

Abstract
Purpose.
Retinal ganglion cell (RGC) death and failed axonal regeneration after trauma or disease, including glaucomatous and mitochondrial optic neuropathies, are linked increasingly to dysfunctional mitochondrial dynamics. However, how mitochondrial dynamics influence axon growth largely is unstudied. We examined intrinsic mitochondrial organization in embryonic and postnatal RGCs and the roles that mitochondrial dynamics have in regulating neurite growth and guidance.
Methods.
RGCs were isolated from embryonic day 20 (E20) or postnatal days 5 to 7 (P5–7) Sprague-Dawley rats by anti-Thy1 immunopanning. After JC-1 loading, mitochondria were analyzed in acutely purified RGCs by flow cytometry and in RGC neurites by fluorescence microscopy. Intrinsic axon growth was modulated by overexpressing Krüppel-like family (KLF) transcription factors, or mitochondrial dynamics were altered by inhibiting dynamin related protein-1 (DRP-1) pharmacologically or by overexpressing mitofusin-2 (Mfn-2). Mitochondrial organization, neurite growth, and growth cone motility and guidance were analyzed.
Results.
Mitochondrial dynamics and function are regulated developmentally in acutely purified RGCs and in nascent RGC neurites. Mitochondrial dynamics are modulated differentially by KLFs that promote or suppress growth. Acutely inhibiting mitochondrial fission reversibly suppressed axon growth and lamellar extension. Inhibiting DRP-1 or overexpressing Mfn-2 altered growth cone responses to chondroitin sulfate proteoglycan, netrin-1, and fibronectin.
Conclusions.
These results support the hypothesis that mitochondria locally modulate signaling in the distal neurite and growth cone to affect the direction and the rate of neurite growth.
Mitochondrial dynamics are differentially regulated in retinal ganglion cells with differing intrinsic growth potentials. Altering mitochondrial dynamics in regenerating RGC neurites alters growth cone motility, decision-making, and growth rate.
Introduction
Central nervous system (CNS) neurons fail to regenerate after injury or disease due to reduced intrinsic axon growth ability,1–4 insufficient neurotrophic factors,5–7 and extrinsic, glial-associated inhibitors.8–10 Increasingly, evidence suggests these factors act in part by altering mitochondrial dynamics and function. Mitochondria can be regulated either directly or indirectly by proteins that are intrinsic neurite growth regulators, like Krüppel-like families (KLFs),11,12 PTEN,13–15 and mTOR16; neurotrophic factors, like BDNF17,18 and NGF19–21; and by extrinsic growth inhibitors, like Nogo,22 semaphorin 3a,23,24 and chondroitin sulfate proteoglycans (CSPGs),25–27 suggesting mitochondria may modulate signaling controlling neurite growth.
Mitochondria are heterogeneous organelles, differing in subcellular distribution, membrane potential (Δψm), and metabolic activity that act individually or in networks regulated by fission, fusion, transport, docking, biogenesis, and mitophagy.28–30 These dynamics are critical to regulating mitochondrial distribution and bioenergetics,31–33 and mutations in proteins regulating mitochondrial dynamics can lead to mitochondrial dysfunction, and poor cellular and organism health,34 including retinal ganglion cell (RGC) degeneration.35,36 Mitochondria are prominent in growth cones in vitro37 and in vivo,38 and mitochondrial organization is influenced by cues19,37 and second messenger systems39,40 that regulate neurite growth. However, largely unstudied is the developmental organization of mitochondria in neurites during growth, and the roles mitochondrial dynamics have in neurite growth and guidance. We demonstrated that Δψm and bioenergetics are regulated developmentally in RGCs with differing intrinsic neurite growth potentials. Altering intrinsic neurite growth ability by KLFs modulates mitochondrial dynamics, and altering mitochondrial fission/fusion dynamics alters neurite growth rate, and growth cone motility and decision-making.
Materials and Methods
Animals
Experiments conformed to the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and were approved by the University of Miami Institutional Biosafety Committee and the Institutional Animal Care and Use Committee.
Cell Culture
RGCs were purified from male and female E20-P7 Sprague-Dawley rats (Harlan Laboratories, Indianapolis, IN) by immunopanning,41 and cultured on poly-D-lysine (70 kilodalton [kDa], 10 μg/mL; Sigma, St. Louis, MO) and laminin coated (2 μg/mL; Sigma) MatTeK dishes (P35G-1.0-2-C) as described.42
Flow Cytometry
Purified embryonic day 20 (E20) or P7 RGCs were equilibrated with the membrane potential–sensitive dye JC-1 (500 nM; 5,5′6,6′-tetrachloro-1,1′,3.3′-tetraethylbenzimidazolcarbocyanine iodide; Invitrogen, Carlsbad, CA) in PBS for 20 minutes with or without FCCP (10 μM; carbonylcyanide-P-trifluoromethoxyphenylhydrazone; Sigma) and 6 to 50 K cells were analyzed (n = 4 experiments) with forward and side scatter in a Becton Dickinson FACScan (Becton Dickinson, San Jose, CA). Data were acquired in list mode, evaluated with WinList software (Verity Software House, Topsham, ME), and data were analyzed statistically by ANOVA here and below. To prevent differences in mitochondrial bioenergetics due to differences in E20 and P7 RGC preparation, E20 and P7 RGCs were dissected, immunopanned, and loaded with JC-1 in parallel under the same conditions and time frame.
JC-1 Loading and Analysis
RGCs, dissected and immunopanned in parallel and cultured as above, were equilibrated with JC-1 (153 nM) for 20 minutes43 before imaging the distal 100 to 150 μm of neurites not longer than 30 minutes post equilibration. For analysis, a single set of DIC, and exposure-matched epi-fluorescent JC-1 monomer and J-aggregate emissions captured with 38 HE and 64 HE excitation and emission filter sets (Zeiss, Thornwood, NY) respectively, was acquired from each neurite. All images were processed identically with subtraction calculated from the mean cytoplasmic fluorescence in FCCP (10 μM) treated controls. JC-1 loading efficiency was optimized, and verified by correlative JC-1 and DIC time-lapse microscopy, and correlative time-lapse and immunocytochemistry as described44 with anti-TOM20 (Abcam, Cambridge, MA). Low (JC-1 monomer) and high (J-aggregate) potential mitochondria were quantified with the ImageJ mitochondrial analysis plug-in (ImageJ; National Institutes of Health, Bethesda, MD). Since individual mitochondria were indiscernible in clusters, clusters were measured as one for analyzing and discussing area and linear density. Relative Δψm was calculated from the ratio of the mean JC-1 monomer and J-aggregate emissions at 530 and 590 nm, respectively.
Time-Lapse Microscopy
After treating cultures with mitochondrial division inhibitor-1 (Mdivi-1; Sigma), time-lapse images were recorded,45 and lamellar and filopodial protrusion frequencies analyzed as described.46 To compare data from growth cones differing in size and activity levels, data were normalized to the pre-addition mean.
Oxygen Consumption and Extracellular Acidification Rates
Oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured using the XF96 extracellular flux analyzer per instruction (Seahorse Bioscience, North Billerica, MA). E20 and P5 RGCs were purified and cultured as above in parallel at approximately 50,000 cells per well in XF96 microplates. At 2 days in vitro (DIV), full-SATO media was replaced with unbuffered Dulbecco's modified Eagle's medium (DMEM; 250 mM glucose, 1 mM sodium pyruvate, and 2 mM GlutaMax, pH 7.4). After incubating for 30 minutes in the XF Prep Station, plates were loaded into the XF96 analyzer. OCR and ECAR were analyzed by XF96 software from four consecutive rate measurements before and during oligomycin (1 μM) and FCCP (300 nM) injections from 10 (P5) and 7 (E20) replicate wells. The number of RGCs per well was normalized by quantifying the total DNA per well (CyQuant; Molecular Probes, Invitrogen).
Mitochondrial DNA (mtDNA) Detection
mtDNA was detected by treating RGC cultures for one hour with 5-ethynyl-2′-deoxyuridine (EdU, 10 μM) and processed as described.47 EdU was amplified with Click-IT Oregon green (C-10083; Invitrogen), then anti-Oregon green-HRP (1:1000, A21253; Invitrogen), and tyramide per instructions (T-20912; Invitrogen). EdU puncta were quantified by the ImageJ particle analysis plug-in (NIH).
Neurite Growth Assay
RGCs were co-transfected by suspending 3-500K RGCs in lipofectamine (27 μL) with 0.3 μg pMax-GFP (Amaxa Biosystems GmbH, Cologne, Germany) and 1.7 μg of KLF4, KLF6, or Mfn-2 plasmid (Open Biosystems, Huntsville, AL) before electroporation (SCN1 program; Amaxa). Electroporated cells were washed once with NB-SATO, centrifuged, resuspended in NB-SATO, and plated as above. Mitochondria labeled with MitoTracker CMXROS (Invitrogen) were incubated for 2 minutes with a 1:10,000 dilution of a MitoTracker CMXROS stock (1 mM in dimethyl sulfoxide [DMSO]; Invitrogen) in NB-SATO and then washed 3 times with NB-SATO. Neurite lengths and MitoTracker-labeled mitochondria were measured with ImageJ (NIH).
Neurite Guidance Assay
CSPG (3 μg/ml; gift from Vance Lemmon), netrin-1, or fibronectin (2 μg/mL; Sigma) stripes were stamped on glass coverslips pretreated with HCl (1N, 3 hours) as described48 and then overlaid with laminin (2 μg/mL) in neural basal (Gibco, Grand Island, NY) at 37°C for 15 minutes before culturing RGCs as above. Neurite crossing and length were analyzed for at least 100 neurites from at least three separate experiments.
Results
Mitochondrial Mass, Δψm, and Bioenergetics Are Developmentally Regulated in RGCs
RGCs' intrinsic axon growth ability decreases developmentally after birth; compared to embryonic day 20 (E20), intrinsic axon growth ability decreases approximately 5-fold by postnatal day 2 (P2) and continues to decrease into adulthood.1 To determine if mitochondrial mass and Δψm also are regulated developmentally in RGCs during this developmental window, acutely purified E20 and P7 RGCs, purified and processed in parallel, were loaded with the Δψm-sensitive dye JC-143,49 and analyzed by flow cytometry (Fig. 1a). JC-1 monomer emissions at 530 nm, representing the total mitochondrial mass, were lower in E20 RGCs than in P7 RGCs, consistent with an increased respiratory capacity in postnatal RGCs (e.g., Figs. 2g, 2i). In contrast, JC-1 emission at 590 nm from JC-1 J-aggregates, which only form in high potential regions within mitochondria and, thus, represent the proportion of the total mitochondria at a high membrane potential, was similar in E20 and P7 RGCs (n = 4, Fig. 1b). The ratio of 590 to 530 nm emissions, which indicates Δψm independent of mitochondrial mass, was higher in E20 RGCs (0.89 ± 0.22) than in P7 RGCs (0.41 ± 0.02, Fig. 1c). Thus, although E20 RGCs have fewer mitochondria, a greater percentage of E20 mitochondria are polarized in acutely purified RGCs, consistent with a greater workload on E20 mitochondria (e.g., Figs. 2g, 2i). Uncoupling with FCCP50,51 reduced the relative Δψm similarly in E20 and P7 RGCs by 89 ± 1.3 and 93 ± 1.5%, respectively (Figs. 1a, 1c). These results indicated mitochondrial mass and Δψm are regulated developmentally in RGCs during the same developmental window as intrinsic RGC axon growth potential.
Figure 1. .

Mitochondrial mass and Δψm are regulated developmentally in embryonic and postnatal RGCs. Mitochondrial mass and Δψm were analyzed by flow cytometry in acutely purified E20 and P7 RGCs loaded with JC-1. (a) Representative dot plots for JC-1 emission at 530 nm (JC-1 green), representing the total mitochondrial mass, and at 590 nm (JC-1 red), representing the polarized or high potential mitochondrial mass in E20 and P7 RGCs without (Control) and with the mitochondrial uncoupler/depolarizer FCCP (10 μM). (b) E20 RGCs (black bars) had a smaller total mitochondrial mass (530 nm) than P7 RGCs (white bars), whereas the polarized (590 nm) mitochondrial mass was similar. (c) E20 RGCs had a higher mean Δψm (590/530 nm ratio) than P7 RGCs. FCCP depolarization reduced Δψm similarly at both ages. N = 4 experiments of 6000 to 50,000 events (cells) per experiment. Error bars: SEM. *P < 0.05.
Figure 2. .

In nascent RGC neurites, mitochondrial Δψ and function are regulated developmentally. (a–c) DIC images of the distal neurite and growth cone (GC). JC-1 monomer emission at 530 nm (green) revealed total mitochondria and JC-1 J-aggregate emission at 590 nm (red) revealed the high potential, polarized regions within mitochondria. Total mitochondria were detected as individuals (arrows) and as complexes (arrowheads) that differed in their degree of polarization ranging from partial (arrows, arrowheads) to complete (open arrowheads). (a) E20 RGC representing the general organization of mitochondria in large complexes (arrowhead) in E20 and P5 RGCs at 1 DIV. (b) P5 RGC representing mitochondrial redistribution to smaller clusters in E20 and P5 RGCs by 2 DIV. This P5 RGC also shows increased polarization seen in P5 RGCs at 2 DIV. (c) FCCP (10 uM) blocked J-aggregates, but not JC-1 monomer uptake. (d-f) Total and polarized mean area (d), LMD (e), and Δψm (f) were measured at 1, 2, and 3 DIV. (d) Mean areas decreased in E20 (black) and P5 (white) neurites by 2 DIV. In contrast, polarized areas were greater in P5 at 1 DIV and increased further by 2 DIV before returning to 1 DIV sizes by 3 DIV. (e) LMDs were similar at 1 DIV, and then decreased similarly in E20 and P5 RGCs by 2 DIV, and then remained similarly sized through 3 DIV. Like mean area, the polarized LMD was initially higher in P5 RGCs at 1 DIV and then increased further by 2 DIV before returning to 1 DIV levels by 3 DIV. (f) Δψm increased in E20 and P5 from 1 to 2 DIV, but to a much greater extent in P5. Δψm remained stable in E20 RGCs through 3 DIV, but reversed in P5 RGCs by 3 to 1 DIV levels, lower than E20. (g-i) Oxidative phosphorylation and glycolysis measured at 2 DIV in E20 and P5 RGCs with Seahorse Bioscience technology. (g). Basal respiration is similar in E20 and P5 RGCs, whereas maximal respiration is greater in P5 RGCs. (h). In contrast, basal and maximal glycolysis is greater in E20 RGCs. (i) The basal and maximal OCR/ECAR ratios are greater in P5 RGCs. Error bars: SEM. Bar: 10 μm, n ≥ 30 neurites per age per DIV. *P < 0.001, **P < 0.05.
In cultured E20 and P5 RGCs, axons initially grow at similar rates during the first DIV. However, by the second DIV, E20 axons increase their growth rate approximately 5-fold, whereas P5 RGCs slow.1 To determine quantitatively if mitochondrial mass and Δψm correlate with this change in growth rate, we analyzed total and polarized (high potential) mitochondrial areas and linear densities, and Δψm in the distal neurites of JC-1–loaded E20 and P5 RGCs at 1, 2, and 3 DIV.
Initially, JC-1 loading specificity and conditions were optimized to reveal accurately and consistently the total mitochondrial population and the proportion of the total population that was polarized. As used in other studies on mitochondria in neurites, short incubation times with JC-1 (e.g., 3–5 minutes)52,53 led to high variability and incomplete equilibration likely due to the slow equilibration kinetics of JC-1.43 High concentrations of JC-1 (e.g., 3–30 μM)52–54 led to apparent toxicity, including gross morphologic changes in neurites, mitochondrial deformation, saturated cytoplasmic JC-1, and non-specific J-aggregates. Thus, we used a loading concentration optimized to 153 nM, approximately 20 to 200 times lower than used previously, with careful pre-equilibration and without JC-1 washout43 to prevent re-equilibration artifacts. Before imaging, JC-1 was equilibrated for 20 minutes at which point J-aggregates reached their maximum, similar to the time course reported in other cell types.55 JC-1 specificity for mitochondria was confirmed with correlative JC-1 imaging, time-lapse DIC microscopy, and immunocytochemistry56 (not shown). JC-1 monomers revealed the total mitochondrial population ranged from round to filamentous organized individually or in larger complexes (Figs. 2a, 2b). J-aggregates ranged from small puncta within mitochondria to entire mitochondria, indicating Δψm heterogeneity within and between mitochondria. Uncoupling mitochondria with FCCP reduced J-aggregates to barely detectable levels (Fig. 2c), indicating proper JC-1 optimization.
To determine if mitochondrial complex size changes during neurite growth, total and polarized mean mitochondrial areas were analyzed in the distal neurites and growth cones of E20 or P5 RGCs over 3 DIV (Figs. 2d–f). At 1 DIV, total mitochondria, detected by JC-1 monomer emissions, were organized in larger complexes at both ages that, by 2 DIV, decreased in area by 75% in E20 RGCs and by 54% in P5 RGCs (Fig. 2d), and then remained similarly sized at both ages through 3 DIV, consistent with early mitochondrial biogenesis in neurites followed by redistribution as neurites elongate. Time-lapse imaging of mitochondrial transport does not support the selective transport of mitochondria from the cell soma to the distal neurite during neurite growth (not shown). Polarized areas within mitochondria, detected by JC-1 J-aggregate emissions, were smaller in E20 RGCs than in P5 RGCs and remained similarly sized over 3 DIV. In contrast, polarized areas in P5 RGCs increased in size 45% between 1 and 2 DIV before decreasing in size 44% by 3 DIV.
To determine if mitochondrial density also changes in the distal neurite, we measured the linear mitochondrial density (LMD; mitochondrial area per unit length of axon) of the total and polarized regions. The total LMD also changed similarly at both ages (Fig. 2e). At 1 DIV, the LMD was similar in both ages before decreasing in E20 RGCs 61% and in P5 RGCs 52% by 2 DIV, and then remaining constant through 3 DIV. Like area, the polarized LMD in E20 neurites was lower than in P5 at 1 DIV and remained stable over 3 DIV. In contrast, the polarized LMD in P5 neurites increased 102% by 2 DIV. Like area, by 3 DIV the polarized LMD in P5 neurites decreased 74% to below 1 DIV levels. Thus, the mean total mitochondrial complex area and the LMD changes similarly as E20 and P5 neurites elongate. However, the polarized mean area and polarized LMD spikes in P5 neurites at 2 DIV during the time frame when E20 neurite growth rate accelerates and P5 neurite growth rate slows.
These differences in polarized LMD translated to differences in the relative Δψm. Compared to 1 DIV, the relative Δψm in E20 RGCs increased 198% by 2 DIV (Fig. 2d) and remained stable through 3 DIV, suggesting E20 mitochondria reached a steady state conducive to supporting sustained neurite growth. In contrast, the relative Δψm in P5 neurites spiked at 2 DIV, increasing 360% before reversing to 1 DIV levels, suggesting potential metabolic or oxidative stress in P5 RGC neurites at 2 DIV. Together, these results indicate Δψm is regulated developmentally in RGC neurites with differing intrinsic neurite growth potentials largely due to developmental differences in the regulation of mitochondrial polarization at 2 DIV.
To determine if these differences in E20 and P5 Δψm at 2 DIV correlate with differences in mitochondrial and cellular bioenergetics, we measured the OCR and the ECAR rate in E20 and in P5 RGCs at 2 DIV using Seahorse Bioscience technology (Figs. 2g–i). At 2 DIV, the basal OCR was similar in E20 and P5 RGCs. However, the maximum OCR was greater in P5 RGCs (Fig. 1g) consistent with increased oxidative phosphorylation (OXPHOS) in P5 RGCs and the increased polarized areas, LMD, and Δψm observed in P5 neurites. In contrast, basal and maximum ECARs were greater E20 RGCs (Fig. 1h), indicating a greater basal glycolytic rate and capacity in E20 RGCs. Finally, the ratio of OCR to ECAR shows that P5 RGC's energy production depends more on OXPHOS than on glycolysis than E20 RGCs. One caveat is that the Seahorse does not distinguish mitochondria in neurites from mitochondria in cell bodies. Nevertheless, together these data indicated E20 and P5 RGCs differ with respect to their bioenergetics at a time point when neurite growth rate is regulated differentially in E20 and P5 RGCs. Specifically, postnatal development is characterized by an increase in neurite mitochondrial polarization and whole cell maximum OCR.
Altering Intrinsic Growth and Mitochondrial Organization
To determine if altering intrinsic neurite growth ability regulates mitochondrial dynamics, we over-expressed KLFs that either suppress (KLF4) or enhance (KLF6) intrinsic neurite growth57 (Fig. 3). KLF4 reduced MitoTracker-labeled mitochondrial complex size 65 ± 0.5% in E20 RGCs (n = 30 neurites) and 65 ± 0.5 % in P5 RGCs (n = 30 neurites), but did not decrease mitochondrial density at either age (Fig. 3a). KLF6 also reduced MitoTracker-labeled mitochondria at both ages, and to a greater extent than KLF4 (Fig. 3a). In contrast to KLF4, KLF6 reduced mitochondrial density in E20 and P5 RGCs. These results showed that KLF family transcription factors regulate mitochondrial size and density, and suggest a hypothesis that the smaller, less dense mitochondria in the distal neurite, seen in embryonic RGCs and after KLF6 overexpression, are associated with or are at least sufficient to support the enhanced neurite growth seen in these two situations.
Figure 3. .
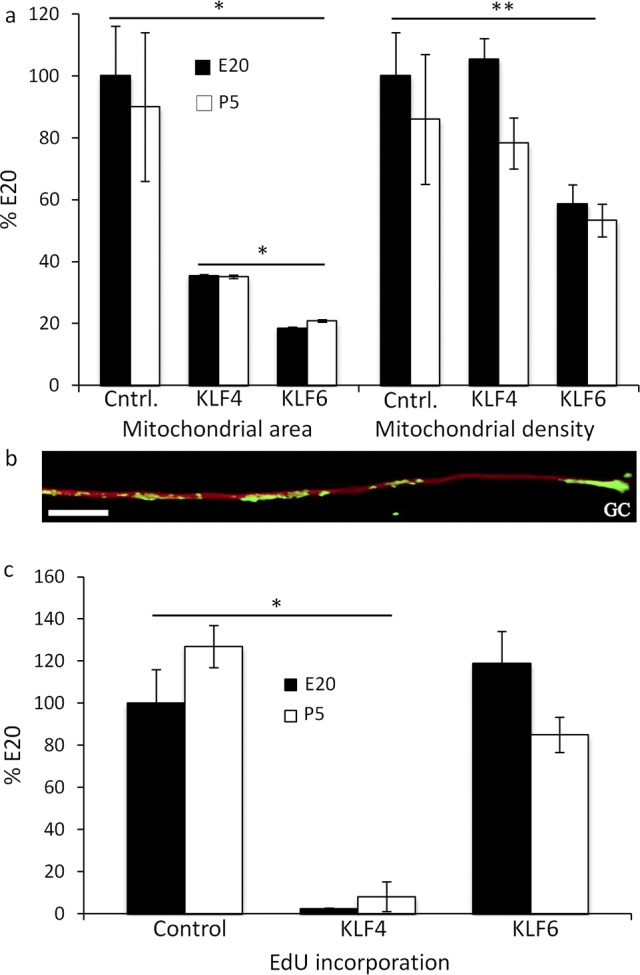
Overexpressing KLF4 and KLF6 differentially regulates mean mitochondrial area, density, and mtDNA replication. (a) Normalized to E20, E20 (black) and P5 (white) control (Cntrl.) RGC mitochondria detected with MitoTracker CMXROS had similar mean areas at similar densities. In contrast, KLF4 reduced the mean area in E20 and P5 RGCs, but not density at 1 DIV. KLF6 reduced the mean area in E20 and P5 RGCs to lower levels than KLF4. KLF6 also reduced the mean density in E20 and P5 RGCs. (b) mtDNA was detected by amplifying incorporated EdU with Click-IT-Oregon green followed by anti-Oregon green-HRP tyramide amplification (green), anti-β3-tubulin (red), GC. (c) KLF4 but not KLF6 reduced EdU incorporation in E20 (black) and P5 (white) RGCs. Error bars: SEM. Bar: 10 μm. n ≥ 30 neurites per condition, *P < 0.0001, **P < 0.05.
To determine if altered mitochondrial dynamics by KLFs reflected reduced mitochondrial biogenesis, mtDNA replication was analyzed in E20 and P5 RGCs, and in KLF4 or KLF6 over-expressing RGC neurites by detecting EdU incorporated into replicating DNA.58,59 After a 1-hour EdU perfusion, EdU was detected at similar levels in E20 and P5 RGCs as individual puncta, and in clusters in neurites and in 60 of 60 growth cones analyzed (e.g., Fig. 3b), similar to the distribution of total mitochondria in E20 and P5 neurites at 1 DIV (e.g., Fig. 1a), supporting the hypothesis that these sites represent local mitochondrial biogenesis in the neurite and in the growth cone. In contrast, KLF4 reduced EdU puncta in E20 neurites by 98% ± 0.4% (n = 30) and in P5 neurites by 92% ± 7% (n = 30), suggesting reduced biogenesis may be associated with the suppressed neurite growth induced by KLF4. In contrast, KLF6 did not significantly lower EdU-positive puncta despite smaller, less dense mitochondria, indicating greater mtDNA replication per mitochondrion (Fig. 3c). Thus, mitochondrial biogenesis is similar in E20 and P5 RGC neurites early when neurite growth rate is similar. However, altering intrinsic growth by KLFs can differentially regulate mtDNA replication and, thus, biogenesis.
Mitochondrial Dynamics Regulate Neurite Growth Rate and Growth Cone Motility
Since altering KLF transcription factors altered mitochondrial dynamics, we asked whether altering mitochondrial dynamics, in turn, affects neurite growth by inhibiting the mitochondrial fission-inducing protein, dynamin related protein-1 (DRP-1) with mitochondrial division inhibitor-1 (Mdivi-1).60,61 Consistent with inhibiting fission, Mdivi-1 increased mitochondrial lengths in E20 and P5 RGC neurites (Fig. 4). Within 20 minutes, total and polarized mitochondria were organized as long filaments (Fig. 4a). Though mitochondrial complex lengths doubled in E20 and P5 RGCs, complex lengths in P5 neurites were approximately 2-fold longer than in E20 neurites, indicating developmental differences in mitochondrial fission/fusion dynamics.
Figure 4. .
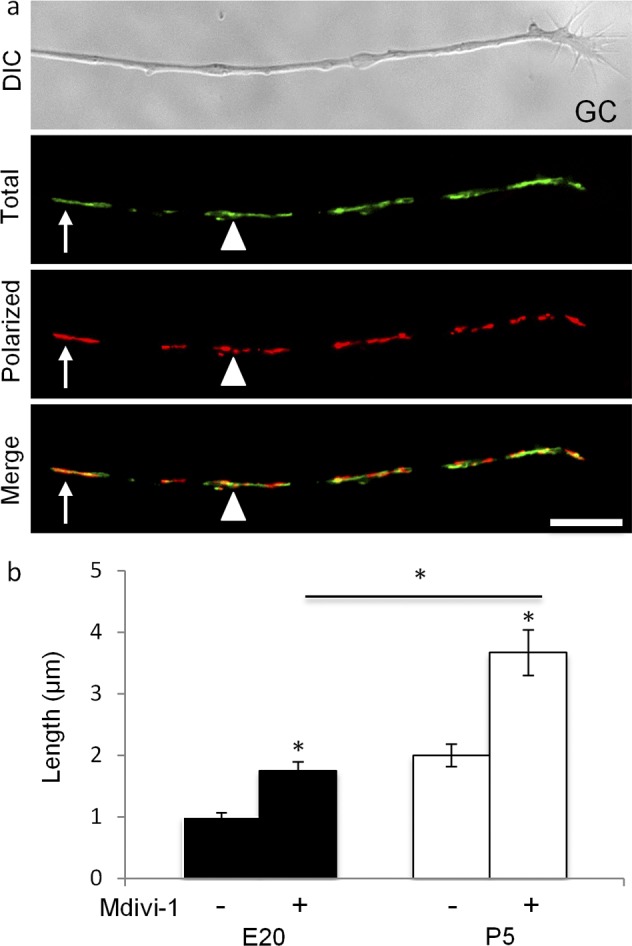
Mdivi-1 increases mitochondrial complex length in E20 and P4 RGC neurites. (a) At 20 minutes after adding Mdivi-1 (20 μM), JC-1 monomers and J-aggregates were organized as long filaments individually (arrow) or in filamentous clusters (arrowhead) with uniform (arrow) and heterogeneous Δψm (arrowhead, see merge). Scale bar: 10 μm. (b) Mdivi-1 increased mitochondrial length in E20 (black) and in P5 (white) RGCs, but to different lengths. N ≥ 30 neurites, n ≥ 100 mitochondrial complexes per age per condition. Error bars: SEM. *P < 0.001.
To determine if altering mitochondrial fission/fusion dynamics elicits changes in neurite growth, we analyzed high-resolution, time-lapse recordings of RGC neurites perfused with Mdivi-1 (20 μM). Coincident with increased mitochondrial length, Mdivi-1 reversibly suppressed neurite growth rate and lamellar, but not filopodial, protrusion, resulting in slow-growing filopodial growth cones (Figs. 5a–c). Mdivi-1 suppression was protracted; neurite growth rate and lamellar protrusion decreased continually over 20 to 40 minutes (e.g., Figs. 5b, 5c) averaging 0.7% ± 22% and 38 ± 5%, respectively (n = 5 neurites, Fig. 5d) of their pre-Mdivi-1 means. In one RGC, the neurite slowed and then retracted after Mdivi-1 perfusion. Neurite growth and lamellar protrusion returned to normal after Mdivi-1 washout over a similar time course (Fig. 5d). Mdivi-1 also slightly reduced filopodial protrusions, though not to statistical significance (Fig. 5d), that returned to the pre-addition protrusion rate after washout. Reperfusing Mdivi-1 after washout again suppressed neurite growth rate and lamellar protrusion (n = 3; e.g., Figs. 5b, 5c), suggesting mitochondrial fission/fusion dynamics regulate neurite growth rate and growth cone lamellar protrusion activity. Moreover, increased mitochondrial length in neurites correlates with reduced neurite growth.
Figure 5. .

Mdivi-1 reversibly reduces neurite growth rate and lamellar, but not filopodial protrusion. (a) A lamellar GC (time t = 0′) protruding broad lamellipodia (l) and filopodia (f) was perfused with Mdivi-1 (20 μM, t = 20′). Over the next 40′, neurite growth rate slowed (compare arrows to reference line at t = 0′–20′ and 20′–60′) and the GC's phenotype changed from lamellar to filopodial (compare t = 20′ and 40′). After washout (t = 60′), neurite growth rate remained unchanged for approximately 20′ (t = 80′) and then increased rapidly (compare arrow, t = 80′ and 120′). Concurrently, the GC's phenotype changed from filopodial back to lamellar. (b, c). Quantification of the GC in the montage in (a). (b) Neurite growth rate and (c) lamellar protrusion were reversibly inhibited by Mdivi-1. Filopodial protrusion rate was unchanged with the initial Mdivi-1 perfusion, but increased after washout. Reperfusion of Mdivi-1 (20 μM) at 140′ (not shown in montage) replicated the initial Mdivi-1 perfusion, reducing neurite growth (b) and lamellar protrusion (c), but not filopodial protrusion frequencies. (d) Mean change in neurite growth, and lamellar and filopodial protrusion rates from five individual experiments, pre-addition (pre, grey), with Mdivi-1 (20 μM, black), and after Mdivi-1 washout (white). (e-g) When Mdivi-1 (20 μM) was added to RGC cultures before neurite initiation, (e) RGCs initiated and extended phenotypically normal neurites at (f) control (vehicle, 0 μM) rates with (g) small, less dense mitochondria. Error bars: SEM. Scale bars: 10 μm. *P < 0.001.
To determine if Mdivi-1 suppresses neurite growth generally, Mdivi-1 was perfused before neurite initiation (Figs. 5e–g). In the presence of Mdivi-1 (0–20 μM), E20 and P5 RGCs extended neurites with phenotypically normal growth cones (Fig. 5e) at rates equal to control RGCs at either age (Fig. 5f; note embryonic RGC axon growth rate advantage compared to postnatal RGCs). However, as detected by MitoTracker, mitochondrial size and density were reduced in E20 neurites 78 and 83%, respectively (n = 30 neurites), and in P5 neurites 86 and 87%, respectively (n = 30 neurites, Fig. 5g) consistent with decreased fission increasing net fusion in the cell body and, thus, reducing mitochondrial transport into nascent neurites.34 The small mitochondria present in nascent neurites in the presence of Mdivi-1 may differ with respect to their fusion machinery or represent incomplete DRP-1 inhibition. These results suggested that acutely increasing mitochondrial fusion in neurites and in growth cones, but not Mdivi-1 toxicity nor inhibiting DRP-1 activity per se, regulates neurite growth rate and lamellar protrusion. Furthermore, like mitochondria in E20 neurites and after KLF6 over-expression, small mitochondria in neurites support growth.
Mitochondrial Dynamics Regulate Growth Cone Decision-Making
Since increasing mitochondrial fusion reduced lamellar protrusion and lamellar protrusion can regulate growth cone steering responses to guidance cues,46 we asked if increasing mitochondrial fusion changes growth cone steering responses to physiologically relevant molecules that either inhibit or permit neurite growth. Mitochondrial fusion was increased either pharmacologically with Mdivi-1 or genetically by overexpressing mitofusin-2 (Mfn-2), which, like Mdivi-1, increased mitochondrial length without altering mitochondrial density (Figs. 6a–c). Growth cone steering responses then were analyzed in an in vitro stripe assay48 pairing laminin stripes against either a stripe with an inhibitory factor, CSPG, or a permissive factor, either netrin-1 or fibronectin. In control cultures, RGCs typically turn and do not cross on to CSPG stripes. However, compared to 6.7% in control cultures (n = 100, Fig. 6e), increasing mitochondrial fusion with Mdivi-1 increased CSPG stripe crossing to 20% (n = 100), whereas Mfn-2 increased stripe crossing to 45% (n = 100), without altering neurite growth rate (Fig. 6f). Increasing mitochondrial fusion also altered growth cone steering responses to stripes with permissive cues. Mdivi-1 decreased crossing from netrin-1 and fibronectin to laminin (Figs. 6g, 6h). These results indicated mitochondrial fission and fusion dynamics regulate growth cone steering responses to inhibitory and permissive cues, suggesting that mitochondrial dynamics have a general role in growth cone decision-making.
Figure 6. .

Altering mitochondrial fission and fusion dynamics alters growth cone decision-making. (a) MitoTracker labeled mitochondria in control (Cntrl), pMax transfected RGCs, and in Mfn-2 transfected RGCs. (b) Mfn-2 overexpression increased the mean mitochondrial size without changing (c) mitochondrial linear density. (d) Control (Cntrl) RGCs (red, phalloidin-Tritc) growing on laminin (black) stripes turn and grow along CSPG (3 μg/mL, green) borders. Increasing mitochondrial fusion by overexpressing Mfn-2 (green, pMax-GFP) increased CSPG stripe crossing (arrows) by P5 RGCs. (e) Compared to untreated RGCs (cntrl), Mdivi-1 (20 μM) and Mfn-2 overexpression increased CSPG stripe crossing. (f) Neither Mdivi-1 nor Mfn-2–transfected RGCs demonstrated altered neurite growth rate between stripe-turning and stripe-crossing RGC subsets. (g) RGCs (red, anti-β3-tubulin) growing on netrin or fibronectin (fibron.) stripes (green) adjacent to laminin (black) stripes. (h) Mdivi-1 reduced crossing from netrin and fibronectin stripes to laminin. n > 100 neurites per condition. Scale bar: 10 μm (a) and 5 μm (d). *P < 0.05, **P < 0.001.
Discussion
These data showed that mitochondrial dynamics regulate neurite growth rate and guidance by documenting several new findings. First, mitochondrial reorganization in embryonic and postnatal RGCs follows a similar pattern during early neurite growth characterized initially by local biogenesis in nascent neurites and growth cones. Consistent with previous studies,62–64 E20 and P5 RGC mitochondria were organized primarily in clusters in nascent neurites and all growth cones during growth consistent with de novo biogenesis in the neurites and growth cones. These clusters contained newly replicated mtDNA and a low Δψm that increased as the clusters reduced in size during neurite elongation, consistent with the hypothesis that maturing mitochondria are transported out of local biogenic clusters in neurites and growth cones to support elongating neurites. This proposition extends the potential for mitochondrial biogenesis described in peripheral neurites58,59 to localized sites in CNS neuron neurites and to growth cones.
Second, optimal mitochondrial dynamics appear to support enhanced neurite growth. In nonneuronal cells, mitochondrial morphology, biogenesis, distribution, and signaling change with the state of the cell.65,66 Our results suggested mitochondrial dynamics also change with the state of neurite growth. Suppressing mitochondrial fission pharmacologically with acute Mdivi-1 (e.g., Figs. 5a–d) or increasing mitochondrial fusion genetically with Mfn-2 overexpression suppressed neurite growth (not shown) in embryonic and postnatal RGCs. Similarly, neurites with enhanced growth potential, for example E20 RGCs1 or KLF6-overexpressing RGCs,57 had smaller, less dense polarized mitochondria, and similar mtDNA replication rates in the distal neurite and growth cone. Conversely, mitochondria in neurites with suppressed neurite growth potential, for example KLF4-overexpressing and P5 RGCs, deviated from these dynamics. In KLF4-overexpressing RGCs, though mitochondria were smaller, mtDNA replication was suppressed in distal neurites, suggesting dysfunctional mitochondrial biogenesis and likely reduced bioenergetics, which depends on proper mtDNA replication in neurons.67 In P5 RGCs, polarized mitochondrial areas, density, and Δψm were greater than in E20 RGCs over the first 2 DIV before decreasing dramatically by 3 DIV. Δψm is dynamic, fluctuating regionally and globally,55 in growth cones,68 in dendrites,52 during axogenesis,63 during mitochondrial transport,53 and in response to synaptic activity,53 and growth factors and guidance cues.68 We extended these findings by showing Δψm also is dynamic in RGCs with differing intrinsic neurite growth potentials.
Altered mitochondrial polarization correlated with differences in embryonic and postnatal bioenergetics. Regenerating axons appear to rely less on OXPHOS and more on glycolysis, analogous to the low oxygen environment during prenatal development.69 This may favor embryonic RGCs and explain the variable Δψm in postnatal RGCs that may be under stress adjusting from OXPHOS to glycolysis to meet the metabolic demands required for neurite regeneration.70,71 Postnatal RGCs exhibited increased respiratory capacity and reduced glycolytic capacity compared to embryonic RGCs, indicating a switch from glycolysis to OXPHOS as the main source of metabolic energy in postnatal RGCs. A switch back to glycolysis, that is the Warburg effect,72 may be necessary to support regenerative neurite growth. In nonneuronal cells, the regulation of glycolysis and OXPHOS is critical to proper progenitor proliferation and biosynthesis, independent of ATP production,73 indicating neurite growth signaling regulated by the balance between glycolysis and OXPHOS warrants further study.
Third, inhibiting DRP-1–mediated mitochondrial fission reversibly suppressed neurite growth rate and lamellar protrusion in RGCs, suggesting mitochondrial fission/fusion dynamics influence cytoskeletal remodeling regulating lamellar protrusion. The temporal and spatial regulation of axon growth rate and growth cone motility is fundamental to patterning in the nervous system,74 neuroplasticity in learning and memory,75 and regeneration.76 Though many cytoskeletal mechanisms underlying neurite growth77,78 and growth cone protrusive initiations44,79 have been described, the control over neurite growth rate, and the location, rate, and turnover of lamellar protrusions in growth cones is unclear. Recently, we showed that altering signaling endosome localization can alter neurite growth rate, and lamellar and filopodial protrusion and turnover.45 Here, we extended those observations by showing that mitochondrial dynamics also may modulate the mechanisms regulating neurite growth rate and lamellar protrusion, supporting the idea that organelle organization and dynamics define subdomains that regulate locally cytoskeletal dynamics underlying axon growth and growth cone protrusive activity. Like signaling endosomes,80 mitochondria also regulate intracellular signaling pathways, including Ca2+ and cAMP signaling, that can regulate neurite growth rate and lamellar protrusion.81,82 Thus, altering mitochondrial organization may alter locally second messenger signaling regulating neurite growth rate and cytoskeletal assembly in growth cones.
Finally, altering mitochondrial fission/fusion dynamics altered RGC growth cone steering responses to guidance cues that steer RGC axons in vitro and in vivo. Growth cones guide axons to their targets by altering their motility in response to intrinsic and extrinsic factors in the developing embryo.83 However, though progress has been made on the temporal and spatial control of second messenger transients during growth cone turning,84,85 how growth cones integrate information from multiple second messengers simultaneously to effect changes in axon growth remains unclear. Here, we showed that mitochondrial fission/fusion dynamics may regulate growth cone signaling generally, since altering mitochondrial dynamics altered growth cone turning responses to inhibitory, CSPG, and permissive, netrin-1, and fibronectin guidance cues. Mitochondria are prominent in all growth cones in vitro86 and in vivo87 during development88 and regeneration,64 and thus mitochondria are positioned temporally and spatially to modulate or integrate signals from multiple sources.
Dysfunctional mitochondrial dynamics and reduced bioenergetics are linked to impaired neuroplasticity and neuronal degeneration in Alzheimer's disease,89 Parkinson's disease,70 diseases of the visual system,90 psychiatric disorders,91 and stroke.92 The development of therapeutics aimed to enhance mitochondrial function in these disorders is a persistent challenge. Thus, understanding how mitochondrial dynamics regulate neurite growth and local protrusive activity may reveal novel strategies for developing therapeutics to treat diseases and injury in the CNS.
Acknowledgments
Vance Lemmon provided CSPG and micro-fluidic stamps; Eleut Hernandez and Kristina Russano provided excellence in animal husbandry; George McNamara and Gabriel Gaidosh provided expert microscopy, and Oliver Umland provided flow cytometry.
Footnotes
Supported by NIH Grants R01-EY020297 (JLG), P30-EY014801 (University of Miami), and NRSA T32-NS007044 (MBS); the American Health Assistance Foundation; a Research to Prevent Blindness Medical Student Fellowship (JEW); as well as an unrestricted grant from Research to Prevent Blindness to the University of Miami. JLG is the Walter G. Ross Distinguished Chair in Ophthalmic Research.
Disclosure: M.B. Steketee, None; S.N. Moysidis, None; J.E. Weinstein, None; A. Kreymerman, None; J.P. Silva, None; S. Iqbal, None; J.L. Goldberg, None.
References
- 1.Goldberg JL, Klassen MP, Hua Y, Barres BA. Amacrine-signaled loss of intrinsic axon growth ability by retinal ganglion cells. Science. 2002;296:1860–1864 [DOI] [PubMed] [Google Scholar]
- 2.Blackmore M, Letourneau PC. Changes within maturing neurons limit axonal regeneration in the developing spinal cord. J Neurobiol. 2006;66:348–360 [DOI] [PubMed] [Google Scholar]
- 3.Chen DF, Jhaveri S, Schneider GE. Intrinsic changes in developing retinal neurons result in regenerative failure of their axons. Proc Natl Acad Sci U S A. 1995;92:7287–7291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dusart I, Airaksinen MS, Sotelo C. Purkinje cell survival and axonal regeneration are age dependent: an in vitro study. J Neurosci. 1997;17:3710–3726 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou LJ, Ren WJ, Zhong Y, et al. Limited BDNF contributes to the failure of injury to skin afferents to produce a neuropathic pain condition. Pain. 2010;148:148–157 [DOI] [PubMed] [Google Scholar]
- 6.Gordon T, Gordon K. Nerve regeneration in the peripheral nervous system versus the central nervous system and the relevance to speech and hearing after nerve injuries. J Commun Disord. 2010;43:274–285 [DOI] [PubMed] [Google Scholar]
- 7.Mansour-Robaey S, Clarke DB, Wang YC, Bray GM, Aguayo AJ. Effects of ocular injury and administration of brain-derived neurotrophic factor on survival and regrowth of axotomized retinal ganglion cells. Proc Natl Acad Sci U S A. 1994;91:1632–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Nash M, Pribiag H, Fournier AE, Jacobson C. Central nervous system regeneration inhibitors and their intracellular substrates. Mol Neurobiol. 2009;40:224–235 [DOI] [PubMed] [Google Scholar]
- 9.Yiu G, He Z. Glial inhibition of CNS axon regeneration. Nat Rev Neurosci. 2006;7:617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cafferty WB, Duffy P, Huebner E, Strittmatter SM. MAG and OMgp synergize with Nogo-A to restrict axonal growth and neurological recovery after spinal cord trauma. J Neurosci. 2010;30:6825–6837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang R, Han M, Zheng B, Li YJ, Shu YN, Wen JK. Kruppel-like factor 4 interacts with p300 to activate mitofusin 2 gene expression induced by all-trans retinoic acid in VSMCs. Acta Pharmacol Sin. 2010;31:1293–1302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Laub F, Lei L, Sumiyoshi H, et al. Transcription factor KLF7 is important for neuronal morphogenesis in selected regions of the nervous system. Mol Cell Biol. 2005;25:5699–5711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson's disease caused by mutations in PINK1. Science. 2004;304:1158–1160 [DOI] [PubMed] [Google Scholar]
- 14.Thomas KJ, Cookson MR. The role of PTEN-induced kinase 1 in mitochondrial dysfunction and dynamics. Int J Biochem Cell Biol. 2009;41:2025–2035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weihofen A, Thomas KJ, Ostaszewski BL, Cookson MR, Selkoe DJ. Pink1 forms a multiprotein complex with Miro and Milton, linking Pink1 function to mitochondrial trafficking. Biochemistry. 2009;48:2045–2052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ramanathan A, Schreiber SL. Direct control of mitochondrial function by mTOR. Proc Natl Acad Sci U S A. 2009;106:22229–22232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Markham A, Cameron I, Franklin P, Spedding M. BDNF increases rat brain mitochondrial respiratory coupling at complex I, but not complex II. Eur J Neurosci. 2004;20:1189–1196 [DOI] [PubMed] [Google Scholar]
- 18.Aguiar AS Jr, Tuon T, Pinho CA, et al. Intense exercise induces mitochondrial dysfunction in mice brain. Neurochem Res. 2008;33:51–58 [DOI] [PubMed] [Google Scholar]
- 19.Chada SR, Hollenbeck PJ. Nerve growth factor signaling regulates motility and docking of axonal mitochondria. Curr Biol. 2004;14:1272–1276 [DOI] [PubMed] [Google Scholar]
- 20.Chada SR, Hollenbeck PJ. Mitochondrial movement and positioning in axons: the role of growth factor signaling. J Exp Biol. 2003;206:1985–1992 [DOI] [PubMed] [Google Scholar]
- 21.Lee CW, Peng HB. Mitochondrial clustering at the vertebrate neuromuscular junction during presynaptic differentiation. J Neurobiol. 2006;66:522–536 [DOI] [PubMed] [Google Scholar]
- 22.Hu WH, Hausmann ON, Yan MS, Walters WM, Wong PK, Bethea JR. Identification and characterization of a novel Nogo-interacting mitochondrial protein (NIMP). J Neurochem. 2002;81:36–45 [DOI] [PubMed] [Google Scholar]
- 23.Mann F, Rougon G. Mechanisms of axon guidance: membrane dynamics and axonal transport in semaphorin signalling. J Neurochem. 2007;102:316–323 [DOI] [PubMed] [Google Scholar]
- 24.Fan J, Mansfield SG, Redmond T, Gordon-Weeks PR, Raper JA. The organization of F-actin and microtubules in growth cones exposed to a brain-derived collapsing factor. J Cell Biol. 1993;121:867–878 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee JK, Geoffroy CG, Chan AF, et al. Assessing spinal axon regeneration and sprouting in Nogo-, MAG-, and OMgp-deficient mice. Neuron. 2010;66:663–670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Giger RJ, Hollis ER II, Tuszynski MH. Guidance molecules in axon regeneration. Cold Spring Harb Perspect Biol. 2010;2:a001867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sandvig A, Berry M, Barrett LB, Butt A, Logan A. Myelin-, reactive glia-, and scar-derived CNS axon growth inhibitors: expression, receptor signaling, and correlation with axon regeneration. Glia. 2004;46:225–251 [DOI] [PubMed] [Google Scholar]
- 28.Youle RJ, Karbowski M. Mitochondrial fission in apoptosis. Nat Rev Mol Cell Biol. 2005;6:657–663 [DOI] [PubMed] [Google Scholar]
- 29.Ventura-Clapier R, Garnier A, Veksler V. Transcriptional control of mitochondrial biogenesis: the central role of PGC-1alpha. Cardiovasc Res. 2008;79:208–217 [DOI] [PubMed] [Google Scholar]
- 30.Chan DC. Dissecting mitochondrial fusion. Dev Cell. 2006;11:592–594 [DOI] [PubMed] [Google Scholar]
- 31.Wakabayashi J, Zhang Z, Wakabayashi N, et al. The dynamin-related GTPase Drp1 is required for embryonic and brain development in mice. J Cell Biol. 2009;186:805–816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Detmer SA, Chan DC. Complementation between mouse Mfn1 and Mfn-2 protects mitochondrial fusion defects caused by CMT2A disease mutations. J Cell Biol. 2007;176:405–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Alavi MV, Bette S, Schimpf S, et al. A splice site mutation in the murine Opa1 gene features pathology of autosomal dominant optic atrophy. Brain. 2007;130:1029–1042 [DOI] [PubMed] [Google Scholar]
- 34.Chen H, Chan DC. Mitochondrial dynamics--fusion, fission, movement, and mitophagy--in neurodegenerative diseases. Hum Mol Genet. 2009;18:R169–R176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Carelli V, Ross-Cisneros FN, Sadun AA. Mitochondrial dysfunction as a cause of optic neuropathies. Prog Retin Eye Res. 2004;23:53–89 [DOI] [PubMed] [Google Scholar]
- 36.Yu-Wai-Man P, Bailie M, Atawan A, Chinnery PF, Griffiths PG. Pattern of retinal ganglion cell loss in dominant optic atrophy due to OPA1 mutations. Eye (Lond). 2011;25:596–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Morris RL, Hollenbeck PJ. The regulation of bidirectional mitochondrial transport is coordinated with axonal outgrowth. J Cell Sci. 1993;104:917–927 [DOI] [PubMed] [Google Scholar]
- 38.Gordon-Weeks PR. Neuronal Growth Cones. New York: Cambridge University Press; 2000:260 [Google Scholar]
- 39.MacAskill AF, Kittler JT. Control of mitochondrial transport and localization in neurons. Trends Cell Biol. 2010;20:102–112 [DOI] [PubMed] [Google Scholar]
- 40.Rui Y, Tiwari P, Xie Z, Zheng JQ. Acute impairment of mitochondrial trafficking by beta-amyloid peptides in hippocampal neurons. J Neurosci. 2006;26:10480–10487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Barres BA, Silverstein BE, Corey DP, Chun LL. Immunological, morphological, and electrophysiological variation among retinal ganglion cells purified by panning. Neuron. 1988;1:791–803 [DOI] [PubMed] [Google Scholar]
- 42.Hu Y, Cho S, Goldberg JL. Neurotrophic effect of a novel TrkB agonist on retinal ganglion cells. Invest Ophthalmol Vis Sci. 2010;51:1747–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chazotte B. Labeling mitochondria with JC-1. Cold Spring Harb Protoc. 2011;pii: pdb.prot065490. doi: 10.1101/pdb.prot065490 [DOI] [PubMed] [Google Scholar]
- 44.Steketee M, Balazovich K, Tosney KW. Filopodial initiation and a novel filament-organizing center, the focal ring. Mol Biol Cell. 2001;12:2378–2395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Steketee MB, Moysidis SN, Jin XL, et al. Nanoparticle-mediated signaling endosome localization regulates growth cone motility and neurite growth. Proc Natl Acad Sci U S A. 2011;108:19042–19047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Steketee MB, Tosney KW. Contact with isolated sclerotome cells steers sensory growth cones by altering distinct elements of extension. J Neurosci. 1999;19:3495–3506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lentz SI, Edwards JL, Backus C, et al. Mitochondrial DNA (mtDNA) biogenesis: visualization and duel incorporation of BrdU and EdU into newly synthesized mtDNA in vitro. J Histochem Cytochem. 2009;58:207–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Knoll B, Weinl C, Nordheim A, Bonhoeffer F. Stripe assay to examine axonal guidance and cell migration. Nat Protoc. 2007;2:1216–1224 [DOI] [PubMed] [Google Scholar]
- 49.Salvioli S, Ardizzoni A, Franceschi C, Cossarizza A. JC-1, but not DiOC6(3) or rhodamine 123, is a reliable fluorescent probe to assess delta psi changes in intact cells: implications for studies on mitochondrial functionality during apoptosis. FEBS Lett. 1997;411:77–82 [DOI] [PubMed] [Google Scholar]
- 50.Nicholls DG, Budd SL. Mitochondria and neuronal survival. Physiol Rev. 2000;80:315–360 [DOI] [PubMed] [Google Scholar]
- 51.Pilatus U, Aboagye E, Artemov D, Mori N, Ackerstaff E, Bhujwalla ZM. Real-time measurements of cellular oxygen consumption, pH, and energy metabolism using nuclear magnetic resonance spectroscopy. Magn Reson Med. 2001;45:749–755 [DOI] [PubMed] [Google Scholar]
- 52.Overly CC, Rieff HI, Hollenbeck PJ. Organelle motility and metabolism in axons vs dendrites of cultured hippocampal neurons. J Cell Sci. 1996;109:971–980 [DOI] [PubMed] [Google Scholar]
- 53.Miller KE, Sheetz MP. Axonal mitochondrial transport and potential are correlated. J Cell Sci. 2004;117:2791–2804 [DOI] [PubMed] [Google Scholar]
- 54.Hillefors M, Gioio AE, Mameza MG, Kaplan BB. Axon viability and mitochondrial function are dependent on local protein synthesis in sympathetic neurons. Cell Mol Neurobiol. 2007;27:701–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Keil VC, Funke F, Zeug A, Schild D, Muller M. Ratiometric high-resolution imaging of JC-1 fluorescence reveals the subcellular heterogeneity of astrocytic mitochondria. Pflugers Arch. 2011;462:693–708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Steketee MB, Tosney KW. Three functionally distinct adhesions in filopodia: shaft adhesions control lamellar extension. J Neurosci. 2002;22:8071–8083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Moore DL, Blackmore MG, Hu Y, et al. KLF family members regulate intrinsic axon regeneration ability. Science. 2009;326:298–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Amiri M, Hollenbeck PJ. Mitochondrial biogenesis in the axons of vertebrate peripheral neurons. Dev Neurobiol. 2008;68:1348–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vincent AM, Edwards JL, McLean LL, et al. Mitochondrial biogenesis and fission in axons in cell culture and animal models of diabetic neuropathy. Acta Neuropathol. 2010;120:477–489 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tanaka A, Youle RJ. A chemical inhibitor of DRP1 uncouples mitochondrial fission and apoptosis. Mol Cell. 2008;29:409–410 [DOI] [PubMed] [Google Scholar]
- 61.Cassidy-Stone A, Chipuk JE, Ingerman E, et al. Chemical inhibition of the mitochondrial division dynamin reveals its role in Bax/Bak-dependent mitochondrial outer membrane permeabilization. Dev Cell. 2008;14:193–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ruthel G, Hollenbeck PJ. Response of mitochondrial traffic to axon determination and differential branch growth. J Neurosci. 2003;23:8618–8624 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dedov VN, Dedova IV, Armati PJ. Transport of mitochondria during axonogenesis. IUBMB Life. 2000;49:549–552 [DOI] [PubMed] [Google Scholar]
- 64.Dedov VN, Armati PJ, Roufogalis BD. Three-dimensional organisation of mitochondrial clusters in regenerating dorsal root ganglion (DRG) neurons from neonatal rats: evidence for mobile mitochondrial pools. J Peripher Nerv Syst. 2000;5:3–10 [DOI] [PubMed] [Google Scholar]
- 65.Prigione A, Adjaye J. Modulation of mitochondrial biogenesis and bioenergetic metabolism upon in vitro and in vivo differentiation of human ES and iPS cells. Int J Devl Biol. 2010;54:1729–1741 [DOI] [PubMed] [Google Scholar]
- 66.Kondoh H, Lleonart ME, Nakashima Y, et al. A high glycolytic flux supports the proliferative potential of murine embryonic stem cells. Antioxid Redox Signal. 2007;9:293–299 [DOI] [PubMed] [Google Scholar]
- 67.Chen H, Vermulst M, Wang YE, et al. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Verburg J, Hollenbeck PJ. Mitochondrial membrane potential in axons increases with local nerve growth factor or semaphorin signaling. J Neurosci. 2008;28:8306–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Rust RS. Energy metabolism of developing brain. Curr Opin Neurol. 1994;7:160–165 [DOI] [PubMed] [Google Scholar]
- 70.Henchcliffe C, Beal MF. Mitochondrial biology and oxidative stress in Parkinson disease pathogenesis. Nat Clin Pract Neurol. 2008;4:600–609 [DOI] [PubMed] [Google Scholar]
- 71.Diaz G, Setzu MD, Zucca A, et al. Subcellular heterogeneity of mitochondrial membrane potential: relationship with organelle distribution and intercellular contacts in normal, hypoxic and apoptotic cells. J Cell Sci. 1999;112:1077–1084 [DOI] [PubMed] [Google Scholar]
- 72.Warburg O. On the origin of cancer cells. Science. 1956;123:309–314 [DOI] [PubMed] [Google Scholar]
- 73.Agathocleous M, Love NK, Randlett O, et al. Metabolic differentiation in the embryonic retina. Nat Cell Biol. 2012;14:859–864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Butler SJ, Tear G. Getting axons onto the right path: the role of transcription factors in axon guidance. Development. 2007;134:439–448 [DOI] [PubMed] [Google Scholar]
- 75.Gutierrez H, Davies AM. Regulation of neural process growth, elaboration and structural plasticity by NF-kappaB. Trends Neurosci. 2011;34:316–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hur EM, Yang IH, Kim DH, et al. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc Natl Acad Sci U S A. 2011;108:5057–5062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Jung H, O'Hare CM, Holt CE. Translational regulation in growth cones. Curr Opin Genet Dev. 2011;21:458–464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Dent EW, Gertler FB. Cytoskeletal dynamics and transport in growth cone motility and axon guidance. Neuron. 2003;40:209–227 [DOI] [PubMed] [Google Scholar]
- 79.Spillane M, Ketschek A, Jones SL, et al. The actin nucleating Arp2/3 complex contributes to the formation of axonal filopodia and branches through the regulation of actin patch precursors to filopodia. Dev Neurobiol. 2011;71:747–758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Cosker KE, Courchesne SL, Segal RA. Action in the axon: generation and transport of signaling endosomes. Curr Opin Neurobiol. 2008;18:270–275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Zhang XF, Forscher P. Rac1 modulates stimulus-evoked Ca(2+) release in neuronal growth cones via parallel effects on microtubule/endoplasmic reticulum dynamics and reactive oxygen species production. Mol Biol Cell. 2009;20:3700–3712 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Tojima T, Hines JH, Henley JR, Kamiguchi H. Second messengers and membrane trafficking direct and organize growth cone steering. Nat Rev Neurosci. 2011;12:191–203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kalil K, Dent EW. Touch and go: guidance cues signal to the growth cone cytoskeleton. Curr Opin Neurobiol. 2005;15:521–526 [DOI] [PubMed] [Google Scholar]
- 84.Nicol X, Hong KP, Spitzer NC. Spatial and temporal second messenger codes for growth cone turning. Proc Natl Acad Sci U S A. 2011;108:13776–13781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Mai J, Fok L, Gao H, Zhang X, Poo MM. Axon initiation and growth cone turning on bound protein gradients. J Neurosci. 2009;29:7450–7458 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Morris RL, Hollenbeck PJ. Axonal transport of mitochondria along microtubules and F-actin in living vertebrate neurons. J Cell Biol. 1995;131:1315–1326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Benshalom G, Reese TS. Ultrastructural observations on the cytoarchitecture of axons processed by rapid-freezing and freeze-substitution. J Neurocytol. 1985;14:943–960 [DOI] [PubMed] [Google Scholar]
- 88.Tennyson VM. The fine structure of the axon and growth cone of the dorsal root neuroblast of the rabbit embryo. J Cell Biol. 1970;44:62–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Moreira PI, Cardoso SM, Santos MS, Oliveira CR. The key role of mitochondria in Alzheimer's disease. J Alzheimers Dis. 2006;9:101–110 [DOI] [PubMed] [Google Scholar]
- 90.Yu-Wai-Man P, Griffiths PG, Chinnery PF. Mitochondrial optic neuropathies -- disease mechanisms and therapeutic strategies. Prog Retin Eye Res. 2011;30:81–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron. 2008;60:748–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Jung JE, Kim GS, Chen H, et al. Reperfusion and neurovascular dysfunction in stroke: from basic mechanisms to potential strategies for neuroprotection. Mol Neurobiol. 2010;41:172–179 [DOI] [PMC free article] [PubMed] [Google Scholar]