

SUMMARY
Friedreich’s ataxia (FRDA) is the most common hereditary ataxia in the caucasian population and is characterized by a mixed spinocerebellar and sensory ataxia, hypertrophic cardiomyopathy and increased incidence of diabetes. FRDA is caused by impaired expression of the FXN gene coding for the mitochondrial protein frataxin. During the past ten years, the development of mouse models of FRDA has allowed better understanding of the pathophysiology of the disease. Among the mouse models of FRDA, the liver conditional mouse model pointed to a tumor suppressor activity of frataxin leading to the hypothesis that individuals with FRDA might be predisposed to cancer. In the present work, we investigated the presence and the incidence of neoplasia in the largest FRDA patient cohorts from the USA, Australia and Europe. As no predisposition to cancer could be observed in both cohorts, we revisited the phenotype of the liver conditional mouse model. Our results show that frataxin-deficient livers developed early mitochondriopathy, iron-sulfur cluster deficits and intramitochondrial dense deposits, classical hallmarks observed in frataxin-deficient tissues and cells. With age, a minority of mice developed structures similar to the ones previously associated with tumor formation. However, these peripheral structures contained dying, frataxin-deficient hepatocytes, whereas the inner liver structure was composed of a pool of frataxin-positive cells, due to inefficient Cre-mediated recombination of the Fxn gene, that contributed to regeneration of a functional liver. Together, our data demonstrate that frataxin deficiency and tumorigenesis are not associated.
INTRODUCTION
Ataxias are a heterogeneous group of neurological disorders characterized by loss of coordination due to alteration of cerebellar functions. Friedreich’s ataxia (FRDA) is the most prevalent hereditary ataxia in Caucasians. FRDA is characterized by progressive spinocerebellar and sensory ataxia with absence of deep tendon reflexes, dysarthria, pyramidal signs, muscular weakness and positive extensor plantar responses (Harding, 1981; Pandolfo and Pastore, 2009). Neurological symptoms result from degeneration of large sensory neurons in the dorsal root ganglia (DRG) and posterior columns in the spinal cord, followed by degeneration of the spinocerebellar and corticospinal tracts and of the dentate nucleus. FRDA is also characterized by primary non-neurological manifestations including hypertrophic cardiomyopathy and increased incidence of diabetes (Harding and Hewer, 1983). The mutated gene in FRDA encodes a small protein called frataxin (Campuzano et al., 1997; Campuzano et al., 1996). All FRDA patients carry at least one expansion of a GAA triplet repeat in the first intron of the FXN gene. Most patients are homozygous for this mutation, but a few patients (4%) are compound heterozygous for the GAA expansion and a different mutation (nonsense, missense, deletions, insertions) leading to loss of frataxin function (Campuzano et al., 1996; Cossee et al., 1999; Gellera et al., 2007). The presence of the GAA expansion leads to transcriptional silencing of FXN, resulting in diminished (but not absent) residual expression of a structurally and functionally normal frataxin protein in patients. The pathophysiology of FRDA in patients is characterized by intracellular iron deposits (Lamarche et al., 1980) and a deficit in mitochondrial iron-sulfur (Fe-S) cluster-containing enzymes (aconitase and respiratory chain complexes I–III) (Rotig et al., 1997). The presence of markers of oxidative damage in blood and urine samples of individuals with FRDA has also been reported (Bradley et al., 2004; Emond et al., 2000; Schulz et al., 2000), but the correlation between these markers and disease features is complicated (Di Prospero et al., 2007; Myers et al., 2008; Schulz et al., 2009).
Frataxin is a small evolutionary conserved protein and is located in mitochondria in eukaryotes (Campuzano et al., 1997; Schmucker et al., 2008). Although the exact function of frataxin is still unclear, there is now clear evidence that frataxin is involved in Fe-S cluster biogenesis through its binding to the core complex of the mitochondrial de novo Fe-S cluster machinery (Schmucker et al., 2011; Tsai and Barondeau, 2010).
TRANSLATIONAL IMPACT.
Clinical issue
Friedreich’s ataxia (FRDA) is the most common hereditary ataxia, affecting 1 in 50,000 individuals in the Caucasian population. FRDA is characterized by a mixed sensory and spinocerebellar ataxia, primarily resulting from the degeneration of sensory neurons in the dorsal root ganglia, and by primary non-neurological manifestations, including hypertrophic cardiomyopathy and glucose intolerance. The disease-causing gene encodes frataxin, a small mitochondrial protein involved in the biogenesis of iron-sulfur clusters, which are vital cellular cofactors. FRDA is caused by an intronic GAA repeat expansion mutation that is present on both alleles in most patients, leading to a drastic decrease of frataxin expression. In the past 10 years, the development of mouse models of FRDA using conditional approaches or GAA-based strategies has advanced our understanding of FRDA genetics and pathophysiology, and of the cellular function of frataxin. Interestingly, the characterization of a liver-specific conditional mouse model (obtained through deletion of the frataxin gene using the albumin-promoter-driven Cre recombinase) showed that frataxin deficiency caused liver tumorigenesis, suggesting that frataxin is a tumor suppressor protein. Combining these results with rare case reports of neoplasia in individuals with FRDA led to the hypothesis that individuals with FRDA might be predisposed to cancer.
Results
This paper combines the clinical investigations on cancer reports carried out on the largest cohorts of individuals with FRDA from the United States, Australia and Europe with a new, more detailed characterization of the liver conditional mouse model. The clinical investigations demonstrate that cancer is not a common finding in individuals with FRDA, and that the incidence of cancer is no higher than in the general population. In addition, a reexamination of the liver conditional mouse model demonstrates that frataxin deficiency triggers early mitochondrial dysfunction, iron-sulfur cluster deficits and cell death, leading to reduced life span in most of the mice. Abnormal peripheral structures, similar to those previously associated with tumor formation, were observed only in a minority of mice with normal life expectancy and were shown to be mainly composed of dying, frataxin-deficient cells, rather than proliferating tumor cells. The majority of the liver in these surviving mice is composed of functional hepatocytes, owing to incomplete deletion of the frataxin gene; these provide a pool of cells for liver regeneration.
Implications and future directions
The data demonstrate that FRDA is not associated with cancer predisposition, unlike other neurodegenerative disorders such as ataxia telangiectasia, a distinct recessive ataxia. The absence of cancer predisposition in FRDA is important for the clinical management of patients and for developing appropriate therapeutic approaches. Furthermore, these findings provide evidence that frataxin is unlikely to be a tumor suppressor and that tumor formation in frataxin-deficient liver was previously reported owing to misinterpretation of an artifact. Although accurate characterization of the liver phenotype underlying frataxin deficiency can be carried out in young mice, these results also illustrate the limitations of the albumin-promoter-driven Cre recombinase system for generating liver-specific conditional mouse models.
To characterize and understand the molecular and genetic basis of FRDA, conditional knockout mice (Puccio et al., 2001; Ristow et al., 2003; Simon et al., 2004; Thierbach et al., 2005) and GAA-based mouse models with partial deficiency of frataxin (Al-Mahdawi et al., 2006; Miranda et al., 2002) were generated. Cardiac and neurological conditional mouse models (Puccio et al., 2001; Simon et al., 2004), as well as mice expressing the human FXN locus containing an expanded GAA repeat (Al-Mahdawi et al., 2006), reproduce clinical and biochemical features observed in FRDA patients, including hypertrophic cardiomyopathy, a mixed sensory and spinocerebellar ataxia, Fe-S cluster enzyme deficit and mitochondrial iron accumulation.
Among the mouse models of FRDA, the conditional Fxn deletion in liver was reported to trigger tumorigenesis (Thierbach et al., 2010; Thierbach et al., 2012; Thierbach et al., 2005), suggesting that frataxin is a tumor suppressor protein and that FRDA might be associated with a predisposition to cancer. At the molecular level, tumorigenesis in frataxin-deficient cells was proposed to be the result of higher cellular sensitivity to oxidative stress and impaired capacity of the cellular DNA repair machinery (Thierbach et al., 2010; Thierbach et al., 2005). Interestingly, Ataxia telangiectasia (AT), a distinct early-onset autosomal recessive ataxia due to mutations in the ATM gene, is biochemically associated with susceptibility to reactive oxygen species and DNA repair defects, and is clinically associated with predisposition to neoplasia (Perlman et al., 2012). To date, no clinical investigation regarding the correlation between FRDA and cancer has ever been pursued, and only rare case reports, which cannot conclusively establish a link between frataxin deficiency and neoplasia in human, are available in the literature (Ackroyd et al., 1996; Barr et al., 1986; De Pas et al., 1999; Kidd et al., 2001; Misiakos et al., 2011).
In the present study, we used natural history data from clinical studies based on the largest cohorts of FRDA patients from the USA, Australia and Europe to investigate predisposition to neoplasia. As no overrepresentation of cancer in individuals with FRDA was observed, we, in parallel, revisited the characterization of the liver-specific conditional mouse model in which the suggestion of a link with tumorigenesis was reported. Our results demonstrate that Fxn deletion in liver triggers early mitochondriopathy, with the classic hallmarks of frataxin deficiency. Only a minority of mice developed abnormal liver structures similar to those previously associated with tumor formation. However, the peripheral abnormal structures were composed mainly of dying, frataxin-deficient hepatocytes whereas the inner liver structure in these mice contained proliferating frataxin-positive hepatocytes that contributed to liver regeneration, increased survival and an increase in body weight. The tumor-like lobules therefore appeared to be degenerating liver masses rather than composed by proliferating cancer cells, as previously suggested by Thierbach and colleagues (Thierbach et al., 2010; Thierbach et al., 2012; Thierbach et al., 2005). Together, our data demonstrate that an association between frataxin deficiency and tumorigenesis should be reconsidered.
RESULTS
Clinical investigations show no higher occurrence of cancer in FRDA patients
To determine the incidence of cancer in individuals with FRDA, we took advantage of ongoing natural history studies based on cohorts from the USA and Australia (FA-COMS cohort), and from Europe (EFACTS cohort). The FA-COMS cohort (n=578) was 50% female with a mean age of 28.1 years (range 7–78). Mean length of the shorter GAA allele was 608 repeats, with 18 people carrying point mutations in conjunction with a single expanded allele. Mean age of onset was 13.8 years. Interim neurologic results from this cohort have previously appeared (Friedman et al., 2010), although none has reported aspects of neoplasia. There were a total of 1694 visits (1115 follow-up visits), including several subjects up to 8 years after the initial visit. The annual follow-up rate at year 1 is 65%, and is largely maintained after that time. Eight neoplasms were identified in seven of the 578 subjects with FRDA (Table 1). These included four dermatologic cancers (two melanomas and two basal cell carcinomas), two breast cancers, one chronic lymphocytic leukemia and one osteosarcoma (Table 1). Excluding the basal cell cancers that are not tracked in the general population statistics, the prevalence of cancer in the FA-COMS cohort was of 1.04% (6/578). Four patients were diagnosed with cancer before their initial study participation, while the remaining cancers were captured during study participation with an incidence of approximately one cancer per 250 patient-years (0.4% per year). Only one patient, with breast cancer, was deceased from the cancer. The EFACTS cohort (with currently 361 patients) was 53.2% females with a mean age of 34.6 (range 7–76). To date, only baseline data are available for this cohort because no follow-up visits have yet started. Molecular diagnosis for FRDA was confirmed in all individuals. Neoplasm was identified in 12 of the 361 individuals with FRDA (Table 2), but only five of them developed cancer: three subjects were diagnosed for breast cancer, one for thyroid carcinoma and one for melanoma (Table 2), therefore giving a prevalence of cancer of 1.38% (5/361).
Table 1.
Clinical investigation of neoplasms in the FA-COMS cohort (n = 578)

Table 2.
Clinical investigation of neoplasms in the EFACTS cohort (n=361)

Together, data obtained with FA-COMS and EFACTS cohorts showed that cancer is not a common finding in the medical history of individuals with FRDA. Furthermore, no high incidence of cancer could be detected after several years of follow-up in the FA-COMS cohort.
Liver-specific deletion of Fxn in mouse triggers early mitochondriopathy
The idea that frataxin might be a tumor suppressor is essentially based on the evidence of tumor-like structure formation in a mouse model in which Fxn was specifically deleted in liver (Alb-Cre-FxnL3/L− mice) (Thierbach et al., 2010; Thierbach et al., 2012; Thierbach et al., 2005). To confirm whether frataxin depletion triggers tumorigenesis in mouse liver, we raised a new generation of liver-specific conditional mice. These mice were obtained as previously described by Thierbach and colleagues (Thierbach et al., 2005), using the same mouse strain carrying the conditional allele that was created in our laboratory (allowing deletion of exon 4 of Fxn) (Puccio et al., 2001), as well as the same mouse strain carrying an albumin promoter-driven Cre transgene (Postic et al., 1999).
The newly generated Alb-Cre-FxnL3/L− mice were born in the expected Mendelian ratio (data not shown). Drastic frataxin depletion in the liver was observed at 2 weeks of age by western blot (Fig. 1A). Both female and male Alb-Cre-FxnL3/L− mice had a normal body weight at birth (Fig. 1B and data not shown), but, starting from 4 weeks on, female and male Alb-Cre-FxnL3/L− mice failed to gain weight (Fig. 1B and data not shown). Life expectancy was also significantly decreased in Alb-Cre-FxnL3/L− mice; 50% died before 8 weeks of age (Fig. 1C). However, approximately 20% of Alb-Cre-FxnL3/L− mice showed a normal life expectancy compared with control mice (Fig. 1C). Interestingly, these surviving Alb-Cre-FxnL3/L− mice (Alb-Cre-FxnL3/L–(S)) went through the same incapacity to thrive as the majority of Alb-Cre-FxnL3/L− mice (Fig. 1B), but then progressively gained weight with age to finally reach levels similar to control levels (Fig. 1B). This phenomenon was observed in both males and females, indicating the absence of a gender bias (data not shown).
Fig. 1.

Conditional deletion of Fxn gene in the liver. A) Representative western blot showing frataxin expression level in liver samples from 2-week-old control and Alb-Cre-FxnL3/L− mice. (B) Weight curves obtained with control (n=12), Alb-Cre-FxnL3/L− with reduced life expectancy (n=10) and surviving Alb-Cre-FxnL3/L− (Alb-Cre-FxnL3/L–(S), n=2) females. Values are shown as the average obtained for each age ± s.d. (C) Survival curves obtained with control (n=16) and Alb-Cre-FxnL3/L− (n=23) mice. (D,E) Photographs of control mouse liver (D) and Alb-Cre-FxnL3/L− mouse liver with strong lipid accumulation (E) at 4 weeks. (F,G) H&E staining of liver sections from control (F) and Alb-Cre-FxnL3/L− (G) mice at 4 weeks. (H,I) Oil Red staining of liver sections from control (H) and Alb-Cre-FxnL3/L− (I) mice. (J) Ultrastructural analysis of Alb-Cre-FxnL3/L− liver at 4 weeks by electron microscopy. m, mitochondria; L, lipid droplets; N, nucleus. Scale bar: 2 μm.
To understand the early consequences of frataxin depletion in liver, mice were dissected at 4 weeks after birth. Macroscopically, steatosis was a constant feature of the liver of Alb-Cre-FxnL3/L− mice (Fig. 1D,E), although the level of lipid accumulation varied between mice (data not shown). On hematoxylin-eosin (H&E) stained sections, the structure of the liver was affected in Alb-Cre-FxnL3/L− mice as compared with control, with larger hepatocytes and the presence of vacuoles characteristic of lipid accumulation (Fig. 1F,G). Steatosis was confirmed by Oil Red staining (Fig. 1H,I). Ultrastructural analysis by electron microscopy showed highly affected hepatocytes in Alb-Cre-FxnL3/L− mice compared with control (Fig. 1J and supplementary material Fig. S1A–C), with the presence of giant mitochondria and abnormal mitochondrial structures showing loss of matrix material and loss of cristae (Fig. 1J and supplementary material Fig. S1C). Lipid droplets were also observed (Fig. 1J). The nuclear structure displayed necrotic features in most hepatocytes (Fig. 1J and supplementary material Fig. S1C). In a few of the affected mitochondria, electron-dense deposits were observed (supplementary material Fig. S1D,E). These aggregates are reminiscent of the characteristic iron deposits identified in patients (Lamarche et al., 1993; Michael et al., 2006) and in the cardiac mouse model of FRDA (Puccio et al., 2001). Together, the results show that the early phenotype is characterized by highly affected hepatocytes with mitochondrial dysfunction and rare but significant electron-dense intramitochondrial deposits, classic hallmarks of frataxin-deficient tissues and cells (Calmels et al., 2009; Michael et al., 2006; Puccio et al., 2001). Furthermore, mitochondrial dysfunction in Alb-Cre-FxnL3/L− mice is associated with liver steatosis.
Lobule structures composed of abnormal hepatocytes progressively form on the liver surface of surviving Alb-Cre-FxnL3/L–(S) mice
The evolution of the liver phenotype with age was analyzed by dissecting the surviving Alb-Cre-FxnL3/L–(S) mice at 15, 20 and 30 weeks. At dissection, livers of Alb-Cre-FxnL3/L–(S) mice were significantly smaller in size (Fig. 2A and supplementary material Fig. S2A), with occasionally one or more lobes of the liver absent (data not shown). Furthermore, progressive formation of abnormal structures on the surface of the liver of Alb-Cre-FxnL3/L–(S) mice was observed (Fig. 2A and supplementary material Fig. S2A). These structures strongly resemble the lobules described by Thierbach and colleagues (Thierbach et al., 2010; Thierbach et al., 2005) that were identified as tumors. No steatosis was observed at these ages, both macroscopically (Fig. 2A) and by Oil Red staining of liver sections (data not shown). H&E stained sections of the lobules revealed that these structures were essentially composed of large cells surrounded by areas of fibrosis (Fig. 2B and supplementary material Fig. S2B,C). Ultrastructural analysis of the lobules showed that the large cells were hepatocytes containing highly proliferating abnormal mitochondria (Fig. 2C–E) with clear matrix and electron-dense deposits (Fig. 2E), the presence of cellular debris, and that some cells had clear features of dying cells (Fig. 2D), thus suggesting that these cells are frataxin-deficient. Large abnormal cells were, however, observed only on the periphery of the liver of Alb-Cre-FxnL3/L–(S) mice within the lobules (supplementary material Fig. 2C). Indeed, H&E stained sections of the inner structure of liver from Alb-Cre-FxnL3/L–(S) mice revealed a normal liver organization, with hepatocytes displaying normal features as compared with control mice (Fig. 2F,G and supplementary material Fig. 2C).
Fig. 2.

Evolution of the liver structure from 15 weeks to 30 weeks. (A) Photographs of livers from control and Alb-Cre-FxnL3/L− mice at 15, 20 and 30 weeks. (B) H&E staining of a lobule of a 20-week-old Alb-Cre-FxnL3/L− mouse. (C-E) Ultrastructural analysis of hepatocytes within a lobule of a 20-week-old Alb-Cre-FxnL3/L− mouse. (F) H&E staining of the liver of a 20-week-old control mouse. (G) H&E staining of the inner structure of a liver in a 20-week-old Alb-Cre-FxnL3/L− mouse. Scale bars: 5 μm (C,D) and 1 μm (E). Arrow, dense intramitochondrial iron deposit; f, fibrosis; m, mitochondria; star, cellular debris; N, nucleus; m/m, dividing mitochondria.
Lobule formation and mouse survival correlate with frataxin re-expression in liver
Frataxin is involved in the early steps of Fe-S cluster biogenesis (Schmucker et al., 2011; Tsai and Barondeau, 2010), and its depletion in mouse tissue leads to Fe-S cluster deficit (Martelli et al., 2007; Puccio et al., 2001; Thierbach et al., 2005). We therefore investigated the impact of Fxn deletion on Fe-S cluster-dependent enzymes in the liver of Alb-Cre-FxnL3/L− mice at different ages. Succinate dehydrogenase (SDH) activity was previously reported to be strongly affected in Alb-Cre-FxnL3/L− mice (Thierbach et al., 2005). Using SDH-staining of liver cryosections, we confirmed a strong deficit of SDH activity at 4 weeks (Fig. 3A,B). However, whereas a deficit was also observed in the lobules of 20-week-old surviving mice (Fig. 3C), the inner liver structure of Alb-Cre-FxnL3/L–(S) mice at 20 weeks displayed normal SDH activity (Fig. 3D). Glutamine phosphoribosylpyrophosphate amidotransferase (GPAT) maturation and xanthine oxidoreductase (XOR) activity, both dependent on Fe-S cluster biogenesis, were also previously reported to be affected in frataxin-deficient mouse livers at 4 weeks (Martelli et al., 2007). GPAT maturation was confirmed to be impaired at 4 weeks in Alb-Cre-FxnL3/L− mice by western blot (Fig. 3E). Whereas the maturation was still impaired at 15 weeks (Fig. 3E), the protein level of the mature form of GPAT (matGPAT) was back to normal at 20 and 30 weeks (Fig. 3E). Similarly, XOR activity in liver of Alb-Cre-FxnL3/L− mice progressively increased with age and no difference could be observed between Alb-Cre-FxnL3/L–(S) and control mice at 30 weeks (Fig. 3F). Interestingly, the increase in Fe-S cluster-dependent activities correlated with the progressive increase of frataxin expression in the liver of Alb-Cre-FxnL3/L− mice, as determined by western blot (Fig. 3E) and quantitative real-time PCR (qRT-PCR) (Fig. 3G).
Fig. 3.

Re-expression of frataxin in surviving Alb-Cre-FxnL3/L− mice. (A–D) Succinate dehydrogenase staining of liver sections from control (A) and Alb-Cre-FxnL3/L− (B) mice at 4 weeks, and of a lobule (C) or the inner liver structure (D) at 20 weeks. (E) Western blot on liver extracts from 4-, 15-, 20- and 30-week-old control (ct) and Alb-Cre-FxnL3/L− (mt) mice using specific antibodies against GPAT, GAPDH and frataxin. matGPAT, mature form of GPAT; *, non-specific band. (F) In-gel xanthine oxido-reductase activity using liver extracts from 4-, 20- and 30-week-old control (ct) and Alb-Cre-FxnL3/L− (mt) mice. (G) Frataxin mRNA levels in liver from mice aged 4 weeks (n=3) and 30 weeks (n=3) as determined by qRT-PCR. Values are given as the mean ± s.d. NS, not significant.
Older surviving mice show liver regeneration
To determine the reason for the expression of frataxin in older surviving Alb-Cre-FxnL3/L–(S) mice, genotyping was performed on liver samples of 4- and 30-week-old mice. The FxnL3 conditional allele that was used to generate the Alb-Cre-FxnL3/L− mice contains three LoxP sites (Puccio et al., 2001): two sites surrounding the Neomycin-resistant cassette (LoxP#1 and LoxP#2), and a third site (LoxP#3) bracketing with LoxP#2 exon 4 of Fxn (supplementary material Fig. S3A). When the Cre recombinase is expressed, three recombination events are possible: the most efficient recombination occurs between LoxP#1 and LoxP#3, therefore leading to the FxnL− allele (supplementary material Fig. S3A); recombination between LoxP#1 and LoxP#2, or between LoxP#2 and LoxP#3 can also occur to give the FxnL2+ or FxnL2− allele, respectively. Whereas the FxnL2− allele leads to the absence of frataxin expression, the FxnL2+ allele allows normal frataxin expression. However, if the Cre recombinase expression is maintained, the FxnL2+ allele can undergo further recombination to give rise to the FxnL− allele (supplementary material Fig. S3A). However, data from our laboratory suggest that this recombination is less efficient than recombination between LoxP#1 and LoxP#3 of the FxnL3 allele (H.P. and L.R., unpublished data).
At 4 weeks, genotyping of liver of Alb-Cre-FxnL3/L− mice revealed the presence of the Cre recombinase (as expected from tail genotyping), the presence of the FxnL− allele and a signal for the FxnL3 allele that is most likely due to the presence of hepatic cells that do not express albumin (Kupffer cells and endothelial cells) (Fig. 4A). Interestingly, at 30 weeks, genotyping of Alb-Cre-FxnL3/L− mice showed the additional presence of a PCR product longer than the product obtained for the wild-type (+) allele (Fig. 4A). This product corresponds to the FxnL2+ allele, as confirmed by DNA sequencing (supplementary material Fig. S3B), thus demonstrating that a significant subset of hepatocytes had only partially recombined and retained exon 4. In addition, quantitative evaluation of the complete deletion of FxnL3 allele in liver using qRT-PCR on genomic DNA showed an underrepresentation of the FxnL− allele in Alb-Cre-FxnL3/L− mice at 30 weeks compared with 4 weeks (Fig. 4B), indicating that liver is composed of a mixed population of deleted and undeleted hepatocytes at 30 weeks.
Fig. 4.
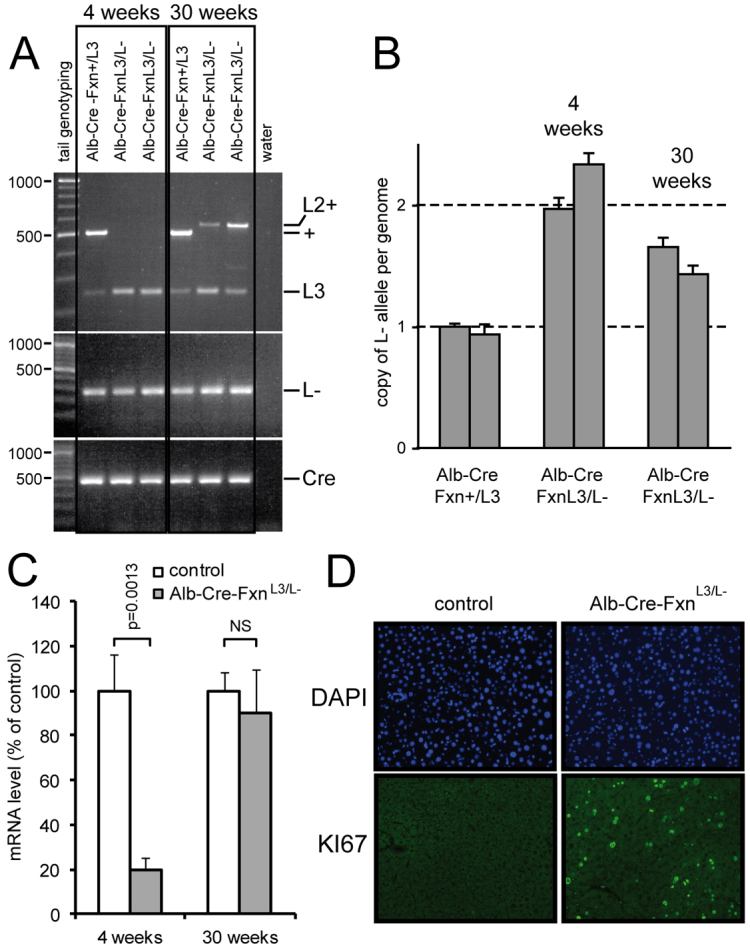
Phenotype of surviving Alb-Cre-FxnL3/L− mice is due to liver regeneration. (A) Genotyping using liver samples from control (Alb-Cre-Fxn+/L3) and Alb-Cre-FxnL3/L− mice at 4 and 30 weeks as indicated. (B) Quantitative evaluation of L-allele copy number in the liver of two controls (Alb-Cre-Fxn+/L3), two Alb-Cre-FxnL3/L− mice at 4 weeks and two Alb-Cre-FxnL3/L− mice at 30 weeks by RT-PCR. Values are given as the mean of triplicates for each mouse ± s.d. (C) Albumin mRNA level in liver from mice aged 4 weeks (n=3) and 30 weeks (n=3) as determined by qRT-PCR. Values are given as the mean ± s.d. (D) Ki67 expression as observed using immunohistochemistry on liver sections of 20-week-old control and Alb-Cre-FxnL3/L− mice.
To assess the impact of frataxin re-expression on liver function, we measured the capacity of liver to synthesized albumin, a capacity that has been previously shown to be impaired in Alb-Cre-FxnL3/L− mice due to liver failure (Thierbach et al., 2005). Whereas the level of mRNA encoding albumin was drastically decreased at 4 weeks (Fig. 4C), the mRNA levels were similar between surviving Alb-Cre-FxnL3/L–(S) and control mice at 30 weeks (Fig. 4C), thus indicating that the liver is functional at 30 weeks.
Liver is an organ that can regenerate in the context of massive hepatocyte death and liver mass loss (for a review see Michalopoulos, 2011). Because liver mass loss was clearly observed during dissection of the surviving Alb-Cre-FxnL3/L–(S) mice (Fig. 2A and supplementary material Fig. S2A), we wondered whether the undeleted hepatocytes identified by genotyping could form a pool of cells for liver regeneration. To address this question, we assessed the capacity of hepatocytes to proliferate by immunohistochemistry using Ki67 expression as a marker (Fig. 4D). Whereas no signal was observed in control mice (Fig. 4D), the liver of Alb-Cre-FxnL3/L–(S) mice displayed numerous nuclei positive for KI67 at 20 weeks (Fig. 4D). From the different sections that were analyzed, only hepatocytes displaying a normal morphology were positive for KI67, and no signal was observed in the large hepatocytes constituting the lobules (data not shown).
Together, the data indicate that older surviving Alb-Cre-FxnL3/L–(S) mice can survive due to liver regeneration from undeleted hepatocytes that gives rise to a mainly functional organ.
DISCUSSION
The present work shows that cancer is not a common finding in the medical history of persons with FRDA. The prevalence determined for the FA-COMS is 1.04% and is clearly below the 3.39% that corresponds to the 19-year prevalence of cancer determined for the general US population (http://seer.cancer.gov/faststats) (supplementary material Table S1). Similarly, in 2008, a 5-year prevalence in the adult population of western Europe was reported to be 1.89% (GLOBOCAN 2008, http://globocan.iarc.fr). If only adults (>20 years) are considered, the EFACTS cohort shows a prevalence of 1.64% (5/304), a value also below the prevalence of cancer for the European population (supplementary material Table S1). The difference between the general population and our cohorts can essentially be explained by the difference in age distribution and the lower mean age of the FRDA cohorts (supplementary material Table S1). It is also worth noting that in both FRDA cohorts, the mean age at cancer diagnosis was over 40 years (supplementary material Table S1), suggesting that, as for the general population, cancer is associated with aging rather than with FRDA. Furthermore, the observed incidence of cancer in the FA-COMS study was less than 0.48% per year, the incidence observed in the US population (supplementary material Table S1). In addition, the neoplasms observed in individuals with FRDA were not typically highly aggressive and were not clustered in any subtype.
Although FRDA shares some of the biochemical features with ataxia telangiectasia (AT), in particular sensitivity to oxidative stress, it does not share the clinical predisposition for tumor susceptibility. Ataxia with oculomotor apraxia type 1, another recessive ataxia, shares the biochemical features of susceptibility to DNA damage with AT (for a review, see Rass et al., 2007), but also has no clear increased risk of neoplasia (Le Ber et al., 2003). Therefore, the clinical data provided herein further suggest that only AT among recessive ataxias is conclusively linked to tumorigenesis.
Our clinical study might be limited by two confounders. The first is that individuals with both FRDA and cancer might be less likely to participate in a natural history study such as those analyzed here. The second is that existing patients who develop serious cancers might be lost to follow-up, and thus the cancer might not be recorded in their updated medical history. Still, the number of identified cases of cancer in our study is so low that it is unlikely that these confounders would fundamentally obscure a susceptibility to tumors in FRDA. Furthermore, previous clinical reports match the findings observed in the natural history cohorts. Cancer was not a major cause of death in a recent study of mortality in FRDA (Tsou et al., 2011), and previous reports of neoplasms in FRDA have mainly been case reports of distinct uncommon tumors with single subjects (Barr et al., 1986; De Pas et al., 1999; Misiakos et al., 2011), or multiple (two) cases of distinct neoplasms in siblings with FRDA (Ackroyd et al., 1996). Such associations are most readily explained by coincidence or the presence of non-frataxin-related genetic susceptibility such as in the case report of two sisters with FRDA presenting breast cancer (Kidd et al., 2001). Thus, although we cannot prove that FRDA has no association with cancer, particularly for rare forms of cancer, the clinical data presented here and published elsewhere (Tsou et al., 2011) do not support such an association. Furthermore, it is worth pointing out that, in particular, no liver cancer was detected in the natural history of the cohorts analyzed herein, despite reports of tumors in the liver conditional mouse model (Thierbach et al., 2010; Thierbach et al., 2012; Thierbach et al., 2005).
To understand how frataxin deficiency could trigger apparent tumorigenesis in liver, we generated a new colony of Alb-Cre-FxnL3/L− mice using the same strategy in a predominantly C57BL/6J genetic background (>80%), as previously described (Thierbach et al., 2005). The general characteristics of the new generation of Alb-Cre-FxnL3/L− mice were similar to those previously reported, if we take into account the fact that weight curves were previously described up to 10 weeks of age and life expectancy was only recorded from 5 weeks of age on (Thierbach et al., 2005). The partial description of survival and weight evolution in Alb-Cre-FxnL3/L− mice made by Thierbach and colleagues prevented two important observations: first, that only 20% of Alb-Cre-FxnL3/L− mice (and not 50% as previously reported) show normal life expectancy; and second, that the Alb-Cre-FxnL3/L− mice that did not show reduced life span progressively gained weight after 10 weeks of age to finally reach levels similar to those of control mice. Furthermore, we showed that only the surviving Alb-Cre-FxnL3/L–(S) mice displayed the progressive formation of abnormal structures on the liver structure, i.e. lobules that were previously identified as tumors (Thierbach et al., 2005). In this context, two main questions had to be addressed: Why do the majority of Alb-Cre-FxnL3/L− mice die before 8 weeks? And, more provocatively, how would tumor formation lead to survival in older Alb-Cre-FxnL3/L− mice?
By characterizing the Alb-Cre-FxnL3/L− mice at 4 weeks of age, we observed a strongly affected liver, displaying the classic hallmarks of frataxin-deficient tissue such as abnormal mitochondrial structures, deficit of Fe-S cluster-dependent activities and intramitochondrial electron-dense deposits reminiscent of iron deposits. The strong liver failure that was observed at this age is the most likely reason for the incapacity to thrive and the early death of Alb-Cre-FxnL3/L− mice. Interestingly, it was recently reported that FRDA patients display subclinical mitochondrial dysfunction in the liver (Stuwe et al., 2011).
The formation of lobule structures previously associated with tumor formation could only be observed in later stages, mainly between 20 and 30 weeks of age. These lobules contained large hepatocytes displaying key features of frataxin-deficient cells: mitochondrial dysfunction (deficient SDH activity, mitochondrial proliferation, clear matrix and loss of cristae), intramitochondrial electron-dense deposits and signs of cell death. However, a normal inner liver structure was observed in Alb-Cre-FxnL3/L–(S) mice at 20 and 30 weeks and correlated with the increase in Fe-S cluster-dependent activities, increased level of frataxin expression and a normal liver function. Genotyping of liver samples further demonstrated that frataxin re-expression was due to the presence of hepatocytes with partially recombined FxnL2+ allele. Together with clear evidence of liver mass loss and cellular proliferation, these results suggest that the phenotype leading to normal liver function, gain of weight and survival involves liver regeneration through proliferation of a significant population of undeleted hepatocytes.
The origin of the FxnL2+ hepatocytes is unknown. One hypothesis is that they might have been selected at an early age of the Alb-Cre-FxnL3/L− mice because FxnL2+ hepatocytes are more resistant to Cre-mediated recombination than FxnL3 hepatocytes (H.P. and L.R., unpublished data), although no signal corresponding to the FxnL2+ allele could be observed by genotyping liver samples at 4 weeks. On the other hand, it has been recently reported that liver regeneration in mice could occur from albumin (Alb)-naive, and thus Cre-naive in our case, cells that progressively differentiate into hepatocytes and other hepatic cells after hepatectomy (Iverson et al., 2011). In this case, FxnL2+ hepatocytes could have been selected after regeneration started, during the progressive differentiation of Alb-naive cells and expression of the Cre recombinase (supplementary material Fig. S4).
Interestingly, liver regeneration has previously been reported to occur in another conditional mouse model in which the Alb-Cre transgene was used to delete the Cox10 gene (Diaz et al., 2008). In this model, undeleted hepatocytes progressively replaced Cox10-deleted hepatocytes that were undergoing apoptosis (Diaz et al., 2008), thus indicating that the Alb-Cre system is not fully efficient. This inefficiency, which was reported to result from defective Cre recombinase expression in a subset of hepatocytes (Diaz et al., 2008), might thus participate in providing a pool of undeleted cells for the liver regeneration observed in Alb-Cre-FxnL3/L− mice. However, no formation of lobules on the surface of the Cox10-deficient liver has been reported (Diaz et al., 2008), suggesting that another phenomenon is taking place in Alb-Cre-FxnL3/L− mice.
As indicated above, the lobules we observed in Alb-Cre-FxnL3/L− mice contained large hepatocytes displaying key features of frataxin-deficient cells. These cells could account for the proportion of deleted cells in livers of surviving Alb-Cre-FxnL3/L− mice, as observed by determination of the copy number of FxnL− allele per genome. The deleted hepatocytes in the periphery of the liver might result from maintained expression of Cre recombinase in FxnL2+ hepatocytes, which would finally trigger deletion of exon 4. Furthermore, cell death was observed within the lobules, indicating that Fxn-deleted hepatocytes are not viable. The absence of a positive signal for KI67 in these large cells further indicated their incapacity to proliferate. Together, these data show that these structures are incompatible with tumor formation. In addition, tumor formation from Fxn-deleted cells is unlikely because previous data demonstrated that complete deletion of Fxn is not compatible with cellular proliferation (Calmels et al., 2009).
Together, our data provide clear evidence that the development of lobules in Alb-Cre-FxnL3/L− mice is associated with artifactual liver regeneration, inherent to the Alb-Cre-based mouse model, and that this phenotype cannot be directly attributed to frataxin deficiency in liver. We showed that Fxn deletion in liver triggers early mitochondriopathy, Fe-S cluster deficiency and liver failure, which lead to death in the majority of mice. While ageing, liver regeneration can eventually take over in some Alb-Cre-FxnL3/L− mice, thus rescuing the phenotype resulting from early Fxn deletion and allowing mice to survive. Therefore, data obtained with Alb-Cre-FxnL3/L− mice should be taken with care if old mice have been used to characterize the consequences of frataxin deficiency, such as in the recently reported reversion of carbohydrate metabolism impairment observed between 5-week-old and 17-month-old Alb-Cre-FxnL3/L− mice (Thierbach et al., 2012).
In conclusion, both clinical investigations and characterization of the liver conditional mouse model described herein provide evidence that previously reported data suggesting that FRDA and frataxin deficiency are associated with predisposition to cancer and tumorigenesis need to be reconsidered.
METHODS
Retrospective clinical analysis
We reviewed anonymized data collected by an ongoing natural history study of FRDA from ten centers in the USA and one in Australia (FA-COMS cohort). All protocols were approved by the CHOP IRB. The medical history of 578 patients was assessed for the type and number of neoplasms recorded since the study’s inception. A total of 1694 visits were analyzed. For purpose of analysis, all cellular dysplasias were excluded. The age of neoplasm diagnosis was also noted.
Data were also obtained from a European natural history study of FRDA, which started in 2011, currently involving eight clinical centers in Austria, Belgium, France, Germany, Italy and Spain (EFACTS cohort). All protocols obtained ethics approval from the European Commission, which supports the project, and from local Ethics Committees. Written informed consent was required from each individual to be included in the study. A detailed medical history that includes any neoplasm ever diagnosed for the patient was obtained. So far, only baseline data are available. As for the FA-COMS study, all cellular dysplasias were excluded from the analysis and the age of neoplasm diagnosis was noted.
Mice
Mice with a specific deletion of the Fxn gene in liver (Alb-Cre-FxnL3/L−) were generated as previously described (Thierbach et al., 2005). Mice were analyzed in 87.5% C57BL/6J and 12.5% 129/Sv mixed background. Genotyping of the mice was carried out as previously described (Puccio et al., 2001). Mice were maintained in a temperature and humidity controlled animal facility, with a 12-hour light and dark cycle and free access to water and a standard rodent chow (D03, SAFE, Villemoisson-sur-Orge, France). Breeding and maintenance of mice were performed according to institutional guidelines. Animals were killed by CO2 inhalation and tissues were immediately collected, weighed and frozen in liquid nitrogen or processed for biochemical and histological analysis.
Histopathology, immunohistochemistry and electron microscopy
Tissues were dissected, fixed overnight in formalin, dried in 70% ethanol, embedded in paraffin and sectioned (5 μm) on a microtome using standard techniques. Some sections were stained with H&E. Histochemical analyses using Oil Red staining and SDH staining were carried out on cryostat sections (10 μm) of unfixed liver as described (Puccio et al., 2001).
Immunohistochemistry was carried out on paraffin-embedded sections. Sections were dewaxed and hydrated using successive bathing in Histosol (2× 3 minutes), ethanol (2× 3 minutes), 90% ethanol (3 minutes), 70% ethanol (3 minutes) and water (5 minutes). Sections were then submitted to microwave treatment (2× 2.5 minutes at 800 W) in 10 mM citrate buffer, pH 6.0. Samples were washed in 0.05% PBS-Tween (PBS-T) (3× 5 minutes) before the addition of saturation buffer (PBS-T + 5% normal goat serum, NGS) for 30 minutes. The anti-KI67 antibody (NCL-Ki67p, Novocastra Laboratories; 1:500 in PBS-T + 5% NGS) was applied overnight at 4°C. Sections were then washed with PBS-T (3× 5 minutes) and exposed to goat anti-rabbit antibody coupled to Alexa Fluor 488 (Molecular Probes; 1:1000 in PBS-T + 5% NGS) for 1 hour at room temperature. Sections were washed with PBS-T (3× 5 minutes) and slides were mounted using Poly-Mount (Polysciences, Warrington, PA) mounting medium.
For electron microscopy, dissected livers were fixed in a freshly made mixture of 2.5% paraformaldehyde and 2.5% glutaraldehyde in cacodylate buffer (0.1 M, pH 7.2), rinsed in cacodylate buffer, postfixed in 0.1 M cacodylate buffer + 1% osmium tetroxide for 1 hour at 4°C, dehydrated and embedded in Epon. Ultrathin sections were cut at 70 nm and contrasted with uranyl acetate and lead citrate and examined with a Morgagni 268D electron microscope.
Immunoblotting
Total liver extracts were prepared as previously described (Martelli et al., 2007). Immunoblotting was carried out using specific antibodies against frataxin (R1270, IGBMC, Illkirch, France; 1:1000), GPAT (R2372, IGBMC; 1:2000), βTUB (2A2, IGBMC; 1:40,000) and GAPDH (MAB374, Chemicon International; 1:20,000). Horseradish peroxidase-coupled secondary antibodies were used at a dilution of 1:5000.
Quantitative real-time PCR
Total RNA extraction, reverse transcription and qRT-PCR were achieved as previously reported (Martelli et al., 2007). The following primers were used: Fxn forward, 5′-ATGG-CGTGCTCACCATTAAG-3′ and reverse, 5′-GGCCAATG-AAGACAAGTCCA-3′; Alb forward, 5′-GACAAGG-AAAGCTGCCTGAC-3′ and reverse, 5′-TTCTGCA-AAGTCAGCATTGG-3′. Hprt was used as a housekeeping gene (Martelli et al., 2007). Quantitative deletion of the FxnL3 allele on genomic DNA was performed using the primers previously described to amplify the deleted allele (Puccio et al., 2001) and primers amplifying the Pepck gene (forward, 5′-TCAACACCGACCTCCCTTAC-3′ and reverse, 5′-CAT-TGTGCCGCTATCTCAAA-3′) as control. Results were analyzed using the ΔCt method.
Xanthine oxido-reductase in-gel activity
Livers were homogenized in extraction buffer (20 mM Tris-HCl, 250 mM saccharose, pH 7.2, 2 mM EDTA, 40 mM KCl and Roche Complete Protease Inhibitor Cocktail) using an Ultraturax homogenizer. The homogenate was then centrifuged at 15,000 g for 15 minutes at 4°C. Supernatant (60 μg) was loaded onto 8% polyacrylamide native gel. After migration using SDS-glycine buffer, the gel was incubated in 250 mM Tris-HCl, pH 8.5, for 10 minutes before incubation in the staining buffer (0.5 mg/ml hypoxanthine, 0.5 mg/ml thiazolyl blue tetrazolium bromide and 50 μg/ml phenazine methosulfate). The reaction was stopped before saturation of the signal by washing several times with water.
Stastistical analyses
Differences between mean values were evaluated using the bilateral Student’s t-test. P<0.05 was considered significant.
Supplementary Material
Acknowledgments
The authors would like to thank all the patients and their families around the world who contributed to this study. We thank Alicia Brocht (University of Rochester) for data extraction from the FA-COMS database, Josiane Hergueux and Jean-Luc Weickert (IGBMC) for technical help. We thank Michel Koenig for careful reading of the manuscript and discussion.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interest.
AUTHOR CONTRIBUTIONS
D.R.L. and S.P. conceived and designed the FA-COMS portion. L.S.F. and D.R.L. extracted and analyzed data of the FA-COMS database. K.F. and J.B.S. extracted and analyzed data of the EFACTS database. A.M. and H.P. conceived and designed the experiments with the mice. A.M., L.R. and N.M. performed the experiments on mouse samples. A.M., H.P. and N.M. analyzed data. A.M., D.R.L., M.P. and H.P. wrote the paper.
FUNDING
This work was supported by the Friedreich’s Ataxia Research Alliance, the Friedreich’s Ataxia Research Alliance New Investigator Grant to A.M., the National Ataxia Foundation and the European Community under the European Research Council [grant number 206634/ISCATAXIA to H.P.] and the 7th Framework Program [242193/EFACTS].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.009829/-/DC1
REFERENCES
- Ackroyd R., Shorthouse A. J., Stephenson T. J. (1996). Gastric carcinoma in siblings with Friedreich’s ataxia. Eur. J. Surg. Oncol. 22, 301–303 [DOI] [PubMed] [Google Scholar]
- Al-Mahdawi S., Pinto R. M., Varshney D., Lawrence L., Lowrie M. B., Hughes S., Webster Z., Blake J., Cooper J. M., King R., et al. (2006). GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 88, 580–590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr H., Page R., Taylor W. (1986). Primary small bowel ganglioneuroblastoma and Friedreich’s ataxia. J. R. Soc. Med. 79, 612–613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley J. L., Homayoun S., Hart P. E., Schapira A. H., Cooper J. M. (2004). Role of oxidative damage in Friedreich’s ataxia. Neurochem. Res. 29, 561–567 [DOI] [PubMed] [Google Scholar]
- Calmels N., Schmucker S., Wattenhofer-Donze M., Martelli A., Vaucamps N., Reutenauer L., Messaddeq N., Bouton C., Koenig M., Puccio H. (2009). The first cellular models based on frataxin missense mutations that reproduce spontaneously the defects associated with Friedreich ataxia. PLoS ONE 4, e6379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campuzano V., Montermini L., Molto M. D., Pianese L., Cossee M., Cavalcanti F., Monros E., Rodius F., Duclos F., Monticelli A., et al. (1996). Friedreich’s ataxia: autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 271, 1423–1427 [DOI] [PubMed] [Google Scholar]
- Campuzano V., Montermini L., Lutz Y., Cova L., Hindelang C., Jiralerspong S., Trottier Y., Kish S. J., Faucheux B., Trouillas P., et al. (1997). Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 6, 1771–1780 [DOI] [PubMed] [Google Scholar]
- Cossee M., Durr A., Schmitt M., Dahl N., Trouillas P., Allinson P., Kostrzewa M., Nivelon-Chevallier A., Gustavson K. H., Kohlschutter A., et al. (1999). Friedreich’s ataxia: point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 45, 200–206 [DOI] [PubMed] [Google Scholar]
- De Pas T., Martinelli G., De Braud F., Peccatori F., Catania C., Aapro M. S., Goldhirsch A. (1999). Friedreich’s ataxia and intrathecal chemotherapy in a patient with lymphoblastic lymphoma. Ann. Oncol. 10, 1393. [DOI] [PubMed] [Google Scholar]
- Di Prospero N. A., Baker A., Jeffries N., Fischbeck K. H. (2007). Neurological effects of high-dose idebenone in patients with Friedreich’s ataxia: a randomised, placebo-controlled trial. Lancet Neurol. 6, 878–886 [DOI] [PubMed] [Google Scholar]
- Diaz F., Garcia S., Hernandez D., Regev A., Rebelo A., Oca-Cossio J., Moraes C. T. (2008). Pathophysiology and fate of hepatocytes in a mouse model of mitochondrial hepatopathies. Gut 57, 232–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emond M., Lepage G., Vanasse M., Pandolfo M. (2000). Increased levels of plasma malondialdehyde in Friedreich ataxia. Neurology 55, 1752–1753 [DOI] [PubMed] [Google Scholar]
- Friedman L. S., Farmer J. M., Perlman S., Wilmot G., Gomez C. M., Bushara K. O., Mathews K. D., Subramony S. H., Ashizawa T., Balcer L. J., et al. (2010). Measuring the rate of progression in Friedreich ataxia: implications for clinical trial design. Mov. Disord. 25, 426–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellera C., Castellotti B., Mariotti C., Mineri R., Seveso V., Didonato S., Taroni F. (2007). Frataxin gene point mutations in Italian Friedreich ataxia patients. Neurogenetics 8, 289–299 [DOI] [PubMed] [Google Scholar]
- Harding A. E. (1981). Friedreich’s ataxia: a clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 104, 589–620 [DOI] [PubMed] [Google Scholar]
- Harding A. E., Hewer R. L. (1983). The heart disease of Friedreich’s ataxia: a clinical and electrocardiographic study of 115 patients, with an analysis of serial electrocardiographic changes in 30 cases. Q. J. Med. 52, 489–502 [PubMed] [Google Scholar]
- Iverson S. V., Comstock K. M., Kundert J. A., Schmidt E. E. (2011). Contributions of new hepatocyte lineages to liver growth, maintenance, and regeneration in mice. Hepatology 54, 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd A., Coleman R., Whiteford M., Barron L. H., Simpson S. A., Haites N. E. (2001). Breast cancer in two sisters with Friedreich’s ataxia. Eur. J. Surg. Oncol. 27, 512–514 [DOI] [PubMed] [Google Scholar]
- Lamarche J., Shapcott D., Côté M., Lemieux B. (1993). Cardiac iron deposits in Friedreich’s ataxia. In Handbook of Cerebellar Diseases (ed. Lechtenberg R.), pp. 453–457 Newark: Dekker, M. [Google Scholar]
- Lamarche J. B., Cote M., Lemieux B. (1980). The cardiomyopathy of Friedreich’s ataxia morphological observations in 3 cases. Can. J. Neurol. Sci. 7, 389–396 [DOI] [PubMed] [Google Scholar]
- Le Ber I., Moreira M. C., Rivaud-Pechoux S., Chamayou C., Ochsner F., Kuntzer T., Tardieu M., Said G., Habert M. O., Demarquay G., et al. (2003). Cerebellar ataxia with oculomotor apraxia type 1, clinical and genetic studies. Brain 126, 2761–2772 [DOI] [PubMed] [Google Scholar]
- Martelli A., Wattenhofer-Donze M., Schmucker S., Bouvet S., Reutenauer L., Puccio H. (2007). Frataxin is essential for extramitochondrial Fe-S cluster proteins in mammalian tissues. Hum. Mol. Genet. 16, 2651–2658 [DOI] [PubMed] [Google Scholar]
- Michael S., Petrocine S. V., Qian J., Lamarche J. B., Knutson M. D., Garrick M. D., Koeppen A. H. (2006). Iron and iron-responsive proteins in the cardiomyopathy of Friedreich’s ataxia. Cerebellum 5, 257–267 [DOI] [PubMed] [Google Scholar]
- Michalopoulos G. K. (2011). Liver regeneration: alternative epithelial pathways. Int. J. Biochem. Cell Biol. 43, 173–179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda C. J., Santos M. M., Ohshima K., Smith J., Li L., Bunting M., Cossee M., Koenig M., Sequeiros J., Kaplan J., et al. (2002). Frataxin knockin mouse. FEBS Lett. 512, 291–297 [DOI] [PubMed] [Google Scholar]
- Misiakos E. P., Siama E., Schizas D., Petropoulos C., Zavras N., Economopoulos N., Charalabopoulos A., Macheras A. (2011). Massive uterine leiomyoma in a patient with Friedreich’s ataxia: is there a possible association? Case Report Med. 2011, 648217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers L. M., Lynch D. R., Farmer J. M., Friedman L. S., Lawson J. A., Wilson R. B. (2008). Urinary isoprostanes in Friedreich ataxia: lack of correlation with disease features. Mov. Disord. 23, 1920–1922 [DOI] [PubMed] [Google Scholar]
- Pandolfo M., Pastore A. (2009). The pathogenesis of Friedreich ataxia and the structure and function of frataxin. J. Neurol. 256 Suppl. 1, 9–17 [DOI] [PubMed] [Google Scholar]
- Perlman S. L., Boder Deceased E., Sedgewick R. P., Gatti R. A. (2012). Ataxia-telangiectasia. Handb. Clin. Neurol. 103, 307–332 [DOI] [PubMed] [Google Scholar]
- Postic C., Shiota M., Niswender K. D., Jetton T. L., Chen Y., Moates J. M., Shelton K. D., Lindner J., Cherrington A. D., Magnuson M. A. (1999). Dual roles for glucokinase in glucose homeostasis as determined by liver and pancreatic beta cell-specific gene knock-outs using Cre recombinase. J. Biol. Chem. 274, 305–315 [DOI] [PubMed] [Google Scholar]
- Puccio H., Simon D., Cossee M., Criqui-Filipe P., Tiziano F., Melki J., Hindelang C., Matyas R., Rustin P., Koenig M. (2001). Mouse models for Friedreich ataxia exhibit cardiomyopathy, sensory nerve defect and Fe-S enzyme deficiency followed by intramitochondrial iron deposits. Nat. Genet. 27, 181–186 [DOI] [PubMed] [Google Scholar]
- Rass U., Ahel I., West S. C. (2007). Defective DNA repair and neurodegenerative disease. Cell 130, 991–1004 [DOI] [PubMed] [Google Scholar]
- Ristow M., Mulder H., Pomplun D., Schulz T. J., Muller-Schmehl K., Krause A., Fex M., Puccio H., Muller J., Isken F., et al. (2003). Frataxin deficiency in pancreatic islets causes diabetes due to loss of beta cell mass. J. Clin. Invest. 112, 527–534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotig A., de Lonlay P., Chretien D., Foury F., Koenig M., Sidi D., Munnich A., Rustin P. (1997). Aconitase and mitochondrial iron-sulphur protein deficiency in Friedreich ataxia. Nat. Genet. 17, 215–217 [DOI] [PubMed] [Google Scholar]
- Schmucker S., Argentini M., Carelle-Calmels N., Martelli A., Puccio H. (2008). The in vivo mitochondrial two-step maturation of human frataxin. Hum. Mol. Genet. 17, 3521–3531 [DOI] [PubMed] [Google Scholar]
- Schmucker S., Martelli A., Colin F., Page A., Wattenhofer-Donze M., Reutenauer L., Puccio H. (2011). Mammalian frataxin: an essential function for cellular viability through an interaction with a preformed ISCU/NFS1/ISD11 iron-sulfur assembly complex. PLoS ONE 6, e16199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz J. B., Dehmer T., Schols L., Mende H., Hardt C., Vorgerd M., Burk K., Matson W., Dichgans J., Beal M. F., et al. (2000). Oxidative stress in patients with Friedreich ataxia. Neurology 55, 1719–1721 [DOI] [PubMed] [Google Scholar]
- Schulz J. B., Di Prospero N. A., Fischbeck K. (2009). Clinical experience with high-dose idebenone in Friedreich ataxia. J. Neurol. 256, 42–45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simon D., Seznec H., Gansmuller A., Carelle N., Weber P., Metzger D., Rustin P., Koenig M., Puccio H. (2004). Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J. Neurosci. 24, 1987–1995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuwe S. H., Goetze O., Arning L., Banasch M., Schmidt W. E., Schols L., Saft C. (2011). Hepatic mitochondrial dysfunction in Friedreich ataxia. BMC Neurol. 11, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierbach R., Schulz T. J., Isken F., Voigt A., Mietzner B., Drewes G., von Kleist-Retzow J. C., Wiesner R. J., Magnuson M. A., Puccio H., et al. (2005). Targeted disruption of hepatic frataxin expression causes impaired mitochondrial function, decreased life span and tumor growth in mice. Hum. Mol. Genet. 14, 3857–3864 [DOI] [PubMed] [Google Scholar]
- Thierbach R., Drewes G., Fusser M., Voigt A., Kuhlow D., Blume U., Schulz T. J., Reiche C., Glatt H., Epe B., et al. (2010). The Friedreich’s ataxia protein frataxin modulates DNA base excision repair in prokaryotes and mammals. Biochem. J. 432, 165–172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thierbach R., Florian S., Wolfrum K., Voigt A., Drewes G., Blume U., Bannasch P., Ristow M., Steinberg P. (2012). Specific alterations of carbohydrate metabolism are associated with hepatocarcinogenesis in mitochondrially impaired mice. Hum. Mol. Genet. 21, 656–663 [DOI] [PubMed] [Google Scholar]
- Tsai C. L., Barondeau D. P. (2010). Human frataxin is an allosteric switch that activates the Fe-S cluster biosynthetic complex. Biochemistry 49, 9132–9139 [DOI] [PubMed] [Google Scholar]
- Tsou A. Y., Paulsen E. K., Lagedrost S. J., Perlman S. L., Mathews K. D., Wilmot G. R., Ravina B., Koeppen A. H., Lynch D. R. (2011). Mortality in Friedreich ataxia. J. Neurol. Sci. 307, 46–49 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}