

SUMMARY
Hmx1 is a homeodomain transcription factor expressed in the developing eye, peripheral ganglia, and branchial arches of avian and mammalian embryos. Recent studies have identified a loss-of-function allele at the HMX1 locus as the causative mutation in the oculo-auricular syndrome (OAS) in humans, characterized by ear and eye malformations. The mouse dumbo (dmbo) mutation, with similar effects on ear and eye development, also results from a loss-of-function mutation in the Hmx1 gene. A recessive dmbo mutation causing ear malformation in rats has been mapped to the chromosomal region containing the Hmx1 gene, but the nature of the causative allele is unknown. Here we show that dumbo rats and mice exhibit similar neonatal ear and eye phenotypes. In midgestation embryos, dumbo rats show a specific loss of Hmx1 expression in neural-crest-derived craniofacial mesenchyme (CM), whereas Hmx1 is expressed normally in retinal progenitors, sensory ganglia and in CM, which is derived from mesoderm. High-throughput resequencing of 1 Mb of rat chromosome 14 from dmbo/dmbo rats, encompassing the Hmx1 locus, reveals numerous divergences from the rat genomic reference sequence, but no coding changes in Hmx1. Fine genetic mapping narrows the dmbo critical region to an interval of ∼410 kb immediately downstream of the Hmx1 transcription unit. Further sequence analysis of this region reveals a 5777-bp deletion located ∼80 kb downstream in dmbo/dmbo rats that is not apparent in 137 other rat strains. The dmbo deletion region contains a highly conserved domain of ∼500 bp, which is a candidate distal enhancer and which exhibits a similar relationship to Hmx genes in all vertebrate species for which data are available. We conclude that the rat dumbo phenotype is likely to result from loss of function of an ultraconserved enhancer specifically regulating Hmx1 expression in neural-crest-derived CM. Dysregulation of Hmx1 expression is thus a candidate mechanism for congenital ear malformation, most cases of which remain unexplained.
INTRODUCTION
Hmx1 is a homeodomain transcription factor expressed in the developing eye, peripheral ganglia and branchial arches of avian and mammalian embryos (Wang et al., 2000; Yoshiura et al., 1998). Relatively little is known about the function of Hmx1, but recently an Hmx1 allele has been identified as the causative genetic defect in a human disorder, oculo-auricular syndrome (OAS) (Schorderet et al., 2008). Patients with OAS exhibit malformations of the outer ear (pinna) and defects of the eye, including microphthalmia, cataract, coloboma and retinal dystrophy (Schorderet et al., 2008; Vaclavik et al., 2011).
Animal models of OAS include the dumbo (dmbo) and misplaced ears (mpe) mutations in mice (Munroe et al., 2009). Mouse dmbo, mouse mpe and the known human Hmx1 allele causing OAS are all coding mutations that affect the Hmx1 DNA-binding homeodomain, and thus are predicted to result in loss of function. In addition to malformed ears, dumbo mice exhibit eye malformations, although less severe than those observed in the OAS patients identified to date. In both mouse and man, hearing is spared. In rats, the dumbo (dmbo) trait identified in animals kept by amateur ‘fancy rat’ breeders has also been mapped to a 5 Mb region encompassing the Hmx1 locus (Kuramoto et al., 2010), but the nature of the causative mutation is unknown.
Recent work in mice has also revealed a role for Hmx1 in the development of sensory neurons. Hmx1 is widely expressed in the sensory peripheral nervous system, including a subset of neurons in the trigeminal ganglion, geniculate ganglion, superior ganglion of the IX–X ganglion complex and dorsal root ganglia (Wang et al., 2000; Yoshiura et al., 1998). In the caudal cranial ganglia, Hmx1 is confined to somatosensory neurons and is not expressed in the distal viscerosensory component of these ganglia. Despite the wide expression of Hmx1 in the sensory system, only the geniculate somatosensory neurons appear to require it for neurogenesis (Quina et al., 2012), whereas early development of the trigeminal and dorsal root ganglia appear normal in dumbo mice.
TRANSLATIONAL IMPACT.
Clinical issue
The oculo-auricular syndrome (OAS) is a rare human craniofacial disorder that involves multiple anomalies of the eyes (retinal dystrophia, microphthalmia, chorioretinal colobomas and cataract) and ears (lobe malformation and simplification of ear morphology). OAS in humans has been linked to a deletion in the gene HMX1, a transcription factor expressed in the developing eye, peripheral nervous system and branchial arches of birds and mammals. However, the function of this gene and the nature of the disrupted embryological processes that result in these anomalies are not known. Moreover, the genetic and non-genetic causes of much more prevalent congenital anomalies of the ear, with and without associated eye malformations, remain largely unknown. Eye and ear anomalies have profound effects on the lives of the affected individuals. The study of animal models that advance our understanding of these conditions, and enable specific diagnosis and ultimately treatment of patients, are an invaluable resource.
Results
In prior work, the dumbo phenotype, named for its characteristic ear malformation, has been linked to the Hmx1 gene in rats and mice. Our results show that neonatal dumbo rats have a similar phenotype to that observed in dumbo mice, with characteristic ventral displacement of the ear and a moderate degree of microphthalmia. Dumbo rats show a specific loss of Hmx1 expression in neural-crest-derived craniofacial mesenchyme, particularly the maxillary component and most of the mandibular component of the first branchial arch, and the distal part of the second branchial arch. Expression of Hmx1 in the early developing retina and peripheral nervous system is intact. Unlike the known OAS HMX1 allele in humans and the dumbo mutation in mice (both of which prevent translation of the Hmx1 homeodomain), sequence analysis of the chromosomal region that encompasses the rat Hmx1 locus reveals no coding changes in the Hmx1 open reading frame. Instead, dumbo rats exhibit a novel 5777 bp deletion 79 kb downstream from Hmx1, which includes an ‘ultraconserved’ region of ∼500 bp that is likely to be crucial for Hmx1 expression in the branchial arches.
Implications and future directions
This detailed genetic analysis of the dumbo rat suggests that the causative mutation is deletion of a conserved enhancer specifically affecting Hmx1 expression in neural-crest-derived mesenchyme, and future studies will focus on unraveling the nature of this regulatory allele. In future studies of individuals with ear malformations, exon sequencing alone will not be sufficient to exclude the Hmx1 locus; regulatory alleles of Hmx1 must also be considered. These results also suggest that Hmx1 lies in the middle of a poorly understood pathway for development of the pinna. Thus, genes both upstream and downstream of Hmx1, as well as the Hmx1 locus itself, are candidates for the site of the causative allele in studies of individuals with syndromic or isolated eye and ear anomalies.
In the present study, we show that dumbo rats and mice exhibit similar ear malformations and extent of microphthalmia. Hmx1 protein expression in dumbo rat embryos shows no change in the early developing eye and sensory ganglia compared with controls, but dumbo embryos exhibit specific loss of Hmx1 expression in the mesenchymal components of branchial arches 1 and 2 (BA1 and BA2), which give rise to the pinna. Fate-mapping of Hmx1-expressing craniofacial mesenchyme (CM) cells in transgenic mice shows that this region of decreased Hmx1 expression in the dumbo rat corresponds to CM that is derived from neural crest, but not from cranial mesoderm. To better understand the genetic basis of this specific loss of Hmx1 expression, we resequenced 1 Mb of rat chromosome 14 encompassing the Hmx1 locus, revealing numerous divergences from the rat genomic reference sequence, but no coding changes in Hmx1. Single nucleotide polymorphisms within this span allowed the dmbo critical region to be narrowed to the interval of ∼410 kb immediately downstream of the Hmx1 transcription unit. Within this region, we identified a 5777-bp deletion that encompasses a ∼500-bp region conserved across all vertebrate species. Together, these results suggest that the rat dmbo mutation is a cis-acting regulatory allele of an ultraconserved enhancer that is necessary for Hmx1 expression in neural-crest-derived CM. Dysregulation of Hmx1 expression is thus a potential causative mechanism in many patients with congenital ear malformations, a common abnormality for which the etiology is largely unknown.
RESULTS
Dumbo rats exhibit congenital malformations of the pinna similar to dumbo mice, and modest reduction in ocular size at birth
In order to compare the phenotypes of dumbo mice and rats, we first examined the morphology of the ear and eye in newborn dumbo animals and controls. The characteristic ventral displacement of the ear in dumbo rats and mice, which gives the mutation its name, has been previously described in adults (Kuramoto et al., 2010; Munroe et al., 2009). To define an endpoint for developmental studies, we examined the morphology of the external ear in neonatal dumbo rats and mice at postnatal day 2 (P2) and P1, respectively. At P2, dumbo rats exhibited marked ventral displacement of the pinna compared with heterozygous controls (Fig. 1A,B). P1 Hmx1dm/dm mice also exhibited ventral displacement and, in addition, the top of the pinna was rotated posteriorly by 33° relative to controls (Fig. 1C,D).
Fig. 1.
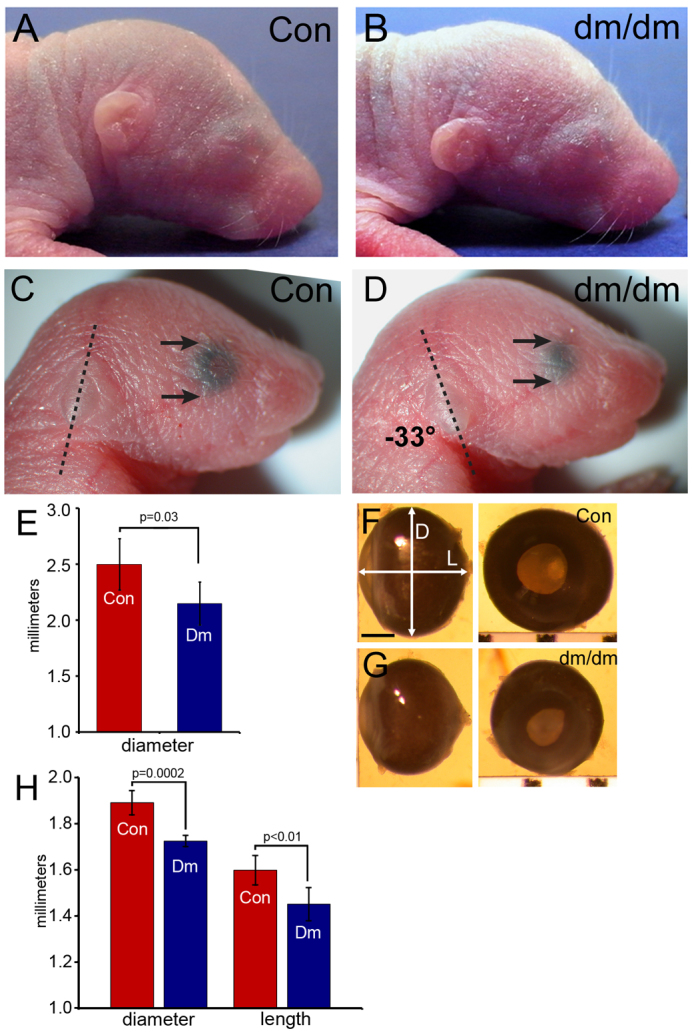
Ear and eye phenotypes in the newborn rat and mouse. Dumbo rat and mouse pups and controls were compared at P2 and P1, respectively. (A) Rat controls (dmbo/+) and (B) rat dumbo (dmbo/dmbo) pups. (C) External features of controls (Hmxdm/+ or Hmx1+/+, which are indistinguishable) and (D) dumbo mice (Hmx1dm/dm), showing characteristic rotation and ventral displacement of the ear (dotted lines). Reduced ocular size is evident through the closed eye (arrows). (E) Measurement of eye diameter in P2 rat embryos (control, mean 2.5 mm, n=6; dumbo, mean 2.15 mm, n=4; P=0.03, unpaired t-test). (F,G) P1 mouse eye, shown in lateral and frontal views for both genotypes. D, diameter; L, length. (H) Measurement of reduced eye diameter and length in the P1 eye of Hmx1dm/dm mice, n=5 of each genotype. Diameter: control, mean=1.89 mm; Dumbo, mean=1.72 mm; P=0.0002 (unpaired t-test). Length: control, mean=1.60 mm; Dumbo, mean=1.45 mm; P=0.009. Error bars show ± s.d.
To determine the extent of congenital microphthalmia in dumbo rats and mice we measured the neonatal eye. The average diameter of the newborn dumbo rat eye was modestly decreased, to 86% that of heterozygous controls (Fig. 1E). However, the eye was of normal size at E14 (data not shown), indicating that the ocular size difference must be generated during the period of extensive eye growth in late gestation. Prior characterization of the Hmx1dm/dm mouse identified a microphthalmia phenotype in adults but did not quantify the reduction in eye size (Munroe et al., 2009). In the present study, comparison of P1 dumbo mice (Hmx1dm/dm) to littermate controls (Hmx1dm/+ and Hmx1+/+) revealed that the mean diameter and length of the orbit in Hmx1dm/dm mice was reduced to 91% that of controls, corresponding to approximately 75% of normal ocular volume (Fig. 1F–H). We then undertook detailed developmental studies to examine the cellular expression of Hmx1 in the developing CM and eye in order to better understand these deformities.
Expression of Hmx1 in the mouse craniofacial mesenchyme
In developing mice, Hmx1 mRNA can be detected in BA2 at E10.5, and Hmx1 protein in the mesenchyme of proximal BA1 at E11.5 (Quina et al., 2012; Wang et al., 2000; Yoshiura et al., 1998). Malformation of the pinna is evident by E14.5 in Hmx1dm/dm embryos (data not shown). Thus the crucial events leading to the dumbo ear phenotype occur in the E10.5–E14.5 developmental interval in mice. To better understand the expression of Hmx1 at the onset of pinna development, we first examined in detail the expression of Hmx1 protein in the CM of E11.5 Hmx+/+ mouse embryos (Fig. 2). Hmx1dm/dm mouse embryos were not examined because we have previously demonstrated that Hmx1 protein is not detectable in these embryos at these stages (Quina et al., 2012).
Fig. 2.
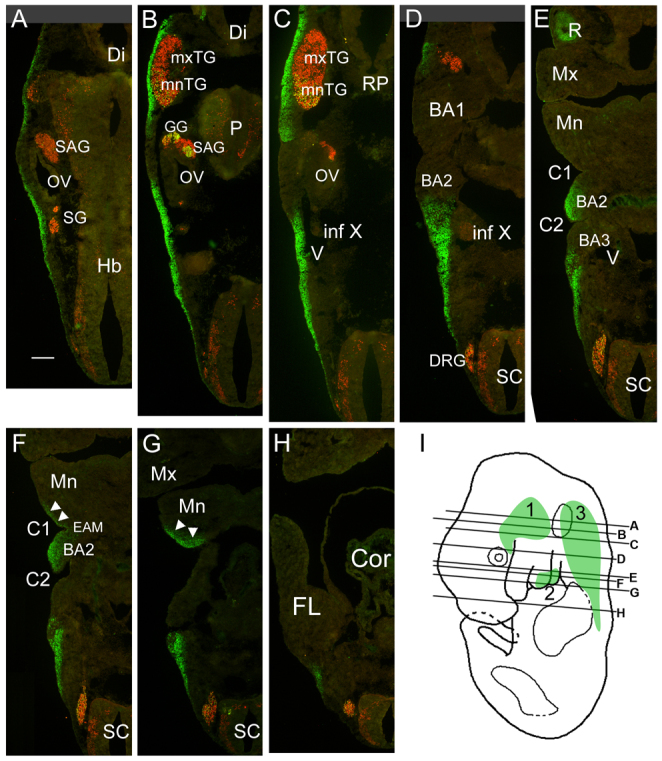
Hmx1 expression in developing mouse craniofacial mesenchyme. (A–H) Serial sections of a wild-type E11.5 mouse embryo showing the expression domains of Hmx1 in the CM. Successively more ventral planes of section show distribution of Hmx1-expressing cells in the CM. Arrows in F and G indicate the region of expression in ventral BA1. (I) Planes of section in A–H. Shaded areas indicate regions of Hmx1 expression: 1, part of proximal BA1 overlying the trigeminal ganglion; 2, the caudal half of distal BA2 and a small region of the caudal/distal tip of the mandibular component of BA1; 3, an extensive region of posterior mesenchyme, caudal to the head vein and branchial arches, ending at the top of the limb bud. BA1, branchial arch 1; BA2, branchial arch 2; BA3, branchial arch 3; C1, brachial (pharyngeal) cleft (groove) 1; C2, brachial cleft 2; Cor, heart; Di, diencephalon; DRG, dorsal root ganglion; EAM, external auditory meatus; FL, forelimb; Hb, hindbrain; GG, geniculate ganglion; Inf X, inferior ganglion of the IX–X ganglion complex (nodose/petrosal ganglion); Mn, mandibular process (of BA1); Mx, maxillary process (of BA1); mnTG, mandibular lobe, trigeminal ganglion; mxTG, maxillary lobe, trigeminal ganglion; OV, otic vesicle; P, pons; RP, Rathke’s pouch; SAG, statoacoustic ganglion; SC, spinal cord; SG, superior ganglion; V, head vein. Scale bar: 200 μm.
In the E11.5 mouse embryo, Hmx1 was expressed in three regions of the CM. Region 1 consisted of the part of proximal BA1 overlying the trigeminal ganglion (Fig. 2A–D). Region 2 comprised the caudal half of distal BA2 (Fig. 2D–F), and a small region of the caudal or distal tip of the mandibular component of BA1 (Fig. 2F,G). Region 3 consisted of an extensive region of posterior mesenchyme, caudal to the head vein and branchial arches, ending at the top of the limb bud (Fig. 2A–H). By contrast, extensive regions of CM, particularly the maxillary component and most of the mandibular component of BA1 and the proximal part of BA2, do not express Hmx1. Based on past studies of the embryological origin of the external ear, only Region 2 CM in the distal part of BA1 and BA2 is likely to contribute directly to the pinna.
Expression of Hmx1 in the dumbo rat embryo
In order to understand the expression of Hmx1 in the dumbo rat embryo, and the basis of the rat dumbo phenotype, we examined Hmx1 expression in E13 rat embryos (equivalent to the E11.5 mouse described above) and in E14 rat embryos (equivalent to an E12.5 mouse). Dumbo rats and embryos were generated by interbreeding KFRS4/Kyo parental rats. Age-matched controls were generated from crosses of the PVG/seac strain (see Methods).
Examination of BA1 in E13 dumbo rat embryos and controls revealed loss of Hmx1 expression in the proximal part of BA1, overlying the trigeminal ganglion, defined in controls as Region 1 (Fig. 3B–I). The thickness of the mesenchyme overlying the trigeminal ganglion, measured as the distance between the trigeminal ganglion itself and the surface ectoderm, was not altered, indicating that there was loss of Hmx1 expression in this domain rather than loss of the CM cells as such. Expression of Hmx1 was also diminished in the region of branchial arches 3 and 4, overlying the superior ganglion (Fig. 3F–G). Remarkably, expression of Hmx1 was not affected in the sensory neurons residing at the same axial levels, including the mandibular lobe of the trigeminal ganglion (Fig. 3H,I), the somatosensory neurons of the geniculate ganglion or the Hmx1-expressing neurons in the statoacoustic ganglion (Fig. 3J,K). The Hmx1 signal in the retina and the characteristic nasotemporal gradient of retinal expression were also unchanged in dumbo embryos compared with controls (Fig. 4B,C).
Fig. 3.
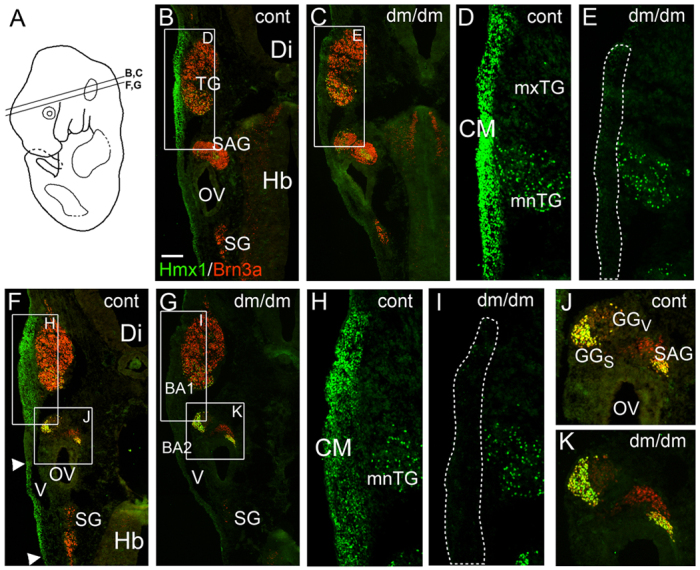
Loss of Hmx1 expression in proximal BA1 in the E13 Dumbo rat. E13 rat embryos, developmentally equivalent to E11.5 mice, were examined using immunofluorescence for Hmx1 and for Brn3a, which identifies somatosensory neurons. (A) Plane of section in subsequent views. (B–E) Control (B,D) and dumbo (C,E) embryos showing loss of expression of Hmx1 in the dorsalmost part of BA1 in the mutant. The CM overlying the TG is present (dashed line in E), but fails to express Hmx1. Expression of Hmx1 is unaffected in the mnTG and SAG. (F–K) Control (F,H,J) and dumbo (G,I,K) embryos showing loss of Hmx1 expression in BA1 in dumbo embryos. Some loss of Hmx1 expression is also noted in posterior mesenchyme overlying the head vein in the mutant (arrowheads, F). CM, craniofacial mesenchyme; Di, diencephalon; GG, geniculate ganglion; GGS, geniculate ganglion, somatosensory component; GGV, geniculate ganglion, viscerosensory component; Hb, hindbrain; mnTG, mandibular lobe, trigeminal ganglion; mxTG, maxillary lobe, trigeminal ganglion; OV, otic vesicle; SAG, statoacoustic ganglion; SG, superior ganglion (of IX–X ganglion complex); TG, trigeminal ganglion, V, head vein. Scale bar: 200 μm.
Fig. 4.
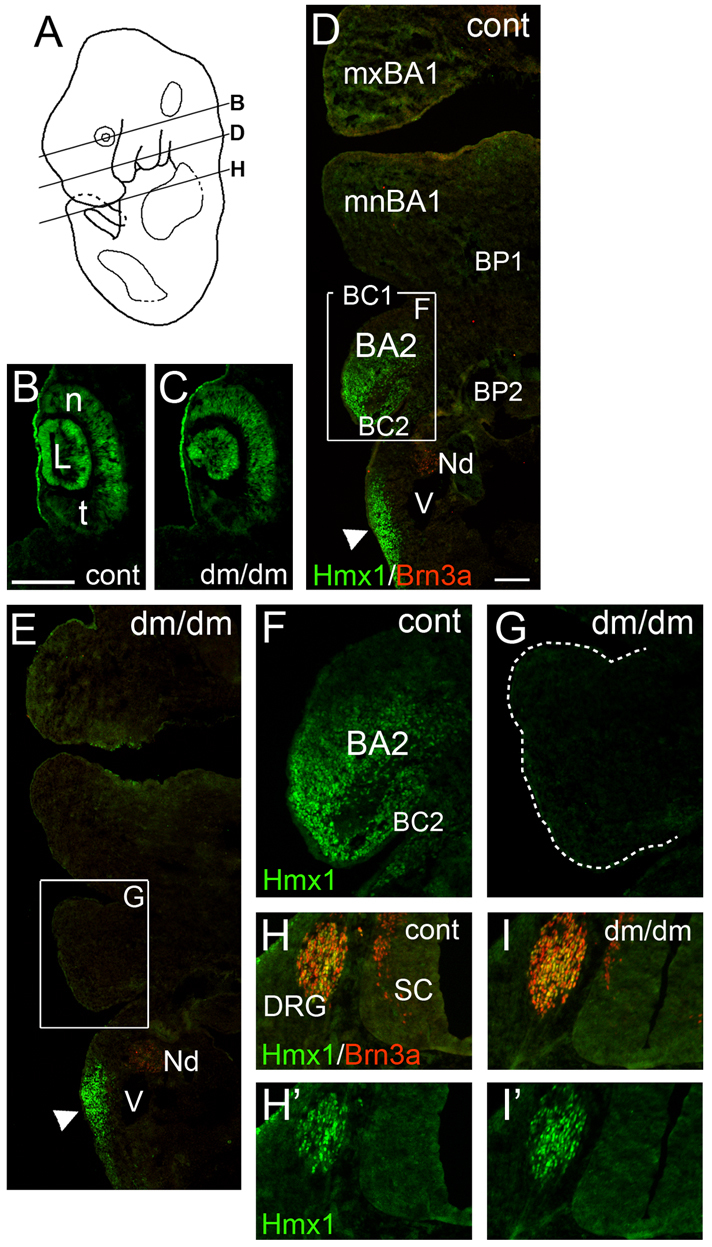
Loss of Hmx1 expression in BA2 in the E13 Dumbo rat. (A) Plane of section in subsequent views. (B,C) Hmx1 is expressed in a nasotemporal gradient in the retina which is unchanged in dumbo embryos. (D–G) Expression of Hmx1 in the distal branchial arches; top of figures is anterior. Hmx1 is not expressed in distal BA1 at this stage. Hmx1 expression in distal BA2 is lost in the dumbo embryo (G). Expression of Hmx1 in posterior mesenchyme is not affected (D,E arrowheads). Some neurons in the inferior part of the tenth ganglion are weakly positive for Brn3a. (H–I′) Expression of Hmx1 in the DRG is unchanged in dumbo embryos. Top of figure is dorsal. BA2, branchial arch 2; BC1, brachial cleft 1; BC2, brachial cleft 2; BP1, brachial pouch 1; BP1, brachial pouch 2; DRG, dorsal root ganglion; L, lens; mxBA1, maxillary component of BA1; mnBA1, mandibular component of BA1; n, nasal (aspect of retina); Nd, inferior tenth (nodose) cranial ganglion; t, temporal (aspect of retina); SC, spinal cord; V, head vein. Scale bars: 200 μm (B) and 100 μm (D).
Similar loss of Hmx1 expression was seen in the ventral branchial arches of E13 dumbo rat embryos, designated as Region 2. Hmx1 was expressed in the caudal part of distal BA2 in control embryos, whereas dumbo embryos revealed a complete loss of Hmx1 expression in this region (Fig. 4D–G). By contrast, expression was unchanged in the posterior mesenchyme designated as Region 3 (Fig. 4D,E). The cervical dorsal root ganglia (DRG) of dumbo embryos also maintained normal Hmx1 expression (Fig. 4H,I), as did the cranial sensory ganglia.
In E14 rat embryos (equivalent to an E12.5 mouse), Hmx1 was prominently expressed in the lateral facial mesenchyme, including a region of expression posterior to the eye, and dorsal to the branchial arches, which was not evident at E13 (Fig. 5). Double-labeling experiments with Hmx1 and Ap2α demonstrated that Hmx1 is expressed in the mesenchyme of this region, but not in the surface ectoderm (supplementary material Fig. S1). To better define the Hmx1 expression differences in dumbo embryos, serial sections were examined throughout this dorsal domain, and the extent of Hmx1 expression was measured using semi-quantitative immunofluorescence (Fig. 5A–C). The overall size of the area of expression was reduced moderately (44%) in the most dorsal region of expression, at the level of the eye and lateral to the hypothalamus (Fig. 5B,C, level 0), and severely (74–92%) in the ventral region of expression, lateral to the mandibular lobe of the trigeminal ganglion and pons (Fig. 5B,C, levels 7–9; P=1.7×10−4 for all ten planes of section). In the intermediate sections, where there was partial loss of Hmx1 expression, the decreased expression occurred mainly rostral to the midpoint of the trigeminal ganglion (Fig. 5C, arrows).
Fig. 5.
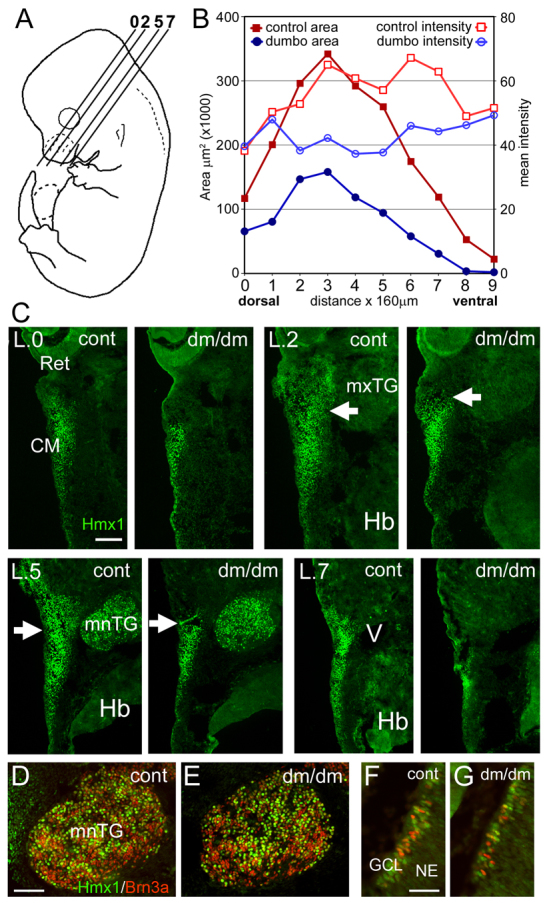
Altered Hmx1 expression in the E14 dumbo rat. Hmx1 expression was examined by immunofluorescence in the lateral cranial mesenchyme of E14 embryos, at which stage the branchial arches are no longer anatomically distinct structures. (A) Planes of section for levels analyzed in subsequent views. (B,C) Semiquantitative analysis of Hmx1 expression in BA1 at E14. The area and intensity of Hmx1 expression was measured at 160-μm intervals encompassing the cranial mesenchyme lateral to the trigeminal ganglion and pons. The overall area of Hmx1 immunofluorescence staining were significantly diminished in the dumbo embryo (paired t-test, n=10, P=0.00017). In the more dorsal sections, the loss of expression occurred mainly rostral to a boundary at about the midpoint of the trigeminal ganglion (arrows). The main effect on Hmx1 expression was to reduce the area of expression, but the intensity of the immunofluorescence signal was also somewhat diminished in the expressing area (paired t-test, n=10, P=0.003). (D,E) Expression of Hmx1 in mandibular lobe of trigeminal ganglion. (F,G) Expression of Hmx1 in differentiating retinal ganglion cells, some of which also express Brn3a. CM, craniofacial mesenchyme; GCL, ganglion cell layer; Hb, hindbrain; mnTG, mandibular lobe, trigeminal ganglion; mxTG, maxillary lobe, trigeminal ganglion; NE, neuroepithelium; Ret, retina; V, head vein. Scale bars: 100 μm (C,D) and 50 μm (F).
By contrast, no difference was observed in Hmx1 expression in the mandibular lobe of the trigeminal ganglion (Fig. 5D,E). In the retina, the overall expression of Hmx1 in the E14 retinal neuroepithelium was reduced compared with E13, but no difference was noted between dumbo embryos and controls. Onset of Hmx1 expression was noted in early differentiating retinal ganglion cells (neurons), which first appear at this stage, some of which also expressed Brn3a (Fig. 5F,G). The normal expression of Hmx1 in the eye at this stage is consistent with our observation that ocular size was also normal at E14. We conclude that the mild microphthalmia observed in neonatal dumbo rats is due to diminished ocular growth in late gestation.
Loss of Hmx1 expression in the Dumbo rat embryo occurs specifically in neural-crest-derived mesenchyme
Detailed examination of E13 and E14 rat embryos shows that the loss of Hmx1 expression in dumbo embryos is not uniform, but instead shows marked regional specificity, with profound loss in CM in the branchial arches and relative sparing of expression in regions of Hmx1 expression in the parietal region adjacent to the hindbrain.
The craniofacial mesenchyme, which gives rise to the bones and cartilage of the head, is known to originate from both the cranial neural crest and mesoderm, depending on location (Le Douarin et al., 1993). Our observation that Hmx1 expression in dumbo rat embryos is lost only in specific regions of CM led us to hypothesize that the effect of the rat dmbo mutation might differ according to the embryological origin of the affected CM cells. In mice, tissue derived from neural crest in the cranium and branchial arches can be identified by embryological fate mapping, using a Wnt1-Cre driver and a LoxP-mediated reporter (Danielian et al., 1998). In mid-gestation embryos, a clear boundary can be shown between neural-crest-derived frontonasal structures and mesoderm-derived parietal structures (Jiang et al., 2002). Because no such approach is available for rats, we chose to determine the embryological origin of the Hmx1-expressing CM in a transgenic mouse model.
To assess the embryological origin of the Hmx1-expressing cells in the CM, we crossed mice carrying Wnt1-Cre with a reporter strain, Ai6, which conditionally expresses the fluorescent reporter ZS-green from a modified Rosa26 locus (Madisen et al., 2010). In E12.5 Wnt1-Cre/Ai6 mouse embryos, expression of the induced marker gene revealed an occult boundary between the neural-crest-derived CM in frontonasal and branchial arch regions and the mesoderm-derived CM in the parietal region (Fig. 6), which was consistent with prior studies (Jiang et al., 2002; Yoshida et al., 2008). Hmx1-expressing CM cells were found in both the neural crest and mesoderm compartments, but the embryological origin of the Hmx1 cells differed markedly according to axial level. In the most dorsal and rostral domain of expression, at the level of the diencephalon (Fig. 6B), Hmx1-expressing CM cells were derived entirely from mesoderm. At the level of the trigeminal and geniculate ganglia and the future pons, an increasing proportion of the Hmx1-expressing CM cells resided in the neural-crest-derived compartment in progressively more ventral and caudal sections (Fig. 6C–E). Caudal to the otic region, in CM overlying the superior ganglion and future medulla, Hmx1 was again expressed predominantly in the mesoderm-derived CM (Fig. 6F).
Fig. 6.
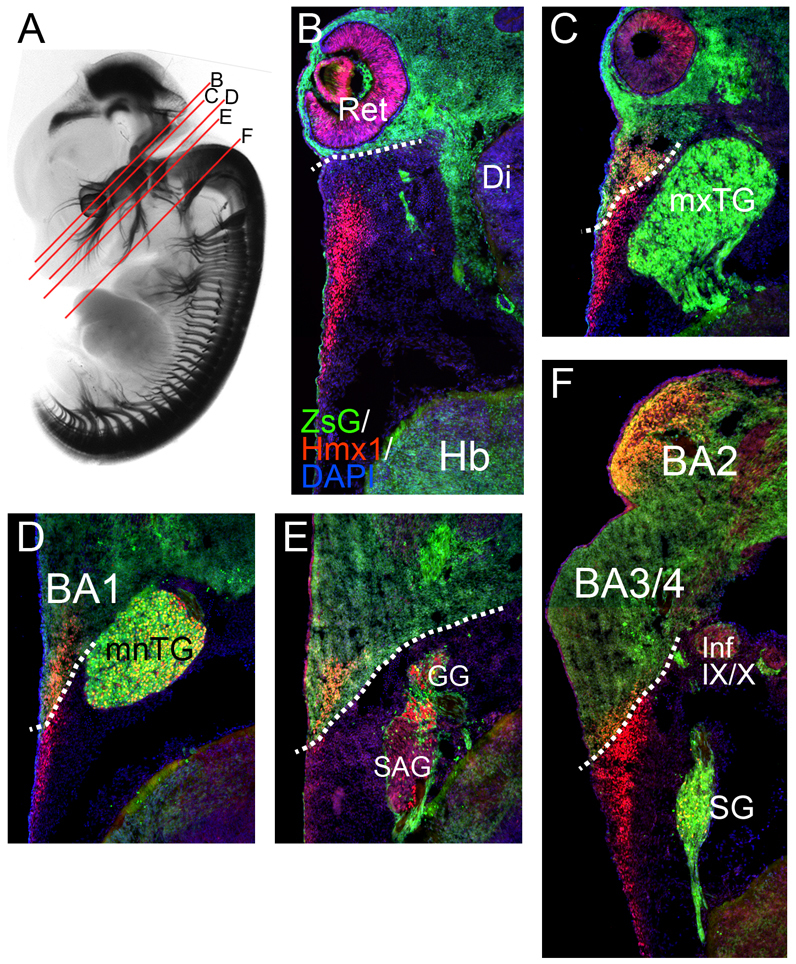
Hmx1 is expressed in CM of neural crest and non-crest origin. A Wnt1-Cre transgenic line was interbred with the reporter strain Ai6, expressing the fluorescent protein ZEG from a modified Rosa26 locus. Mouse embryos were analyzed at E12.5. (A) Whole-mount E12.5 embryo showing planes of section for levels analyzed in subsequent views. The ganglia and projections of the sensory peripheral nervous system are marked by expression of a LacZ transgene targeted to the Brn3a locus. (B–F) Progressively more ventral and caudal sections showing expression of Hmx1 in crest-derived and mesoderm-derived CM. The endogenous fluorescence of the ZEG reporter appears in green. Dashed lines demarcate the border of the cranial neural-crest-derived tissue. BA1-4, branchial arch 1–4; Di, diencephalon; GG, geniculate ganglion; Hb, hindbrain; inf IX/X, inferior part of IX–X ganglion complex (nodose and petrosal ganglia); mnTG, mandibular lobe, trigeminal ganglion; mxTG, maxillary lobe, trigeminal ganglion; Ret, retina; SAG, statoacoustic ganglion; SG, superior ganglion (of the IX–X ganglion complex).
Although it is not possible to directly fate-map the neural crest and parietal mesoderm in rat embryos, the loss of Hmx1 expression in the dumbo rat appears to be largely confined to the neural crest compartment, with persistence of Hmx1 expression in the mesoderm-derived CM (compare Fig. 5, L5 with Fig. 6D and Fig. 5, L7 with Fig. 6E). However, the Hmx1 expression is not affected in the trigeminal or dorsal root ganglia, which are also largely neural-crest-derived. Thus, it appears that the dumbo rat phenotype results from loss of Hmx1 expression specifically within the neural-crest-derived CM, suggesting a tissue-specific defect in the regulation of Hmx1 expression.
Sequence of the rat dumbo critical region reveals deletion of a unique distal conserved region
Tissue-specific loss of Hmx1 expression in neural-crest-derived CM could in principle result from the loss of a trans-acting regulatory factor governing expression in this domain, or from the disruption of a cis-acting neural-crest- or CM-specific regulatory element at the Hmx1 locus. However, prior mapping of the rat dmbo allele to a critical region of ∼5 Mb encompassing the Hmx1 locus suggested a cis-acting mechanism (Kuramoto et al., 2010).
In an effort to determine the nature of the dmbo mutation, we performed resequencing of a 1.0-Mb region of the dmbo/dmbo rat strain KFRS4, spanning the interval from 80.5 to 81.5 Mb of chromosome 14, which includes the core of the dmbo critical region (see Methods). Comparison of this region with the Brown Norway (BN) reference sequence revealed 31 single nucleotide polymorphisms (SNPs) residing in the coding sequences of nine of the ten annotated genes in this interval (supplementary material Table S1), as well as numerous SNPs in the intergenic regions. Eight of the SNPs resulted in conservative amino acid changes, and 23 were synonymous. The single SNP identified within the Hmx1 coding sequence was silent (L251L).
The identification of additional SNPs distinguishing KFRS4 from the BN reference sequence allowed further refinement of the genetic map of the dmbo critical region. To fine-map the dmbo locus, we assessed the progeny of a (BN/SsNSlc × KFRS4/Kyo)F1 × KFRS4/Kyo backcross. Transmission or non-transmission of the dmbo allele was determined by the appearance of the pinna at 3 weeks of age. A total of 614 backcross progeny were examined, of which 322 expressed the dumbo ear phenotype and 292 had normal ears. Recombination between the chromosomal markers D14Rat10 and D14Rat57 was observed in 31 of the backcross progeny, and these animals were used for fine mapping (Fig. 7). By genotyping of recombinant progeny with Ablim2_SNP(H42Y) and Hmx1_SNP(L251L), the minimal dmbo critical region was refined to a 411.5-kb segment between 80.58 and 81.0 Mb of Chr14 (Fig. 7A). The haplotypes of animals used to define the critical region appear in supplementary material Fig. S2.
Fig. 7.
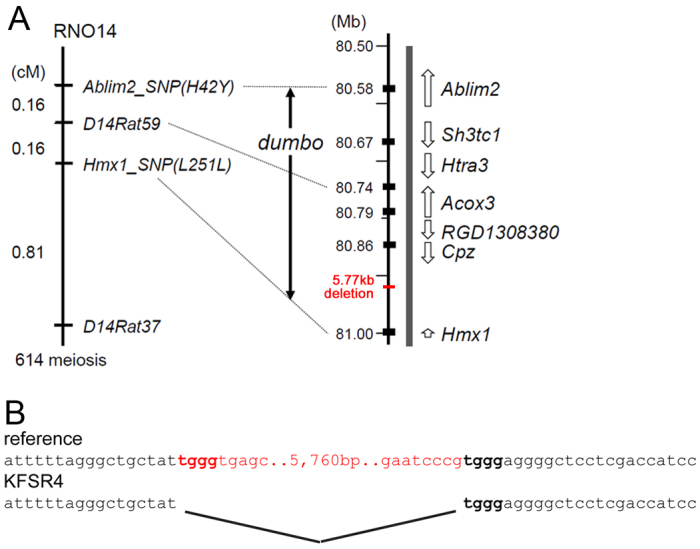
Structure of the rat dmbo critical region. (A) Genetic linkage map (left) and physical map (right) of the region of rat chromosome 14 encompassing the dmbo allele. The linkage map was derived from examination of 614 progeny of a (BN/SsNSlc × KFRS4/Kyo)F1 × KFRS4/Kyo backcross. Recombination events between the markers Ablim2_SNP(H42Y) and Hmx1_SNP(L251L) place the dmbo allele critical region within a 410-kb interval. Resequencing data revealed a novel 5777-bp deletion between the Cpz and Hmx1 genes (red). (B) Structure of the KFRS4/Kyo deletion breakpoint.
Only two mis-sense mutations were localized to this refined dmbo critical region (supplementary material Table S1), Cpz_S23P and Acox3_N287H. However, these SNPs are not specific to the KFRS4/Kyo strain. Cpz_S23P has been identified as a homozygous SNP (rs64860291) between the BN reference strain and the Sprague Dawley (SD) strain, which does not have dumbo ears. Acox3_N287H has been identified by our group as a homozygous mutation in the NER rat strain (data not shown), an inbred line derived from Crj:Wistar rats, which does not exhibit dumbo ears.
Although resequencing provided ∼99% coverage of the dmbo critical region, we wished to determine whether small discontinuities detected in the sequence data represented genomic rearrangements. To search for deletions or insertions, we used long PCR to amplify segments of DNA from dumbo and control rats spanning the 120-kb intergenic region from the 3′ end of the Hmx1 gene to the 3′ end of the Cpz gene. One primer set spanning the region 80–90 kb downstream of the Hmx1 gene yielded a ∼6 kb shorter product in dumbo samples than in controls, and a mixed product in heterozygotes (supplementary material Table S2). Sequencing of the PCR product from this region revealed a 5777-bp deletion in KFRS4/Kyo genomic sequence relative to the reference strain (Fig. 7B). The deletion was not detected in a panel of 137 other rat strains, strongly suggesting that it is the causative allele for the rat dumbo phenotype. To further verify the relationship between this deletion and the dumbo phenotype, we amplified the breakpoint region in DNA samples obtained from four dumbo rats with diverse genetic backgrounds, obtained from an enthusiast breeder in California. Each of these animals exhibited the same 5777-bp deletion 3′ to the Hmx1 transcribed region observed in KFRS4/Kyo (supplementary material Fig. S3), and no coding changes in the Hmx1 exonic sequence.
Because the 5.8-kb dmbo deletion lies in a presumed intergenic region, we used BLAST and VISTA genomic search tools to determine whether the deletion region contained sequence elements conserved across species. A BLAST search revealed a high degree of similarity between the rat dmbo deletion region and sequences in the Hmx1-Cpz intergenic region in the mouse (Chr:5) and human (Chr:4) genomes. The BLAST search did not reveal significant homology to identified transcripts or expressed sequence tags in any species, confirming the intergenic nature of the deletion region and suggesting instead a regulatory function for the conserved sequence.
Because of the limited annotation of the rat genome, we used the homologous region of the mouse genome to generate VISTA alignments (Dubchak et al., 2009) of conserved sequences in the dmbo deletion region across multiple species. VISTA analysis revealed a highly conserved region of ∼500 bp within the deletion region that is present in all mammalian species (Fig. 8A). MultiZ alignment revealed that a core sequence is conserved across vertebrate classes, including in the chicken, frog and zebrafish genomes (Fig. 8B). This Hmx distal conserved region (DCR) occurs consistently in a syntenic chromosomal region including the Hmx locus or loci and the Cpz locus in all species for which complete data are available. In chicken and zebrafish, two closely related Hmx-class genes are present in tandem in this region. Although the genomic distance between the Hmx transcribed region and the DCR varies from 45 kb (zebrafish) to 326 kb (opossum), no intervening identified transcripts occur between the Hmx loci and the DCR in any species. We conclude that the Hmx DCR is a strong candidate for an ancient distal enhancer that regulates Hmx expression in the branchial arch CM.
Fig. 8.
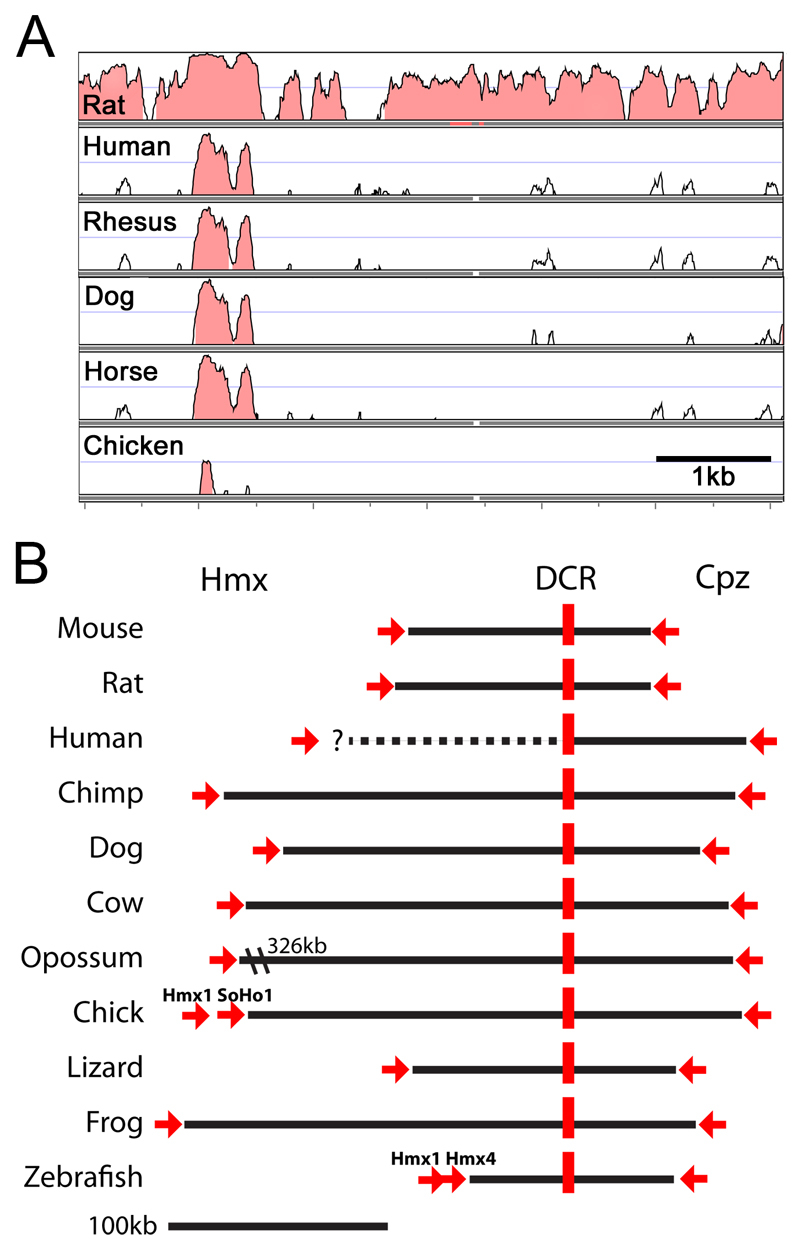
Comparative genomics of the Hmx1 distal conserved region. (A) VISTA alignment of the dmbo deletion region. The reference sequence for the alignment is chr5:35,807,810-35,813,977 (version NCBI37/mm9) of the mouse genome. A highly conserved region of ∼500 bp is detected across all mammalian species, and a core sequence is conserved across vertebrate classes. (B) Map of the relationship of the DCR to Hmx and Cpz genes for all vertebrate species for which contiguous genomic sequence is available. Red arrows indicate the 3× end of the actual or predicted Hmx1 and Cpz transcripts, and the direction of transcription. The distance from Hmx1 to the DCR in the human genome is unknown because of a gap in genomic coverage. The chick genome contains two Hmx1 homologues, Hmx1 and SOHo1, transcribed on the same strand; similarly, the zebrafish genome contains two Hmx1 homologues, Hmx1 and Hmx4. For each species, the genome build version and genomic coordinates of the core conserved sequence (∼300 bp) are as follows: Mouse, mm9, chr5:35,808,825-35,809,130; Rat, rn4, chr14:80,916,323-80,916,629; Human, hg18, chr4:8,753,059-8,753,365; Chimp, panTro2, chr4_random:6,195,662-6,195,968; Dog, canFam2, chr3:63,192,444-63,192,750; Cow, bosTau3, chr6:108,192,356-108,192,675; Opossum, monDom4, chr5:228,001,978-228,002,289; Chick, galGal3, chr4:84,267,370-84,267,675; Lizard, anoCar1, scaffold_326:1,558,135-1,558,406; Frog, xenTro2, scaffold_583:238,403-238,706; Zebrafish, danRer7, chr1:41,246,062-41,246,105.
DISCUSSION
The dumbo mouse and rat strains were named for their most obvious physical characteristic, displaced and malformed ears, prior to any knowledge of the genetic mechanism underlying these malformations. The dumbo mouse phenotype was identified in a mutagenesis screen (Wilson et al., 2005), whereas the dumbo rat mutation arose spontaneously among ‘fancy’ rats kept by enthusiast breeders, probably in the western United States, and has become a popular pet strain on the basis of its novel appearance. Given the large number of genes that are likely to play a role in the morphogenesis of the pinna, it is remarkable that both the mouse and rat dumbo strains converged on the same gene locus. The appearance of the mouse and rat dumbo ears are quite similar, but at the cellular level the phenotypes of the mouse and rat mutants reveal other striking differences. In recent work, we have shown that Hmx1dm/dm mice exhibit marked loss of geniculate ganglion neurons and the associated posterior auricular nerve (Quina et al., 2012). The cranial nerve defects in dumbo mice were identified using transgenic tract-tracing, and this method cannot not be used in the rat, but the geniculate ganglion itself appears normal in dumbo rat embryos. Furthermore, Hmx1 expression is normal in dumbo rats during the critical developmental period in which the geniculate ganglion defects appear in mice. In general, we infer that the sensory ganglia develop normally in dumbo rats, because sensory expression of Hmx1 does not appear to be affected.
Even without the sequence of the dumbo rat Hmx1 locus, it is clear that the fundamental genetic mechanism of the dmbo mutation is distinctive in the mouse and rat. The mouse Hmx1dm allele is a nonsense mutation preceding the Hmx1 homeodomain (Munroe et al., 2009), and Hmx1 protein is undetectable in Hmx1dm/dm mice, probably because the truncated protein is rapidly degraded (Quina et al., 2012). Similarly, the causative allele in human oculoauricular syndrome is predicted to result in loss of protein function (Schorderet et al., 2008). By contrast, in dumbo rat embryos, Hmx1 immunoreactivity appears normal in the retina, sensory ganglia and the part of the CM that is not derived from neural crest, findings that are not compatible with a nonsense mutation. Sequence data confirm that the dumbo rat Hmx1 open reading frame and splice junctions are intact. Instead, the KFRS4 strain of dumbo rats exhibits a 5777-bp deletion approximately 79 kb distal to the end of the final Hmx1 exon that is not found in any of 137 other rat strains tested. This, combined with the identification of a highly conserved region in the dumbo deletion region, strongly suggests that the functional rat dmbo allele consists of the loss of an enhancer that regulates expression in neural-crest-derived CM.
The physical distance between the Hmx1 transcription unit and the Hmx1 DCR, from ∼80 kb in mouse and rat up to 326 kb in the opossum, is remarkable. However, there are examples of verified enhancer function at even greater distances. One example is a highly conserved cis-acting element that regulates Shh expression in the developing limb and is the site of mutations resulting in limb abnormalities in humans and mice, despite the fact that it is located ∼1 Mb from the Shh transcription unit, in an intron of an upstream gene (Lettice and Hill, 2005; Lettice et al., 2002; Sagai et al., 2005; Sagai et al., 2004). Although not identified in prior studies, the Hmx1 DCR has key characteristics of the class of regulatory sequences that have been called ‘conserved non-coding elements’ (Woolfe et al., 2005), and ‘ultraconserved enhancers’ (Pennacchio et al., 2006). In addition to recognizable sequence conservation between higher and lower vertebrates, these elements are often located adjacent to developmental regulators, such as tissue-specific transcription factors. Although in some cases the targeted deletion of ultraconserved regions has failed to show an obvious phenotype (Ahituv et al., 2007), in the case of the rat Hmx1 DCR the loss-of-function phenotype is clearly highly penetrant, resulting in characteristic ear deformities in the large range of genetic backgrounds maintained by the rat-enthusiast breeder community.
Prior to the mapping of the rat dmbo mutation to the Hmx1 locus, dysregulation of the homeobox genes Msx1 and Dlx1 was proposed as a causative mechanism for the rat dumbo phenotype (Katerji et al., 2009). Loss-of-function mutations in Msx1/2 and Dlx1/2 result in much more extensive defects in craniofacial development than the phenotypes observed in the dumbo rat and mouse (Ishii et al., 2005; Qiu et al., 1997; Qiu et al., 1995). Clearly, the causative mutation does not reside in these genes, but they are potential upstream regulators of Hmx1.
The phenotypes of the dumbo rat and mouse help to clearly distinguish the role of Hmx1 from that of the structurally related factors Hmx2 and Hmx3 (Wang et al., 2000; Yoshiura et al., 1998), which are highly coexpressed, and have well-characterized and partially redundant roles in vestibular and hypothalamic development in mice (Wang et al., 2001; Wang et al., 2004; Wang et al., 1998). Two Hmx1 homologues have been identified in the chicken, GH6/Hmx1 and SOHo1. The expression of genes encoding these factors in the branchial arches, retina and cranial and spinal sensory ganglia suggest that they are the functional homologues of mouse and rat Hmx1, not Hmx2/3 (Deitcher et al., 1994; Stadler and Solursh, 1994). The DCR identified in the dumbo rat is located downstream of the chick Hmx1 and SOHo1 transcription units, providing further confirmation that they are the functional homologues of mammalian Hmx1. Like mouse Hmx1, GH6 and SOHo1 are expressed in a nasal-temporal gradient in the eye, and misexpression of these factors alters the topographic mapping of retinal axons onto their midbrain targets (Schulte and Cepko, 2000). However, these findings do not explain the ocular defects observed in OAS patients (coloboma) or dumbo rats and mice (microphthalmia), and further studies of the role of Hmx1 in retinal development are necessary.
The distinctive nature of the Hmx1 mutant phenotypes, resulting either from the loss of protein function or defective regulation, is underscored by comparison with the effects of other genes that regulate morphogenesis of the pinna. One example is Hoxa2, which affects ear development through specification of segmental identity in the branchial arches, a classic homeotic function. BA2 development is particularly influenced by the loss of Hoxa2, which results in transformation of BA2-derived structures into BA1 phenotypes and absence of the pinna (Santagati et al., 2005). Although Hmx1 is a homeodomain protein, its late expression in a restricted set of neural and mesenchymal cell types across all cranial segments excludes a Hox-like role in the assignment of the segmental identity. Consistent with this, Hmx1 mutants do not show the extensive changes in multiple structures derived from BA2, such as the inner ear and brainstem, that are observed in Hoxa2 mutants. In chick embryos, mis-expression of Hoxa2 in BA1 can induce ectopic expression of the chick Hmx1-class factor SOHo1, suggesting that Hmx1 might lie downstream of this Hox gene (Grammatopoulos et al., 2000). However, this cannot reflect an exclusive requirement for Hoxa2 because the normal boundaries of Hmx1 expression extend beyond the Hoxa2 expression domain.
An Hmx gene is also present in the Drosophila genome [H6-like-homeobox, FlyBase ID FBgn0264005 (Wang et al., 2000)]. The regulatory function of the Drosophila Hmx protein overlaps that of mouse Hmx2/3, because Drosophila Hmx can partially rescue the hypothalamic and inner ear phenotypes observed in Hmx2/3 double mutant mouse embryos (Wang et al., 2004). The ‘non-homeotic homeobox’ role of the Hmx family is underscored by the fact that although insertional mutants of Drosophila Hmx are known (http://flybase.org/reports/FBgn0264005.html), they apparently do not exhibit homeotic transformations and there are no published reports of Hmx function in flies.
Mutations affecting development of the external ear might also result from defective neural crest development, a mechanism exemplified by the role of Tcof1/Treacle in Treacher Collins Syndrome (TCS), an autosomal dominant disorder that frequently includes hypoplasia of the facial bones, cleft palate and middle ear defects in addition to malformation of the pinna (Dixon et al., 2007). Treacle is expressed throughout the dorsal neural tube and early neural crest. Mice haplo-insufficient for Tcof1 replicate some of the core features of TCS and demonstrate a crucial role for Tcof1 in the craniofacial neural crest (Dixon et al., 2006). By contrast, Hmx1 is not expressed in the early neural crest, and our fate-mapping studies have shown that Hmx1 is expressed in the CM without respect to the crest-derived compartments in the developing head. Thus the developmental roles of Tcof1 and Hmx1 might intersect but are not analogous.
These findings on the unique developmental role of Hmx1 have significant implications for understanding human congenital disorders that affect the ear and lateral face and that have a major impact on individuals affected by these disorders. Abnormal morphology and malpositioning of the ear (typically described as ‘low-set’ and ‘posteriorly rotated’) are among the most common malformations seen in these patients. Ear anomalies might occur in dysmorphic syndromes, where they represent one of a multitude of abnormally developed structures, or in isolation (Calzolari et al., 1999; Carey et al., 2006; Suutarla et al., 2007). It is also not surprising that the stigma associated with ear malformation and the burden of one or more corrective surgeries can have significant psychosocial sequelae (Steffen et al., 2010).
Much progress has been made in the identification of genes responsible for many dysmorphic syndromes, including TCS, Miller syndromes (DHODH, Ng et al., 2010), CHARGE (CHD7, Lalani et al., 2004; Vissers et al., 2004), the 22q11.2 deletion syndrome (TBX1, Chieffo et al., 1997) and Townes-Brock syndrome (SALL1, Kohlhase et al., 1998). However, malformation of the ear is more commonly observed as an isolated finding (Canfield et al., 2009; Carey et al., 2006; Forrester and Merz, 2005; Harris et al., 1996; Mastroiacovo et al., 1995; Shaw et al., 2004; Suutarla et al., 2007). To our knowledge, the Hmx1-linked allele responsible for the rat dumbo phenotype is the first genetic variant to be identified in any species with such a high degree of specificity for malformation of the pinna. It suggests that neural-crest-specific regulatory alleles at the Hmx1 locus could underlie isolated malformations of the pinna in humans, as a variant of the OAS that does not include severe eye deformities. It also provides a causative model for common ear malformations and is potentially the key to a developmental pathway for pinna morphogenesis that underlies these disorders.
METHODS
Animals and genotyping
Mice bearing the dmbo allele (Hmx1dm), strain B6;C3Fe-Hmx1dmbo/Rw/JcsJKjn, were obtained from Jackson Laboratories (Stock #008677). The Hmx1dm allele was originally the result of ENU mutagenesis on a C57BL/6 genetic background (Wilson et al., 2005). Mice were crossed to C57BL/6N (Charles River) for two to five additional generations prior to the experiments described. Experiments were performed with littermate controls. Genotyping of the Hmx1dm and wild-type Hmx1 (Hmx1+) alleles was performed by real-time PCR using oligonucleotide primers having 3′ termini at the point mutation that characterizes the dmbo mutation, as previously described (Quina et al., 2012).
The founder rat expressing the dumbo ear phenotype [SRR04, (Kuramoto et al., 2010)] was obtained from a commercial breeder in the United States and crossed with the PVG/seac strain of Black Hooded rats to generate the strain KFRS4, which is homozygous for the recessive alleles dumbo (dmbo) and the coat color allele head spot (hs). In prior work, the dmbo locus was mapped to the 5.7-Mb region defined by D14Rat10 and D14Rat57 (Kuramoto et al., 2010).
To further refine the dmbo locus, we produced 614 (BN/SsNSlc × KFRS4/Kyo)F1 × KFRS4/Kyo backcross progeny. The BN/SsNSlc rat strain was purchased from Japan SLC (Hamamatsu, Japan). Genotypes of the dmbo locus were determined by the appearance of the outer ear (dumbo malformation or wild-type) at 3 weeks of age. Animals with recombination events between D14Rat10 and D14Rat57 were used for the fine mapping of the dmbo critical region.
Newborn dumbo rats and E13 and E14 dumbo embryos were generated by interbreeding KFRS4/Kyo parental rats. For Hmx1 expression studies, age-matched wild-type controls were generated from PVG/seac rats. For measurements of ocular diameter, littermate controls were used. Mouse embryos were staged according to a published method (Theiler, 1972) and the corresponding rat embryo stages were identified according to Witschi (Altman and Katz, 1962).
Resequencing of the rat dmbo critical region
Regions for targeted resequencing spanned from 80.5 to 81.5 Mb of the rat chromosome 14 and contained the dmbo critical region. The 1.0-Mb target region of KFRS4/Kyo DNA was enriched using the SureSelect oligonucleotide hybridization solution-based capture technique (Agilent). The enriched fragment library was then subjected to PCR amplification, followed by sequencing on an Illumina GAIIx platform. Coverage of 40× or greater was obtained for 98.86% of the target region. The reads generated were mapped to the rat genome reference sequence (Baylor HGSC v3.4/rn4) using Burrows-Wheeler alignment (Li and Durbin, 2009). The known variants between KFRS4 and the rat genome reference sequence were identified using the Ensembl BioMart search function from the Ensembl variation 66 database. Further resequencing of the dmbo critical region was performed using long PCR using PrimeSTAR GXL DNA polymerase (Takara, Otsu, Japan). VISTA alignment of the dmbo deletion region was performed using the Lawrence Berkeley National Laboratory and US Department of Energy Joint Genome Institute VISTA browser http://genome.lbl.gov/vista/index.shtml (Dubchak et al., 2009). Identification of conserved sequences across mammalian and vertebrate species was performed using MultiZ alignment using the UCSC genome browser http://genome.ucsc.edu/ (Kent et al., 2002).
Immunofluorescence
Embryos for immunofluorescence were fixed in 4% paraformaldehyde in phosphate-buffered saline, equilibrated in 15% then 30% sucrose, embedded in OCT medium and cryo-sectioned at 14–20 μm, depending on stage. Antisera to Hmx1 was prepared in rabbits to a glutathione-S-transferase (GST)/Hmx1 fusion protein containing amino acids 2–188 of the Hmx1 protein, excluding the conserved homeodomain, as previously described (Quina et al., 2012). The Hmx1 antiserum shows no reactivity to CM in Hmx1dm/dm mice. Polyclonal rabbit and guinea pig antisera against Brn3a have also been previously described (Quina et al., 2005). Mouse monoclonal anti-Ap2α antibody (3B5) was obtained from the Developmental Studies Hybridoma Bank. Alexa-Fluor-conjugated species-specific secondary antibodies were obtained from Life Technologies (Grand Island, NY).
For semiquantitative immunofluorescence measurement of Hmx1 expression, serial sections of dumbo and control rat embryos were processed, immunostained and imaged in parallel, with the same imaging parameters and no post-processing. Areas of interest in the CM were defined manually, and the area, total fluorescence signal and mean fluorescence signal were calculated using ImageJ.
Acknowledgments
We would like to thank Janell McMorran of Ratterfly Rattery, Bonita CA, as well as Sondra Truffat and Madeleine Truffat for introducing E.E.T. to dumbo rats, and Nicole Fung for initial work on the dumbo rat coding sequence. We would also like to thank Mayuko Yokoe for her technical assistance and the National BioResource Project – Rat for providing the KFRS4/Kyo strain (NBRP#0572).
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
L.Q., E.E.T., T.K. and T.S. conceived and designed the experiments. L.Q. and T.K. performed the experiments. L.Q., E.E.T., T.K. and T.C.C. analyzed the data. L.Q., E.E.T., T.K., D.V.L. and T.C.C. wrote the paper.
FUNDING
Supported by Department of Veterans Affairs MERIT funding, and the National Institutes of Health [grant numbers HD33442, MH065496 and NS064933, to E.E.T.]. E.E.T. is a NARSAD Investigator. Also supported by grants-in-aid for Scientific Research from the Japan Society for the Promotion of Science [grant number 21300153 to T.K.] and a grant-in-aid for Cancer Research from the Ministry of Health, Labour and Welfare.
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009910/-/DC1
REFERENCES
- Ahituv N., Zhu Y., Visel A., Holt A., Afzal V., Pennacchio L. A., Rubin E. M. (2007). Deletion of ultraconserved elements yields viable mice. PLoS Biol. 5, e234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman P. L., Katz D. D. (1962). Growth, including reproduction and morphological development (ed. Altman P. L., Dittmer D. S.). Washington: Federation of American Societies for Experimental Biology [Google Scholar]
- Calzolari F., Garani G., Sensi A., Martini A. (1999). Clinical and radiological evaluation in children with microtia. Br. J. Audiol. 33, 303–312 [DOI] [PubMed] [Google Scholar]
- Canfield M. A., Langlois P. H., Nguyen L. M., Scheuerle A. E. (2009). Epidemiologic features and clinical subgroups of anotia/microtia in Texas. Birth Defects Res. A Clin. Mol. Teratol. 85, 905–913 [DOI] [PubMed] [Google Scholar]
- Carey J. C., Park A. H., Muntz H. R. (2006). External Ear. In Human Malformations and Related Anomalies (ed. Stevenson R. E.), pp. 329–338 Oxford: Oxford University Press [Google Scholar]
- Chieffo C., Garvey N., Gong W., Roe B., Zhang G., Silver L., Emanuel B. S., Budarf M. L. (1997). Isolation and characterization of a gene from the DiGeorge chromosomal region homologous to the mouse Tbx1 gene. Genomics 43, 267–277 [DOI] [PubMed] [Google Scholar]
- Danielian P. S., Muccino D., Rowitch D. H., Michael S. K., McMahon A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326 [DOI] [PubMed] [Google Scholar]
- Deitcher D. L., Fekete D. M., Cepko C. L. (1994). Asymmetric expression of a novel homeobox gene in vertebrate sensory organs. J. Neurosci. 14, 486–498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J., Jones N. C., Sandell L. L., Jayasinghe S. M., Crane J., Rey J. P., Dixon M. J., Trainor P. A. (2006). Tcof1/Treacle is required for neural crest cell formation and proliferation deficiencies that cause craniofacial abnormalities. Proc. Natl. Acad. Sci. USA 103, 13403–13408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon J., Trainor P., Dixon M. J. (2007). Treacher Collins syndrome. Orthod. Craniofac. Res. 10, 88–95 [DOI] [PubMed] [Google Scholar]
- Dubchak I., Poliakov A., Kislyuk A., Brudno M. (2009). Multiple whole-genome alignments without a reference organism. Genome Res. 19, 682–689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forrester M. B., Merz R. D. (2005). Descriptive epidemiology of anotia and microtia, Hawaii, 1986–2002. Congenit. Anom. 45, 119–124 [DOI] [PubMed] [Google Scholar]
- Grammatopoulos G. A., Bell E., Toole L., Lumsden A., Tucker A. S. (2000). Homeotic transformation of branchial arch identity after Hoxa2 overexpression. Development 127, 5355–5365 [DOI] [PubMed] [Google Scholar]
- Harris J., Kallen B., Robert E. (1996). The epidemiology of anotia and microtia. J. Med. Genet. 33, 809–813 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishii M., Han J., Yen H. Y., Sucov H. M., Chai Y., Maxson R. E., Jr (2005). Combined deficiencies of Msx1 and Msx2 cause impaired patterning and survival of the cranial neural crest. Development 132, 4937–4950 [DOI] [PubMed] [Google Scholar]
- Jiang X., Iseki S., Maxson R. E., Sucov H. M., Morriss-Kay G. M. (2002). Tissue origins and interactions in the mammalian skull vault. Dev. Biol. 241, 106–116 [DOI] [PubMed] [Google Scholar]
- Katerji S., Vanmuylder N., Svoboda M., Rooze M., Louryan S. (2009). Expression of Msx1 and Dlx1 during Dumbo rat head development: Correlation with morphological features. Genet. Mol. Biol. 32, 399–404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent W. J., Sugnet C. W., Furey T. S., Roskin K. M., Pringle T. H., Zahler A. M., Haussler D. (2002). The human genome browser at UCSC. Genome Res. 12, 996–1006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlhase J., Wischermann A., Reichenbach H., Froster U., Engel W. (1998). Mutations in the SALL1 putative transcription factor gene cause Townes-Brocks syndrome. Nat. Genet. 18, 81–83 [DOI] [PubMed] [Google Scholar]
- Kuramoto T., Yokoe M., Yagasaki K., Kawaguchi T., Kumafuji K., Serikawa T. (2010). Genetic analyses of fancy rat-derived mutations. Exp. Anim. 59, 147–155 [DOI] [PubMed] [Google Scholar]
- Lalani S. R., Safiullah A. M., Molinari L. M., Fernbach S. D., Martin D. M., Belmont J. W. (2004). SEMA3E mutation in a patient with CHARGE syndrome. J. Med. Genet. 41, e94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Douarin N. M., Ziller C., Couly G. F. (1993). Patterning of neural crest derivatives in the avian embryo: in vivo and in vitro studies. Dev. Biol. 159, 24–49 [DOI] [PubMed] [Google Scholar]
- Lettice L. A., Hill R. E. (2005). Preaxial polydactyly: a model for defective long-range regulation in congenital abnormalities. Curr. Opin. Genet. Dev. 15, 294–300 [DOI] [PubMed] [Google Scholar]
- Lettice L. A., Horikoshi T., Heaney S. J., van Baren M. J., van der Linde H. C., Breedveld G. J., Joosse M., Akarsu N., Oostra B. A., Endo N., et al. (2002). Disruption of a long-range cis-acting regulator for Shh causes preaxial polydactyly. Proc. Natl. Acad. Sci. USA 99, 7548–7553 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L., Zwingman T. A., Sunkin S. M., Oh S. W., Zariwala H. A., Gu H., Ng L. L., Palmiter R. D., Hawrylycz M. J., Jones A. R., et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mastroiacovo P., Corchia C., Botto L. D., Lanni R., Zampino G., Fusco D. (1995). Epidemiology and genetics of microtia-anotia: a registry based study on over one million births. J. Med. Genet. 32, 453–457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munroe R. J., Prabhu V., Acland G. M., Johnson K. R., Harris B. S., O’Brien T. P., Welsh I. C., Noden D. M., Schimenti J. C. (2009). Mouse H6 Homeobox 1 (Hmx1) mutations cause cranial abnormalities and reduced body mass. BMC Dev. Biol. 9, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng S. B., Buckingham K. J., Lee C., Bigham A. W., Tabor H. K., Dent K. M., Huff C. D., Shannon P. T., Jabs E. W., Nickerson D. A., et al. (2010). Exome sequencing identifies the cause of a mendelian disorder. Nat. Genet. 42, 30–35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennacchio L. A., Ahituv N., Moses A. M., Prabhakar S., Nobrega M. A., Shoukry M., Minovitsky S., Dubchak I., Holt A., Lewis K. D., et al. (2006). In vivo enhancer analysis of human conserved non-coding sequences. Nature 444, 499–502 [DOI] [PubMed] [Google Scholar]
- Qiu M., Bulfone A., Martinez S., Meneses J. J., Shimamura K., Pedersen R. A., Rubenstein J. L. (1995). Null mutation of Dlx-2 results in abnormal morphogenesis of proximal first and second branchial arch derivatives and abnormal differentiation in the forebrain. Genes Dev. 9, 2523–2538 [DOI] [PubMed] [Google Scholar]
- Qiu M., Bulfone A., Ghattas I., Meneses J. J., Christensen L., Sharpe P. T., Presley R., Pedersen R. A., Rubenstein J. L. (1997). Role of the Dlx homeobox genes in proximodistal patterning of the branchial arches: mutations of Dlx-1, Dlx-2, and Dlx-1 and -2 alter morphogenesis of proximal skeletal and soft tissue structures derived from the first and second arches. Dev. Biol. 185, 165–184 [DOI] [PubMed] [Google Scholar]
- Quina L. A., Pak W., Lanier J., Banwait P., Gratwick K., Liu Y., Velasquez T., O’Leary D. D., Goulding M., Turner E. E. (2005). Brn3a-expressing retinal ganglion cells project specifically to thalamocortical and collicular visual pathways. J. Neurosci. 25, 11595–11604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quina L. A., Tempest L., Hsu Y. W., Cox T. C., Turner E. E. (2012). Hmx1 is required for the normal development of somatosensory neurons in the geniculate ganglion. Dev. Biol. 365, 152–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagai T., Masuya H., Tamura M., Shimizu K., Yada Y., Wakana S., Gondo Y., Noda T., Shiroishi T. (2004). Phylogenetic conservation of a limb-specific, cis-acting regulator of Sonic hedgehog (Shh). Mamm. Genome 15, 23–34 [DOI] [PubMed] [Google Scholar]
- Sagai T., Hosoya M., Mizushina Y., Tamura M., Shiroishi T. (2005). Elimination of a long-range cis-regulatory module causes complete loss of limb-specific Shh expression and truncation of the mouse limb. Development 132, 797–803 [DOI] [PubMed] [Google Scholar]
- Santagati F., Minoux M., Ren S. Y., Rijli F. M. (2005). Temporal requirement of Hoxa2 in cranial neural crest skeletal morphogenesis. Development 132, 4927–4936 [DOI] [PubMed] [Google Scholar]
- Schorderet D. F., Nichini O., Boisset G., Polok B., Tiab L., Mayeur H., Raji B., de la Houssaye G., Abitbol M. M., Munier F. L. (2008). Mutation in the human homeobox gene NKX5-3 causes an oculo-auricular syndrome. Am. J. Hum. Genet. 82, 1178–1184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulte D., Cepko C. L. (2000). Two homeobox genes define the domain of EphA3 expression in the developing chick retina. Development 127, 5033–5045 [DOI] [PubMed] [Google Scholar]
- Shaw G. M., Carmichael S. L., Kaidarova Z., Harris J. A. (2004). Epidemiologic characteristics of anotia and microtia in California, 1989–1997. Birth Defects Res. A Clin. Mol. Teratol. 70, 472–475 [DOI] [PubMed] [Google Scholar]
- Stadler H. S., Solursh M. (1994). Characterization of the homeobox-containing gene GH6 identifies novel regions of homeobox gene expression in the developing chick embryo. Dev. Biol. 161, 251–262 [DOI] [PubMed] [Google Scholar]
- Steffen A., Wollenberg B., Konig I. R., Frenzel H. (2010). A prospective evaluation of psychosocial outcomes following ear reconstruction with rib cartilage in microtia. J. Plast. Reconstr. Aesthet. Surg. 63, 1466–1473 [DOI] [PubMed] [Google Scholar]
- Suutarla S., Rautio J., Ritvanen A., Ala-Mello S., Jero J., Klockars T. (2007). Microtia in Finland: comparison of characteristics in different populations. Int. J. Pediatr. Otorhinolaryngol. 71, 1211–1217 [DOI] [PubMed] [Google Scholar]
- Theiler K. (1972). The house mouse; development and normal stages from fertilization to 4 weeks of age. Berlin, New York: Springer-Verlag [Google Scholar]
- Vaclavik V., Schorderet D. F., Borruat F. X., Munier F. L. (2011). Retinal dystrophy in the oculo-auricular syndrome due to HMX1 mutation. Ophthalmic Genet. 32, 114–117 [DOI] [PubMed] [Google Scholar]
- Vissers L. E., van Ravenswaaij C. M., Admiraal R., Hurst J. A., de Vries B. B., Janssen I. M., van der Vliet W. A., Huys E. H., de Jong P. J., Hamel B. C., et al. (2004). Mutations in a new member of the chromodomain gene family cause CHARGE syndrome. Nat. Genet. 36, 955–957 [DOI] [PubMed] [Google Scholar]
- Wang W., Van De Water T., Lufkin T. (1998). Inner ear and maternal reproductive defects in mice lacking the Hmx3 homeobox gene. Development 125, 621–634 [DOI] [PubMed] [Google Scholar]
- Wang W., Lo P., Frasch M., Lufkin T. (2000). Hmx: an evolutionary conserved homeobox gene family expressed in the developing nervous system in mice and Drosophila. Mech. Dev. 99, 123–137 [DOI] [PubMed] [Google Scholar]
- Wang W., Chan E. K., Baron S., Van de Water T., Lufkin T. (2001). Hmx2 homeobox gene control of murine vestibular morphogenesis. Development 128, 5017–5029 [DOI] [PubMed] [Google Scholar]
- Wang W., Grimmer J. F., Van De Water T. R., Lufkin T. (2004). Hmx2 and Hmx3 homeobox genes direct development of the murine inner ear and hypothalamus and can be functionally replaced by Drosophila Hmx. Dev. Cell 7, 439–453 [DOI] [PubMed] [Google Scholar]
- Wilson L., Ching Y. H., Farias M., Hartford S. A., Howell G., Shao H., Bucan M., Schimenti J. C. (2005). Random mutagenesis of proximal mouse chromosome 5 uncovers predominantly embryonic lethal mutations. Genome Res. 15, 1095–1105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woolfe A., Goodson M., Goode D. K., Snell P., McEwen G. K., Vavouri T., Smith S. F., North P., Callaway H., Kelly K., et al. (2005). Highly conserved non-coding sequences are associated with vertebrate development. PLoS Biol. 3, e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida T., Vivatbutsiri P., Morriss-Kay G., Saga Y., Iseki S. (2008). Cell lineage in mammalian craniofacial mesenchyme. Mech. Dev. 125, 797–808 [DOI] [PubMed] [Google Scholar]
- Yoshiura K., Leysens N. J., Reiter R. S., Murray J. C. (1998). Cloning, characterization, and mapping of the mouse homeobox gene Hmx1. Genomics 50, 61–68 [DOI] [PubMed] [Google Scholar]