

Abstract
Dysregulation of cholesterol homeostasis in the brain is increasingly being linked to chronic neurodegenerative disorders, including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), Niemann-Pick type C (NPC) disease and Smith-Lemli Opitz syndrome (SLOS). However, the molecular mechanisms underlying the correlation between altered cholesterol metabolism and the neurological deficits are, for the most part, not clear. NPC disease and SLOS are caused by mutations in genes involved in the biosynthesis or intracellular trafficking of cholesterol, respectively. However, the types of neurological impairments, and the areas of the brain that are most affected, differ between these diseases. Some, but not all, studies indicate that high levels of plasma cholesterol correlate with increased risk of developing AD. Moreover, inheritance of the E4 isoform of apolipoprotein E (APOE), a cholesterol-carrying protein, markedly increases the risk of developing AD. Whether or not treatment of AD with statins is beneficial remains controversial, and any benefit of statin treatment might be due to anti-inflammatory properties of the drug. Cholesterol balance is also altered in HD and PD, although no causal link between dysregulated cholesterol homeostasis and neurodegeneration has been established. Some important considerations for treatment of neurodegenerative diseases are the impermeability of the blood-brain barrier to many therapeutic agents and difficulties in reversing brain damage that has already occurred. This article focuses on how cholesterol balance in the brain is altered in several neurodegenerative diseases, and discusses some commonalities and differences among the diseases.
Introduction
Dysregulation of cholesterol homeostasis in the brain has been linked to chronic neurodegenerative disorders, including Alzheimer’s disease (AD), Huntington’s disease (HD), Parkinson’s disease (PD), Niemann-Pick type C (NPC) disease and Smith-Lemli Opitz syndrome (SLOS), as well as to acute neuronal injuries such as stroke and brain trauma. Thus, the mechanisms underlying the association between altered cholesterol metabolism and neurodegeneration are being actively investigated, particularly in mouse models of these diseases. This article focuses on how normal cholesterol balance is maintained in the brain, and how this balance is altered in these disorders. In addition, some commonalities and differences in the dysregulation of cholesterol homeostasis in the brain in these neurodegenerative diseases are discussed.
Cholesterol dynamics in the brain
Distribution
In mammalian cells, cholesterol is synthesized from the two-carbon molecule acetyl-CoA via a complex pathway that involves over 30 enzymatic steps (summarized in Fig. 1). Cholesterol is required by mammals for the synthesis of steroid hormones and bile acids, for the organization of cell membranes, and for the formation and maintenance of lipid rafts, which are implicated in many aspects of brain function such as growth factor signaling, axon guidance and synaptic transmission. Thus, a deficiency or excess of cholesterol in the brain might be expected to have profound consequences. The biosynthesis of cholesterol is tightly regulated by the abundance of cholesterol (Brown and Goldstein, 1986). In tissues outside the central nervous system (CNS), cholesterol is acquired both from endogenous synthesis and from exogenous lipoproteins delivered from the circulation. However, because plasma lipoproteins do not cross the intact blood-brain barrier, nearly all cholesterol in the brain is synthesized in situ (Dietschy and Turley, 2001). This compartmentalization of cholesterol metabolism in the body explains why cholesterol homeostasis in the CNS is regulated independently of that in the peripheral circulation.
Fig. 1.

Cholesterol biosynthesis. Cholesterol is synthesized from acetyl-CoA. A key intermediate in the pathway, mevalonic acid, is produced from 3-hydroxy-3-methylglutaryl-CoA (HMG-CoA) by the rate-limiting enzyme of the pathway, HMG-CoA reductase; this enzyme is inhibited by the cholesterol-lowering drugs, the statins. In the final step of the pathway, 7-dehydrocholesterol is converted to cholesterol by the enzyme 7-dehydrocholesterol reductase, the defective enzyme in SLOS. Hydroxylated derivatives of cholesterol, such as 24-, 25- and 27-hydroxycholesterol, are produced from cholesterol by specific hydroxylases, such as CYP46.
Although the brain makes up only 2–5% of body mass, approximately 25% of total body cholesterol resides in the brain (Dietschy and Turley, 2001). Thus, the brain is highly enriched in cholesterol compared with other mammalian tissues: whereas the cholesterol concentration in most animal tissues is ∼2 mg/g tissue, the cholesterol concentration in the CNS is 15–20 mg/g tissue (Dietschy and Turley, 2004). The majority (70–90%) of cholesterol in the CNS is in the myelin that surrounds axons and facilitates the transmission of electrical signals. Consequently, cholesterol synthesis in the brain is highest in oligodendrocytes during active myelination and decreases by ∼90% in adults after myelination has been completed (Dietschy and Turley, 2004; Quan et al., 2003). Nevertheless, cholesterol synthesis continues at a low rate in the mature brain, particularly in astrocytes; in the adult brain, the rate of cholesterol biosynthesis is higher in astrocytes than in neurons (Nieweg et al., 2009).
Transport
The brain operates its own lipoprotein transport system, independent of that in the peripheral circulation (Fig. 2). Astrocytes produce cholesterol and apolipoprotein E (APOE) that, together with phospholipids, generate lipoproteins that are similar in size to plasma high-density lipoproteins (Boyles et al., 1985). The secreted APOE acquires cholesterol and phospholipids via the efflux of cellular lipid in a process mediated by one or more of the ATP-binding cassette (ABC) transporters such as ABCA1, ABCG1 and/or ABCG4. The uptake of these lipoproteins by neurons is mediated by receptors of the low-density lipoprotein (LDL) receptor family, such as the LDL receptor, LDL-receptor-related protein (LRP) and APOE receptor 2 (APOER2), that are expressed in neurons and can endocytose the astrocyte-derived APOE-containing lipoprotein particles (Boyles et al., 1989; Herz, 2001b; Posse de Chaves et al., 2000) (Fig. 2). In this manner, cholesterol is shuttled from astrocytes to neurons (Mauch et al., 2001; Michikawa et al., 2000; Vance and Hayashi, 2010). The interaction between APOE-containing lipoproteins and these neuronal receptors seems to be crucial for normal neuronal function: the APOE-containing lipoproteins stimulate synaptogenesis (Mauch et al., 2001), enhance axonal growth (Hayashi et al., 2004) and prevent neuron death (Hayashi et al., 2009; Hayashi et al., 2007). Moreover, a role for APOE in nerve repair has been indicated, because APOE synthesis in glial cells increases by up to 150-fold after a nerve injury (Ignatius et al., 1986). APOE therefore seems to be a key player in regulating cholesterol homeostasis and distribution among cells of the brain.
Fig. 2.

APOE-containing lipoproteins transport cholesterol from astrocytes to neurons. Glial cells, primarily astrocytes, but also microglia, secrete APOE, which acquires cholesterol and phospholipids, thereby forming APOE-containing lipoproteins. These are delivered to neurons where they are endocytosed via cell surface receptors (members of the LDL receptor family). Consequently, cholesterol is delivered to the neurons. Some APOE receptors also function as signaling receptors.
Cholesterol has a remarkably long half-life in the brain (4–6 months in rodents and up to 5 years in humans) (Dietschy and Turley, 2001). There is a low rate of cholesterol synthesis in the adult brain, and cholesterol cannot be degraded in the CNS, but a steady-state level of cholesterol is maintained in the CNS because a small fraction (0.02–0.4%) of the cholesterol pool is excreted from the brain each day (Dietschy and Turley, 2004). The conversion of cholesterol to 24-hydroxycholesterol (Fig. 1), by the enzyme cholesterol 24-hydroxylase (CYP46) that is expressed in a subset of neurons (but not in astrocytes) (Russell et al., 2009), represents a major mechanism by which excess cholesterol is eliminated from the brain. In contrast to cholesterol, 24-hydroxycholesterol can cross the blood-brain barrier, enter the peripheral circulation and be eliminated from the body in bile (Russell et al., 2009). Studies in CYP46-deficient mice show that at least 40% of the cholesterol that is excreted from the brain is in the form of 24-hydroxycholesterol. Interestingly, however, in CYP46-deficient mice, cholesterol does not accumulate because cholesterol synthesis is reduced by ∼40% as a compensatory mechanism (Russell et al., 2009).
Cholesterol and NPC disease
A direct association between impaired cholesterol metabolism in the brain and neurodegeneration has been clearly demonstrated in NPC disease. This disease is a relatively rare (1/150,000 live births) autosomal recessive inherited disorder that causes progressive neurodegeneration and premature death, and is often accompanied by hepatosplenomegaly and lung disease (Vanier and Millat, 2003). A characteristic histological feature of brains of individuals with NPC disease is a massive loss of neurons, particularly Purkinje cells in the cerebellum, consistent with the impairment of motor function in these individuals (Sarna et al., 2003), although neurons in other parts of the brain are probably also affected. NPC disease is caused by mutations in either the NPC1 or NPC2 gene. Because the NPC1 and NPC2 proteins are ubiquitously expressed in animal tissues, it is not clear why the brain is the most severely affected tissue in NPC disease. NPC1 and NPC2 each bind to cholesterol and act in tandem in late endosomes and/or lysosomes to mediate the egress of unesterified cholesterol derived from endocytosed lipoproteins (Fig. 3) (Vance, 2010; Wang et al., 2010). Consequently, in NPC1- or NPC2-deficient cells, including neurons (Karten et al., 2002) and glial cells (Karten et al., 2005; Peake et al., 2011), unesterified cholesterol and other lipids become sequestered in late endosomes and/or lysosomes. Accordingly, the amount of cholesterol in the plasma membrane and endoplasmic reticulum (the cellular site at which cholesterol homeostasis is regulated) is reduced (Liscum et al., 1989). In Npc1−/− neurons, this defect in cholesterol export from late endosomes and/or lysosomes results in a higher-than-normal cholesterol content of neuronal cell bodies and a reduced cholesterol content of distal axons (Karten et al., 2002; Karten et al., 2003). It is possible, therefore, that some of the neurological deficits in NPC disease might be attributable to a deficiency, rather than an excess, of cholesterol in axons. Consistent with this idea, NPC1 and NPC2 are present in recycling endosomes in pre-synaptic nerve terminals, and synaptic vesicle morphology and composition are altered by NPC1 deficiency (Karten et al., 2006). Furthermore, reduction in the amount of cholesterol in cultured Npc1−/− neurons attenuates the exocytosis of synaptic vesicles, so that synaptic function is likely to be impaired in NPC disease (Hawes et al., 2010).
Fig. 3.

NPC1 and NPC2 mediate cholesterol export from late endosomes and/or lysosomes. Cholesterol (blue oval) is released from endocytosed LDLs and binds to NPC2 (red), a soluble protein in the lysosomal lumen.
Subsequently, NPC2 directly transfers the cholesterol to NPC1 (yellow), located in the limiting membrane of the lysosome, so that the hydrophobic cholesterol molecule does not have to travel through the aqueous milieu of the lysosomal lumen. Ultimately, the cholesterol is exported from the lysosomal membrane and is transported via the cytosol to the endoplasmic reticulum and plasma membrane by unknown mechanisms.
As is the case in many other neurodegenerative disorders, neuroinflammation is pronounced in the brains of both individuals with NPC disease and mouse models of the disease. Microglia, the resident immune cells of the brain, were proposed to contribute to the neurodegeneration because microglial activation was observed prior to any evidence of neurodegeneration (Baudry et al., 2003). However, recent studies in cultured cells and in cell-type-specific NPC1 knockout mice have demonstrated that the primary cause of neurodegeneration in NPC disease is a deficiency of NPC1 specifically in neurons, rather than in astrocytes or microglia (Lopez et al., 2011; Peake et al., 2011; Yu et al., 2011).
No effective treatment is currently available for NPC disease. In addition to the sequestration of unesterified cholesterol, glycosphingolipids also accumulate in late endosomes and/or lysosomes of NPC-deficient cells. Therefore, substrate reduction therapy using miglustat, an inhibitor of glycosphingolipid synthesis, is currently being tested in individuals with NPC disease, but with limited success. In an exciting development, however, recent experiments have suggested a novel therapeutic approach for the treatment of NPC disease. A single subcutaneous injection of the cholesterol-binding compound cyclodextrin into 7-day-old Npc1−/− mice significantly slowed the neurodegeneration and extended the lifespan of the mice by ∼50% (Liu et al., 2009). Although cyclodextrin does not cross the blood-brain barrier efficiently, small amounts of cyclodextrin can enter the brain from the peripheral circulation. High doses of cyclodextrin (5–10 mM) are commonly used experimentally to extract cholesterol from cell membranes, but these doses are highly toxic to mouse neurons (Peake et al., 2011). In contrast, lower doses of cyclodextrin (for example ∼0.1 mM) seem not to be neurotoxic but are endocytosed by Npc1−/− cells and release cholesterol trapped in late endosomes and/or lysosomes without extracting significant amounts of cholesterol from the cells (Aqul et al., 2011; Rosenbaum et al., 2010). Consequently, the sequestered cholesterol is mobilized from late endosomes and/or lysosomes of NPC1-deficient cells and is transported to the endoplasmic reticulum (Radhakrishnan et al., 2008). Consistent with this mechanism, my laboratory has shown that 0.1 mM cyclodextrin mobilizes stored cholesterol from late endosomes and/or lysosomes of Npc1−/− neurons, astrocytes and microglia, and normalizes cholesterol homeostasis at the endoplasmic reticulum (Peake and Vance, 2012). A similar conclusion regarding the mechanism of action of cyclodextrin was also reached from studies in intact mice (Aqul et al., 2011). Moreover, the direct intra-thecal delivery of cyclodextrin into the CNS of Npc1−/− mice resulted in a concentration of cyclodextrin of 0.1–0.2 mM in the brain (Aqul et al., 2011), and this concentration produced the same beneficial effects on cholesterol homeostasis as we observed in neurons and glial cells isolated from Npc1−/− mice (Peake and Vance, 2012). Because cyclodextrin is relatively non-toxic, and has been approved as a drug delivery vehicle in humans, this compound is currently being used to treat a limited number of individuals with NPC disease.
Cholesterol and SLOS
Whereas NPC disease is caused by a defect in the intracellular trafficking of cholesterol, several other rare human diseases that cause neurodegeneration and developmental malformations are caused by direct impairment of the cholesterol biosynthetic pathway (reviewed by Porter, 2006; Porter and Herman, 2011). These diseases include lathosterolosis (a defect in 3β-hydroxysteroid-5 desaturase, which converts lathosterol to 7-dehydrocholesterol), desmosterolosis (a defect in reduction of the 24-double bond of the sterol side-chain of cholesterol by 3β-hydroxysterol-24 reductase), cerebrotendinous xanthomatosis [a defect in cholesterol 27-hydroxylase (reviewed by Björkhem and Hansson, 2010)] and congenital hemidysplasia with ichthyosiform erythroderma and limb defects (CHILD) syndrome (a defect in demethylation of the C-4 methyl group of lanosterol). SLOS is the most common of this class of disorders (∼1/20,000 live births) and was first described in 1964 (Smith et al., 1964). SLOS is an autosomal recessive, neurodegenerative and developmental disease caused by mutations in the gene encoding 7-dehydrocholesterol reductase, the enzyme that catalyzes the final step in the cholesterol biosynthetic pathway (Fig. 1). Mutations that reduce the activity of 7-dehydrocholesterol reductase cause elevations in the levels of 7-dehydrocholesterol and abnormally low levels of cholesterol in cells, plasma and the brain (reviewed by Porter, 2006; Porter and Herman, 2011). SLOS is characterized by multiple developmental abnormalities, such as distinctive facial features, limb malformations, microcephaly, cleft palate, polydactyly and holoprosencephaly, as well as severe intellectual impairment (reviewed by Porter, 2006; Porter and Herman, 2011).
Because plasma cholesterol levels are very low in individuals with SLOS, a treatment currently used for the disease is based on the idea of ‘normalizing’ the level of cholesterol in plasma by feeding affected individuals a cholesterol-rich diet (reviewed by Porter and Herman, 2011). Although studies in which this protocol was used involved only small numbers of patients, some anecdotal reports suggest that this treatment might be beneficial. It is not clear, however, why increasing the level of plasma cholesterol would improve the neurological phenotype, because plasma lipoproteins, and the cholesterol carried therein, do not cross the blood-brain barrier or enter the CNS (Dietschy and Turley, 2001). It is also unclear whether the developmental and neurological abnormalities in individuals with SLOS are caused by the low level of cholesterol in the brain or by the abnormal accumulation of the potentially toxic cholesterol precursor, 7-dehydrocholesterol. In support of the latter, mice that lack the cholesterol regulatory protein INSIG exhibit severe facial clefting and have developmental abnormalities that are very similar to those that occur in individuals with SLOS. Because INSIG knockout mice have uncontrolled, high rates of cholesterol synthesis, and a cellular build-up of cholesterol precursors such as 7-dehydrocholesterol, these data suggest that the accumulation of 7-dehydrocholesterol, rather than the deficiency of cholesterol, is the cause of at least some of the pathophysiological abnormalities in SLOS. The developmental abnormalities in individuals with SLOS might also be caused by reduced functioning of the sonic hedgehog (SHH) signaling pathway, which is required for normal embryonic development of the CNS, limbs and facial features (Maity et al., 2005). An important component of this pathway is the SHH protein, whose activity requires the covalent attachment of a cholesterol molecule to the protein (Mann and Beachy, 2000; Porter et al., 1996). The attenuation of cholesterol levels in individuals with SLOS and/or the increased amounts of 7-dehydrocholesterol might, therefore, reduce the activity of SHH, perhaps by altering membrane fluidity.
Cholesterol and AD
Cholesterol levels and AD
AD is a common progressive neurodegenerative disorder in which affected individuals suffer from memory loss and cognitive decline late in life. The brains of individuals with AD contain extracellular deposits of β-amyloid (Aβ) plaques, as well as intracellular neurofibrillary tangles that contain hyperphosphorylated tau, a microtubule-associated protein. The accumulation of Aβ plaques and the loss of neurons, particularly in the hippocampus, are thought to be central events in the development of AD (Selkoe, 2001; Selkoe, 2002). The deposition of Aβ reflects the balance between the production and removal of Aβ peptides from the brain. Consequently, either overproduction or impaired clearance of Aβ, or a combination of both processes, probably plays a key role in the pathophysiology of AD. The Aβ peptides are generated by the proteolytic cleavage of the transmembrane protein amyloid precursor protein (APP) (Fig. 4). lnitially, the APP protein is proteolytically cleaved by either α- or β-secretase. When APP is cleaved by α-secretase, non-amyloidogenic products are generated that do not cause abnormal brain pathology. In contrast, when the C-terminal fragment generated by β-secretase is further cleaved by γ-secretase, Aβ peptides are formed that contain 40 or 42 amino acids, and these peptides are deposited in the pathogenic Aβ plaques. Some of the mechanisms proposed for Aβ degradation are discussed below (reviewed by Grimm et al., 2012; Lee et al., 2012; Tan and Evin, 2012).
Fig. 4.
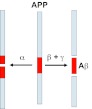
Processing of APP. In the non-amyloidogenic pathway of APP processing, APP is proteolytically cleaved within the Aβ region (red box) by α-secretase (α). In the amyloidogenic pathway, APP is cleaved first by β-secretase (β) and subsequently by γ-secretase (γ) to generate the pathological Aβ fragments that accumulate in brains of individuals with AD.
In contrast to NPC disease and SLOS, no direct causal relationship has been established between AD and the dysregulation of cholesterol metabolism. Thus, although some experimental evidence indicates that alterations in cholesterol metabolism in the brain might contribute to the pathogenesis of AD (Kojro et al., 2001; Simons et al., 1998), it is not clear whether the modification of cholesterol homeostasis in AD brains is a cause or consequence of the disease. The β- and γ-secretases that generate the Aβ peptides from APP are predominantly localized to cholesterol-enriched microdomains of the plasma membrane (Ehehalt et al., 2003; Simons et al., 1998). Several in vitro and in vivo experiments have demonstrated that the cellular concentration of cholesterol can regulate the production and amount of the Aβ peptides. For example, a decreased level of cellular cholesterol increases α-secretase cleavage of APP, thereby decreasing the processing of APP into the toxic Aβ peptides that accumulate in amyloid plaques (Björnsson et al., 1994; Bodovitz and Klein, 1996; Kojro et al., 2001; Simons et al., 1998). Interestingly, the extracellular N-terminus of APP contains a cholesterol-binding site (Barrett et al., 2012). Optimal brain function might also be compromised if the level of cholesterol in neurons drops below a threshold level, because when the cholesterol content of hippocampal neurons was decreased by statin treatment, synaptic density was reduced and synaptic vesicle release was impaired (Mailman et al., 2011).
Some (Jick et al., 2000; Wolozin et al., 2000), but not all (Fassbender et al., 2002; Longenberger and Shah, 2011; Shepardson et al., 2011b), epidemiological studies suggest that increased levels of plasma cholesterol, particularly during mid-life, are a risk factor for the development of AD (Leoni et al., 2010; Umeda et al., 2012). For example, a cholesterol-rich diet in rabbits increased Aβ production (Sparks et al., 1994), but the mechanism by which dietary cholesterol and high levels of plasma cholesterol could contribute to the deposition of Aβ plaques remains unclear, because plasma lipoproteins do not normally cross the blood-brain barrier (Dietschy and Turley, 2001). One suggested explanation for the association between high plasma levels of cholesterol and AD is that the impermeability of the blood-brain barrier might be compromised in individuals with AD so that cholesterol can be transported into the brain from the plasma (Ujiie et al., 2003). However, more recent studies have shown that the levels of plant sterols (the majority of which circulate in plasma) in the brains of individuals with AD are not significantly different from those in non-AD controls, suggesting that the blood-brain barrier remains intact in AD (Shafaati et al., 2011).
In support of the idea that high levels of plasma cholesterol contribute to AD, several studies have suggested that statins, some of which can cross the blood-brain barrier and are widely used cholesterol-lowering drugs, protect against AD (Fassbender et al., 2002; Shepardson et al., 2011a; Wolozin et al., 2000). However, randomized double-blind placebo-controlled studies have shown no beneficial effect of statins on the progression of symptoms in individuals with AD despite significantly lowering plasma cholesterol (Sano et al., 2011; Shepardson et al., 2011b). Clearly, additional studies are required to determine whether high plasma cholesterol directly contributes to the onset and progression of AD (Shepardson et al., 2011a; Shepardson et al., 2011b). One possible mechanism underlying the proposed neuroprotection against AD by statins might be attributable to the anti-inflammatory and/or antioxidant properties of the statins, rather than directly to their cholesterol-lowering effects (reviewed by Vaughan and Gotto, 2004). Thus, the idea that statins are beneficial in AD remains controversial.
APOE and AD
Despite the controversy surrounding the association between plasma cholesterol levels and AD, a well-established link between AD and cholesterol metabolism is the cholesterol-carrying protein APOE, which is highly expressed in the brain. ApoE−/− mice exhibit impaired clearance of degenerating nerves (Fagan et al., 1998), as well as learning defects (Masliah et al., 1995; Oitzl et al., 1997; Veinbergs et al., 1998), although nerve regeneration appears to be normal (Popko et al., 1993). As discussed above, and as depicted in Fig. 2, APOE is secreted by astrocytes and generates cholesterol-carrying lipoproteins that are delivered to, and endocytosed by, neurons. Low levels of APOE in the brain correlate with an increased risk of AD (Bales et al., 2009), but whether or not the cholesterol-carrying function of APOE is involved in AD remains unclear, as discussed above. APOE associates with Aβ peptides (Sanan et al., 1994), thereby enhancing their degradation. A recent study (Cramer et al., 2012) reported that the administration of bexarotene (an agonist of the RXR nuclear receptor) robustly increased the production of APOE, as well as the levels of APOE-containing lipoproteins, in the hippocampus and cortex of a mouse model of AD. Moreover, this agonist enhanced Aβ degradation and markedly reduced Aβ plaque area. Remarkably, bexarotene also rapidly improved memory and cognition in the AD mice. Importantly, none of these beneficial effects occurred in mice that lacked APOE. Thus, the benefits of bexarotene in AD mice were attributed to the increased amount of APOE in the brain and the role of APOE in stimulating Aβ degradation (Cramer et al., 2012). However, in contrast to this report, another recent study has suggested that reducing rather than increasing APOE levels reduces the amount of Aβ in the brain (Bien-Ly et al., 2012). Thus, whether or not the demonstrated benefit of bexarotene in mice can be translated into a treatment for humans with AD remains to be determined, particularly because mice express only one isoform of APOE, whereas humans express three common, alternative APOE isoforms (APOE2, APOE3 and APOE4).
The three human APOE isoforms differ from each other in only a single amino acid. The most common isoform allele is APOE3, whereas only ∼15% of the population carries the APOE4 allele. Importantly, inheritance of the APOE4 allele is the strongest known genetic risk factor for the development of late-onset AD (Corder et al., 1993). Thus, individuals who have a single copy of the APOE4 allele have ∼four times the likelihood of developing AD compared with carriers of the APOE3 allele, whereas inheritance of two APOE4 alleles increases the risk of developing AD by 12- to 20-fold (reviewed by Bu, 2009). In contrast, inheritance of the APOE2 allele seems to protect against AD (Corder et al., 1994). Although the association between the APOE4 isoform and the predisposition of individuals to AD is well established, the reason why APOE4 is a risk factor for AD is still not clear. One possible explanation is that APOE4 supports axonal growth less efficiently than does APOE3 (Hayashi et al., 2004; Nathan et al., 1994). Another possible link between the APOE genotype and AD is that APOE3-containing lipoproteins derived from astrocytes more strongly promote neuron survival than do APOE4-containing lipoproteins (Hayashi et al., 2007). Nevertheless, the most attractive hypothesis is probably that the clearance of Aβ deposits depends on the isoform of APOE, because APOE3 binds to Aβ peptides more strongly than does APOE4 (Castellano et al., 2011; Koistinaho et al., 2004; Strittmatter et al., 1993). APOE2 and APOE3, but not APOE4, form dimers that might contribute to the regulation of Aβ degradation (Xue et al., 2011), and APOE levels in the plasma and brain of humans carrying the APOE4 allele are lower than in APOE3 carriers (Gupta et al., 2011). In support of this finding, more amyloid is deposited in the brains of mice expressing human APOE4 compared with those expressing APOE3 or APOE2 (Bales et al., 2009; Castellano et al., 2011). Thus, APOE seems to regulate Aβ levels in an APOE-isoform-dependent manner, probably because of isoform-specific differences in the capacity of APOE to modulate Aβ degradation.
Association between AD and other genes of cholesterol metabolism
Several other genes involved in cholesterol metabolism and transport have been implicated as risk factors for AD. For example, polymorphisms in two receptors of the LDL receptor family – LRP and the APOER2 receptor, both of which are expressed in neurons (Herz, 2001a; Posse de Chaves et al., 1997) – have been linked to an increased incidence of AD (Elder et al., 2007; Fryer et al., 2005; Kim et al., 2009). APOE and the APOE-containing lipoproteins are ligands for this family of receptors, some of which function both in endocytosis and in signaling pathways (Beffert et al., 2004). LRP seems to participate in the uptake and clearance of Aβ, because these processes are decreased when LRP1 is eliminated in mouse forebrain neurons (reviewed by Bu, 2009).
The expression of the ABC transporter ABC1 has also been linked to AD, and to the regulation of cholesterol and APOE homeostasis in the brain (reviewed by Hirsch-Reinshagen and Wellington, 2007; Vance and Hayashi, 2010). ABCA1 is required for the normal acquisition of lipids by APOE, as demonstrated by the presence of lipid-poor APOE-containing lipoprotein particles in ABCA1 knockout mice (Hirsch-Reinshagen et al., 2004; Wahrle et al., 2004). In addition, the level of APOE in the brain was 80% lower in these mice than in wild-type control mice. When ABCA1 knockout mice were crossed with mouse models of AD, the lack of ABCA1 did not affect Aβ production. However, the poorly lipidated APOE particles that were generated in the ABCA1 knockout mice seemed to be responsible for increasing the amyloid load (Hirsch-Reinshagen et al., 2005; Wahrle et al., 2005). Consistent with these findings, overexpression of ABCA1 in AD mice reduced amyloid deposition (Wahrle et al., 2008).
Overall, the available data indicate that profound alterations in cholesterol metabolism occur in AD, but whether these changes are the cause of the neurodegeneration remains uncertain.
Cholesterol and HD
HD is a progressive, autosomal dominant neurodegenerative disease that is characterized by cognitive and motor deficits. This disorder is caused by a polyglutamate expansion (>36 copies) in the huntingtin protein (Rigamonti et al., 2000). The neurons in the brain that die when exposed to the toxic mutant huntingtin protein are primarily the striatal and cortical neurons (Reiner et al., 1988). The cholesterol biosynthetic pathway is markedly altered in the brains of humans and mice with HD (Valenza et al., 2005) (reviewed by Karasinska and Hayden, 2011; Valenza et al., 2010). For example, expression of mutant huntingtin in brain-derived cells reduced the processing of the sterol response-element binding protein (SREBP; the master regulator of the cholesterol biosynthetic pathway) (Valenza et al., 2005). In addition, the expression of SREBP-1 target genes was downregulated, and cholesterol biosynthesis and cellular cholesterol levels were reduced (Valenza et al., 2005). Consistent with these observations, the level of cholesterol in the striatum and cortex of a mouse model of HD is lower than in non-HD mice (Valenza et al., 2005). Furthermore, the addition of cholesterol to striatal neurons expressing mutant huntingtin increased their survival, suggesting that the deficit in cholesterol contributes directly to the neurological phenotype of HD (Valenza et al., 2005). Nevertheless, no clear molecular mechanism has yet emerged that would directly relate alterations in cholesterol biosynthesis in the brain to the neuronal dysfunction that characterizes HD.
Cholesterol and PD
PD is the second most prevalent progressive neurodegenerative disorder in humans and is characterized by tremor, slowness of movement, rigidity and eventual cognitive impairment. The pathological hallmarks of PD are the neuronal accumulation of α-synuclein in inclusions in Lewy bodies, as well as the loss of dopamine-generating neurons in the substantia nigra region of the brain. The mechanisms responsible for these alterations are not yet entirely clear but are being actively investigated. The administration of L-DOPA, which is converted into dopamine, temporarily reduces the motor symptoms and is commonly used to treat the disease.
A role for cholesterol in the neurodegenerative pathology of PD remains controversial. Intriguingly, however, the α-synuclein protein contains two cholesterol-binding domains (Fantini et al., 2011), and cholesterol seems to modulate α-synuclein aggregation. Some studies indicate that a higher level of plasma cholesterol correlates with a lower incidence of PD (de Lau et al., 2006; Huang et al., 2007), whereas other studies have reported that individuals with a high plasma cholesterol level have an increased risk of developing PD (Hu et al., 2008). In addition, the amount of several hydroxylated cholesterol derivatives produced non-enzymatically by reactive oxygen species is higher in the brains of individuals with PD than in controls (Bosco et al., 2006). Moreover, the cholesterol derivative 27-hydroxycholesterol increases α-synuclein levels and reduces dopamine synthesis, probably by activating the liver X receptor (Marwarha et al., 2011; Rantham Prabhakara et al., 2008). Statins strongly reduce the aggregation of α-synuclein in cultured neurons, whereas supplementation of the neurons with exogenous cholesterol increases α-synuclein aggregation and reduces neuron growth (Bar-On et al., 2008). Furthermore, statin treatment of a transgenic mouse model of PD reduced α-synuclein aggregation (Crews et al., 2008). Taken together, these studies suggest that reduction of plasma cholesterol in individuals with PD by statin treatment (reviewed by Roy and Pahan, 2011) might attenuate the deposition of α-synuclein aggregates in the brain. Nevertheless, it is also possible that some of the beneficial effects of the statins in individuals with PD are due to the anti-inflammatory properties of these drugs (reviewed by Vaughan and Gotto, 2004).
Oxysterols and neurodegenerative diseases
Abnormal levels of oxygenated derivatives of cholesterol (oxysterols) have also been linked to several of the neurodegenerative diseases described above (reviewed by Leoni and Caccia, 2011). For example, the concentration of 24-hydroxycholesterol is increased in cerebrospinal fluid in both AD and HD, and correlates with the level of APOE, cholesterol and tau in this fluid (Björkhem et al., 2006; Leoni and Caccia, 2011). Interestingly, adenovirus-mediated overexpression of cholesterol 24-hydroxylase in young AD mice reduces amyloid deposits and limits cognitive decline (Hudry et al., 2010). Moreover, when a mouse model of AD was cross-bred with mice lacking acyl-CoA:cholesterol acyltransferase-1 (ACAT-1; the enzyme that esterifies cholesterol), the amount of 24-hydroxycholesterol in the brain increased and amyloid pathology was attenuated (Bryleva et al., 2010). In addition, a positive correlation was observed between AD and plasma levels of another oxysterol, 27-hydroxycholesterol. This oxysterol is produced outside the brain but crosses the blood-brain barrier from the plasma when this barrier is compromised. The amount of plasma 27-hydroxycholesterol correlates with the amount of cholesterol in the circulation. Furthermore, the level of 27-hydroxycholesterol in plasma, brain and cerebrospinal fluid is markedly increased in individuals with AD (Shafaati et al., 2011). These observations might, therefore, provide a possible link between high levels of plasma cholesterol and AD. However, in the disease cerebrotendinous xanthomatosis, the enzyme that generates 27-hydroxycholesterol from cholesterol (cholesterol 27-hydroxylase) is defective, so the amount of 27-hydroxycholesterol in plasma is abnormally low – yet neurodegeneration is prevalent. In individuals with SLOS, in addition to high levels of 7-dehydrocholesterol in plasma and brain (reviewed by Porter and Herman, 2011), the plasma also contains reduced amounts of 24-hydroxycholesterol and increased amounts of 27-hydroxycholesterol, compared with levels in unaffected individuals (Björkhem et al., 2011); the mechanism responsible for these differences is not clear. Finally, the levels of several non-enzymatically generated oxidation products of cholesterol are markedly increased in the plasma of individuals with NPC disease compared with non-affected controls (Jiang et al., 2011). Although alterations in the amount of oxygenated derivatives of cholesterol have not been shown to be directly responsible for the neurodegeneration in any of these diseases, the presence of abnormal amounts of these sterols in plasma might provide useful diagnostic biomarkers for the evaluation of disease progression (Leoni and Caccia, 2011).
Commonalities and differences in cholesterol metabolism in neurodegenerative diseases
As discussed above, abnormalities have been reported in cholesterol metabolism in several different neurodegenerative diseases. An important question that arises from these observations is: does a common molecular mechanism link the neurodegenerative phenotype to altered cholesterol metabolism in the brain? If so, does this link mean that a common therapeutic approach for these diseases might be possible? From the information currently available the answer to these questions is probably ‘no’.
In most cases, a causal link between cholesterol metabolism and neurodegeneration has not been established. The exceptions are SLOS and NPC disease, which result from mutations in genes directly involved in the biosynthesis and intracellular trafficking of cholesterol, respectively. SLOS and NPC disease are caused by distinct alterations in cholesterol metabolism, and the two diseases exhibit different neurological impairments. In NPC disease, cholesterol accumulates in the lysosomal pathway of neurons and glial cells, whereas the cholesterol concentration of the plasma membrane and endoplasmic reticulum is reduced; importantly, the endoplasmic reticulum is the site at which cholesterol synthesis and acquisition are exquisitely feedback regulated by cholesterol. Thus, in NPC disease, the primary reason for neuron death is generally considered to be the sequestration of cholesterol in late endosomes and/or lysosomes. SLOS, in contrast, is caused by mutations in the final enzyme of the cholesterol biosynthetic pathway, resulting not only in abnormally low amounts of cholesterol but also in the accumulation of the cholesterol precursor 7-dehydrocholesterol in plasma and cells of the brain. It remains unclear whether the neurological problems in this disease can be attributed to the deficiency of cholesterol or to the accumulation of the potentially toxic 7-dehydrocholesterol. The reason why developmental abnormalities occur in SLOS has not been completely elucidated. Consequently, even for these two cholesterol-linked diseases, the mechanism by which dysregulation of cholesterol homeostasis leads to neurodegeneration has not been established.
The situation is even more complex for AD. Although high plasma levels of cholesterol in some instances correlate with an increased incidence of AD, there is no direct evidence that alterations in cholesterol homeostasis are responsible for the defects in memory and cognition. Some intriguing similarities have been noted, however, between NPC disease and AD. For example, both diseases exhibit endosomal and/or lysosomal abnormalities. In addition, higher levels of Aβ peptides are present in the brains of very young Npc1−/− mice compared with Npc1+/+ mice (Burns et al., 2003). Furthermore, neurofibrillary tangles containing hyperphosphorylated tau are abundant in the brains of individuals with either NPC disease or AD (Distl et al., 2003). However, despite the well-established association between APOE4 and AD, a molecular mechanism that directly connects cholesterol metabolism with the neurological impairments of AD is lacking. Indeed, it is possible that the observed defects in cholesterol metabolism in AD are the consequence, rather than the cause, of the disease. In HD, cholesterol metabolism is clearly altered in neurons expressing mutant huntingtin, but any causal link between cholesterol imbalance and the neurodegeneration that is characteristic of this disease is tenuous. A clear link has also not been established between cholesterol metabolism and the neurodegenerative phenotype of PD, although in vitro experiments indicate that high levels of cholesterol promote the aggregation of α-synuclein.
An interesting point to consider when comparing cholesterol metabolism in these neurodegenerative diseases is the region of the brain that is primarily affected. In NPC disease, the type of neurons that are most severely affected are the Purkinje cells of the cerebellum, but how cholesterol accumulation in the late endosomes and/or lysosomes preferentially causes the death of Purkinje neurons, rather than neurons in other parts of the brain, is not yet understood. AD, HD and PD are also characterized by the loss of neurons from distinct regions of the brain. In AD, the neurons that fail to survive are primarily in the hippocampus, whereas in HD the survival of striatal neurons is most severely impaired and in PD the dopaminergic neurons in the substantia nigra are the most affected. These observations raise the question of whether there are fundamental differences in the mechanisms that link cholesterol homeostasis and neuronal survival in these different types of neurons.
More information is clearly required to determine whether the neurodegenerative phenotype of these diseases can be attributed to impaired cholesterol metabolism. Nevertheless, possible therapeutic strategies for altering cholesterol metabolism in the brains of individuals suffering from these diseases are currently being considered. As discussed above, the prospect of delivering cyclodextrin intra-thecally for the treatment of individuals with NPC disease is very promising, but this would be a drastic treatment. It is unlikely that this agent would be useful for treating any of the other diseases discussed above. The use of statins is being considered as a therapy for AD given that high levels of plasma cholesterol have been proposed to correlate with the incidence of AD, and some evidence suggests that statins might be beneficial for this disease (Fassbender et al., 2002; Shepardson et al., 2011a; Wolozin et al., 2000). However, these conclusions are controversial, and the reason why any correlation would exist between high plasma cholesterol and impaired brain function in AD is not clear because plasma lipoproteins do not cross the blood-brain barrier. Furthermore, whether any of the potential benefits of the statins for treatment of AD, and perhaps also of PD, are due to their cholesterol-lowering effects or to their anti-inflammatory or antioxidant properties remains to be determined. If the statins proved beneficial for AD and/or PD, use of only the class of statins that can cross the blood-brain barrier would be indicated. Indeed, an important consideration when devising therapeutic strategies that target any metabolic pathway in the brain is that many molecules do not freely cross the blood-brain barrier. Thus, delivery of a drug into the brain is often problematic, as is the use of gene therapy for neurodegenerative diseases. Another problem in devising treatments for neurodegenerative diseases is the difficulty of reversing brain damage that has already occurred.
Many of the questions raised above should stimulate research questions regarding the fundamental molecular mechanisms that regulate cholesterol homeostasis in different types of cells in the brain, and determine how alterations in cholesterol balance affect the survival and function of neurons and glial cells. It is hoped that further studies on the role of cholesterol metabolism in neurodegeneration will facilitate the search for new approaches to treat these diseases.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any financial or competing interests.
FUNDING
This work was supported by grants from the Canadian Institutes for Health Research and the Ara Parseghian Medical Research Foundation.
REFERENCES
- Aqul A., Liu B., Ramirez C. M., Pieper A. A., Estill S. J., Burns D. K., Liu B., Repa J. J., Turley S. D., Dietschy J. M. (2011). Unesterified cholesterol accumulation in late endosomes/lysosomes causes neurodegeneration and is prevented by driving cholesterol export from this compartment. J. Neurosci. 31, 9404–9413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bales K. R., Liu F., Wu S., Lin S., Koger D., DeLong C., Hansen J. C., Sullivan P. M., Paul S. M. (2009). Human APOE isoform-dependent effects on brain beta-amyloid levels in PDAPP transgenic mice. J. Neurosci. 29, 6771–6779 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-On P., Crews L., Koob A. O., Mizuno H., Adame A., Spencer B., Masliah E. (2008). Statins reduce neuronal alpha-synuclein aggregation in in vitro models of Parkinson’s disease. J. Neurochem. 105, 1656–1667 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett P. J., Song Y., Van Horn W. D., Hustedt E. J., Schafer J. M., Hadziselimovic A., Beel A. J., Sanders C. R. (2012). The amyloid precursor protein has a flexible transmembrane domain and binds cholesterol. Science 336, 1168–1171 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baudry M., Yao Y., Simmons D., Liu J., Bi X. (2003). Postnatal development of inflammation in a murine model of Niemann-Pick type C disease: immunohistochemical observations of microglia and astroglia. Exp. Neurol. 184, 887–903 [DOI] [PubMed] [Google Scholar]
- Beffert U., Stolt P. C., Herz J. (2004). Functions of lipoprotein receptors in neurons. J. Lipid Res. 45, 403–409 [DOI] [PubMed] [Google Scholar]
- Bien-Ly N., Gillespie A. K., Walker D., Yoon S. Y., Huang Y. (2012). Reducing human apolipoprotein E levels attenuates age-dependent Aβ accumulation in mutant human amyloid precursor protein transgenic mice. J. Neurosci. 32, 4803–4811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björkhem I., Hansson M. (2010). Cerebrotendinous xanthomatosis: an inborn error in bile acid synthesis with defined mutations but still a challenge. Biochem. Biophys. Res. Commun. 396, 46–49 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Heverin M., Leoni V., Meaney S., Diczfalusy U. (2006). Oxysterols and Alzheimer’s disease. Acta Neurol. Scand. Suppl. 114, 43–49 [DOI] [PubMed] [Google Scholar]
- Björkhem I., Lövgren-Sandblom A., Piehl F., Khademi M., Pettersson H., Leoni V., Olsson T., Diczfalusy U. (2011). High levels of 15-oxygenated steroids in circulation of patients with multiple sclerosis: fact or fiction? J. Lipid Res. 52, 170–174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björnsson O. G., Sparks J. D., Sparks C. E., Gibbons G. F. (1994). Regulation of VLDL secretion in primary culture of rat hepatocytes: involvement of cAMP and cAMP-dependent protein kinases. Eur. J. Clin. Invest. 24, 137–148 [DOI] [PubMed] [Google Scholar]
- Bodovitz S., Klein W. L. (1996). Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J. Biol. Chem. 271, 4436–4440 [DOI] [PubMed] [Google Scholar]
- Bosco D. A., Fowler D. M., Zhang Q., Nieva J., Powers E. T., Wentworth P., Jr, Lerner R. A., Kelly J. W. (2006). Elevated levels of oxidized cholesterol metabolites in Lewy body disease brains accelerate alpha-synuclein fibrilization. Nat. Chem. Biol. 2, 249–253 [DOI] [PubMed] [Google Scholar]
- Boyles J. K., Pitas R. E., Wilson E., Mahley R. W., Taylor J. M. (1985). Apolipoprotein E associated with astrocytic glia of the central nervous system and with nonmyelinating glia of the peripheral nervous system. J. Clin. Invest. 76, 1501–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyles J. K., Zoellner C. D., Anderson L. J., Kosik L. M., Pitas R. E., Weisgraber K. H., Hui D. Y., Mahley R. W., Gebicke-Haerter P. J., Ignatius M. J., et al. (1989). A role for apolipoprotein E, apolipoprotein A-I, and low density lipoprotein receptors in cholesterol transport during regeneration and remyelination of the rat sciatic nerve. J. Clin. Invest. 83, 1015–1031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown M. S., Goldstein J. L. (1986). A receptor-mediated pathway for cholesterol homeostasis. Science 232, 34–47 [DOI] [PubMed] [Google Scholar]
- Bryleva E. Y., Rogers M. A., Chang C. C., Buen F., Harris B. T., Rousselet E., Seidah N. G., Oddo S., LaFerla F. M., Spencer T. A., et al. (2010). ACAT1 gene ablation increases 24(S)-hydroxycholesterol content in the brain and ameliorates amyloid pathology in mice with AD. Proc. Natl. Acad. Sci. USA 107, 3081–3086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu G. (2009). Apolipoprotein E and its receptors in Alzheimer’s disease: pathways, pathogenesis and therapy. Nat. Rev. Neurosci. 10, 333–344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burns M., Gaynor K., Olm V., Mercken M., LaFrancois J., Wang L., Mathews P. M., Noble W., Matsuoka Y., Duff K. (2003). Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J. Neurosci. 23, 5645–5649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castellano J. M., Kim J., Stewart F. R., Jiang H., DeMattos R. B., Patterson B. W., Fagan A. M., Morris J. C., Mawuenyega K. G., Cruchaga C., et al. (2011). Human apoE isoforms differentially regulate brain amyloid-β peptide clearance. Sci. Transl. Med. 3, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corder E. H., Saunders A. M., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Small G. W., Roses A. D., Haines J. L., Pericak-Vance M. A. (1993). Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer’s disease in late onset families. Science 261, 921–923 [DOI] [PubMed] [Google Scholar]
- Corder E. H., Saunders A. M., Risch N. J., Strittmatter W. J., Schmechel D. E., Gaskell P. C., Jr, Rimmler J. B., Locke P. A., Conneally P. M., Schmader K. E., et al. (1994). Protective effect of apolipoprotein E type 2 allele for late onset Alzheimer disease. Nat. Genet. 7, 180–184 [DOI] [PubMed] [Google Scholar]
- Cramer P. E., Cirrito J. R., Wesson D. W., Lee C. Y., Karlo J. C., Zinn A. E., Casali B. T., Restivo J. L., Goebel W. D., James M. J., et al. (2012). ApoE-directed therapeutics rapidly clear β-amyloid and reverse deficits in AD mouse models. Science 335, 1503–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crews L., Mizuno H., Desplats P., Rockenstein E., Adame A., Patrick C., Winner B., Winkler J., Masliah E. (2008). Alpha-synuclein alters Notch-1 expression and neurogenesis in mouse embryonic stem cells and in the hippocampus of transgenic mice. J. Neurosci. 28, 4250–4260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Lau L. M., Koudstaal P. J., Hofman A., Breteler M. M. (2006). Serum cholesterol levels and the risk of Parkinson’s disease. Am. J. Epidemiol. 164, 998–1002 [DOI] [PubMed] [Google Scholar]
- Dietschy J. M., Turley S. D. (2001). Cholesterol metabolism in the brain. Curr. Opin. Lipidol. 12, 105–112 [DOI] [PubMed] [Google Scholar]
- Dietschy J. M., Turley S. D. (2004). Thematic review series: brain lipids. Cholesterol metabolism in the central nervous system during early development and in the mature animal. J. Lipid Res. 45, 1375–1397 [DOI] [PubMed] [Google Scholar]
- Distl R., Treiber-Held S., Albert F., Meske V., Harzer K., Ohm T. G. (2003). Cholesterol storage and tau pathology in Niemann-Pick type C disease in the brain. J. Pathol. 200, 104–111 [DOI] [PubMed] [Google Scholar]
- Ehehalt R., Keller P., Haass C., Thiele C., Simons K. (2003). Amyloidogenic processing of the Alzheimer beta-amyloid precursor protein depends on lipid rafts. J. Cell Biol. 160, 113–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elder G. A., Cho J. Y., English D. F., Franciosi S., Schmeidler J., Sosa M. A., Gasperi R. D., Fisher E. A., Mathews P. M., Haroutunian V., et al. (2007). Elevated plasma cholesterol does not affect brain Abeta in mice lacking the low-density lipoprotein receptor. J. Neurochem. 102, 1220–1231 [DOI] [PubMed] [Google Scholar]
- Fagan A. M., Murphy B. A., Patel S. N., Kilbridge J. F., Mobley W. C., Bu G., Holtzman D. M. (1998). Evidence for normal aging of the septo-hippocampal cholinergic system in apoE (−/−) mice but impaired clearance of axonal degeneration products following injury. Exp. Neurol. 151, 314–325 [DOI] [PubMed] [Google Scholar]
- Fantini J., Carlus D., Yahi N. (2011). The fusogenic tilted peptide (67–78) of α-synuclein is a cholesterol binding domain. Biochim. Biophys. Acta 1808, 2343–2351 [DOI] [PubMed] [Google Scholar]
- Fassbender K., Stroick M., Bertsch T., Ragoschke A., Kuehl S., Walter S., Walter J., Brechtel K., Muehlhauser F., Von Bergmann K., et al. (2002). Effects of statins on human cerebral cholesterol metabolism and secretion of Alzheimer amyloid peptide. Neurology 59, 1257–1258 [DOI] [PubMed] [Google Scholar]
- Fryer J. D., Demattos R. B., McCormick L. M., O’Dell M. A., Spinner M. L., Bales K. R., Paul S. M., Sullivan P. M., Parsadanian M., Bu G., et al. (2005). The low density lipoprotein receptor regulates the level of central nervous system human and murine apolipoprotein E but does not modify amyloid plaque pathology in PDAPP mice. J. Biol. Chem. 280, 25754–25759 [DOI] [PubMed] [Google Scholar]
- Grimm M. O., Rothhaar T. L., Hartmann T. (2012). The role of APP proteolytic processing in lipid metabolism. Exp. Brain Res. 217, 365–375 [DOI] [PubMed] [Google Scholar]
- Gupta V. B., Laws S. M., Villemagne V. L., Ames D., Bush A. I., Ellis K. A., Lui J. K., Masters C., Rowe C. C., Szoeke C., et al. (2011). Plasma apolipoprotein E and Alzheimer disease risk: the AIBL study of aging. Neurology 76, 1091–1098 [DOI] [PubMed] [Google Scholar]
- Hawes C. M., Wiemer H., Krueger S. R., Karten B. (2010). Pre-synaptic defects of NPC1-deficient hippocampal neurons are not directly related to plasma membrane cholesterol. J. Neurochem. 114, 311–322 [DOI] [PubMed] [Google Scholar]
- Hayashi H., Campenot R. B., Vance D. E., Vance J. E. (2004). Glial lipoproteins stimulate axon growth of central nervous system neurons in compartmented cultures. J. Biol. Chem. 279, 14009–14015 [DOI] [PubMed] [Google Scholar]
- Hayashi H., Campenot R. B., Vance D. E., Vance J. E. (2009). Protection of neurons from apoptosis by apolipoprotein E-containing lipoproteins does not require lipoprotein uptake and involves activation of phospholipase Cgamma1 and inhibition of calcineurin. J. Biol. Chem. 284, 29605–29613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi H. H., Campenot R. B., Vance D. E., Vance J. E. (2007). Apolipoprotein E-containing lipoproteins protect neurons from apoptosis via a signaling pathway involving low-density lipoprotein receptor-related protein-1. J. Neurosci. 27, 1933–1941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herz J. (2001a). Lipoprotein receptors: beacons to neurons? Trends Neurosci. 24, 193–195 [DOI] [PubMed] [Google Scholar]
- Herz J. (2001b). The LDL receptor gene family: (un)expected signal transducers in the brain. Neuron 29, 571–581 [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V., Wellington C. L. (2007). Cholesterol metabolism, apolipoprotein E, adenosine triphosphate-binding cassette transporters, and Alzheimer’s disease. Curr. Opin. Lipidol. 18, 325–332 [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V., Zhou S., Burgess B. L., Bernier L., McIsaac S. A., Chan J. Y., Tansley G. H., Cohn J. S., Hayden M. R., Wellington C. L. (2004). Deficiency of ABCA1 impairs apolipoprotein E metabolism in brain. J. Biol. Chem. 279, 41197–41207 [DOI] [PubMed] [Google Scholar]
- Hirsch-Reinshagen V., Maia L. F., Burgess B. L., Blain J. F., Naus K. E., McIsaac S. A., Parkinson P. F., Chan J. Y., Tansley G. H., Hayden M. R., et al. (2005). The absence of ABCA1 decreases soluble ApoE levels but does not diminish amyloid deposition in two murine models of Alzheimer disease. J. Biol. Chem. 280, 43243–43256 [DOI] [PubMed] [Google Scholar]
- Hu G., Antikainen R., Jousilahti P., Kivipelto M., Tuomilehto J. (2008). Total cholesterol and the risk of Parkinson disease. Neurology 70, 1972–1979 [DOI] [PubMed] [Google Scholar]
- Huang X., Chen H., Miller W. C., Mailman R. B., Woodard J. L., Chen P. C., Xiang D., Murrow R. W., Wang Y. Z., Poole C. (2007). Lower low-density lipoprotein cholesterol levels are associated with Parkinson’s disease. Mov. Disord. 22, 377–381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E., Van Dam D., Kulik W., De Deyn P. P., Stet F. S., Ahouansou O., Benraiss A., Delacourte A., Bougnères P., Aubourg P., et al. (2010). Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol. Ther. 18, 44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ignatius M. J., Gebicke-Härter P. J., Skene J. H., Schilling J. W., Weisgraber K. H., Mahley R. W., Shooter E. M. (1986). Expression of apolipoprotein E during nerve degeneration and regeneration. Proc. Natl. Acad. Sci. USA 83, 1125–1129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X., Sidhu R., Porter F. D., Yanjanin N. M., Speak A. O., te Vruchte D. T., Platt F. M., Fujiwara H., Scherrer D. E., Zhang J., et al. (2011). A sensitive and specific LC-MS/MS method for rapid diagnosis of Niemann-Pick C1 disease from human plasma. J. Lipid Res. 52, 1435–1445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jick H., Zornberg G. L., Jick S. S., Seshadri S., Drachman D. A. (2000). Statins and the risk of dementia. Lancet 356, 1627–1631 [DOI] [PubMed] [Google Scholar]
- Karasinska J. M., Hayden M. R. (2011). Cholesterol metabolism in Huntington disease. Nat. Rev. Neurol. 7, 561–572 [DOI] [PubMed] [Google Scholar]
- Karten B., Vance D. E., Campenot R. B., Vance J. E. (2002). Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J. Neurochem. 83, 1154–1163 [DOI] [PubMed] [Google Scholar]
- Karten B., Vance D. E., Campenot R. B., Vance J. E. (2003). Trafficking of cholesterol from cell bodies to distal axons in Niemann Pick C1-deficient neurons. J. Biol. Chem. 278, 4168–4175 [DOI] [PubMed] [Google Scholar]
- Karten B., Hayashi H., Francis G. A., Campenot R. B., Vance D. E., Vance J. E. (2005). Generation and function of astroglial lipoproteins from Niemann-Pick type C1-deficient mice. Biochem. J. 387, 779–788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karten B., Campenot R. B., Vance D. E., Vance J. E. (2006). The Niemann-Pick C1 protein in recycling endosomes of presynaptic nerve terminals. J. Lipid Res. 47, 504–514 [DOI] [PubMed] [Google Scholar]
- Kim J., Castellano J. M., Jiang H., Basak J. M., Parsadanian M., Pham V., Mason S. M., Paul S. M., Holtzman D. M. (2009). Overexpression of low-density lipoprotein receptor in the brain markedly inhibits amyloid deposition and increases extracellular A beta clearance. Neuron 64, 632–644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koistinaho M., Lin S., Wu X., Esterman M., Koger D., Hanson J., Higgs R., Liu F., Malkani S., Bales K. R., et al. (2004). Apolipoprotein E promotes astrocyte colocalization and degradation of deposited amyloid-beta peptides. Nat. Med. 10, 719–726 [DOI] [PubMed] [Google Scholar]
- Kojro E., Gimpl G., Lammich S., Marz W., Fahrenholz F. (2001). Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha–secretase ADAM 10. Proc. Natl. Acad. Sci. USA 98, 5815–5820 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C. Y., Tse W., Smith J. D., Landreth G. E. (2012). Apolipoprotein E promotes β-amyloid trafficking and degradation by modulating microglial cholesterol levels. J. Biol. Chem. 287, 2032–2044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leoni V., Caccia C. (2011). Oxysterols as biomarkers in neurodegenerative diseases. Chem. Phys. Lipids 164, 515–524 [DOI] [PubMed] [Google Scholar]
- Leoni V., Solomon A., Kivipelto M. (2010). Links between ApoE, brain cholesterol metabolism, tau and amyloid beta-peptide in patients with cognitive impairment. Biochem. Soc. Trans. 38, 1021–1025 [DOI] [PubMed] [Google Scholar]
- Liscum L., Ruggiero R. M., Faust J. R. (1989). The intracellular transport of low density lipoprotein-derived cholesterol is defective in Niemann-Pick type C fibroblasts. J. Cell Biol. 108, 1625–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B., Turley S. D., Burns D. K., Miller A. M., Repa J. J., Dietschy J. M. (2009). Reversal of defective lysosomal transport in NPC disease ameliorates liver dysfunction and neurodegeneration in the npc1−/− mouse. Proc. Natl. Acad. Sci. USA 106, 2377–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longenberger J., Shah Z. A. (2011). Simvastatin and other HMG-CoA reductase inhibitors on brain cholesterol levels in Alzheimer’s disease. Curr. Alzheimer Res. 8, 434–442 [DOI] [PubMed] [Google Scholar]
- Lopez M. E., Klein A. D., Dimbil U. J., Scott M. P. (2011). Anatomically defined neuron-based rescue of neurodegenerative Niemann-Pick type C disorder. J. Neurosci. 31, 4367–4378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mailman T., Hariharan M., Karten B. (2011). Inhibition of neuronal cholesterol biosynthesis with lovastatin leads to impaired synaptic vesicle release even in the presence of lipoproteins or geranylgeraniol. J. Neurochem. 119, 1002–1015 [DOI] [PubMed] [Google Scholar]
- Maity T., Fuse N., Beachy P. A. (2005). Molecular mechanisms of Sonic hedgehog mutant effects in holoprosencephaly. PNAS 102, 17026–17031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann R. K., Beachy P. A. (2000). Cholesterol modification of proteins. Biochim. Biophys. Acta 1529, 188–202 [DOI] [PubMed] [Google Scholar]
- Marwarha G., Rhen T., Schommer T., Ghribi O. (2011). The oxysterol 27-hydroxycholesterol regulates α-synuclein and tyrosine hydroxylase expression levels in human neuroblastoma cells through modulation of liver X receptors and estrogen receptors – relevance to Parkinson’s disease. J. Neurochem. 119, 1119–1136 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Masliah E., Mallory M., Ge N., Alford M., Veinbergs I., Roses A. D. (1995). Neurodegeneration in the central nervous system of apoE-deficient mice. Exp. Neurol. 136, 107–122 [DOI] [PubMed] [Google Scholar]
- Mauch D. H., Nägler K., Schumacher S., Göritz C., Müller E. C., Otto A., Pfrieger F. W. (2001). CNS synaptogenesis promoted by glia-derived cholesterol. Science 294, 1354–1357 [DOI] [PubMed] [Google Scholar]
- Michikawa M., Fan Q. W., Isobe I., Yanagisawa K. (2000). Apolipoprotein E exhibits isoform-specific promotion of lipid efflux from astrocytes and neurons in culture. J. Neurochem. 74, 1008–1016 [DOI] [PubMed] [Google Scholar]
- Nathan B. P., Bellosta S., Sanan D. A., Weisgraber K. H., Mahley R. W., Pitas R. E. (1994). Differential effects of apolipoproteins E3 and E4 on neuronal growth in vitro. Science 264, 850–852 [DOI] [PubMed] [Google Scholar]
- Nieweg K., Schaller H., Pfrieger F. W. (2009). Marked differences in cholesterol synthesis between neurons and glial cells from postnatal rats. J. Neurochem. 109, 125–134 [DOI] [PubMed] [Google Scholar]
- Oitzl M. S., Mulder M., Lucassen P. J., Havekes L. M., Grootendorst J., de Kloet E. R. (1997). Severe learning deficits in apolipoprotein E-knockout mice in a water maze task. Brain Res. 752, 189–196 [DOI] [PubMed] [Google Scholar]
- Peake K. B., Vance J. E. (2012). Normalization of cholesterol homeostasis by 2-hydroxypropyl-β-cyclodextrin in neurons and glia from Niemann-Pick C1 (NPC1)-deficient mice. J. Biol. Chem. 287, 9290–9298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peake K. B., Campenot R. B., Vance D. E., Vance J. E. (2011). Niemann-Pick Type C1 deficiency in microglia does not cause neuron death in vitro. Biochim. Biophys. Acta 1812, 1121–1129 [DOI] [PubMed] [Google Scholar]
- Popko B., Goodrum J. F., Bouldin T. W., Zhang S. H., Maeda N. (1993). Nerve regeneration occurs in the absence of apolipoprotein E in mice. J. Neurochem. 60, 1155–1158 [DOI] [PubMed] [Google Scholar]
- Porter F. D. (2006). Cholesterol precursors and facial clefting. J. Clin. Invest. 116, 2322–2325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter F. D., Herman G. E. (2011). Malformation syndromes caused by disorders of cholesterol synthesis. J. Lipid Res. 52, 6–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter J. A., Young K. E., Beachy P. A. (1996). Cholesterol modification of hedgehog signaling proteins in animal development. Science 274, 255–259 [DOI] [PubMed] [Google Scholar]
- Posse de Chaves E. I., Rusiñol A. E., Vance D. E., Campenot R. B., Vance J. E. (1997). Role of lipoproteins in the delivery of lipids to axons during axonal regeneration. J. Biol. Chem. 272, 30766–30773 [DOI] [PubMed] [Google Scholar]
- Posse de Chaves E. I., Vance D. E., Campenot R. B., Kiss R. S., Vance J. E. (2000). Uptake of lipoproteins for axonal growth of sympathetic neurons. J. Biol. Chem. 275, 19883–19890 [DOI] [PubMed] [Google Scholar]
- Quan G., Xie C., Dietschy J. M., Turley S. D. (2003). Ontogenesis and regulation of cholesterol metabolism in the central nervous system of the mouse. Brain Res. Dev. Brain Res. 146, 87–98 [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A., Goldstein J. L., McDonald J. G., Brown M. S. (2008). Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab. 8, 512–521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rantham Prabhakara J. P., Feist G., Thomasson S., Thompson A., Schommer E., Ghribi O. (2008). Differential effects of 24-hydroxycholesterol and 27-hydroxycholesterol on tyrosine hydroxylase and alpha-synuclein in human neuroblastoma SH-SY5Y cells. J. Neurochem. 107, 1722–1729 [DOI] [PubMed] [Google Scholar]
- Reiner A., Albin R. L., Anderson K. D., D’Amato C. J., Penney J. B., Young A. B. (1988). Differential loss of striatal projection neurons in Huntington disease. Proc. Natl. Acad. Sci. USA 85, 5733–5737 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigamonti D., Bauer J. H., De-Fraja C., Conti L., Sipione S., Sciorati C., Clementi E., Hackam A., Hayden M. R., Li Y., et al. (2000). Wild-type huntingtin protects from apoptosis upstream of caspase-3. J. Neurosci. 20, 3705–3713 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenbaum A. I., Zhang G., Warren J. D., Maxfield F. R. (2010). Endocytosis of beta-cyclodextrins is responsible for cholesterol reduction in Niemann-Pick type C mutant cells. Proc. Natl. Acad. Sci. USA 107, 5477–5482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy A., Pahan K. (2011). Prospects of statins in Parkinson disease. Neuroscientist 17, 244–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell D. W., Halford R. W., Ramirez D. M., Shah R., Kotti T. (2009). Cholesterol 24-hydroxylase: an enzyme of cholesterol turnover in the brain. Annu. Rev. Biochem. 78, 1017–1040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanan D. A., Weisgraber K. H., Russell S. J., Mahley R. W., Huang D., Saunders A., Schmechel D., Wisniewski T., Frangione B., Roses A. D., et al. (1994). Apolipoprotein E associates with beta amyloid peptide of Alzheimer’s disease to form novel monofibrils. Isoform apoE4 associates more efficiently than apoE3. J. Clin. Invest. 94, 860–869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano M., Bell K. L., Galasko D., Galvin J. E., Thomas R. G., van Dyck C. H., Aisen P. S. (2011). A randomized, double-blind, placebo-controlled trial of simvastatin to treat Alzheimer disease. Neurology 77, 556–563 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarna J. R., Larouche M., Marzban H., Sillitoe R. V., Rancourt D. E., Hawkes R. (2003). Patterned Purkinje cell degeneration in mouse models of Niemann-Pick type C disease. J. Comp. Neurol. 456, 279–291 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2001). Clearing the brain’s amyloid cobwebs. Neuron 32, 177–180 [DOI] [PubMed] [Google Scholar]
- Selkoe D. J. (2002). Alzheimer’s disease is a synaptic failure. Science 298, 789–791 [DOI] [PubMed] [Google Scholar]
- Shafaati M., Marutle A., Pettersson H., Lövgren-Sandblom A., Olin M., Pikuleva I., Winblad B., Nordberg A., Björkhem I. (2011). Marked accumulation of 27-hydroxycholesterol in the brains of Alzheimer’s patients with the Swedish APP 670/671 mutation. J. Lipid Res. 52, 1004–1010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepardson N. E., Shankar G. M., Selkoe D. J. (2011a). Cholesterol level and statin use in Alzheimer disease: I. Review of epidemiological and preclinical studies. Arch. Neurol. 68, 1239–1244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepardson N. E., Shankar G. M., Selkoe D. J. (2011b). Cholesterol level and statin use in Alzheimer disease: II. Review of human trials and recommendations. Arch. Neurol. 68, 1385–1392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons M., Keller P., De Strooper B., Beyreuther K., Dotti C. G., Simons K. (1998). Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 95, 6460–6464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. W., Lemli L., Opitz J. M. (1964). A newly recognized syndrome of multiple congenital anomalies. J. Pediatr. 64, 210–217 [DOI] [PubMed] [Google Scholar]
- Sparks D. L., Scheff S. W., Hunsaker J. C., 3rd, Liu H., Landers T., Gross D. R. (1994). Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp. Neurol. 126, 88–94 [DOI] [PubMed] [Google Scholar]
- Strittmatter W. J., Weisgraber K. H., Huang D. Y., Dong L.-M., Salvesen G. S., Pericak-Vance M., Schmechel D., Saunders A. M., Goldgaber D., Roses A. D. (1993). Binding of human apolipoprotein E to synthetic amyloid beta peptide: isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 90, 8098–8102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan J., Evin G. (2012). B-site APP-cleaving enzyme 1 trafficking and Alzheimer’s disease pathogenesis. J. Neurochem. 120, 869–880 [DOI] [PubMed] [Google Scholar]
- Ujiie M., Dickstein D. L., Carlow D. A., Jefferies W. A. (2003). Blood-brain barrier permeability precedes senile plaque formation in an Alzheimer disease model. Microcirculation 10, 463–470 [DOI] [PubMed] [Google Scholar]
- Umeda T., Tomiyama T., Kitajima E., Idomoto T., Nomura S., Lambert M. P., Klein W. L., Mori H. (2012). Hypercholesterolemia accelerates intraneuronal accumulation of Aβ oligomers resulting in memory impairment in Alzheimer’s disease model mice. Life Sci .(in press). [DOI] [PubMed] [Google Scholar]
- Valenza M., Rigamonti D., Goffredo D., Zuccato C., Fenu S., Jamot L., Strand A., Tarditi A., Woodman B., Racchi M., et al. (2005). Dysfunction of the cholesterol biosynthetic pathway in Huntington’s disease. J. Neurosci. 25, 9932–9939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valenza M., Leoni V., Karasinska J. M., Petricca L., Fan J., Carroll J., Pouladi M. A., Fossale E., Nguyen H. P., Riess O., et al. (2010). Cholesterol defect is marked across multiple rodent models of Huntington’s disease and is manifest in astrocytes. J. Neurosci. 30, 10844–10850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance J. E. (2010). Transfer of cholesterol by the NPC team. Cell Metab. 12, 105–106 [DOI] [PubMed] [Google Scholar]
- Vance J. E., Hayashi H. (2010). Formation and function of apolipoprotein E-containing lipoproteins in the nervous system. Biochim. Biophys. Acta 1801, 806–818 [DOI] [PubMed] [Google Scholar]
- Vanier M. T., Millat G. (2003). Niemann-Pick disease type C. Clin. Genet. 64, 269–281 [DOI] [PubMed] [Google Scholar]
- Vaughan C. J., Gotto A. M., Jr (2004). Update on statins: 2003. Circulation 110, 886–892 [DOI] [PubMed] [Google Scholar]
- Veinbergs I., Jung M. W., Young S. J., Van Uden E., Groves P. M., Masliah E. (1998). Altered long-term potentiation in the hippocampus of apolipoprotein E-deficient mice. Neurosci. Lett. 249, 71–74 [DOI] [PubMed] [Google Scholar]
- Wahrle S. E., Jiang H., Parsadanian M., Legleiter J., Han X., Fryer J. D., Kowalewski T., Holtzman D. M. (2004). ABCA1 is required for normal central nervous system ApoE levels and for lipidation of astrocyte-secreted apoE. J. Biol. Chem. 279, 40987–40993 [DOI] [PubMed] [Google Scholar]
- Wahrle S. E., Jiang H., Parsadanian M., Hartman R. E., Bales K. R., Paul S. M., Holtzman D. M. (2005). Deletion of Abca1 increases Abeta deposition in the PDAPP transgenic mouse model of Alzheimer disease. J. Biol. Chem. 280, 43236–43242 [DOI] [PubMed] [Google Scholar]
- Wahrle S. E., Jiang H., Parsadanian M., Kim J., Li A., Knoten A., Jain S., Hirsch-Reinshagen V., Wellington C. L., Bales K. R., et al. (2008). Overexpression of ABCA1 reduces amyloid deposition in the PDAPP mouse model of Alzheimer disease. J. Clin. Invest. 118, 671–682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M. L., Motamed M., Infante R. E., Abi-Mosleh L., Kwon H. J., Brown M. S., Goldstein J. L. (2010). Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 12, 166–173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolozin B., Kellman W., Ruosseau P., Celesia G. G., Siegel G. (2000). Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch. Neurol. 57, 1439–1443 [DOI] [PubMed] [Google Scholar]
- Xue Y., Lee S., Ha Y. (2011). Crystal structure of amyloid precursor-like protein 1 and heparin complex suggests a dual role of heparin in E2 dimerization. Proc. Natl. Acad. Sci. USA 108, 16229–16234 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu T., Shakkottai V. G., Chung C., Lieberman A. P. (2011). Temporal and cell-specific deletion establishes that neuronal Npc1 deficiency is sufficient to mediate neurodegeneration. Hum. Mol. Genet. 20, 4440–4451 [DOI] [PMC free article] [PubMed] [Google Scholar]