c.1085dupT) we studied are known to be present on the same haplotype in the Hutterites and in populations from which some of the Hutterite founders might have originated.26,41,42,44 We hypothesize that as more human genomes are sequenced, many mutations currently thought to be private to the Hutterites will be discovered at low frequencies in other European populations.
Consequences of Short Consensus Haplotypes on Disease Mapping
The possibility that some undiscovered AR mutations in the Hutterites might be present on haplotypes that have very short regions of overlap could confound mapping efforts that utilize homozygosity and linkage analysis. As an example worst-case scenario, some carriers of TRIM32 p.Asp487Asn only share a ∼449 kb segment, so an affected child born to two such carriers would carry the mutation within an extremely short run of homozygosity. If the child were genotyped on the Illumina HumanCytoSNP-12 array (median intermarker distance of 6.2 kb), which is often used in Mendelian disease mapping, the child would have as few as 72 consecutive homozygous SNPs around the disease locus. Although the literature contains numerous examples of successful homozygosity-mapping efforts, many more mutations might have thus far eluded identification by being carried on short haplotypes.
Carrier Burden in a Founder Population
Early theoretical studies of lethal mutation burden in humans estimated that each person is a carrier of 3–5 recessive lethal mutations,54–56 whereas a recent study of 448 disease mutations in 104 individuals from outbred populations reported a mean carrier burden of 2.8 per genome.47 Although we cannot compare these values against the mean carrier burden in this study because we only studied 14 mutations, our simulations of carrier burden demonstrate that the Hutterites are expected to carry fewer AR disease-causing mutations per individual than what is reported for individuals from large, outbred populations. Although it seems counterintuitive that the Hutterites have the same or even less of a carrier burden per individual in light of the relatively high incidence of AR disease in the Hutterites, the fact that each Hutterite is more likely to marry someone who carries the same mutation results in a higher incidence of homozygous children, albeit for a smaller number of diseases. This scenario will also result in recessive mutations being removed from the population at a greater rate than in an outbred population.49 This was evidenced by the simulation showing an 8% decrease in carrier burden for lethal recessive mutation over just 13 generations.
Unexpectedly, we found that even if the 64 Hutterite founders carried only three unique AR mutations each, 73 mutations (95% confidence interval = 61–85) are expected to be segregating in the population today; this leaves a large (≥2.5×) discrepancy between the numbers of expected and observed AR diseases and mutations in the Hutterites. Some of these as yet undiscovered diseases might be mild, have onset late in life, or have variable penetrance or expressivity: one of the TRIM32 p.Asp487Asn homozygotes is 43 years old and is currently asymptomatic, and we have also previously reported an asymptomatic SMA homozygote. Other mutations are expected to be sufficiently rare that homozygosity for these mutations might not yet have occurred. An alternative (or complementary) explanation is that because some of the founders were most likely related, the same mutation might have been introduced into the population by multiple founders. The truth probably lies between these extremes—there are perhaps 30 or more disease mutations yet to be discovered in this well-studied population.
Acknowledgments
The authors thank Gerald Salen (University of Medicine and Dentistry of NJ) and David Mymin (University of Manitoba, Winnipeg) for conducting clinical studies in the individuals with sitosterolemia; Soma Das for providing PCR primers for GJB2; Mark Abney, Graeme Bell, Nancy Cox, and Lawrence Uricchio for helpful discussions; Brent Werness for mathematical assistance and suggestions; and the Hutterites for their continued participation in our studies. This research was supported in part by grants R01 HD21244 and R01 HL085197 and the National Heart, Lung, and Blood Institute (NHLBI)-funded Exome Sequencing Service at the Broad Institute.
Contributor Information
Jessica X. Chong, Email: jxchong@uchicago.edu.
Carole Ober, Email: c-ober@bsd.uchicago.edu.
Supplemental Data
Web Resources
The URLs for data presented herein are as follows:
Code for simulations from the Ober Lab, http://ober.bsd.uchicago.edu
Online Mendelian Inheritance in Man (OMIM), http://www.omim.org
References
- 1.Peltonen L. Molecular background of the Finnish disease heritage. Ann. Med. 1997;29:553–556. doi: 10.3109/07853899709007481. [DOI] [PubMed] [Google Scholar]
- 2.Sheffield V.C., Stone E.M., Carmi R. Use of isolated inbred human populations for identification of disease genes. Trends Genet. 1998;14:391–396. doi: 10.1016/s0168-9525(98)01556-x. [DOI] [PubMed] [Google Scholar]
- 3.Arcos-Burgos M., Muenke M. Genetics of population isolates. Clin. Genet. 2002;61:233–247. doi: 10.1034/j.1399-0004.2002.610401.x. [DOI] [PubMed] [Google Scholar]
- 4.McKusick V.A. Genetic studies in American inbred populations with particular reference to the Old Order Amish. Isr. J. Med. Sci. 1973;9:1276–1284. [PubMed] [Google Scholar]
- 5.Ruiz-Perez V.L., Ide S.E., Strom T.M., Lorenz B., Wilson D., Woods K., King L., Francomano C., Freisinger P., Spranger S. Mutations in a new gene in Ellis-van Creveld syndrome and Weyers acrodental dysostosis. Nat. Genet. 2000;24:283–286. doi: 10.1038/73508. [DOI] [PubMed] [Google Scholar]
- 6.Höglund P., Haila S., Socha J., Tomaszewski L., Saarialho-Kere U., Karjalainen-Lindsberg M.L., Airola K., Holmberg C., de la Chapelle A., Kere J. Mutations of the Down-regulated in adenoma (DRA) gene cause congenital chloride diarrhoea. Nat. Genet. 1996;14:316–319. doi: 10.1038/ng1196-316. [DOI] [PubMed] [Google Scholar]
- 7.Pastinen T., Perola M., Ignatius J., Sabatti C., Tainola P., Levander M., Syvänen A.C., Peltonen L. Dissecting a population genome for targeted screening of disease mutations. Hum. Mol. Genet. 2001;10:2961–2972. doi: 10.1093/hmg/10.26.2961. [DOI] [PubMed] [Google Scholar]
- 8.Myerowitz R., Costigan F.C. The major defect in Ashkenazi Jews with Tay-Sachs disease is an insertion in the gene for the alpha-chain of beta-hexosaminidase. J. Biol. Chem. 1988;263:18587–18589. [PubMed] [Google Scholar]
- 9.Engert J.C., Bérubé P., Mercier J., Doré C., Lepage P., Ge B., Bouchard J.-P., Mathieu J., Melançon S.B., Schalling M. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000;24:120–125. doi: 10.1038/72769. [DOI] [PubMed] [Google Scholar]
- 10.Zielenski J., Fujiwara T.M., Markiewicz D., Paradis A.J., Anacleto A.I., Richards B., Schwartz R.H., Klinger K.W., Tsui L.C., Morgan K. Identification of the M1101K mutation in the cystic fibrosis transmembrane conductance regulator (CFTR) gene and complete detection of cystic fibrosis mutations in the Hutterite population. Am. J. Hum. Genet. 1993;52:609–615. [PMC free article] [PubMed] [Google Scholar]
- 11.Hostetler J.A. Johns Hopkins University Press; Baltimore: 1974. Hutterite society. [Google Scholar]
- 12.Steinberg, A., Bleibtreu, H., Kurczynski, T., Martin, A., and Kurczynski, E. (1967). Genetic studies in an inbred human isolate. Proceedings of the Third International Congress of Human Genetics, 267–290.
- 13.Boycott K.M., Parboosingh J.S., Chodirker B.N., Lowry R.B., McLeod D.R., Morris J., Greenberg C.R., Chudley A.E., Bernier F.P., Midgley J. Clinical genetics and the Hutterite population: A review of Mendelian disorders. Am. J. Med. Genet. A. 2008;146A:1088–1098. doi: 10.1002/ajmg.a.32245. [DOI] [PubMed] [Google Scholar]
- 14.Ober C., Abney M., McPeek M.S. The genetic dissection of complex traits in a founder population. Am. J. Hum. Genet. 2001;69:1068–1079. doi: 10.1086/324025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Martin A.O. The founder effect in a human isolate: Evolutionary implications. Am. J. Phys. Anthropol. 1970;32:351–367. doi: 10.1002/ajpa.1330320305. [DOI] [PubMed] [Google Scholar]
- 16.Kosova G., Pickrell J.K., Kelley J.L., McArdle P.F., Shuldiner A.R., Abney M., Ober C. The CFTR Met 470 allele is associated with lower birth rates in fertile men from a population isolate. PLoS Genet. 2010;6:e1000974. doi: 10.1371/journal.pgen.1000974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ober C., Aldrich C.L., Chervoneva I., Billstrand C., Rahimov F., Gray H.L., Hyslop T. Variation in the HLA-G promoter region influences miscarriage rates. Am. J. Hum. Genet. 2003;72:1425–1435. doi: 10.1086/375501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ober C., Tan Z., Sun Y., Possick J.D., Pan L., Nicolae R., Radford S., Parry R.R., Heinzmann A., Deichmann K.A. Effect of variation in CHI3L1 on serum YKL-40 level, risk of asthma, and lung function. N. Engl. J. Med. 2008;358:1682–1691. doi: 10.1056/NEJMoa0708801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Abney M., McPeek M.S., Ober C. Broad and narrow heritabilities of quantitative traits in a founder population. Am. J. Hum. Genet. 2001;68:1302–1307. doi: 10.1086/320112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cusanovich D.A., Billstrand C., Zhou X., Chavarria C., De Leon S., Michelini K., Pai A.A., Ober C., Gilad Y. The combination of a genome-wide association study of lymphocyte count and analysis of gene expression data reveals novel asthma candidate genes. Hum. Mol. Genet. 2012;21:2111–2123. doi: 10.1093/hmg/dds021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Ober C., Nord A.S., Thompson E.E., Pan L., Tan Z., Cusanovich D., Sun Y., Nicolae R., Edelstein C., Schneider D.H. Genome-wide association study of plasma lipoprotein(a) levels identifies multiple genes on chromosome 6q. J. Lipid Res. 2009;50:798–806. doi: 10.1194/jlr.M800515-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Çalışkan M., Chong J.X., Uricchio L., Anderson R., Chen P., Sougnez C., Garimella K., Gabriel S.B., dePristo M.A., Shakir K. Exome sequencing reveals a novel mutation for autosomal recessive non-syndromic mental retardation in the TECR gene on chromosome 19p13. Hum. Mol. Genet. 2011;20:1285–1289. doi: 10.1093/hmg/ddq569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chong J.X., Oktay A.A., Dai Z., Swoboda K.J., Prior T.W., Ober C. A common spinal muscular atrophy deletion mutation is present on a single founder haplotype in the US Hutterites. Eur. J. Hum. Genet. 2011;19:1045–1051. doi: 10.1038/ejhg.2011.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ober C., Bombard A., Dhaliwal R., Elias S., Fagan J., Laffler T.G., Martin A.O., Rosinsky B. Studies of cystic fibrosis in Hutterite families by using linked DNA probes. Am. J. Hum. Genet. 1987;41:1145–1151. [PMC free article] [PubMed] [Google Scholar]
- 25.Huang L., Szymanska K., Jensen V.L., Janecke A.R., Innes A.M., Davis E.E., Frosk P., Li C., Willer J.R., Chodirker B.N. TMEM237 is mutated in individuals with a Joubert syndrome related disorder and expands the role of the TMEM family at the ciliary transition zone. Am. J. Hum. Genet. 2011;89:713–730. doi: 10.1016/j.ajhg.2011.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Loucks C., Parboosingh J.S., Chong J.X., Ober C., Siu V.M., Hegele R.A., Rupar C.A., McLeod D.R., Pinto A., Chudley A.E., Innes A.M. A shared founder mutation underlies restrictive dermopathy in Old Colony (Dutch-German) Mennonite and Hutterite patients in North America. Am. J. Med. Genet. A. 2012;158A:1229–1232. doi: 10.1002/ajmg.a.35302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Innes A.M., Boycott K.M., Puffenberger E.G., Redl D., MacDonald I.M., Chudley A.E., Beaulieu C., Perrier R., Gillan T., Wade A., Parboosingh J.S. A founder mutation in BBS2 is responsible for Bardet-Biedl syndrome in the Hutterite population: utility of SNP arrays in genetically heterogeneous disorders. Clin. Genet. 2010;78:424–431. doi: 10.1111/j.1399-0004.2010.01481.x. [DOI] [PubMed] [Google Scholar]
- 28.Davey K.M., Parboosingh J.S., McLeod D.R., Chan A., Casey R., Ferreira P., Snyder F.F., Bridge P.J., Bernier F.P. Mutation of DNAJC19, a human homologue of yeast inner mitochondrial membrane co-chaperones, causes DCMA syndrome, a novel autosomal recessive Barth syndrome-like condition. J. Med. Genet. 2006;43:385–393. doi: 10.1136/jmg.2005.036657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frosk P., Weiler T., Nylen E., Sudha T., Greenberg C.R., Morgan K., Fujiwara T.M., Wrogemann K. Limb-girdle muscular dystrophy type 2H associated with mutation in TRIM32, a putative E3-ubiquitin-ligase gene. Am. J. Hum. Genet. 2002;70:663–672. doi: 10.1086/339083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wang J., Joy T., Mymin D., Frohlich J., Hegele R.A. Phenotypic heterogeneity of sitosterolemia. J. Lipid Res. 2004;45:2361–2367. doi: 10.1194/jlr.M400310-JLR200. [DOI] [PubMed] [Google Scholar]
- 31.Alagramam K.N., Yuan H., Kuehn M.H., Murcia C.L., Wayne S., Srisailpathy C.R., Lowry R.B., Knaus R., Van Laer L., Bernier F.P. Mutations in the novel protocadherin PCDH15 cause Usher syndrome type 1F. Hum. Mol. Genet. 2001;10:1709–1718. doi: 10.1093/hmg/10.16.1709. [DOI] [PubMed] [Google Scholar]
- 32.Mymin D., Wang J., Frohlich J., Hegele R.A. Image in cardiovascular medicine. Aortic xanthomatosis with coronary ostial occlusion in a child homozygous for a nonsense mutation in ABCG8. Circulation. 2003;107:791. doi: 10.1161/01.cir.0000050545.21826.ad. [DOI] [PubMed] [Google Scholar]
- 33.Uricchio L.H., Chong J.X., Ross K.D., Ober C., Nicolae D.L. Accurate imputation of rare and common variants in a founder population from a small number of sequenced individuals. Genet. Epidemiol. 2012;36:312–319. doi: 10.1002/gepi.21623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Armistead J., Khatkar S., Meyer B., Mark B.L., Patel N., Coghlan G., Lamont R.E., Liu S., Wiechert J., Cattini P.A. Mutation of a gene essential for ribosome biogenesis, EMG1, causes Bowen-Conradi syndrome. Am. J. Hum. Genet. 2009;84:728–739. doi: 10.1016/j.ajhg.2009.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kenneson A., Van Naarden Braun K., Boyle C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002;4:258–274. doi: 10.1097/00125817-200207000-00004. [DOI] [PubMed] [Google Scholar]
- 36.Bourgain C., Abney M., Schneider D., Ober C., McPeek M.S. Testing for Hardy-Weinberg equilibrium in samples with related individuals. Genetics. 2004;168:2349–2361. doi: 10.1534/genetics.104.031617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.O’Connell J.R., Weeks D.E. PedCheck: A program for identification of genotype incompatibilities in linkage analysis. Am. J. Hum. Genet. 1998;63:259–266. doi: 10.1086/301904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Heyer E. One founder/one gene hypothesis in a new expanding population: Saguenay (Quebec, Canada) Hum. Biol. 1999;71:99–109. [PubMed] [Google Scholar]
- 39.Abney M., Ober C., McPeek M.S. Quantitative-trait homozygosity and association mapping and empirical genomewide significance in large, complex pedigrees: Fasting serum-insulin level in the Hutterites. Am. J. Hum. Genet. 2002;70:920–934. doi: 10.1086/339705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kosova G., Abney M., Ober C. Colloquium papers: Heritability of reproductive fitness traits in a human population. Proc. Natl. Acad. Sci. USA. 2010;107(Suppl 1):1772–1778. doi: 10.1073/pnas.0906196106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Stuhrmann M., Dörk T., Frühwirth M., Golla A., Skawran B., Antonin W., Ebhardt M., Loos A., Ellemunter H., Schmidtke J. Detection of 100% of the CFTR mutations in 63 CF families from Tyrol. Clin. Genet. 1997;52:240–246. doi: 10.1111/j.1399-0004.1997.tb02555.x. [DOI] [PubMed] [Google Scholar]
- 42.Schoser B.G.H., Frosk P., Engel A.G., Klutzny U., Lochmüller H., Wrogemann K. Commonality of TRIM32 mutation in causing sarcotubular myopathy and LGMD2H. Ann. Neurol. 2005;57:591–595. doi: 10.1002/ana.20441. [DOI] [PubMed] [Google Scholar]
- 43.Hendrickson B.C., Donohoe C., Akmaev V.R., Sugarman E.A., Labrousse P., Boguslavskiy L., Flynn K., Rohlfs E.M., Walker A., Allitto B. Differences in SMN1 allele frequencies among ethnic groups within North America. J. Med. Genet. 2009;46:641–644. doi: 10.1136/jmg.2009.066969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Frosk P., Greenberg C.R., Tennese A.A.P., Lamont R., Nylen E., Hirst C., Frappier D., Roslin N.M., Zaik M., Bushby K. The most common mutation in FKRP causing limb girdle muscular dystrophy type 2I (LGMD2I) may have occurred only once and is present in Hutterites and other populations. Hum. Mutat. 2005;25:38–44. doi: 10.1002/humu.20110. [DOI] [PubMed] [Google Scholar]
- 45.Morais P., Magina S., Ribeiro Mdo.C., Rodrigues M., Lopes J.M., Thanh Hle.T., Wehnert M., Guimarães H. Restrictive dermopathy—a lethal congenital laminopathy. Case report and review of the literature. Eur. J. Pediatr. 2009;168:1007–1012. doi: 10.1007/s00431-008-0868-x. [DOI] [PubMed] [Google Scholar]
- 46.Strom C.M., Crossley B., Buller-Buerkle A., Jarvis M., Quan F., Peng M., Muralidharan K., Pratt V., Redman J.B., Sun W. Cystic fibrosis testing 8 years on: Lessons learned from carrier screening and sequencing analysis. Genet. Med. 2011;13:166–172. doi: 10.1097/GIM.0b013e3181fa24c4. [DOI] [PubMed] [Google Scholar]
- 47.Bell C.J., Dinwiddie D.L., Miller N.A., Hateley S.L., Ganusova E.E., Mudge J., Langley R.J., Zhang L., Lee C.C., Schilkey F.D. Carrier testing for severe childhood recessive diseases by next-generation sequencing. Sci. Transl. Med. 2011;3:ra4. doi: 10.1126/scitranslmed.3001756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pichler I., Fuchsberger C., Platzer C., Calişkan M., Marroni F., Pramstaller P.P., Ober C. Drawing the history of the Hutterite population on a genetic landscape: Inference from Y-chromosome and mtDNA genotypes. Eur. J. Hum. Genet. 2010;18:463–470. doi: 10.1038/ejhg.2009.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimura M., Maruyama T., Crow J.F. The mutation load in small populations. Genetics. 1963;48:1303–1312. doi: 10.1093/genetics/48.10.1303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Durst R., Colombo R., Shpitzen S., Avi L.B., Friedlander Y., Wexler R., Raal F.J., Marais D.A., Defesche J.C., Mandelshtam M.Y. Recent origin and spread of a common Lithuanian mutation, G197del LDLR, causing familial hypercholesterolemia: Positive selection is not always necessary to account for disease incidence among Ashkenazi Jews. Am. J. Hum. Genet. 2001;68:1172–1188. doi: 10.1086/320123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Risch N., Tang H., Katzenstein H., Ekstein J. Geographic distribution of disease mutations in the Ashkenazi Jewish population supports genetic drift over selection. Am. J. Hum. Genet. 2003;72:812–822. doi: 10.1086/373882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wagener D., Cavalli-Sforza L.L., Barakat R. Ethnic variation of genetic disease: Roles of drift for recessive lethal genes. Am. J. Hum. Genet. 1978;30:262–270. [PMC free article] [PubMed] [Google Scholar]
- 53.Chase G.A., McKusick V.A. Controversy in human genetics: Founder effect in Tay-Sachs disease. Am. J. Hum. Genet. 1972;24:339–340. [PMC free article] [PubMed] [Google Scholar]
- 54.Morton N.E., Crow J.F., Muller H.J. An estimate of the mutational damage in man from data on consanguineous marriages. Proc. Natl. Acad. Sci. USA. 1956;42:855–863. doi: 10.1073/pnas.42.11.855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Morton N.E. The mutational load due to detrimental genes in man. Am. J. Hum. Genet. 1960;12:348–364. [PMC free article] [PubMed] [Google Scholar]
- 56.Muller H.J. Our load of mutations. Am. J. Hum. Genet. 1950;2:111–176. [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.