

SUMMARY
Subarachnoid haemorrhage (SAH) is a major contributor to the burden of stroke on society. Treatment options are limited and animal models of SAH do not always mimic key pathophysiological hallmarks of the disease, thus hindering development of new therapeutics. Inflammation is strongly associated with brain injury after SAH in animals and patients, and inhibition of the pro-inflammatory cytokine interleukin-1 (IL-1) represents a possible therapeutic target. Here we report that a rupture of the middle cerebral artery in the rat produces heterogeneous infarct patterns similar to those observed in human SAH. Administration of the IL-1 receptor antagonist (IL-1Ra) reduced blood-brain barrier breakdown, and the extent of breakdown correlated with brain injury. After SAH, haem oxygenase-1 (HO-1) was strongly expressed around the bleed site and in the cortex and striatum, indicating the presence of free haem, a breakdown product of haemoglobin. HO-1 expression was also found in the same regions as microglial/macrophage expression of IL-1α. The direct effect of haem on IL-1α expression was confirmed in vitro using organotypic slice culture (OSC). Haem-induced cell death was dependent on IL-1 signalling, with IL-1Ra completely blocking cellular injury. Furthermore, stimulation of mouse primary mixed glial cells with haem induced the release of IL-1α, but not IL-1β. Thus, we suggest that haem, released from lysed red blood cells (RBCs) in the subarachnoid space, acts as a danger-associated molecular pattern (DAMP) driving IL-1-dependent inflammation. These data provide new insights into inflammation after SAH-induced brain injury and suggest IL-1Ra as a candidate therapeutic for the disease.
INTRODUCTION
Subarachnoid haemorrhage (SAH) is an acute injury to the brain with devastating consequences. Its occurrence at a comparatively young age (compared with ischaemic stroke), poor outcome and high mortality rate mean that SAH is a major burden on society (Feigin et al., 2003; Feigin et al., 2005; Nieuwkamp et al., 2009). Inflammation is known to contribute to the worsening of acute brain injuries and to chronic brain diseases (e.g. Alzheimer’s disease) and might thus represent a therapeutic target (del Zoppo, 2010; Denes et al., 2010; Jin et al., 2010; Wyss-Coray, 2006). Indeed, inflammation is strongly associated with brain injury after SAH: increases in leukocytes, platelets and serum C-reactive protein (CRP) levels correlate with poorer outcome (Frijns et al., 2006; Kasius et al., 2010; Rothoerl et al., 2006). Furthermore, pro-inflammatory cytokines such as tumour necrosis factor-α (TNFα), interleukin-8 (IL-8), high mobility group box-1 (HMGB-1) and IL-6 are elevated in cerebral spinal fluid (CSF) of patients with a worse clinical grade (Fassbender et al., 2001; Nakahara et al., 2009; Schoch et al., 2007).
The pro-inflammatory cytokine IL-1 is a key mediator of neuronal injury after acute brain injury (Allan et al., 2005), and inhibition of IL-1 actions represents a viable therapeutic strategy (Brough et al., 2011). The two most well characterised pro-inflammatory members of the IL-1 family are IL-1α and IL-1β, and production of these by macrophages and microglia is induced by activation of pattern recognition receptors (PRRs) such as those of the Toll-like receptor (TLR) family (Netea et al., 2008). During infection these PRRs are activated by pathogen-associated molecular patterns (PAMPs). It is not yet clear how they are activated during sterile inflammation (in the absence of infection), but endogenous molecules that are modified during disease or released by dead cells, known as danger-associated molecular patterns (DAMPs), might also activate PRRs (Chen and Nuñez, 2010).
SAH is considered a sterile injury and therefore might elicit a DAMP-mediated inflammatory response. The inflammatory response to sterile injury is similar to microbial infection, and the host receptors that mediate both could be the same (Chen and Nuñez, 2010). However, recent literature highlights sterile-specific mechanisms in the response to injury (Chen and Nuñez, 2010; Rock et al., 2010). Efforts are now being made to elucidate the specific DAMPs that elicit the inflammatory response in disease. Uric acid, released from dying cells, and its derivative, monosodium urate crystals, which cause the inflammatory disease gout, have been identified as molecules that initiate the sterile inflammatory response (Chen et al., 2006; Kono et al., 2010a). Furthermore, crystalline cholesterol found in atherosclerotic plaques, and serum amyloid A, an acute phase response protein in mice, have been recognised as DAMPs that exacerbate inflammation (Duewell et al., 2010; Niemi et al., 2011). The injection of necrotic cells into the peritoneal cavity of mice elicits an inflammatory response that is dependent on IL-1α and IL-1R1 (Chen et al., 2007).
Both IL-1α and IL-1β act at the type I IL-1 receptor (IL-1RI), initiating signalling cascades that result in the expression of inflammatory genes (Korherr et al., 1997; Smith et al., 2009). The endogenous IL-1 receptor antagonist (IL-1Ra) completely blocks signalling at the receptor, inhibiting inflammatory effects of IL-1α and IL-1β (Hannum et al., 1990). IL-1Ra is currently licensed as a therapy for rheumatoid arthritis (as Anakinra) and is the standard treatment for autoinflammatory diseases (Dinarello, 2011). IL-1Ra is also showing promise as an anti-inflammatory for the treatment of central nervous system (CNS) disease: a Phase 2 trial in acute stroke patients showed a reduction in inflammatory markers and improved clinical outcome at 3 months in patients receiving IL-1Ra compared with placebo control (Emsley et al., 2005).
The aim of this study was to investigate the role of IL-1 in brain inflammation in response to experimental SAH, to identify possible DAMPs as drivers of IL-1 expression and release, and test whether inhibiting endogenous IL-1 with recombinant IL-1Ra could improve outcome. We report that IL-1Ra, administered after SAH, was beneficial by reducing blood-brain barrier (BBB) breakdown, and the extent of this breakdown correlated with neuronal damage. Microglia were the major source of IL-1 following SAH. IL-1α expression was dominant compared with that of IL-1β, and was expressed as early as 12 hours after the insult. Haem oxygenase-1 (HO-1) expression after SAH suggested the presence of free haem throughout the brain. Furthermore, haem induced the release of IL-1α in primed mixed glial cells, enhanced IL-1α expression in organotypic slices culture (OSC) and increased cell death, an effect that was blocked by IL-Ra. Thus, we propose that haem, a breakdown product of red blood cells (RBCs), contributes to IL-1α production after SAH and that IL-1Ra could be beneficial in improving patient outcome.
RESULTS
IL-1α is produced by microglia/macrophages early after experimental SAH
To assess the role that IL-1 played in driving acute brain injury after SAH we initially investigated whether it was produced after SAH in the rat. Animals underwent a modified version of the intraluminal filament model of SAH (Park et al., 2008) and were sacrificed 12 hours after haemorrhage. Sham animals underwent identical surgery without perforation of the cerebral artery. Immunofluorescence on coronal sections from injured brains revealed that SAH induced the expression of IL-1α throughout the brain, including in the cortex and striatum (Fig. 1), in the hippocampus and especially in basal structures adjacent to the site of perforation (Fig. 1), where 46% of microglia were found to express IL-1α. Images shown are representative of immunofluorescent-positive cells seen in all animals 12 hours after SAH. IL-1β was also observed but to a much lesser extent and was restricted to a few cells in the striatum. IL-1α was expressed primarily by microglial/macrophage cells, because it colocalised with the ionised calcium binding adaptor molecule 1 (Iba1), which is expressed exclusively by microglia/macrophages (Ito et al., 1998). Microglia dynamically survey their environment (Nimmerjahn et al., 2005), but become activated in response to damage, changing their morphology from long, ramified protrusions to shorter, hyper-ramified branches (Streit et al., 1999). Our results indicate that both resting and fully activated microglia express IL-1α, because expression was observed in cells of different morphology (Fig. 1).
Fig. 1.
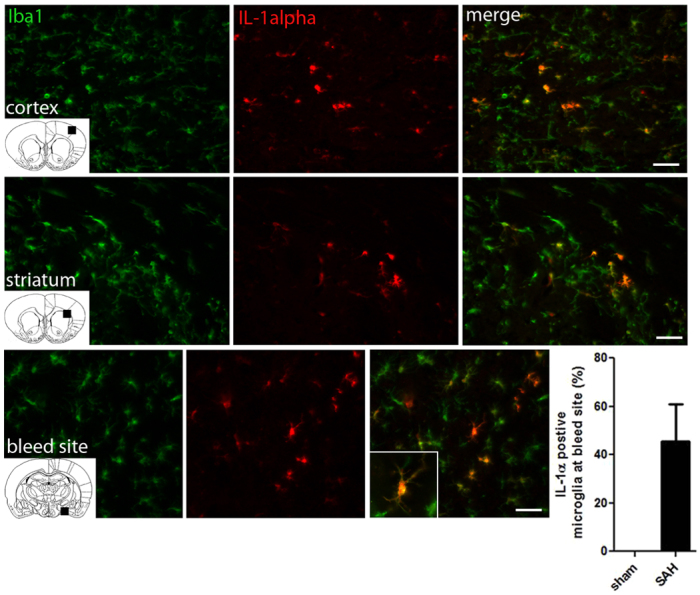
IL-1α is expressed by microglia/macrophages early after experimental SAH. Panels show coronal brain sections of SAH animals 12 hours after bleed. Images are representative of the cortex, striatum and adjacent area to the bleed site after SAH. IL-1α-positive fluorescent cells (red); Iba1-positive fluorescent cells show microglia/macrophages (green). Images are merged to reveal IL-1α colocalisation with microglia/macrophages. Graph shows the percentage of IL-1α-positive microglia cells adjacent to the bleed site (n=5). No IL-1α-positive cells were seen in sham controls. Coronal inserts indicate the area that the image was taken from and insert shows high magnification of IL-1α/Iba1 colocalisation. Scale bars: 50 μm.
HO-1 is expressed throughout the brain early after SAH, in regions of IL-1α expression
During SAH there is an extravasation RBCs that subsequently lyse, releasing haemoglobin (Hgb) and its oxidation product, haem. Haem is converted in brain by haem oxygenase (HO) into carbon monoxide, biliverdin and iron (Ascenzi et al., 2005). HO-1 is expressed primarily by glial cells and is inducible by RBC lysis products (Matz et al., 1997; Turner et al., 1998). Given that HO-1 knockout (KO) mice are more susceptible to sepsis-induced lethality, with free haem contributing to tissue damage (Larsen et al., 2010), we hypothesised that haem might contribute to brain inflammation and damage after SAH. At 12 hours after SAH, HO-1 was upregulated in the cortex, striatum, adjacent to the bleed site (Fig. 2), throughout the meninges and in the choroid plexus, when compared with sham-treated animals, in which it was not detectable. HO-1 is thought to be expressed primarily by glial cells and is induced by haem (Wagner et al., 2003). Previous studies have shown that haem is present throughout the brain after SAH (Lee et al., 2010; Turner et al., 1998), and the upregulation of HO-1 has been suggested as a marker of the presence of haem, although heat shock proteins and a variety of oxidants are also known to induce its expression (Lee et al., 2010; Turner et al., 1998). At 12 hours after SAH, HO-1 is localised to areas of microglial activation and IL-1α expression, but does not colocalise to microglial cells (Fig. 2).
Fig. 2.

Early after SAH, HO-1 is found throughout the brain localised to brain regions expressing IL-1α. Panels show representative coronal brain sections of SAH animals 12 hours after bleed (n=5). Representative images from the area adjacent to the bleed site, the cortex and the striatum are shown. HO-1-positive fluorescent cells (green); IL-1α-positive fluorescent cells (red). Areas of HO-1 immunofluorescence correspond to areas of microglial activation and IL-1α expression seen in Fig. 1. In sham animals, HO-1-positive fluorescence was undetectable. Scale bars: 100 μm.
BBB breakdown is correlated with brain damage and is reduced by IL-1Ra
Experimental studies of SAH in rodents generally do not report any substantial neuronal cell death. This is either because the intervention does not produce infarcts, or because this was not reported as an outcome measure (Lee et al., 2009; Park et al., 2008; Prunell et al., 2003). We found previously, using the modified perforation model, that large blood loads produced focal yet heterogeneous infarct patterns, as seen in human patients (Greenhalgh et al., 2012; Rabinstein et al., 2005). Furthermore, there is a positive correlation between blood load and neuronal damage, where animals with a blood load grade ≥7 incur neuronal loss (Greenhalgh et al., 2012). Variability in blood load is high in this model (Sugawara et al., 2008). Therefore, to investigate the role of inflammation on brain injury after SAH effectively, we decided a priori to only assess animals that had a blood load grade ≥7 (Fig. 3).
Fig. 3.

Blood load grades 48 hours after SAH. (A) Photomicrographs showing examples of blood load grades 48 hours after SAH. (B) Blood load grades were highly variable between animals; as expected, there was no significant difference between groups that went on to receive IL-1Ra (n=15) or placebo (n=14). (C) Blood load grades ≥7 were eligible for analysis.
Mortality after SAH was not affected by treatment: 21% (4/18) of placebo- and 20% (4/19) of IL-1Ra-treated animals died before 48 hours. There were no mortalities in the sham-operated group. Animals were randomly assigned to treatment groups, but not included in the study if they did not survive the initial haemorrhage. As a result, ten placebo and nine IL-1Ra-assigned (but not treated) animals died within moments of the perforation and were therefore not included in the mortality data.
BBB breakdown leads to extravasation of blood-borne protein and cells (vasogenic oedema), which can exacerbate inflammation and contribute to cellular damage (Simard et al., 2007; Simard et al., 2008). To test whether the IL-1 expressed 12 hours after SAH contributed to brain injury, animals underwent sham or SAH surgery and received either IL-1Ra (75 mg/kg) or placebo subcutaneously 15 minutes after haemorrhage. The effect of IL-1Ra on BBB breakdown after SAH was measured at 48 hours by quantifying the infiltration of endogenous rat IgG, a molecule normally excluded from the brain parenchyma by an intact BBB. In animals that received IL-1Ra there was significantly reduced BBB breakdown compared with placebo (Fig. 4A), with no IgG infiltration in sham animals.
Fig. 4.

BBB breakdown correlates with neuronal damage and is significantly reduced by IL-1Ra. (A) Volume of BBB breakdown at 48 hours, as measured by IgG infiltration, is reduced by administration of IL-1Ra (s.c. 75 mg/kg) 15 minutes after bleed. Top panels show representative coronal sections from placebo (n=8) and IL-1Ra (n=8) groups. (B) Linear regression analysis shows positive correlation between IgG infiltration and neuronal damage (r2=0.612). (C) Neuronal damage 48 hours after SAH; there is no significant difference between IL-1Ra (n=8) and placebo (n=8) groups (P=0.059). Inserts show representative coronal sections from placebo and IL-1Ra groups and coronal map indicating the area imaged. (A,C) Unpaired Student’s t-tests; P<0.05 considered significant.
Because neuronal damage in this experimental model of SAH is heterogeneous in its location and size, affecting areas such as the hippocampus, striatum and cortex (individually or in combination), we devised a neuronal damage scoring system to evaluate damage across all areas. Using this score we found that BBB breakdown correlated strongly with neuronal loss (r2=0.612) (Fig. 4B). Although the reduction in brain damage in the IL-1Ra group was not significant at the P<0.05 level (P=0.059) compared with placebo (Fig. 4C), this might be due to higher blood load grades in the IL-1Ra group (Fig. 3). These data suggest that IL-1 is a driver of BBB breakdown after SAH, and its expression correlates with worsening injury.
Endovascular perforation model of SAH does not induce inflammatory changes in the periphery or CSF
Peripheral inflammation can contribute to brain injury in ischaemic stroke, and animal models of focal ischaemia reflect this (Denes et al., 2010; McColl et al., 2009; Wang et al., 2007). Less is known about the role of peripheral inflammation in SAH. To test the hypothesis that SAH increases peripheral inflammation that in turn could contribute to brain injury, we measured the levels of inflammatory mediators in blood before, and then at 3, 24 and 48 hours after, experimental SAH. The cytokines IL-1α, IL-6, TNFα, IL-10, interferon-γ and CXCL1 were all measured and did not change at any time point, apart from CXCL1, which showed a transient increase at 3 hours (Fig. 5A). However, this increase was due to surgical intervention, because SAH did not induce changes above those of sham animals.
Fig. 5.

Experimental SAH does not induce inflammatory changes in the periphery or CSF. Log transformed CXCL-1 concentrations in plasma (A) and IL-6 concentrations in CSF (B). Two-way ANOVA showed a significant effect of time after bleed in CXCL-1 plasma concentrations, with no difference between sham or SAH groups. One-way ANOVA showed there was no significant difference in CSF IL-6 concentrations between groups (n=7–10).
SAH did not induce any changes in IL-1α, IL-6, TNFα, IL-10 and interferon-γ in CSF 48 hours after sham surgery or experimental SAH with IL-1Ra or placebo. This might be due to the single time point sampled or the sensitivity of the assay. Together, these findings indicate that, in this model of SAH, IL-1-mediated brain injury acts at the parenchymal level with limited input from circulating factors.
Haem, a product of RBC lysis, acts as a DAMP to induce expression and release of IL-1α
Hgb and its oxidation product, haem (Ascenzi et al., 2005; Rother et al., 2005), are rapidly distributed throughout brain tissue after SAH in rat (Turner et al., 1998). We therefore tested the hypothesis that free haem can act as a DAMP and drive inflammation after SAH. We applied haemin (the oxidation product of haem) to mouse hippocampal OSCs and mixed glial cultures to measure the direct effect of haem on neuronal death [using propidium iodide (PI) incorporation in OSCs] and IL-1α and IL-1β expression by ELISA. Haemin application led to neuronal death in the OSCs, as seen by the increased PI staining, which was protected by the application of IL-1Ra (500 ng/ml) (Fig. 6). Haemin also induced increased IL-1α expression, and to a greater extent than it did the expression of IL-1β (Fig. 6). This increased expression of IL-1α was similar to the effect of the PAMP lipopolysaccharide (LPS), which was used as a positive control. These data suggest a direct relationship between haemin-induced expression of IL-1α and cell death. In mouse primary mixed glial cultures the production of mature IL-1α and IL-1β is thought to occur in two stages. The first stage requires the expression of the IL-1 precursors (pro-IL-1α and pro-IL-1β). The second stage involves the primed macrophage/microglia (i.e. activated to express pro-IL-1) encountering a further stimulus that results in the activation of multimeric complexes called inflammasomes, which lead to the secretion of both IL-1α and IL-1β (Brough et al., 2011; Franchi et al., 2009). Therefore, in this system, cultured cells must be primed to assess IL-1 release. As expected, haemin did not induce any expression or release of IL-1α or IL-1β without prior LPS priming. However, when applied to glial cells that had received prior stimulation with LPS, haemin induced a significant increase in release of IL-1α 2, 4 and 6 hours after haemin stimulation, but did not affect IL-1β release (Fig. 7). ATP induces large amounts of IL-1 release from LPS-primed cells via stimulation of the P2X7 receptor and was used as a positive control in all IL-1 release experiments. Western blot analysis of supernatants revealed that haemin induced the cleavage of pro-IL-1α (31 kDa) to its mature form (17 kDa) through a calpain-1-dependent mechanism: release of mature IL-1α was blocked by the calpain inhibitor MDL28170 (100 μM) (Fig. 7). These results reveal haem as a potential DAMP in OSCs and mixed glial cells in vitro, and as a specific regulator of IL-1α expression and release. Recently, IL-1α has emerged as a key inflammatory mediator of sterile inflammatory responses (Chen et al., 2007; Kono et al., 2010b; Rock et al., 2010). This is the first report, to our knowledge, to show that haem can act as a DAMP in brain-derived cells, and is the first report of a DAMP that is selective for IL-1α over IL-1β.
Fig. 6.

Haemin-induced neuronal death is IL-1 dependent in OSCs. Treatment of OSCs with haemin (30 μM; oxidation product of haem) induced neuronal death, as seen by PI staining, mainly in the dentate gyrus (DG) and CA1 regions of the hippocampus (A). This effect was abolished by co-treatment with IL-1Ra (500 ng/ml). (B) Representative images of PI staining in OSCs after treatment (note that the image intensity of the haemin + IL-1Ra example has been increased in order to visualise the hippocampus). Haemin treatment induced the expression of IL-1α (C) and IL-1β (D), measured by ELISA, with IL-1α being the predominant isoform induced (n=3–5, P=0.0037 between IL-1α and IL-1β after 6 hours). *P<0.05, **P<0.01, ***P<0.001.
Fig. 7.
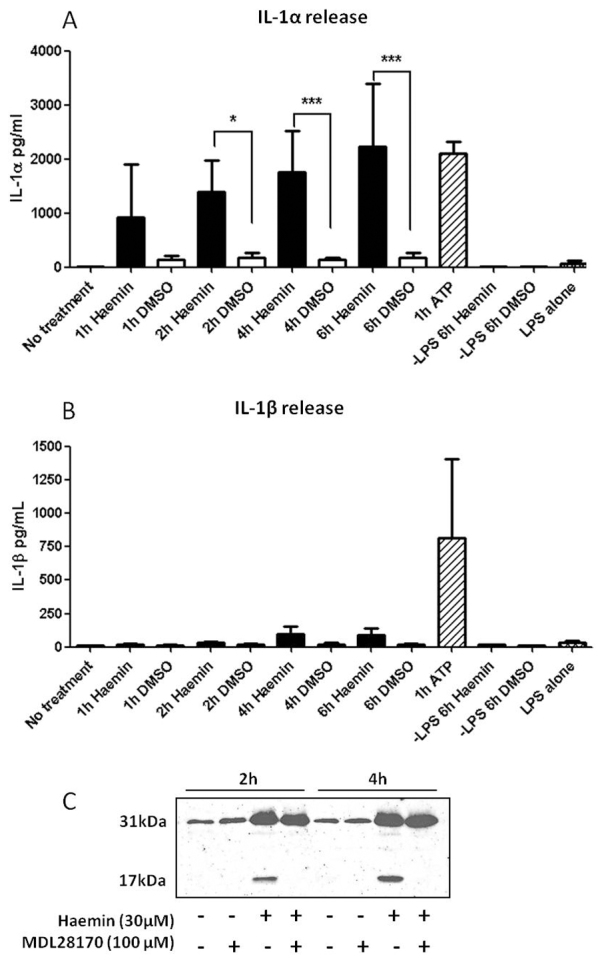
Haemin induces the release of IL-1α but not IL-1β in primary mixed glia. Graphs show the release of IL-1α and IL-1β in LPS-treated mouse primary mixed glial cells after haemin (30 μM; black bars) or vehicle (DMSO; white bars) for 1, 2, 4 and 6 hours. ATP (5 μM) stimulation for 1 hour was used as a positive control (striped bar). IL-1α release was significantly increased after 2, 4 and 6 hours of haem stimulation compared with vehicle (A). Haem stimulation had no effect on release of IL-1β (B). n=4, one-way ANOVA followed by Student’s t-test with Bonferroni’s correction; *P<0.05, **P<0.01, ***P<0.001. Western blot analysis revealed that haemin (30 μM) induced cleavage of pro-IL-1α (31 kDa) to the mature form of IL-1α (17 kDa) through a calpain-1-dependent mechanism, because release of mature IL-1α was blocked by the calpain-1 inhibitor MDL28170 (100 μM) (C).
DISCUSSION
Despite an aging population, mortality rates of SAH patients have declined over the past three decades owing to improved diagnostics and surgical interventions (Nieuwkamp et al., 2009). However, pharmacological treatment options for SAH are limited. The only licensed drug to treat SAH patients is nimodipine, a calcium channel antagonist, of which the benefit to patients is not universal (Dorhout Mees et al., 2008). Here, we report for the first time that IL-1Ra, an emerging CNS therapeutic, is beneficial in a clinically relevant animal model of SAH.
We have recently performed a dose-ranging study that shows that IL-1Ra given intravenously is safe in SAH patients and can achieve experimentally effective concentrations (for the treatment of ischaemic stroke) in the CNS, within a therapeutic time window (Galea et al., 2010). This study adds to the current promise of IL-1Ra as a neuroprotectant in stroke (Greenhalgh et al., 2011) and provides new evidence that brain injury after SAH could be reduced with IL-1Ra treatment. We are currently performing a double-blinded, randomised, placebo-controlled Phase 2 clinical trial investigating inflammatory markers in plasma and CSF after IL-1Ra treatment in SAH (N. Singh, N.J.R., S.M.A. et al., unpublished data) that will provide further information on the potential of IL-1Ra as a treatment for this disease.
Clinically relevant animal models of disease are essential for the development of therapeutics, and guidelines set out by the stroke research community aim to improve the translation from preclinical research to clinical trials (Fisher et al., 2009). The severity of SAH and the heterogeneous outcome after a bleed have resulted in the implementation of many different animal models of SAH. Rodents are widely used in these models, for a variety of reasons, and these are reviewed and compared elsewhere (Gules et al., 2002; Lee et al., 2009; Prunell et al., 2003; Prunell et al., 2005; Titova et al., 2009). Of these models, it is generally accepted that intraluminal perforation of the internal carotid bifurcation most closely mimics clinical SAH; however, a high mortality rate and variation in blood loads are limitations of its use (Lee et al., 2009). Therefore, in the present study we used a modified intraluminal perforation model first described by Park et al. that employs a hollow polytetrafluoroethylene (PTFE) tube concealing a tungsten wire for a cleaner puncture of the artery wall, which reduces mortality (Park et al., 2008). This model faithfully represents human SAH, in the respect that brain injury is diffuse and highly heterogeneous between animals. Although representative of the human disease, the large inter-animal variation makes it difficult to assess outcome after SAH. To reduce variation between groups we only assessed animals with large blood loads for analysis, because these animals are more likely to have brain injury (Greenhalgh et al., 2012). Previous studies have often focussed on vasospasm, by measurement of basilar artery narrowing, as an outcome measure in animal models of SAH (Titova et al., 2009), despite there being little evidence that this correlates with neuronal cell loss in these models. Recent clinical evidence also shows that reduced vasospasm does not improve patient outcome (Vergouwen et al., 2011). Therefore, we believe that the current model represents a viable option for the investigation of neuroprotective drugs in SAH.
The mechanisms underlying brain injury and poor clinical outcome after SAH are complex. A severe complication for survivors of the initial bleed is delayed cerebral ischaemia (DCI) as defined by cerebral infarction identified on computed tomography (CT) or magnetic resonance imaging (MRI), and clinical deterioration (Vergouwen et al., 2010). The cause of DCI is unknown, but vasospasm (Macdonald et al., 2007), microemboli (Vergouwen et al., 2008), spreading depression (Dreier et al., 2009) and inflammation (Chaichana et al., 2010) have all been proposed to play a role, and it is likely that they act in combination.
A recent proposal is that extravasated RBCs in the subarachnoid space are the underlying driver of vasospasm and/or DCI. These RBCs are phagocytosed by infiltrating macrophages and neutrophils, and are then trapped, die and degranulate, releasing endothelins and oxygen free radicals, which result in inflammatory-induced arterial vasoconstriction (Chaichana et al., 2010). We hypothesised that the extravasated RBCs also provide haem, which acts as a DAMP to microglia and macrophages and exacerbates the inflammatory response via the production of IL-1α, worsening brain injury. We show that expression of HO-1 is upregulated throughout the brain after SAH and that IL-1α, but not IL-1β, is produced by glial cells in vivo after SAH and in vitro after haem stimulation. This evidence correlates well with important studies on sterile inflammation in the periphery in mice; these studies identify macrophages as the key cell type sensing sterile injury, and provide evidence that they produce IL-1α to elicit the inflammatory response (Chen et al., 2007; Kono et al., 2010b; Rock et al., 2010). The present study is the first to suggest that microglia/macrophages in the CNS sense sterile inflammation through the same IL-1α-mediated mechanism. It is also the first to identify haem as a DAMP that contributes to IL-1-mediated inflammation in SAH. Furthermore, we have recently shown that IL-1α expression is localised to areas of focal neuronal loss and penumbral tissues after experimental stroke (Luheshi et al., 2011). The present study provides further evidence that IL-1α is involved in cellular injury after a brain insult, because haem induced IL-1α expression and cell death in OSCs, an effect that was blocked with IL-Ra treatment, and inhibiting IL-1α with IL-1Ra reduced measures of brain injury after in vivo SAH.
The release of haem after SAH is a result of RBC lysis in the subarachnoid space. The current work highlights the inflammatory actions of haem before it is metabolised by the HO enzymes to carbon monoxide, biliverdin and iron (Ascenzi et al., 2005), and other work describes the toxicity of haem independent of its iron metabolite (Dang et al., 2011). However, haem metabolism and the release of iron are also implicated in the pathogenesis of SAH (Borsody et al., 2006; Chaichana et al., 2010; Lee et al., 2010). Hgb released from RBCs is highly toxic but is scavenged by macrophages after it has been bound by the serum protein haptoglobin (Ascenzi et al., 2005). Humans express only one of three types of haptoglobin (α1-α1, α1-α2 or α2-α2) and the α2 subunit is associated with a lower Hgb binding affinity. SAH patients expressing the α2 subunit are more likely to suffer from vasospasm than those expressing haptoglobin α1-α1 (Borsody et al., 2006). Oxidation of un-scavenged Hgb releases prosthetic haem groups, which we propose act as DAMPs to exacerbate inflammation and brain injury. Expression of HO-1 and/or the iron handling proteins transferrin (Tf), Tf receptor and ferritin is associated with experimental SAH brain injury, and iron chelation with deferoxamine (DFX) reduces injury (Lee et al., 2010). Furthermore, HO-1 increases brain injury after intracerebral haemorrhage, and bilirubin (a product of haem breakdown) and its oxidation products, known as BOXes, have been implicated in SAH pathogenesis (Clark and Sharp, 2006; Isikay et al., 2011; Wang and Dore, 2007). Thus, a picture is now emerging that several stages of RBC lysis (including activation of cellular processes that are involved in managing its products) might contribute to brain injury after SAH. As such, combinatorial approaches to target each step of this process could prove beneficial after SAH. DFX is safe in patients and is licensed to treat iron toxicity in haemochromatosis (Kontoghiorghes et al., 2005). In SAH, DFX is proposed to target the toxic end product of RBC lysis, iron, and could act synergistically with IL-1Ra, which we propose targets the inflammatory effects of haem. Thus, a dual therapy combing two clinically approved drugs could show increased efficacy and is an attractive option for future study.
In summary, IL-1Ra improves measures of brain injury in a clinically relevant model of SAH by inhibiting IL-1α-driven inflammation. We report haem as a newly identified inducer of IL-1α-mediated sterile inflammation after SAH, implicating it as a DAMP. The products of RBC lysis are abundant in the SAH brain and represent a major target for therapeutic intervention. This is the first report to highlight IL-1Ra as a possible treatment option for SAH and ongoing clinical trials will investigate this further.
METHODS
Animals
Studies were conducted on male Wistar rats (Charles River, UK) weighing 350–500 g under the UK Animals (Scientific Procedures) Act 1986. The animals were kept under a 12-hour light-dark cycle with free access to food and water.
Surgical procedures
Anaesthesia was induced (3%) and maintained (2%) by inhalation of isoflurane, 70% N2O and 30% O2, and core body temperature was maintained throughout the procedure at 37.0±0.5°C. For cerebral blood flow (CBF) monitoring using Laser Doppler (Oxford Optronix, UK), a midline scalp incision was made revealing the skull and bregma. A small burr hole was drilled to the last compact bone layer (5 mm lateral and 1 mm posterior to bregma). A drop of mineral oil was placed in the cavity to aid optical connectivity and the glass optical probe was placed into the cavity and fixed using Loctite superglue (Henkel, UK). Animals were then rotated to the supine position in preparation for the induction of SAH.
SAH
Experimental SAH was induced using a modification of the endovascular perforation model described previously (Park et al., 2008) and further characterised by our research group (Greenhalgh et al., 2012). Briefly, animals were laid supine and a midline neck incision was made, followed by blunt dissection and retraction of superficial tissue and muscle. The carotid arteries were visualised and the external carotid artery (ECA) was coagulated, cut and reflected inferiorly. A hollow PTFE tube (Braintree Scientific, SUBL-120; I.D.: 150 μm.; O.D.: 300 μm), concealing a tungsten wire [75 μm diameter (Advent, UK)], was inserted into the right ECA and advanced ∼18 mm along the internal carotid artery (ICA) until it reached the middle cerebral artery (MCA)-anterior cerebral artery (ACA) bifurcation, as verified by a drop in Laser Doppler signal. The tungsten wire was then advanced through the tubing to protrude 2 mm, perforating the MCA-ACA bifurcation; the wire and tube then were withdrawn immediately. A drop in blood flow that was maintained after withdrawal indicated successful perforation of the artery. Animals that did not survive the first 10 minutes after haemorrhage were not included in the study. In sham animals, the PTFE tube was advanced to MCA-ACA bifurcation and then withdrawn without perforation. Animals were then immediately allowed to recover.
CSF sampling
Rats were anaesthetised and secured in a stereotaxic frame (Stoelting Co., IL). A midline incision was made between the ears, ending approximately 2 cm caudally. The fascia was retracted and muscles bluntly dissected to reveal atlanto-occipital membrane. A 28 gauge needle attached to a 1 ml syringe was used to pierce the membrane and aspirate CSF.
Experimental groups
To assess acute brain inflammation after SAH, rats underwent SAH (n=6) or sham (n=3) surgery and were allowed to recover for 12 hours. To investigate the inflammatory response after experimental SAH, and in particular the role of IL-1, animals were randomised to three groups; sham (n=9), SAH receiving IL-1Ra (n=19) or SAH receiving placebo (n=18). SAH groups received a single subcutaneous (s.c.) dose of human-IL-1Ra (r-met-huIL-1RA: Kineret; Amgen, Thousand Oaks, CA) or placebo (Amgen), at 75 mg/kg, 15 minutes after SAH; experimenter was blinded to treatment group. Blood samples were taken from the tail vein immediately prior to surgery and 3 and 24 hours after SAH. At 48 hours, animals were re-anaesthetised and CSF and cardiac blood were sampled. Rats were perfused transcardially with 120 ml of saline solution (0.9%) and liver was harvested before perfusion of cold paraformaldehyde solution (PFA; 4%). Brains were removed and post-fixed in PFA (4%) for 24 hours before cryoprotection in sucrose solution (20%) for a further 24 hours.
Assessment of blood load after SAH
After 24 hours cryoprotection, brains were photographed at high resolution (Qimaging, Canada). Images of the brains were then analysed using a recently characterised grading system for bleed scale after SAH in rats (Sugawara et al., 2008). Briefly, the basal cistern was divided into six segments. Each segment was allotted a grade from 0 to 3 depending on the amount of subarachnoid blood clot in the segment as follows – Grade 0: no subarachnoid blood; Grade 1: minimal subarachnoid blood; Grade 2: moderate blood clot with recognisable arteries; Grade 3: blood clot obliterating all arteries within the segment. The animals received a total score ranging from 0 to 18 after adding the scores from all six segments. A previous study from our laboratory showed positive correlation between blood load and neuronal damage (Greenhalgh et al., 2012), and only animals with a blood grade ≥7 incur neuronal loss. Variability in blood load grade is high in this model, which has been recognised by various authors (Sugawara et al., 2008). To investigate the role of inflammation in brain injury after SAH we therefore decided a priori to only assess animals that had a blood load grade ≥7.
Histological and immunohistochemical analysis
Coronal brain sections were cut 30 μm thick using a freezing sledge microtome and stored free floating in cryoprotectant solution (6.80 g Na2HPO4 × 2H2O, 0.79 g NaH2PO4 × H2O, 500 ml dH2O, 200 ml glycerol, 300 ml ethylene glycol). Brain sections were mounted and the Nissl stain cresyl violet (Sigma, UK) was used to assess cell morphology and infarct location. Brain sections were analysed under a light microscope and areas of neuronal cell death recorded.
Double immunofluorescence was used with the following antibodies: anti-IL-1α or anti-IL-1β raised in goat (1:100; R&D Systems, UK); microglial marker anti-Iba1, raised in rabbit (1:1000; Wako Chemicals, Germany); anti-HO-1 raised in rabbit (1:400; Enzo Life Sciences, UK). After blocking in 2% normal donkey serum (Vector Laboratories, CA) sections were incubated overnight at 4°C in primary antibodies, which were then visualised with the appropriate fluorochrome-conjugated secondary antisera [Alexa-Fluor-594 (1:500) or Alexa-Fluor-488 (1:500); Molecular Probes). Mounting medium Prolong Gold with DAPI (Invitrogen, UK) was used to mount sections. For immunohistochemistry, endogenous peroxidise activity was blocked with 0.3% hydrogen peroxide (Sigma, UK) in dH2O and sections were treated with 2% normal serum (Vector Laboratories, CA) for 1 hour at room temperature. Biotinylated rabbit anti-rat IgG antibody (1:500; Vector Laboratories, UK) was visualised with the avidin-biotinylated peroxidase complex (Vectastain ABC elite kit) and its substrate diaminobenzidine (DAB; Vector Laboratories, UK). The same procedures were followed using tissue sections without adding primary antibodies and were used as controls for nonspecific secondary antibody labelling. Brightfield images were collected on an Axiocam colour CCD camera (Zeiss, Germany) upright microscope through AxioVision software (Zeiss, Germany). Fluorescent images were collected on an Olympus BX51 upright microscope and captured using a CoolSNAP ES camera (Photometrics, UK) through MetaVue software (Molecular Devices, UK). Specific band-pass filter sets for DAPI, FITC and Texas Red were used to prevent bleed-through from one channel to the next. For quantification of IL-1α-positive microglial cells, total Iba1-positive cells were counted at 20× magnification, immediately adjacent to the site of perforation (n=5). The number of these cells that were also IL-1α positive was expressed as a percentage of the total number of microglial cells.
Assessment of neuronal damage
We devised a semi-quantitative measure of neuronal damage that assesses cell death across brain regions as follows: brains were divided into three regions (hippocampus, cortex and striatum) and scored using the following criteria: 0, no neuronal damage; 1, slight histological abnormalities; 2, focal areas of neuronal loss; and 3, majority of neurons in the region lost. Animals received a total score from all three regions.
Assessment of BBB damage
To assess BBB damage, after immunohistochemical staining for endogenous rat IgG, nine coronal sections were selected that neuroanatomically corresponded to the following coordinates in the rat atlas (Paxinos and Watson, 1998) with respect to bregma (4.2, 3.2, 1.2, −0.26, −1.3, −1.8, −3.14, −4.8, −6.8 mm). This was based on the initial report, which showed that eight sections were sufficient to accurately calculate volumes in the damaged hemisphere (Osborne et al., 1987). Because the region of IgG infiltration was almost exclusively in the ipsilateral hemisphere, the extent of BBB breakdown was corrected for oedema as described previously (Lin et al., 1993). Areas of BBB damage were determined and the total volume was quantified by creating a curve using GraphPad Prism of immunopositive area against distance between coronal levels. The area under the curve represented the volume of BBB breakdown. Blinding to treatment groups was carried out during histological, immunofluorescent and immunohistochemical analysis.
Plasma, cerebrospinal fluid and liver cytokine quantification
The concentrations of IL-1α, IL-6, TNFα, IL-10 and IFNγ were quantified using cytometric bead array (CBA) Flex Sets (BD Biosciences) according to the manufacturer’s protocol in: plasma, prior to surgery and 3, 24 and 48 hours after SAH; and in CSF and liver 48 hours after SAH. CXCL1 was measured, at the above times, using an ELISA (enzyme-linked immunosorbent assay) DuoSet kit (R&D Systems, Abingdon, UK).
OSCs
For hippocampal OSC, brains of C57BL/6J mice (postnatal day 6–7) were prepared according to the protocol described previously (Stoppini et al., 1991). Briefly, 300-μm transverse sections were cut using a vibrating microtome (Leica Microsystems, UK). Hippocampi were dissected out and transferred to 0.4-μm porous membrane inserts (Millipore, Watford, UK). Four hippocampal sections were plated on each 30 mm insert in a six-well plate containing 1 ml of medium [50% HEPES buffered-MEM, 25% heat-inactivated horse serum, 24% HBSS with 2 mM glutamine and 1% penicillin/streptomycin (P/S)]. OSCs were maintained at 37°C, 5% CO2. Full media change was made the next day and every other day until treatment. On day 6, OSCs were transferred to serum-free media containing either vehicle (DMSO) or haemin (30 μM) for 2, 4 or 6 hours with or without recombinant human IL-1Ra (100 or 500 ng/ml). LPS (1 μg/ml) was used as a positive control. For cell death analysis, PI was added to the media and the OSCs were returned to the incubator for 30 minutes. Afterwards, OSCs were washed with PBS and fixed with PFA 4% for 10 minutes, washed and mounted using Prolong Gold (Invitrogen, UK). PI intensity was analysed using ImageJ (NIH Image). For protein analysis, OSC lysates were prepared using PBS containing 0.1% Triton X-100 and protease inhibitor cocktail (Calbiochem, UK).
Preparation of cell cultures
Mixed glia were cultured from C57BL/6J mice at postnatal day 1–4 as previously described (Pinteaux et al., 2002). Briefly, cerebral cortices were stripped of their meninges and dissociated by trituration in DMEM containing 10% fetal bovine serum (FBS) and 1% P/S. Cells were maintained in humidified 95% air/5% CO2, at 37°C, in DMEM containing 10% FBS and 1% P/S for 14–26 days prior to use.
Cultures were treated with or without LPS (1 μg/ml) for 24 hours prior to haemin (30 μM) or vehicle (DMSO) stimulation for 1, 2, 4 and 6 hours. Incubation (1 hour) of LPS-treated mixed glia with ATP (5 μM) was used as a positive control. In some experiments, cultures were treated with the calpain inhibitor MDL28170 (15 minutes, 100 μM) before the addition of haem. Untreated cultures were used as a negative control.
ELISA
Murine IL-1α and IL-1β were quantified by ELISA (DuoSet, R&D Systems, UK) according to the manufacturer’s instructions. Cell lysates were prepared by incubating cultured cells on ice for 10 minutes in 0.1% Triton in PBS with protease inhibitors (Calbiochem, UK).
TRANSLATIONAL IMPACT.
Clinical issue
Subarachnoid haemorrhage (SAH), a form of stroke that can occur spontaneously or as a result of a head injury, is a major burden on society. The loss of productive life years from SAH is comparable to that of cerebral infarction, the most common form of stroke. Despite this, pharmacological treatment options for SAH are restricted to nimodipine, a calcium channel agonist with limited efficacy. Appropriate animal models of SAH mimicking human pathophysiology are essential for the development of neuroprotective drugs, but the neuronal loss observed in human SAH is generally not seen in experimental models.
Results
The authors used a recently described protocol to induce SAH in rats that causes widespread neuronal loss. They show that blocking the pro-inflammatory cytokine interleukin-1 (IL-1) using its endogenous receptor antagonist, IL-1Ra, provides benefits in this model by reducing blood-brain-barrier breakdown, a feature that is correlated with neuronal damage. In addition, they found that haem oxygenase-1 (HO-1) was expressed around the bleed site and in the cortex and striatum, indicating the presence of free haem, a breakdown product of haemoglobin that is released from lysed red blood cells in the subarachnoid space. A direct link between haem-induced cell death and IL-1 was confirmed using organotypic slice culture, which showed that haemin-induced neuronal death was dependent on IL-1, and that cells predominantly expressed the IL-1α isoform. These results suggest that haem acts as a danger-associated molecular pattern (DAMP) driving inflammation in SAH.
Implications and future directions
IL-1Ra is a licensed therapy for rheumatoid arthritis, and is the standard treatment for auto-inflammatory diseases. It is also being tested as treatment for stroke and other disorders. Using an animal model that is appropriate for translating novel therapeutics, this study provides the first preclinical evidence that IL-1Ra might also be beneficial in SAH. These new findings also outline a mechanism of action that implicates haem as a driver of central nervous system (CNS) inflammation. Notably, haem has recently been implicated as a key mediator of severe systemic inflammation in sepsis. This and the data presented here lead the authors to propose that free haem acts as a DAMP that initiates inflammation predominantly through the expression and release of IL-1α (as described for other sterile insults). Thus, IL-1α seems to be a key pro-inflammatory cytokine in acute brain injury after SAH, indicating that inhibition of the IL-1 pathway through blocking IL-1β alone might not be an effective treatment. Furthermore, IL-1α should now be considered a potential player in other forms of brain injury, including stroke. Ongoing studies are investigating CNS and peripheral inflammation in SAH patients being treated with IL-1Ra or placebo, and assessing the effects of treatment on outcome. Future work should consider the prophylactic use of IL-1Ra (in SAH patients with delayed cerebral ischaemia) and investigate targeting other products of the red blood cell lysis pathway in combination with IL-1Ra; for example, with the iron chelator deferoxamine, which is also beneficial in experimental SAH.
Western blot
Mixed glial supernatants were harvested and prepared in sample buffer containing 1% β-mercaptoethanol. Samples were boiled and then electrophoresed on 12% SDS-acrylamide gels. Proteins were transferred to a nitrocellulose membrane and blotted with a goat anti-mouse IL-1α polyclonal antibody (R&D Systems, UK) followed by a HRP-conjugated anti-goat antibody, and subsequently detected using enhanced chemiluminescence reagents (ECL, GE Healthcare, UK).
Statistical analysis
Data were analysed using Student’s t-test for comparing two groups; one-way ANOVAs followed by Student’s t-test with Bonferroni’s correction for comparing two or more groups; two-way ANOVAs followed by Student’s t-test with Bonferroni’s correction for experiments with two independent variables; and, for relationships, linear regression analysis. Data are expressed as the mean ± s.d. ***P<0.001, **P<0.01, *P<0.05.
Acknowledgments
The authors thank Peter March and Robert Fernandez (Bioimaging Facility, Faculty of Life Sciences, University of Manchester, UK) for their help with the microscopy.
Footnotes
COMPETING INTERESTS
N.J.R. is a non-executive Director of AstraZeneca but no part of this work involved AstraZeneca.
AUTHOR CONTRIBUTIONS
A.D.G., D.B., S.G., N.J.R. and S.M.A. conceived and designed the experiments. A.D.G., E.M.R. and S.G. performed the experiments and analysed the data. D.B., N.J.R. and S.M.A. contributed reagents/materials/analysis tools. A.D.G., D.B., N.J.R. and S.M.A. wrote the paper.
FUNDING
This work was supported by the Integrative Mammalian Biology (IMB) initiative (A.D.G. and E.M.R.); the Medical Research Council (MRC) UK (G0801040; N.J.R. and S.M.A.); Wellcome Trust (083485/Z/07/Z; D.B.); and Canadian Institute for Health Research (CIHR) (S.G.).
REFERENCES
- Allan S. M., Tyrrell P. J., Rothwell N. J. (2005). Interleukin-1 and neuronal injury. Nat. Rev. Immunol. 5, 629–640 [DOI] [PubMed] [Google Scholar]
- Ascenzi P., Bocedi A., Visca P., Altruda F., Tolosano E., Beringhelli T., Fasano M. (2005). Hemoglobin and heme scavenging. IUBMB Life 57, 749–759 [DOI] [PubMed] [Google Scholar]
- Borsody M., Burke A., Coplin W., Miller-Lotan R., Levy A. (2006). Haptoglobin and the development of cerebral artery vasospasm after subarachnoid hemorrhage. Neurology 66, 634–640 [DOI] [PubMed] [Google Scholar]
- Brough D., Tyrrell P. J., Allan S. M. (2011). Regulation of interleukin-1 in acute brain injury. Trends Pharmacol. Sci. 32, 617–622 [DOI] [PubMed] [Google Scholar]
- Chaichana K. L., Pradilla G., Huang J., Tamargo R. J. (2010). Role of inflammation (leukocyte-endothelial cell interactions) in vasospasm after subarachnoid hemorrhage. World Neurosurg. 73, 22–41 [DOI] [PubMed] [Google Scholar]
- Chen C.-J., Shi Y., Hearn A., Fitzgerald K., Golenbock D., Reed G., Akira S., Rock K. L. (2006). MyD88-dependent IL-1 receptor signaling is essential for gouty inflammation stimulated by monosodium urate crystals. J. Clin. Invest. 116, 2262–2271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C.-J., Kono H., Golenbock D., Reed G., Akira S., Rock K. L. (2007). Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med. 13, 851–856 [DOI] [PubMed] [Google Scholar]
- Chen G. Y., Nuñez G. (2010). Sterile inflammation: sensing and reacting to damage. Nat. Rev. Immunol. 10, 826–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark J. F., Sharp F. R. (2006). Bilirubin oxidation products (BOXes) and their role in cerebral vasospasm after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 26, 1223–1233 [DOI] [PubMed] [Google Scholar]
- Dang T. N., Bishop G. M., Dringen R., Robinson S. R. (2011). The metabolism and toxicity of hemin in astrocytes. Glia 59, 1540–1550 [DOI] [PubMed] [Google Scholar]
- del Zoppo G. J. (2010). Acute anti-inflammatory approaches to ischemic stroke. Ann. N. Y. Acad. Sci. 1207, 143–148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denes A., Thornton P., Rothwell N. J., Allan S. M. (2010). Inflammation and brain injury: Acute cerebral ischaemia, peripheral and central inflammation. Brain Behav. Immun. 24, 708–723 [DOI] [PubMed] [Google Scholar]
- Dinarello C. A. (2011). Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorhout Mees S. M., Rinkel G. J. E., Feigin V. L., Algra A., van den Bergh W. M., Vermeulen M., van Gijn J. (2008). Calcium antagonists for aneurysmal subarachnoid hemorrhage. Stroke 39, 514–515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier J. P., Major S., Manning A., Woitzik J., Drenckhahn C., Steinbrink J., Tolias C., Oliveira-Ferreira A. I., Fabricius M., Hartings J. A., et al. (2009). Cortical spreading ischaemia is a novel process involved in ischaemic damage in patients with aneurysmal subarachnoid haemorrhage. Brain 132, 1866–1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duewell P., Kono H., Rayner K. J., Sirois C. M., Vladimer G., Bauernfeind F. G., Abela G. S., Franchi L., Nunez G., Schnurr M., et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley H. C. A., Smith C. J., Georgiou R. F., Vail A., Hopkins S. J., Rothwell N. J., Tyrrell P. J., the Acute Stroke Investigators (2005). A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J. Neurol. Neurosurg. Psychiatry 76, 1366–1372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fassbender K., Hodapp B., Rossol S., Bertsch T., Schmeck J., Schütt S., Fritzinger M., Horn P., Vajkoczy P., Kreisel S., et al. (2001). Inflammatory cytokines in subarachnoid haemorrhage: association with abnormal blood flow velocities in basal cerebral arteries. J. Neurol. Neurosurg. Psychiatry 70, 534–537 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feigin V. L., Lawes C. M. M., Bennett D. A., Anderson C. S. (2003). Stroke epidemiology: a review of population-based studies of incidence, prevalence, and case-fatality in the late 20th century. Lancet Neurol. 2, 43–53 [DOI] [PubMed] [Google Scholar]
- Feigin V. L., Rinkel G. J. E., Lawes C. M. M., Algra A., Bennett D. A., van Gijn J., Anderson C. S. (2005). Risk factors for subarachnoid hemorrhage: An updated systematic review of epidemiological studies. Stroke 36, 2773–2780 [DOI] [PubMed] [Google Scholar]
- Fisher M., Feuerstein G., Howells D. W., Hurn P. D., Kent T. A., Savitz S. I., Lo E. H. (2009). Update of the stroke therapy academic industry roundtable preclinical recommendations. Stroke 40, 2244–2250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L., Eigenbrod T., Munoz-Planillo R., Nunez G. (2009). The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 10, 241–247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frijns C. J. M., Kasius K. M., Algra A., Fijnheer R., Rinkel G. J. E. (2006). Endothelial cell activation markers and delayed cerebral ischaemia in patients with subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 77, 863–867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galea J., Ogungbenro K., Hulme S., Greenhalgh A., Aarons L., Scarth S., Hutchinson P., Grainger S., King A., Hopkins S. J., et al. (2010). Intravenous anakinra can achieve experimentally effective concentrations in the central nervous system within a therapeutic time window: results of a dose-ranging study. J. Cereb. Blood Flow Metab. 2, 439–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenhalgh A. D., Ogungbenro K., Rothwell N. J., Galea J. P. (2011). Translational pharmacokinetics: challenges of an emerging approach to drug development in stroke. Expert Opin. Drug Metab. Toxicol. 7, 681–695 [DOI] [PubMed] [Google Scholar]
- Greenhalgh A. D., Rothwell N. J., Allan S. M. (2012). An Endovascular Perforation Model of Subarachnoid Haemorrhage in Rat Produces Heterogeneous Infarcts that Increase with Blood Load. Transl. Stroke Res. 3, 164–172 [DOI] [PubMed] [Google Scholar]
- Gules I., Satoh M., Clower B. R., Nanda A., Zhang J. H. (2002). Comparison of three rat models of cerebral vasospasm. Am. J. Physiol. Heart Circ. Physiol. 283, H2551–H2559 [DOI] [PubMed] [Google Scholar]
- Hannum C. H., Wilcox C. J., Arend W. P., Joslin F. G., Dripps D. J., Heimdal P. L., Armes L. G., Sommer A., Eisenberg S. P., Thompson R. C. (1990). Interleukin-1 receptor antagonist activity of a human interleukin-1 inhibitor. Nature 343, 336–340 [DOI] [PubMed] [Google Scholar]
- Isikay I., Bilginer B., Narin F., Soylemezoglu F., Akalan N. (2011). The effect of intracisternal Zn (II) protoporphyrin IX on vasospasm process in the experimental subarachnoid hemorrhage model. Acta Neurochir. Suppl. 110, 33–37 [DOI] [PubMed] [Google Scholar]
- Ito D., Imai Y., Ohsawa K., Nakajima K., Fukuuchi Y., Kohsaka S. (1998). Microglia-specific localisation of a novel calcium binding protein, Iba1. Mol. Brain Res. 57, 1–9 [DOI] [PubMed] [Google Scholar]
- Jin R., Yang G., Li G. (2010). Inflammatory mechanisms in ischemic stroke: role of inflammatory cells. J. Leukoc. Biol. 87, 779–789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasius K. M., Frijns C. J. M., Algra A., Rinkel G. J. E. (2010). Association of platelet and leukocyte counts with delayed cerebral ischemia in aneurysmal subarachnoid hemorrhage. Cerebrovasc. Dis. 29, 576–583 [DOI] [PubMed] [Google Scholar]
- Kono H., Chen C.-J., Ontiveros F., Rock K. L. (2010a). Uric acid promotes an acute inflammatory response to sterile cell death in mice. J. Clin. Invest. 120, 1939–1949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kono H., Karmarkar D., Iwakura Y., Rock K. L. (2010b). Identification of the cellular sensor that stimulates the inflammatory response to sterile cell death. J. Immunol. 184, 4470–4478 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kontoghiorghes G., Eracleous E., Economides C., Kolnagou A. (2005). Advances in iron overload therapies. prospects for effective use of deferiprone (L1), deferoxamine, the new experimental chelators ICL670, GT56-252, L1NA11 and their combinations. Curr. Med. Chem. 12, 2663–2681 [DOI] [PubMed] [Google Scholar]
- Korherr C., Hofmeister R., Wesche H., Falk W. (1997). A critical role for interleukin-1 receptor accessory protein in interleukin-1 signaling. Eur. J. Immunol. 27, 262–267 [DOI] [PubMed] [Google Scholar]
- Larsen R., Gozzelino R., Jeney V., Tokaji L. S., Bozza F. A., Japiasse A. M., Bonaparte D., Cavalcante M. M., Chora A., Ferreira A., et al. (2010). A central role for free heme in the pathogenesis of severe sepsis. Sci. Transl. Med. 2, 51–71 [DOI] [PubMed] [Google Scholar]
- Lee J., Sagher O., Keep R., Hua Y., Xi G. (2009). Comparison of experimental rat models of early brain injury after subarachnoid hemorrhage. Neurosurgery 65, 331–343 [DOI] [PubMed] [Google Scholar]
- Lee J.-Y., Keep R. F., He Y., Sagher O., Hua Y., Xi G. (2010). Hemoglobin and iron handling in brain after subarachnoid hemorrhage and the effect of deferoxamine on early brain injury. J. Cereb. Blood Flow Metab. 30, 1793–1803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin T. N., He Y. Y., Wu G., Khan M., Hsu C. Y. (1993). Effect of brain edema on infarct volume in a focal cerebral ischemia model in rats. Stroke 24, 117–121 [DOI] [PubMed] [Google Scholar]
- Luheshi N., Kovacs K., Lopez-Castejon G., Brough D., Denes A. (2011). Interleukin-1alpha expression precedes IL-1beta after ischemic brain injury and is localised to areas of focal neuronal loss and penumbral tissues. J. Neuroinflammation 8, 186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald R. L., Pluta R. M., Zhang J. H. (2007). Cerebral vasospasm after subarachnoid hemorrhage: the emerging revolution. Nat. Clin. Pract. Neuro. 3, 256–263 [DOI] [PubMed] [Google Scholar]
- Matz P. G., Weinstein P. R., Sharp F. R. (1997). Heme oxygenase-1 and heat shock protein 70 induction in glia and neurons throughout rat brain after experimental intracerebral hemorrhage. Neurosurgery 40, 152–162 [DOI] [PubMed] [Google Scholar]
- McColl B. W., Allan S. M., Rothwell N. J. (2009). Systemic infection, inflammation and acute ischemic stroke. Neuroscience 158, 1049–1061 [DOI] [PubMed] [Google Scholar]
- Nakahara T., Tsuruta R., Kaneko T., Yamashita S., Fujita M., Kasaoka S., Hashiguchi T., Suzuki M., Maruyama I., Maekawa T. (2009). High-mobility group box 1 protein in CSF of patients with subarachnoid hemorrhage. Neurocrit. Care 11, 362–368 [DOI] [PubMed] [Google Scholar]
- Netea M. G., van de Veerdonk F. L., Kullberg B. J., Van der Meer J. W. M., Joosten L. A. B. (2008). The role of NLRs and TLRs in the activation of the inflammasome. Expert Opin. Biol. Ther. 8, 1867–1872 [DOI] [PubMed] [Google Scholar]
- Niemi K., Teirila L., Lappalainen J., Rajamaki K., Baumann M. H., Oorni K., Wolff H., Kovanen P. T., Matikainen S., Eklund K. K. (2011). Serum amyloid A activates the NLRP3 inflammasome via P2X7 Receptorrand a cathepsin B-sensitive pathway. J. Immunol. 186, 6119–6128 [DOI] [PubMed] [Google Scholar]
- Nieuwkamp D. J., Setz L. E., Algra A., Linn F. H. H., de Rooij N. K., Rinkel G. J. E. (2009). Changes in case fatality of aneurysmal subarachnoid haemorrhage over time, according to age, sex, and region: a meta-analysis. Lancet Neurol. 8, 635–642 [DOI] [PubMed] [Google Scholar]
- Nimmerjahn A., Kirchhoff F., Helmchen F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 308, 1314–1318 [DOI] [PubMed] [Google Scholar]
- Osborne K. A., Shigeno T., Balarsky A. M., Ford I., McCulloch J., Teasdale G. M., Graham D. I. (1987). Quantitative assessment of early brain damage in a rat model of focal cerebral ischaemia. J. Neurol. Neurosurg. Psychiatry 50, 402–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park I.-S., Meno J. R., Witt C. E., Suttle T. K., Chowdhary A., Nguyen T.-S., Ngai A. C., Britz G. W. (2008). Subarachnoid hemorrhage model in the rat: Modification of the endovascular filament model. J. Neurosci. Methods 172, 195–200 [DOI] [PubMed] [Google Scholar]
- Paxinos G., Watson C. (1998). The rat brain in stereotaxic coordinates. New York: Academic Press; [DOI] [PubMed] [Google Scholar]
- Pinteaux E., Parker L. C., Rothwell N. J., Luheshi G. N. (2002). Expression of interleukin-1 receptors and their role in interleukin-1 actions in murine microglial cells. J. Neurochem. 83, 754–763 [DOI] [PubMed] [Google Scholar]
- Prunell G., Mathiesen T., Diemer N., Svendgaard N. (2003). Experimental subarachnoid hemorrhage: subarachnoid blood volume, mortality rate, neuronal death, cerebral blood flow, and perfusion pressure in three different rat models. Neurosurgery 52, 165–175 [DOI] [PubMed] [Google Scholar]
- Prunell G. F., Svendgaard N.-A., Alkass K., Mathiesen T. (2005). Delayed cell death related to acute cerebral blood flow changes following subarachnoid hemorrhage in the rat brain. J. Neurosurg. 102, 1046–1054 [DOI] [PubMed] [Google Scholar]
- Rabinstein A. A., Weigand S., Atkinson J. L. D., Wijdicks E. F. M. (2005). Patterns of cerebral infarction in aneurysmal subarachnoid hemorrhage. Stroke 36, 992–997 [DOI] [PubMed] [Google Scholar]
- Rock K. L., Latz E., Ontiveros F., Kono H. (2010). The sterile inflammatory response. Ann. Rev. Immunol. 28, 321–342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rother R. P., Bell L., Hillmen P., Gladwin M. T. (2005). The clinical sequelae of intravascular hemolysis and extracellular plasma hemoglobin. JAMA 293, 1653–1662 [DOI] [PubMed] [Google Scholar]
- Rothoerl R. D., Axmann C., Pina A.-L., Woertgen C., Brawanski A. (2006). Possible role of the C-Reactive protein and white blood cell count in the pathogenesis of cerebral vasospasm following aneurysmal subarachnoid hemorrhage. J. Neurosurg. Anesthesiol. 18, 68–72 [DOI] [PubMed] [Google Scholar]
- Schoch B., Regel J. P., Wichert M., Gasser T., Volbracht L., Stolke D. (2007). Analysis of intrathecal interleukin-6 as a potential predictive factor for vasospasm in subarachnoid hemorrhage. Neurosurgery 60, 828–836 [DOI] [PubMed] [Google Scholar]
- Simard J. M., Kent T. A., Chen M., Tarasov K. V., Gerzanich V. (2007). Brain oedema in focal ischaemia: molecular pathophysiology and theoretical implications. Lancet Neurol. 6, 258–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simard M., Zhihua G., Woo K., Svetlana I., Cigdem T., Ludmila M., Volodymyr G. (2008). Glibenclamide reduces inflammation, vasogenic edema, and caspase-3 activation after subarachnoid hemorrhage. J. Cereb. Blood Flow Metab. 29, 317–330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith D. E., Lipsky B. P., Russell C., Ketchem R. R., Kirchner J., Hensley K., Huang Y., Friedman W. J., Boissonneault V., Plante M.-M., et al. (2009). A central nervous system-restricted isoform of the interleukin-1 receptor accessory protein modulates neuronal responses to interleukin-1. Immunity 30, 817–831 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoppini L., Buchs P. A., Muller D. (1991). A simple method for organotypic cultures of nervous tissue. J. Neurosci. Methods 37, 173–182 [DOI] [PubMed] [Google Scholar]
- Streit W. J., Walter S. A., Pennell N. A. (1999). Reactive microgliosis. Prog. Neurobiol. 57, 563–581 [DOI] [PubMed] [Google Scholar]
- Sugawara T., Ayer R., Jadhav V., Zhang J. H. (2008). A new grading system evaluating bleeding scale in filament perforation subarachnoid hemorrhage rat model. J. Neurosci. Meth. 167, 327–334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titova E., Ostrowski R., Zhang J., Tang J. (2009). Experimental models of subarachnoid hemorrhage for studies of cerebral vasospasm. Neurol. Res. 31, 568–581 [DOI] [PubMed] [Google Scholar]
- Turner C. P., Bergeron M., Matz P., Zegna A., Noble L. J., Panter S. S., Sharp F. R. (1998). Heme oxygenase-1 is induced in glia throughout brain by subarachnoid hemoglobin. J. Cereb. Blood Flow Metab. 18, 257–273 [DOI] [PubMed] [Google Scholar]
- Vergouwen M. D., Vermeulen M., Coert B. A., Stroes E. S., Roos Y. B. (2008). Microthrombosis after aneurysmal subarachnoid hemorrhage: an additional explanation for delayed cerebral ischemia. J. Cereb. Blood Flow Metab. 28, 1761–1770 [DOI] [PubMed] [Google Scholar]
- Vergouwen M. D. I., Vermeulen M., van Gijn J., Rinkel G. J. E., Wijdicks E. F., Muizelaar J. P., Mendelow A. D., Juvela S., Yonas H., Terbrugge K. G., et al. (2010). Definition of delayed cerebral ischemia after aneurysmal subarachnoid hemorrhage as an outcome event in clinical trials and observational studies: proposal of a multidisciplinary research group. Stroke 41, 2391–2395 [DOI] [PubMed] [Google Scholar]
- Vergouwen M. D., Ilodigue D., Macdonald R. L. (2011). Cerebral infarction after subarachnoid hemorrhage contributes to poor outcome by vasospasm-dependent and independent effects. Stroke 42, 924–929 [DOI] [PubMed] [Google Scholar]
- Wagner K. R., Sharp F. R., Ardizzone T. D., Lu A., Clark J. F. (2003). Heme and iron metabolism: role in cerebral hemorrhage. J. Cereb. Blood Flow Metab. 23, 629–652 [DOI] [PubMed] [Google Scholar]
- Wang J., Dore S. (2007). Heme oxygenase-1 exacerbates early brain injury after intracerebral haemorrhage. Brain 130, 1643–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q., Tang X. N., Yenari M. A. (2007). The inflammatory response in stroke. J. Neuroimmunol. 184, 53–68 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wyss-Coray T. (2006). Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat. Med. 12, 1005–1015 [DOI] [PubMed] [Google Scholar]