

SUMMARY
Spinocerebellar ataxia type 13 (SCA13) is an autosomal dominant disease caused by mutations in the Kv3.3 voltage-gated potassium (K+) channel. SCA13 exists in two forms: infant onset is characterized by severe cerebellar atrophy, persistent motor deficits and intellectual disability, whereas adult onset is characterized by progressive ataxia and progressive cerebellar degeneration. To test the hypothesis that infant- and adult-onset mutations have differential effects on neuronal development that contribute to the age at which SCA13 emerges, we expressed wild-type Kv3.3 or infant- or adult-onset mutant proteins in motor neurons in the zebrafish spinal cord. We characterized the development of CaP (caudal primary) motor neurons at ∼36 and ∼48 hours post-fertilization using confocal microscopy and 3D digital reconstruction. Exogenous expression of wild-type Kv3.3 had no significant effect on CaP development. In contrast, CaP neurons expressing the infant-onset mutation made frequent pathfinding errors, sending long, abnormal axon collaterals into muscle territories that are normally innervated exclusively by RoP (rostral primary) or MiP (middle primary) motor neurons. This phenotype might be directly relevant to infant-onset SCA13 because interaction with inappropriate synaptic partners might trigger cell death during brain development. Importantly, pathfinding errors were not detected in CaP neurons expressing the adult-onset mutation. However, the adult-onset mutation tended to increase the complexity of the distal axonal arbor. From these results, we speculate that infant-onset SCA13 is associated with marked changes in the development of Kv3.3-expressing cerebellar neurons, reducing their health and viability early in life and resulting in the withered cerebellum seen in affected children.
INTRODUCTION
Spinocerebellar ataxia type 13 (SCA13) is a human autosomal dominant disease caused by mutations in the KCNC3 gene, which encodes the Kv3.3 voltage-gated K+ channel (Waters et al., 2006). Kv3.3 is prominently expressed in the cerebellum and in spinal cord motor neurons, and is also found in the hippocampus, cerebral cortex and brainstem (Brooke et al., 2004; Brooke et al., 2006; Chang et al., 2007). SCA13 exists in distinct neurodegenerative and neurodevelopmental forms depending on which KCNC3 mutation is present (Waters et al., 2006). In one form, SCA13 emerges in adulthood with progressive degeneration of the cerebellum accompanied by progressive ataxia. In the other form, SCA13 is evident in early childhood with motor delay, persistent motor deficits and severe atrophy of the cerebellum (Herman-Bert et al., 2000). Intellectual disability and seizures have also been reported in affected individuals, suggesting that infant-onset mutations detrimentally affect Kv3.3-expressing neurons outside of the cerebellum. The infant- and adult-onset phenotypes are strongly correlated with the causative mutation in unrelated affected families, indicating that they do not reflect differences in genetic background (Figueroa et al., 2010; Figueroa et al., 2011).
How KCNC3 mutations give rise to distinct forms of SCA13 that are associated with early cerebellar atrophy or age-related degeneration is unknown. We hypothesize that infant- and adult-onset mutations have differential effects on neuronal development that could contribute to the age at which SCA13 emerges. To test this idea, we expressed wild-type Kv3.3 and infant- and adult-onset mutant proteins in zebrafish to determine their effects on the development of spinal cord motor neurons. Zebrafish primary motor neurons, which endogenously express Kv3.3, provide substantial advantages for this analysis because they have stereotypic developmental schedules, highly reproducible morphologies and exclusive, non-overlapping post-synaptic territories (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986; Issa et al., 2011). In addition, they can be imaged in living animals at different developmental times.
We report that the infant-onset mutation caused frequent axonal pathfinding errors, with the extension of abnormally long collaterals that invaded inappropriate synaptic territory. In contrast, the adult-onset mutation did not result in pathfinding errors, although it tended to increase the complexity of the distal axonal arbor. Importantly, expression of wild-type Kv3.3 had no significant effect on neuronal development. Based on the differential severity of the developmental effects of the early- and late-onset mutations, we speculate that, in SCA13, infant-onset mutations markedly disrupt the development of cerebellar neurons, increasing their susceptibility to cell death early in life and resulting in the severely shrunken cerebellum observed in affected children.
TRANSLATIONAL IMPACT.
Clinical issue
Spinocerebellar ataxia type 13 (SCA13) is a human autosomal dominant disease caused by mutations in KCNC3, which encodes Kv3.3, a voltage-gated K+ channel. Depending on which KCNC3 mutation is present, SCA13 exists in one of two clinical forms that differ in the age of onset. Onset in early childhood is characterized by severe cerebellar atrophy and persistent locomotor deficits, whereas onset during adulthood is characterized by progressive cerebellar degeneration and progressive ataxia. What controls the age of SCA13 onset is unknown. One possibility is that infant-onset mutations markedly disrupt the development of cerebellar neurons, increasing their susceptibility to atrophy and/or cell death early in life. In this study, the authors tested the hypothesis that SCA13 mutations associated with early or late onset have differential effects on neuronal development that could underlie the age at which disease emerges.
Results
The authors studied the development of Kv3.3-expressing motor neurons in the zebrafish spinal cord after exogenously expressing the wild-type Kv3.3 protein, or a mutant protein associated with early-onset disease, or a mutant protein associated with late-onset disease. The development of caudal primary (CaP) motor neurons was characterized at ∼36 and ∼48 hours post-fertilization using confocal microscopy and 3D digital reconstruction. CaP neurons expressing an infant-onset SCA13 mutation made frequent pathfinding errors, sending long, abnormal axon collaterals into muscle territories that are normally innervated exclusively by rostral primary (RoP) or middle primary (MiP) motor neurons. This phenotype could be directly relevant to infant-onset SCA13 because interaction with inappropriate synaptic partners can trigger cell death during brain development. Importantly, pathfinding errors were not detected in CaP neurons expressing exogenous wild-type Kv3.3 or the adult-onset mutation. However, the adult-onset mutation tended to increase the complexity of the distal axonal arbor. These results support the hypothesis that SCA13 mutations have differential effects on neuronal development that could underlie the age of disease onset.
Implications and future directions
This study indicates that Kv3.3 channels play a previously unappreciated role in neuronal development. The authors speculate that early-onset SCA13 is associated with marked changes in the development of Kv3.3-expressing cerebellar neurons, reducing their health and viability early in life and resulting in the withered cerebellum seen in affected children. Future work will focus on identifying cellular mechanisms that translate distinct changes in Kv3.3 function into differential effects on neuronal development. Such mechanisms are likely to contribute to pathogenesis in human SCA13 because Kv3.3 channels play the same physiological roles in zebrafish and mammalian neurons.
RESULTS
Exogenous expression of wild-type Kv3.3 has no significant effect on CaP development
The primary motor neurons of the zebrafish spinal cord are fast-spiking cells that endogenously express Kv3.3 (Issa et al., 2011) and innervate fast twitch muscle (Wen and Brehm, 2005). There are three primary motor neurons per hemisegment, RoP (rostral primary), MiP (middle primary) and CaP (caudal primary), named according to the relative positions of their cell bodies in the spinal cord (Myers et al., 1986). A fourth, variable primary (VaP) motor neuron is occasionally born, but it typically dies by 36 hours post-fertilization (hpf) (Eisen and Melançon, 2001; Eisen et al., 1990). Each of these neurons has a unique, reproducible morphology and exclusively innervates a distinct region of the myotome (Fig. 1A) (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). During development, the CaP axon is the first to exit the spinal cord, followed by the MiP and then the RoP axons (Eisen et al., 1986; Myers et al., 1986; Eisen, 1994). Pioneered by the CaP axon, the primary motor axons follow a common path down the middle of the myotome to the horizontal myoseptum, where they form their first synapses (Melançon et al., 1997). This is called the choice point because it is here that the CaP, MiP and RoP axons diverge to innervate their cell-specific territories of the myotome, developing their distinctive morphologies in the process (Fig. 1A) (Eisen et al., 1986; Myers et al., 1986).
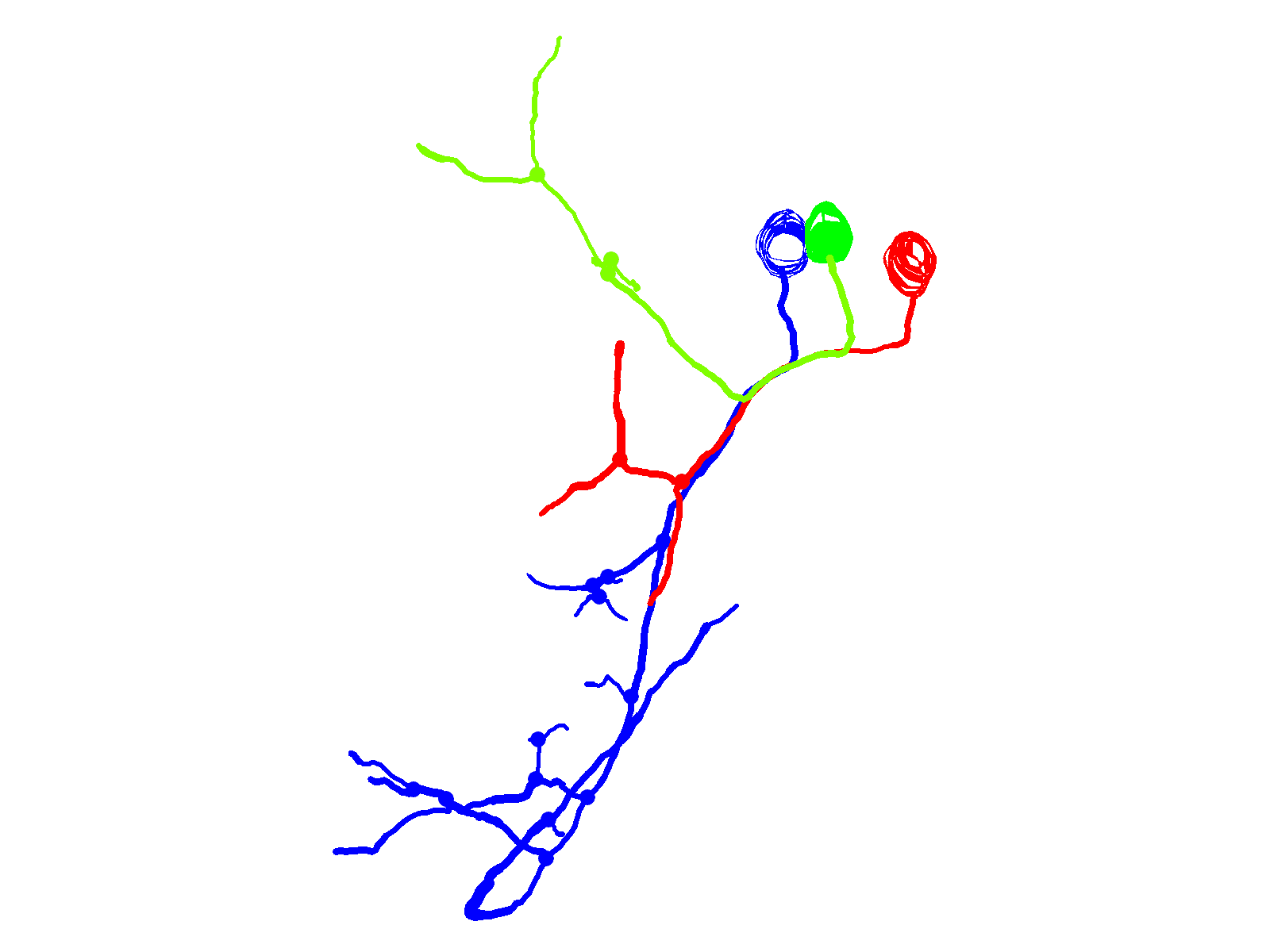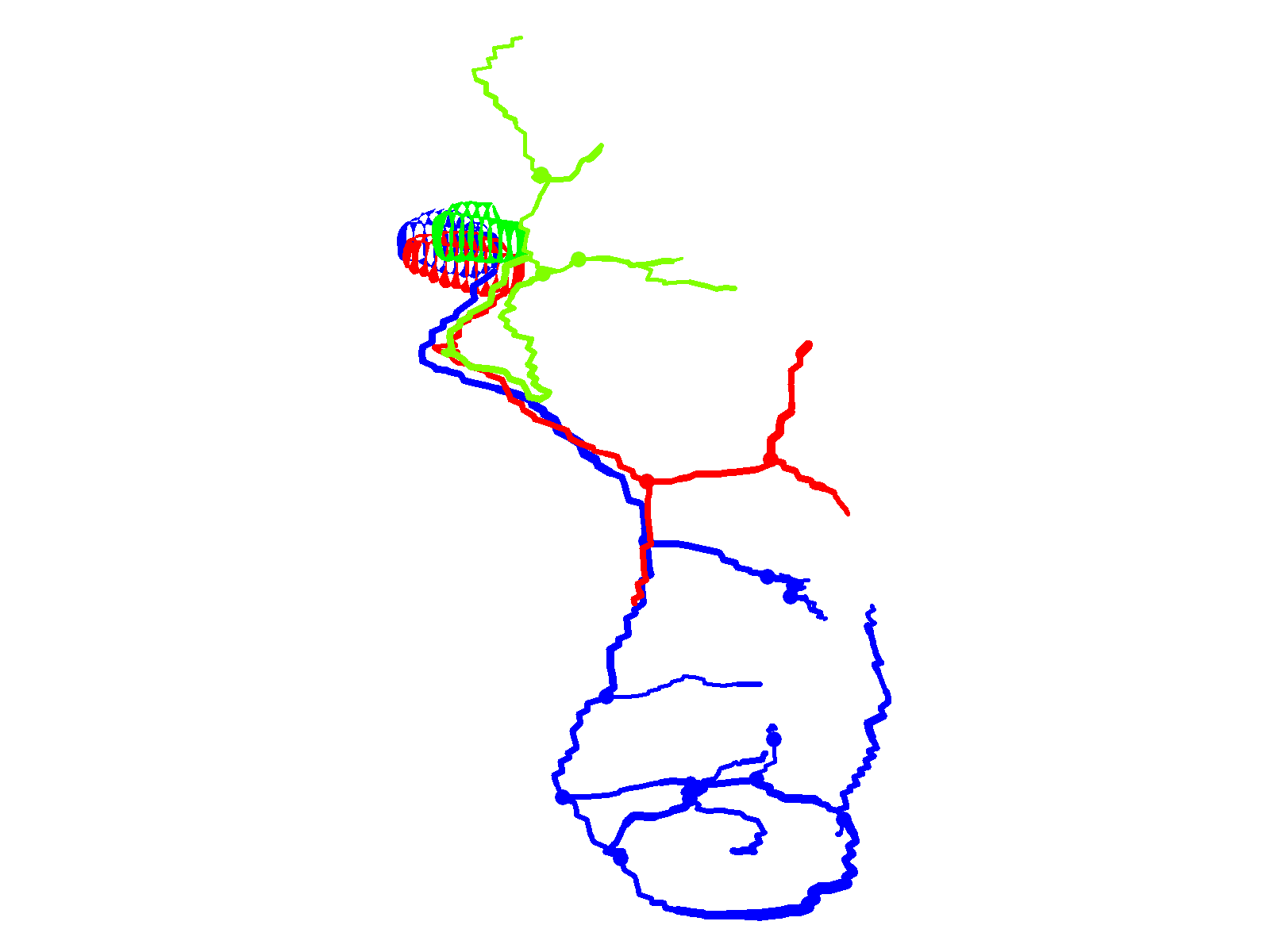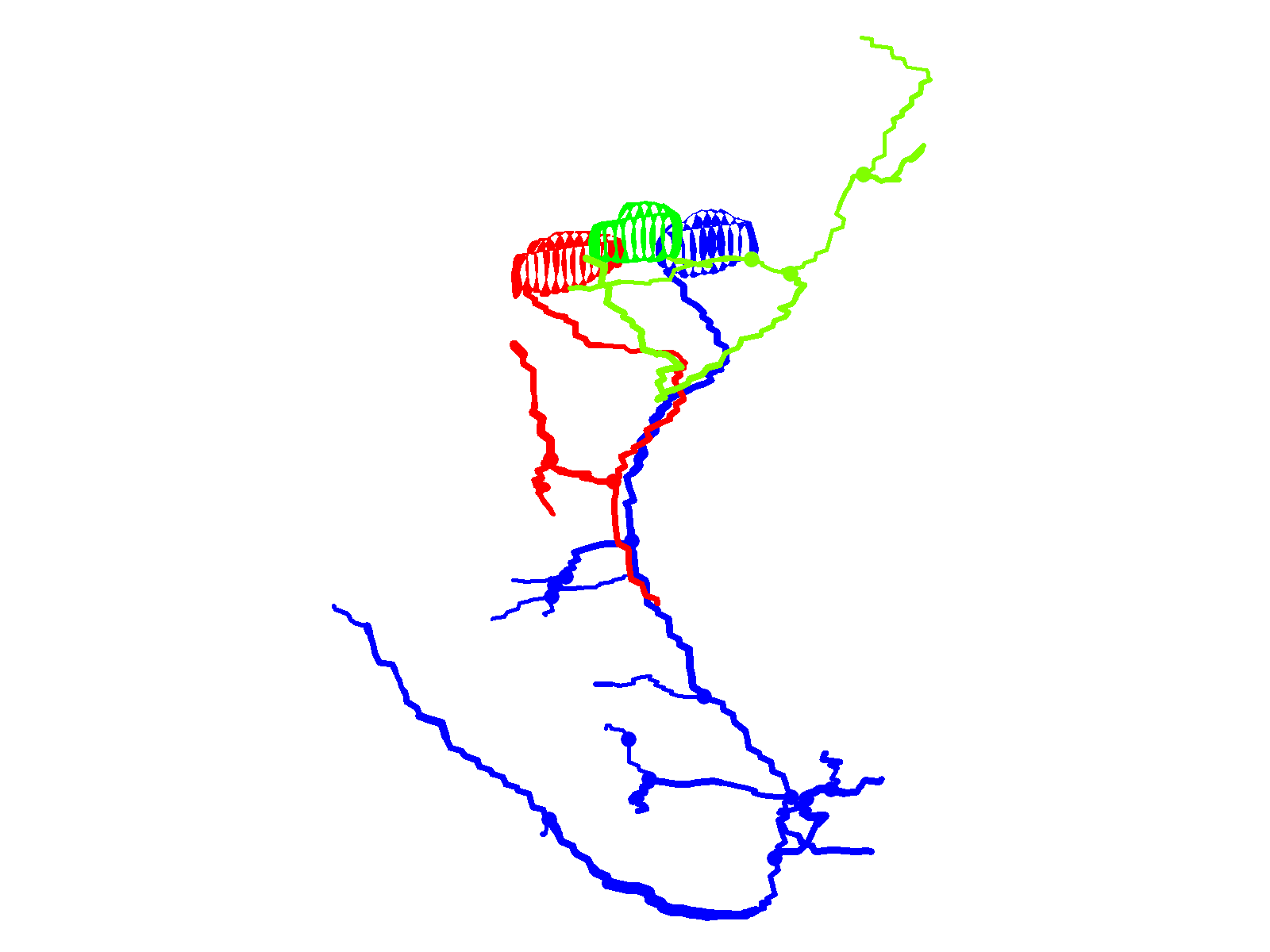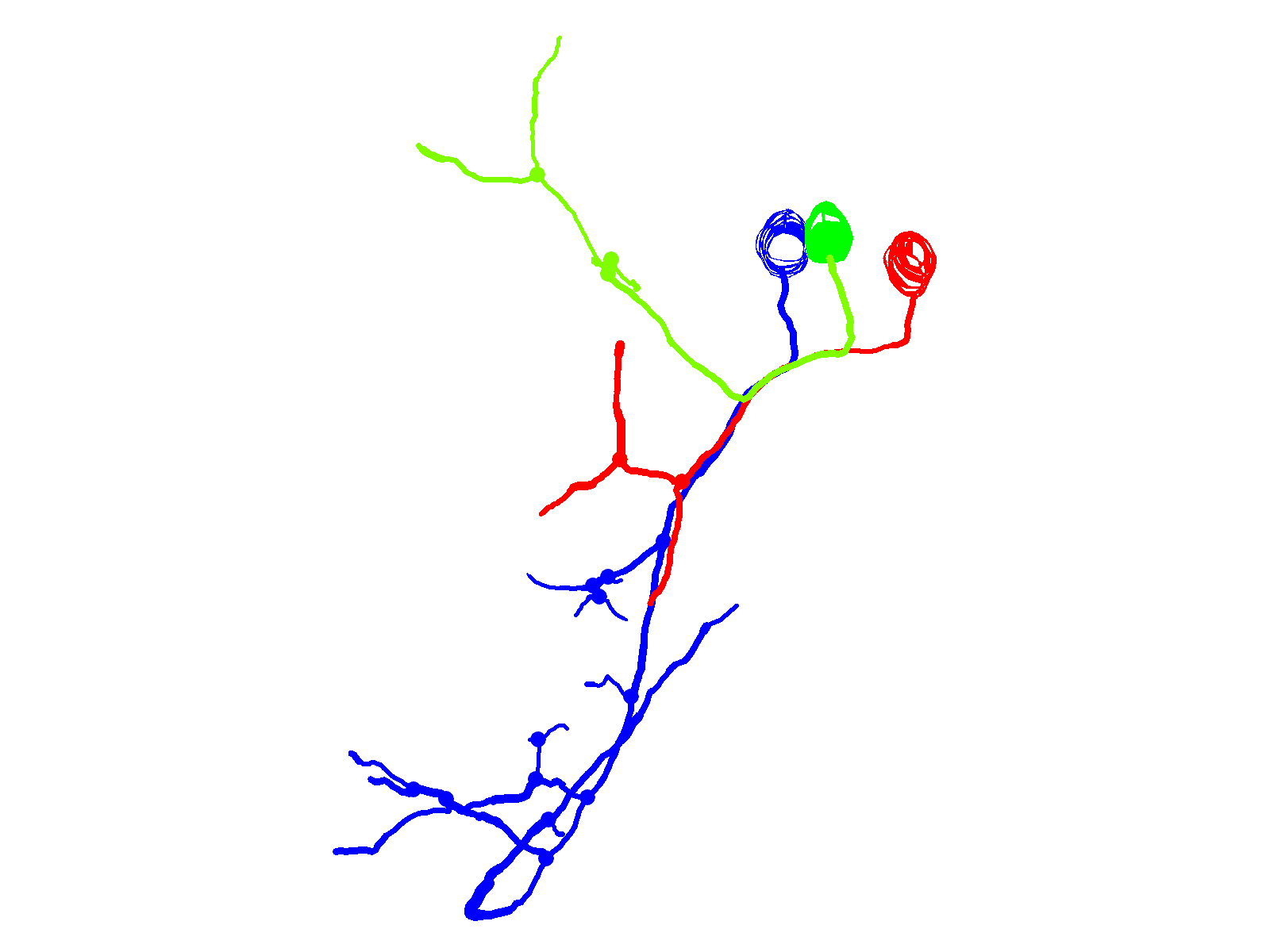
Fig. 1.
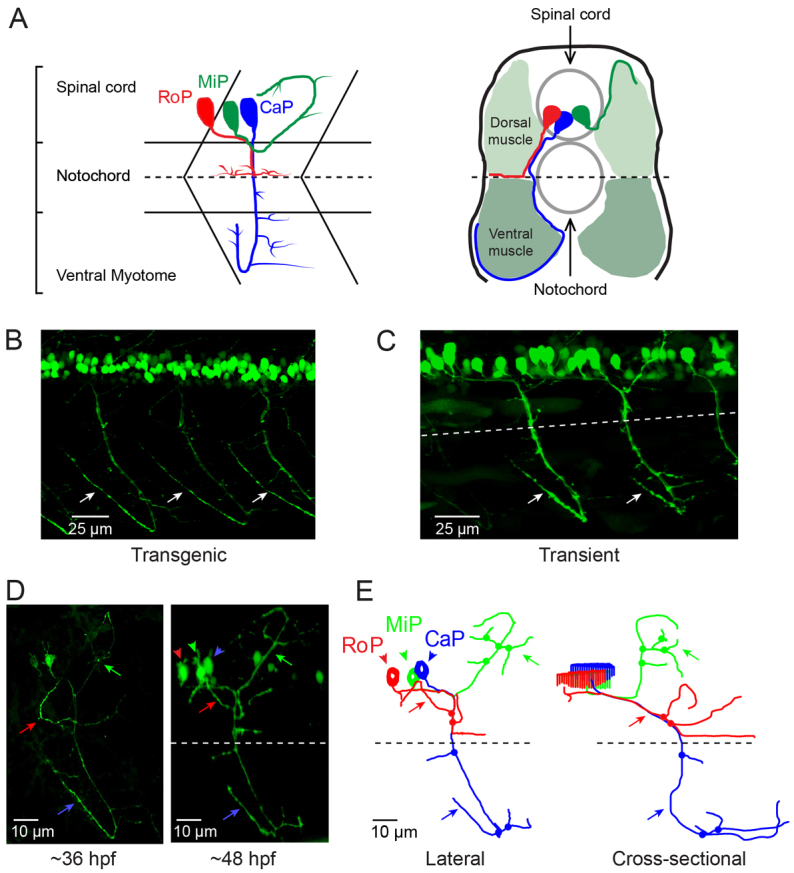
CaP morphology is extremely reproducible. (A) The schematics illustrate the stereotypical morphology and axonal trajectories of the RoP (red), MiP (green) and CaP (blue) primary motor neurons at ∼48 hpf from lateral (left) –and cross-sectional (right) perspectives (Myers et al., 1986). Dashed horizontal lines correspond to the horizontal myoseptum (choice point). Left: chevrons show the hemisegment boundaries. Anterior is to the left and dorsal is up. Right: Dorsal is up. (B) Three adjacent hemisegments from a germline transgenic animal [Tg(Mmu.MNX1-MNE:EGFP)] that expresses EGFP in motor neurons were imaged at ∼48 hpf. Arrows indicate the characteristic loop of the CaP axon under and around the ventral muscle. In this and subsequent figures, confocal images were projected and lateral or quasi-lateral views are shown. Anterior is to the left and dorsal is up. (C) Two adjacent hemisegments from an animal that was injected with the EGFP plasmid for transient expression in motor neurons were imaged at ∼48 hpf. Arrows indicate the characteristic loop of the CaP axon under and around the ventral muscle. In this and subsequent figures, the dashed line indicates the location of the horizontal myoseptum (choice point). (D) Projected confocal images were acquired from an animal transiently expressing EGFP in all three primary motor neurons in one hemisegment at ∼36 hpf (left) and ∼48 hpf (right). (E) Using the confocal image stack acquired at ∼48 hpf, each neuron was traced using Neurolucida. Projected traces are shown from lateral (left) and cross-sectional (right) perspectives. In D and E, and in subsequent figures unless otherwise noted, arrowheads indicate cell bodies and arrows indicate axons: red, RoP; green, MiP; blue, CaP. In E and in subsequent Neurolucida traces, circles indicate branch points.
To test the hypothesis that infant- and adult-onset SCA13 mutations have differential effects on neuronal development, we transiently expressed C-terminal Kv3.3-EGFP fusion proteins or EGFP alone under the control of the motor neuron enhancer from the mouse Mnx1 (Hb9) gene in zebrafish (Nakano et al., 2005). The fusion proteins encoded wild-type Kv3.3 or an adult- or infant-onset SCA13 mutation and were based on the zebrafish Kv3.3 ortholog, Kv3.3a, which is adapted for zebrafish physiology and body temperature (28°C) (Mock et al., 2010). The R335H mutation, which corresponds to the adult-onset mutation R420H in human Kv3.3, substitutes a histidine residue for the third arginine in the S4 transmembrane segment in the voltage sensor domain (Mock et al., 2010; Waters et al., 2006). This mutation will be referred to as aoR3H, where ao indicates adult onset. The F363L mutation, which is equivalent to the infant-onset mutation F448L in human Kv3.3, replaces a phenylalanine at the cytoplasmic end of the S5 transmembrane segment with leucine (Mock et al., 2010; Waters et al., 2006). This mutation will be referred to as ioFL, where io indicates infant onset. Importantly, we have shown that the effects of the SCA13 mutations on the function of human Kv3.3 are well conserved in the zebrafish channel (Mock et al., 2010).
Plasmid DNA encoding EGFP or fusion proteins of wild-type Kv3.3 or the aoR3H or ioFL mutant subunits was injected into the single cells of wild-type (AB) zebrafish embryos at the one-cell stage. This resulted in mosaic expression of the transgene in a variable subset of motor neurons. Animals expressing exogenous EGFP or a Kv3.3-EGFP fusion protein were selected at 30 hpf. Neurons were imaged in living animals using a confocal microscope at ∼36 hpf and again at ∼48 hpf. The trajectory of individual EGFP-expressing neurons was traced through each confocal section using Neurolucida, followed by three-dimensional (3D) digital reconstruction.
We focused on the development of CaP motor neurons. CaP extends its axon deep into the ventral muscle where it is well isolated from other neuronal processes, facilitating developmental analysis (Fig. 1A) (Eisen et al., 1986; Myers et al., 1986; Eisen, 1994). A low level of endogenous Kv3.3 expression can first be detected in CaP cells at 24 hpf (Issa et al., 2011). Kv3.3 expression increases dramatically over the next 24 hours, corresponding to the developmental window analyzed in our experiments. Furthermore, Kv3.3 channel activity controls the excitability of CaP neurons during this period (Issa et al., 2011). Although MiP and probably RoP express Kv3.3 early in development, little is known about the time course of Kv3.3 expression or its role in the electrical activity of these cells (Issa et al., 2011). Therefore, we have not analyzed in detail the effects of SCA13 mutations on MiP and RoP development.
At ∼48 hpf, confocal imaging of a live embryo from a germline transgenic line [Tg(Mmu.MNX1-MNE:EGFP)] that expresses EGFP in motor neurons shows the stereotypical trajectory of the CaP axon reliably reproduced in three neighboring hemisegments (Fig. 1B) (Issa et al., 2011). In this lateral view of projected optical sections, the CaP axon can be seen extending down the middle of the myotome and curving around the ventral aspect of the ventral muscle. Transient expression of EGFP in motor neurons revealed the same morphology (Fig. 1C).
Fig. 1D shows a hemisegment in which all three primary motor neurons expressed EGFP. At ∼36 hpf, the cell-specific trajectories of CaP, MiP and RoP were evident. By ∼48 hpf, the axons had increased in length and extended additional collaterals and branches. Expression of EGFP in all three cells allowed us to establish that Neurolucida tracing and 3D reconstruction can map CaP, MiP and RoP independently of each other. Neurolucida analysis of the confocal image stack acquired at ∼48 hpf provided separate, continuous traces of CaP, MiP and RoP that conformed to the expected cell-specific morphology of each cell type (Fig. 1E) (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). The CaP axon projected to the ventral myotome, whereas the RoP axon migrated along the horizontal myoseptum into the medial myotome. The MiP axon had retracted from the choice point and grown into the dorsal myotome. The 3D reconstruction demonstrates that the MiP, RoP and CaP axons, despite being closely apposed before the choice point, can be unambiguously identified (supplementary material Movie 1).
Prior to investigating the effects of the SCA13 mutations on CaP development, it is essential to determine whether exogenous expression of wild-type Kv3.3 generates a developmental phenotype. The wild-type Kv3.3-EGFP fusion protein was transiently expressed in motor neurons. EGFP fluorescence was detected at ∼30 hpf. Confocal microscopy was performed at ∼36 and ∼48 hpf. Fig. 2A shows a hemisegment in which all three primary motor neurons expressed exogenous wild-type Kv3.3. Neurolucida tracing and 3D reconstruction revealed normal CaP, MiP and RoP morphologies (Fig. 2B; supplementary material Movie 2). No differences from control (EGFP-expressing) neurons were apparent.
Fig. 2.

CaP neurons that express exogenous wild-type Kv3.3 develop normally. (A) Shown are projected confocal images of a hemisegment in which all three primary motor neurons express the wild-type Kv3.3-EGFP fusion protein: left, ∼36 hpf; right, ∼48 hpf. The images are offset from true lateral views. (B) Projected Neurolucida traces obtained from the image stack acquired at ∼48 hpf are shown from the original quasi-lateral (left), corrected lateral (center) and cross-sectional (right) perspectives. See Fig. 1 legend for color code, etc.
CaP neurons expressing the infant-onset mutation extend long, proximal collaterals into inappropriate synaptic territory
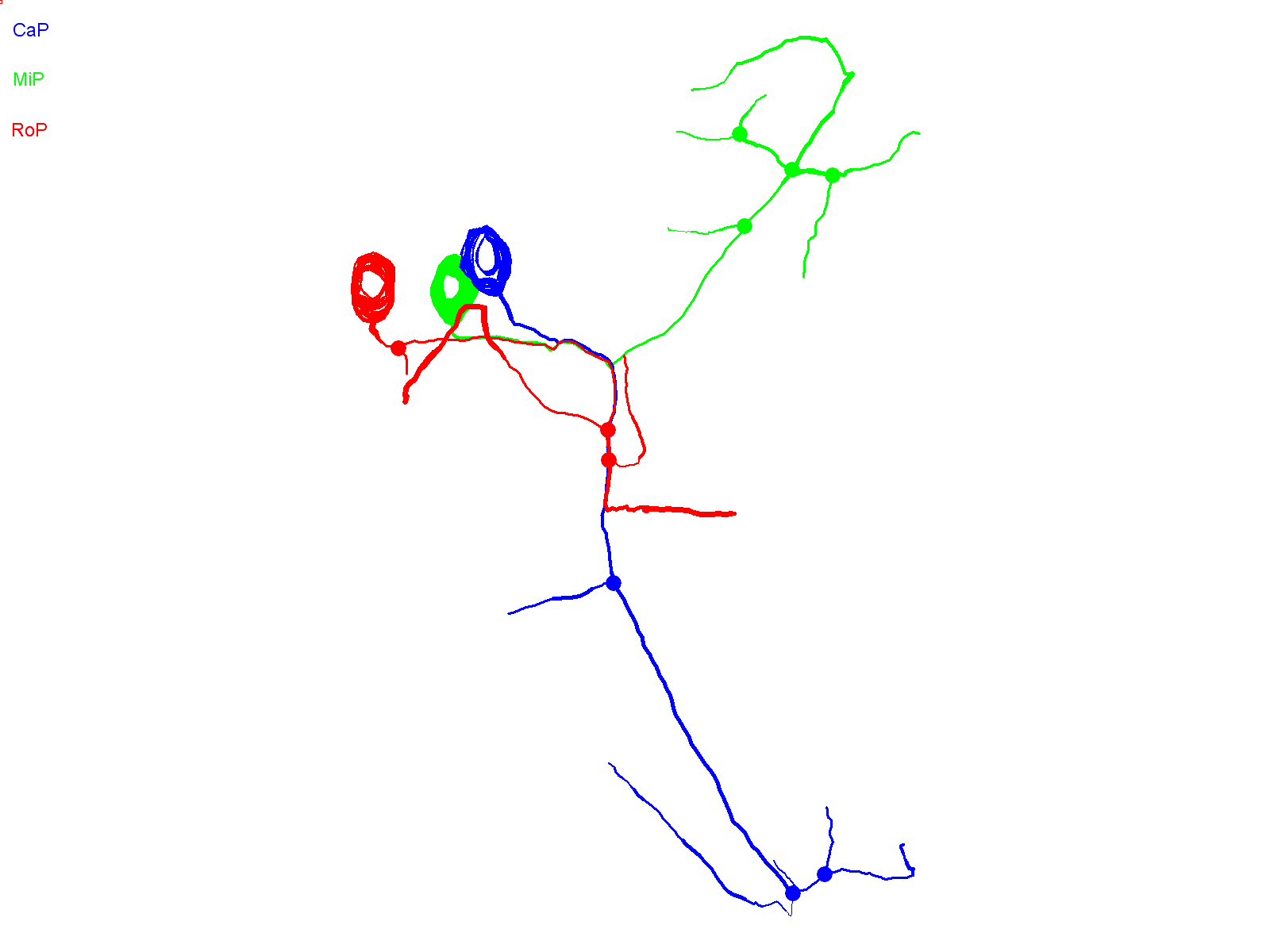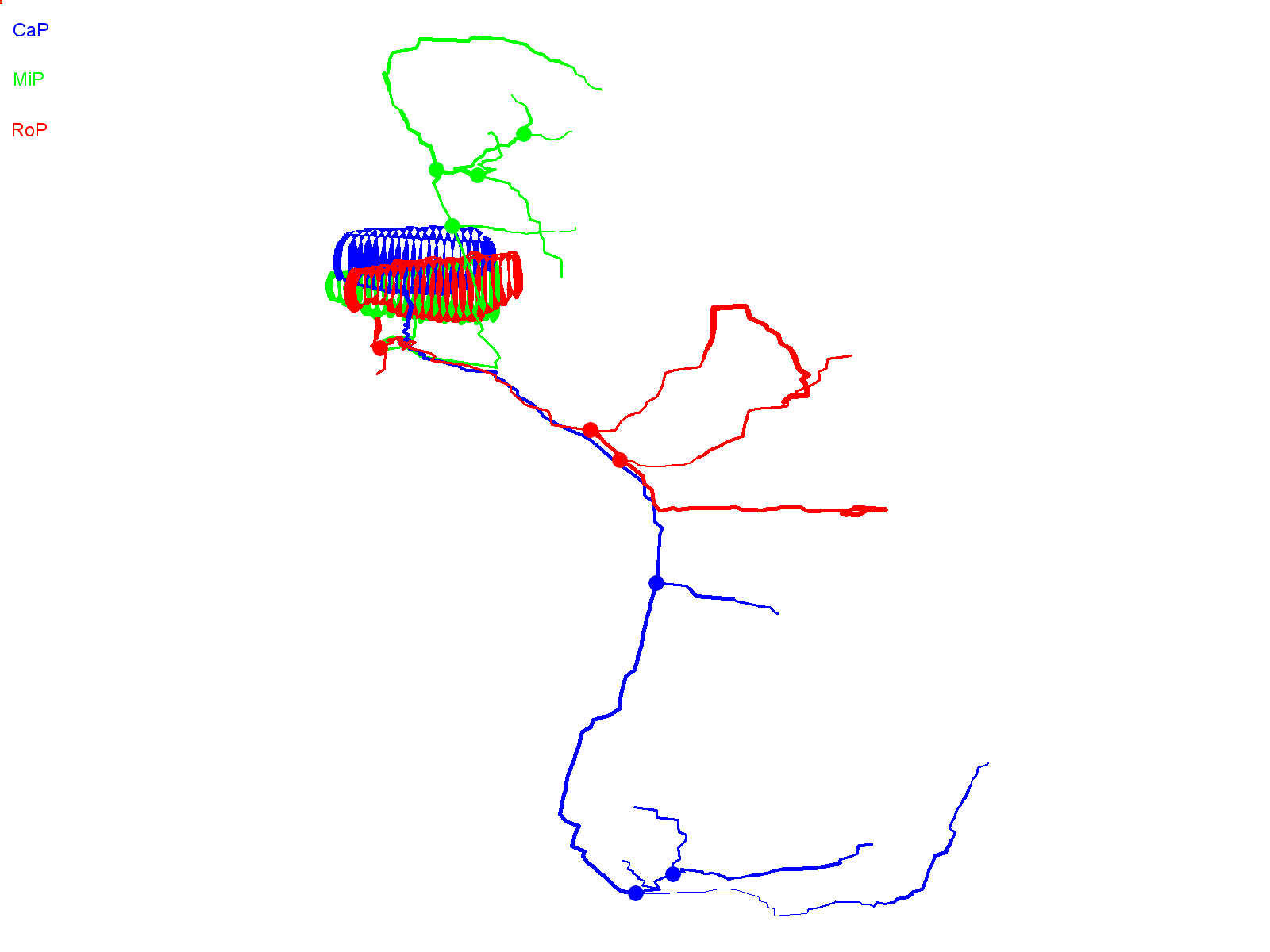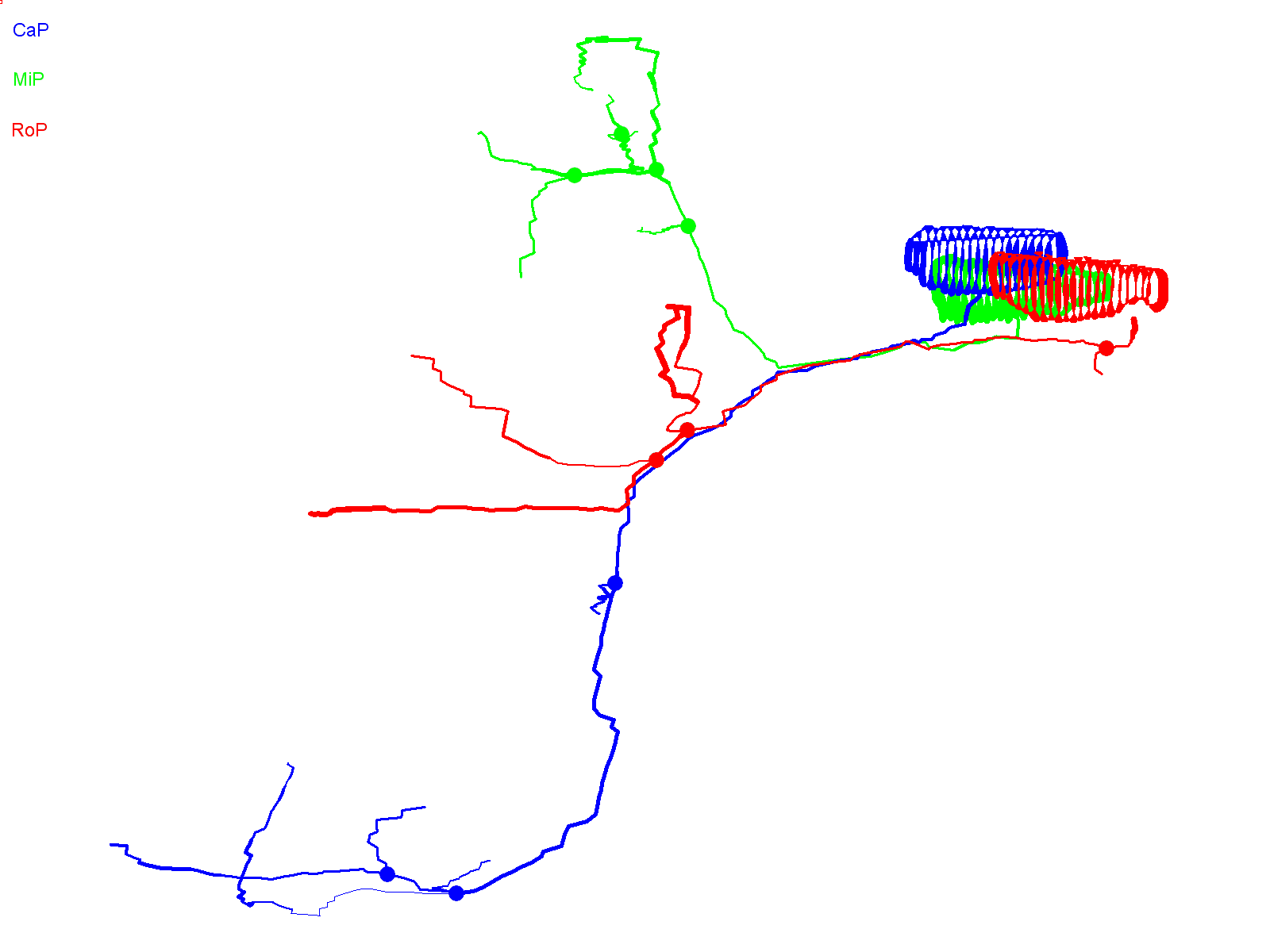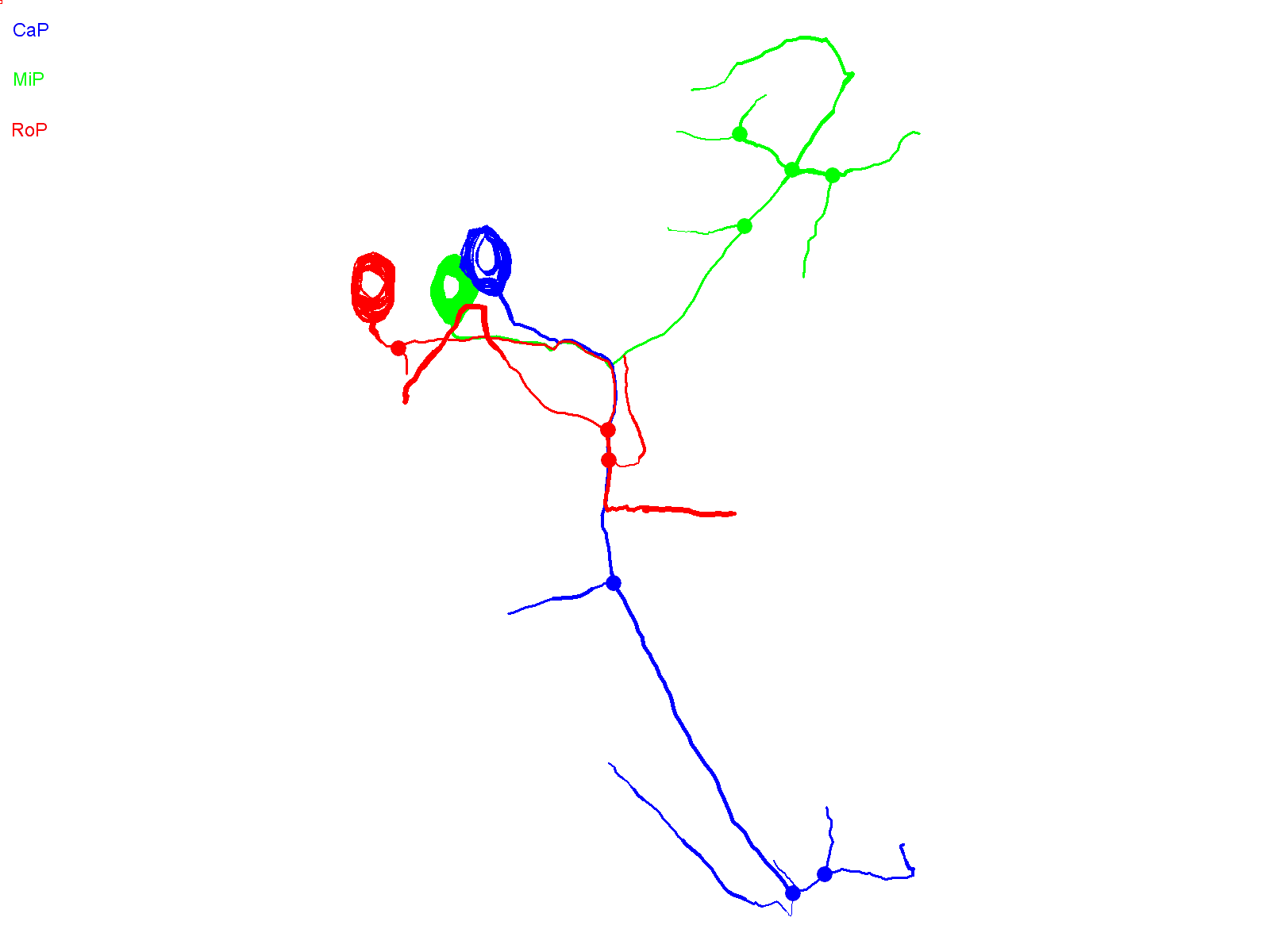
In contrast to wild-type Kv3.3-expressing CaP cells, axons in CaP neurons expressing the ioFL mutation frequently made pathfinding errors, straying from the normal, highly stereotypical trajectory and entering inappropriate postsynaptic territory. Fig. 3A shows two adjacent hemisegments containing ioFL-expressing CaP neurons. By ∼48 hpf, neuron 1 (Fig. 3A,B, right) had extended an aberrant, abnormally long collateral from the vicinity of the choice point into the medial myotome, following a trajectory similar to that of a RoP axon, before turning and invading the dorsal myotome (Fig. 3A,B, arrows; supplementary material Movie 3). In contrast, the distal part of the axon (ventral to the choice point) was confined to the proper ventral muscle territory. Occasionally, the distal axons of ioFL-expressing CaP cells did follow abnormal trajectories. In neuron 2 (Fig. 3A,B, left), the distal end of the main axon wrapped completely around the ventral muscle and then grew laterally across its dorsal region, which is outside its normal target area (Fig. 3A,B, arrowheads; supplementary material Movie 4) (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). However, long axons in ioFL-expressing CaP cells did not cross the hemisegment boundary.
Fig. 3.

Infant-onset FL mutation causes axonal pathfinding errors in CaP neurons. (A) Projected quasi-lateral image acquired at ∼48 hpf shows ioFL-expressing CaP neurons with different axonal abnormalities in adjacent hemisegments. In the right-hand hemisegment, neuron 1 has extended an aberrant collateral into the dorsal myotome (arrow). In the left-hand hemisegment, the main axon shaft of neuron 2 has wrapped around the ventral muscle and then grown into abnormal territory in the dorsal region of the ventral muscle (arrowhead). Note that the other primary motor neurons in the left-hand hemisegment also expressed the ioFL fusion protein. (B) Projected Neurolucida traces obtained from neurons 1 and 2 are shown from quasi-lateral (left) and cross-sectional (right) perspectives. Arrowheads and arrows have the same meaning as in A. (C) Shown are projected confocal images of a hemisegment in which all three primary motor neurons expressed the ioFL-EGFP fusion protein by ∼48 hpf: left, ∼36 hpf; right, ∼48 hpf. Right: the white arrow indicates an aberrant collateral extending from the CaP axon into MiP territory. (D) Projected Neurolucida traces obtained from the ∼48 hpf image in C are shown from lateral (left) and cross-sectional (right) perspectives. The black arrow indicates aberrant collateral extending from the CaP axon into MiP territory. (E) Projected confocal images acquired at ∼48 hpf show examples of ioFL-expressing neurons with an abnormally truncated axon (left) or axons that could not be traced by Neurolucida (right). See Fig. 1 legend for color code, etc.
Aberrant proximal collaterals diverging from the CaP axon could be unambiguously identified in hemisegments in which all three primary motor neurons expressed the ioFL protein (Fig. 3C). In an image taken at ∼36 hpf, the trajectories of the main axonal shafts of the CaP, RoP and MiP neurons appeared normal, having grown into the ventral myotome, along the horizontal myoseptum, and into the dorsal myotome, respectively. However, by ∼48 hpf, CaP had sent an additional, abnormal process into the dorsal myotome (Fig. 3C, white arrow). As shown by Neurolucida tracing and 3D digital reconstruction, the aberrant CaP process could be readily distinguished from the MiP and RoP axons (Fig. 3D; supplementary material Movie 5).
In contrast, pathfinding defects were not evident in the ioFL-expressing MiP and RoP neurons in the same hemisegment (Fig. 3C,D). That two primary motor neurons can develop normally in the presence of a third with pathfinding errors is consistent with published reports showing that the axon of a primary motor neuron adopts its cell-specific morphology independently of the other two primary motor neurons in the same hemisegment (Eisen et al., 1989; Liu et al., 1990; Pike and Eisen, 1990). Importantly, the CaP pathfinding defect emerged between ∼36 and ∼48 hpf (Fig. 3C). The development of MiP and RoP lags behind that of CaP (Eisen et al., 1986; Myers et al., 1986). Therefore, it remains possible that the ioFL mutation alters the pathfinding of MiP and RoP axons at a later developmental time.
Some ioFL-expressing CaP axons were prematurely truncated or took such highly aberrant pathways that they could not be traced using Neurolucida (Fig. 3E). Such cells (5 of 21) were not included in our data set. These five cells came from four different embryos that did not have any obvious anatomical defects. Therefore, the strange morphologies of these CaP neurons did not result from general developmental defects in these animals. Thus, our analysis might underestimate the variability and severity of the pathfinding defects caused by the ioFL mutation. In contrast, we did not exclude any cells that expressed EGFP, wild-type Kv3.3 or aoR3H from our analysis.
Transmitted light images did not reveal any obvious defects in muscle structure in territories contacted by CaP neurons with pathfinding errors. Muscle striations and the structures of the horizontal and vertical myosepta and notochord seemed to be normal (supplementary material Movie 6).
CaP neurons expressing the adult-onset mutation have extensive distal branching
In contrast to CaP neurons that expressed the ioFL mutation, cells expressing the aoR3H mutation did not exhibit pathfinding errors and did not invade inappropriate territories of the musculature. At ∼36 hpf, the axonal trajectory of an aoR3H-expressing cell appeared normal (Fig. 4A). At ∼48 hpf, we noted that the distal axonal arbors of aoR3H-expressing cells tended to branch excessively (Fig. 4A,B; supplementary material Movie 7). Fig. 4 shows a pronounced example.
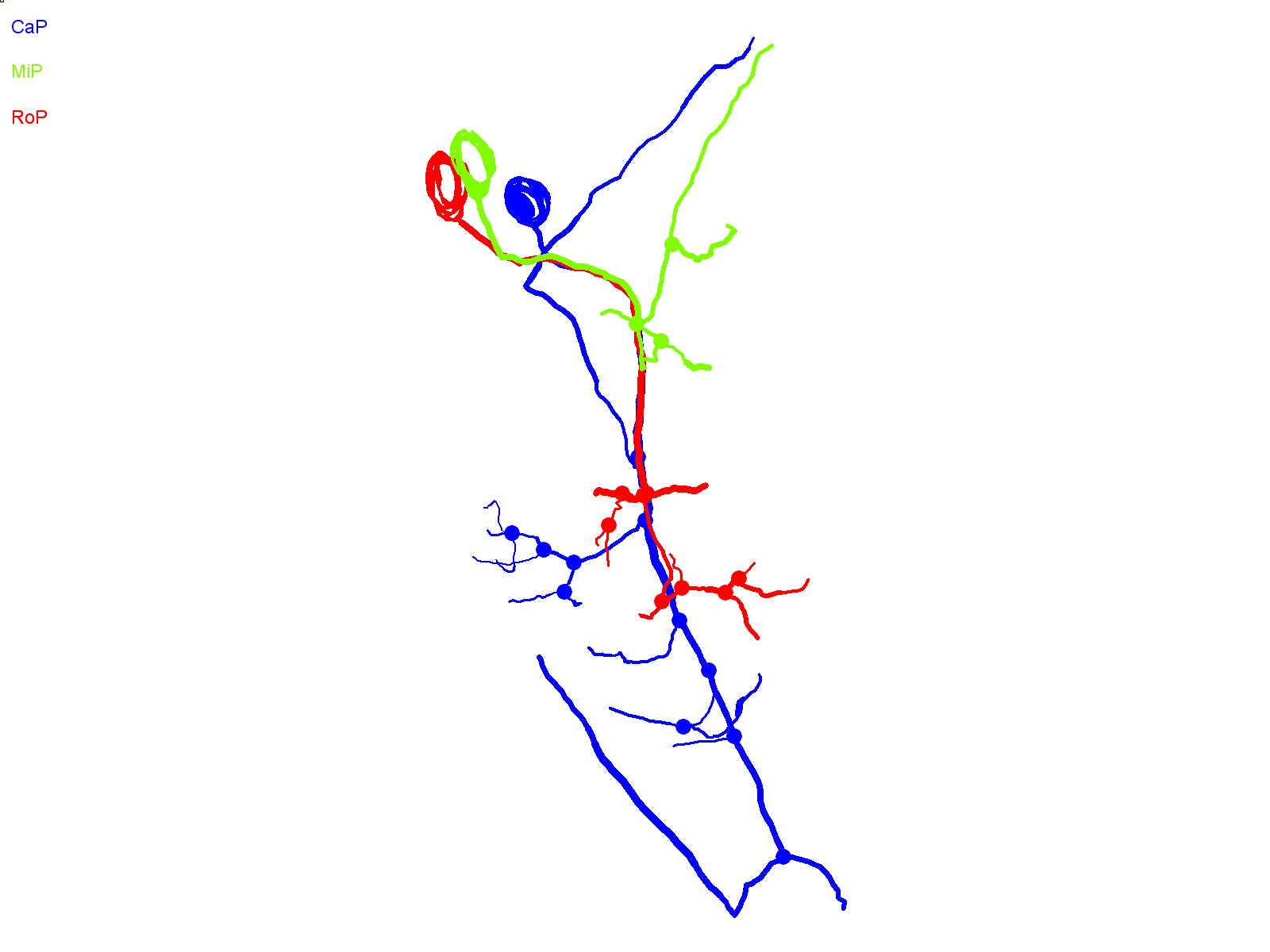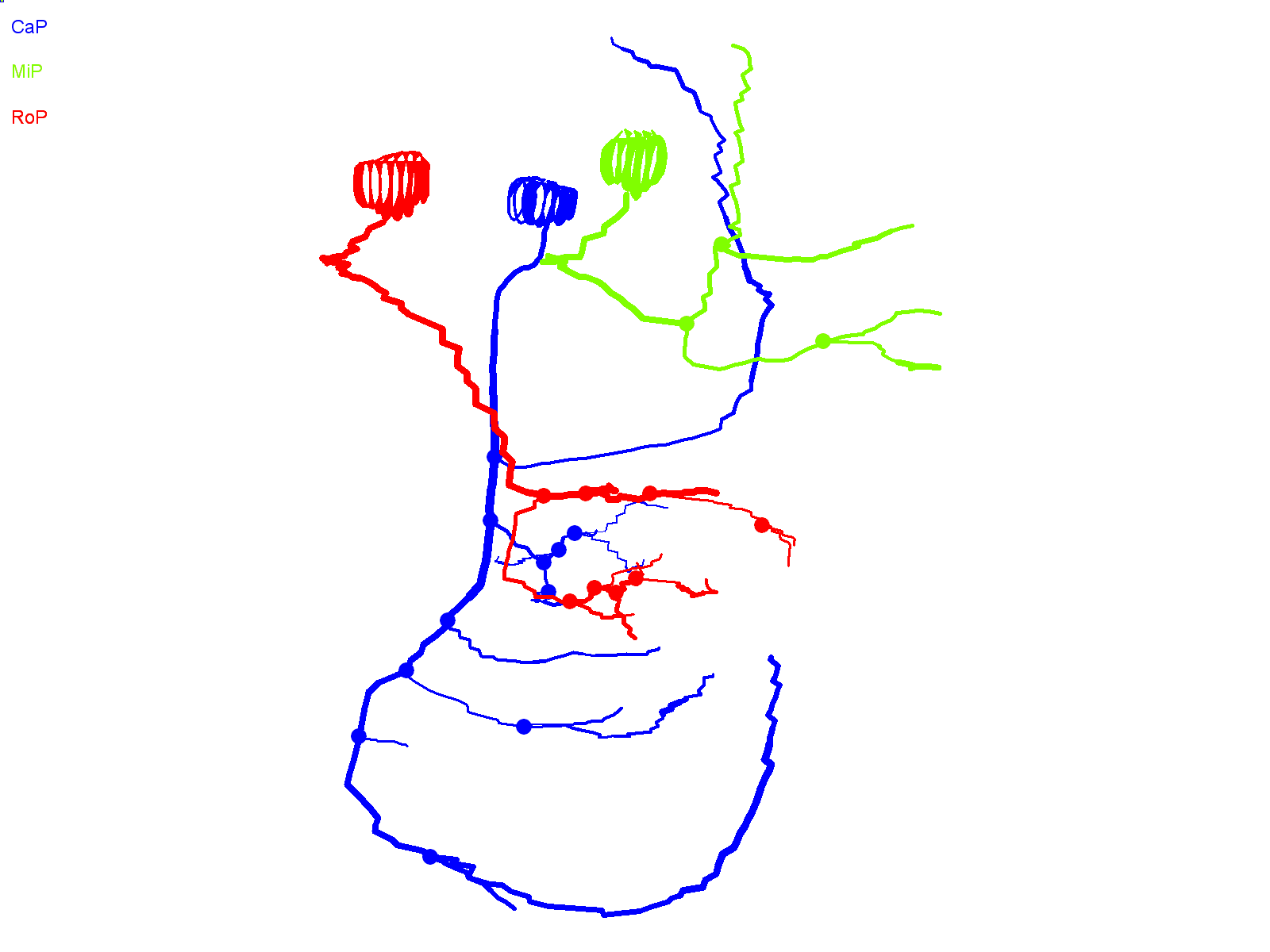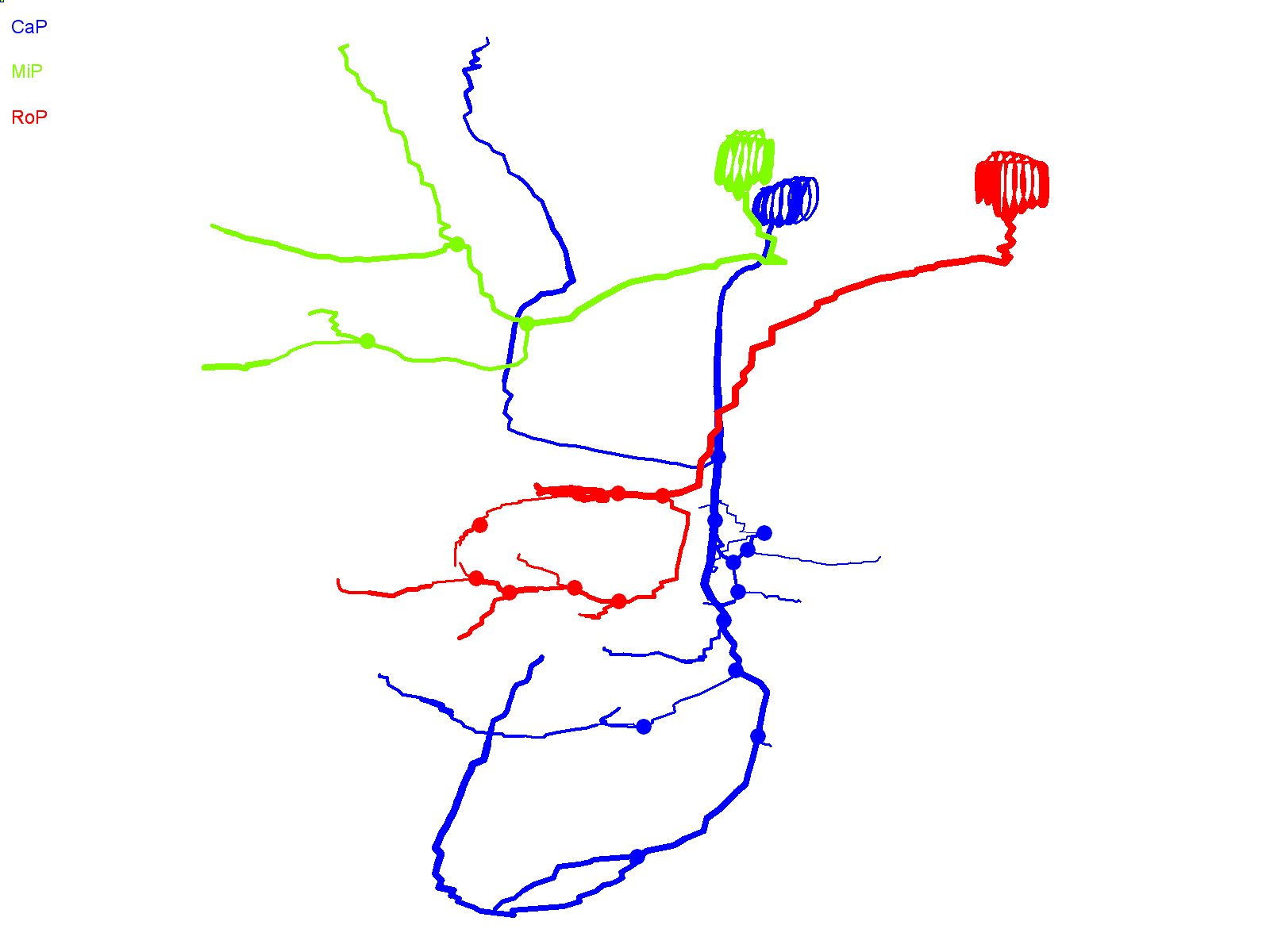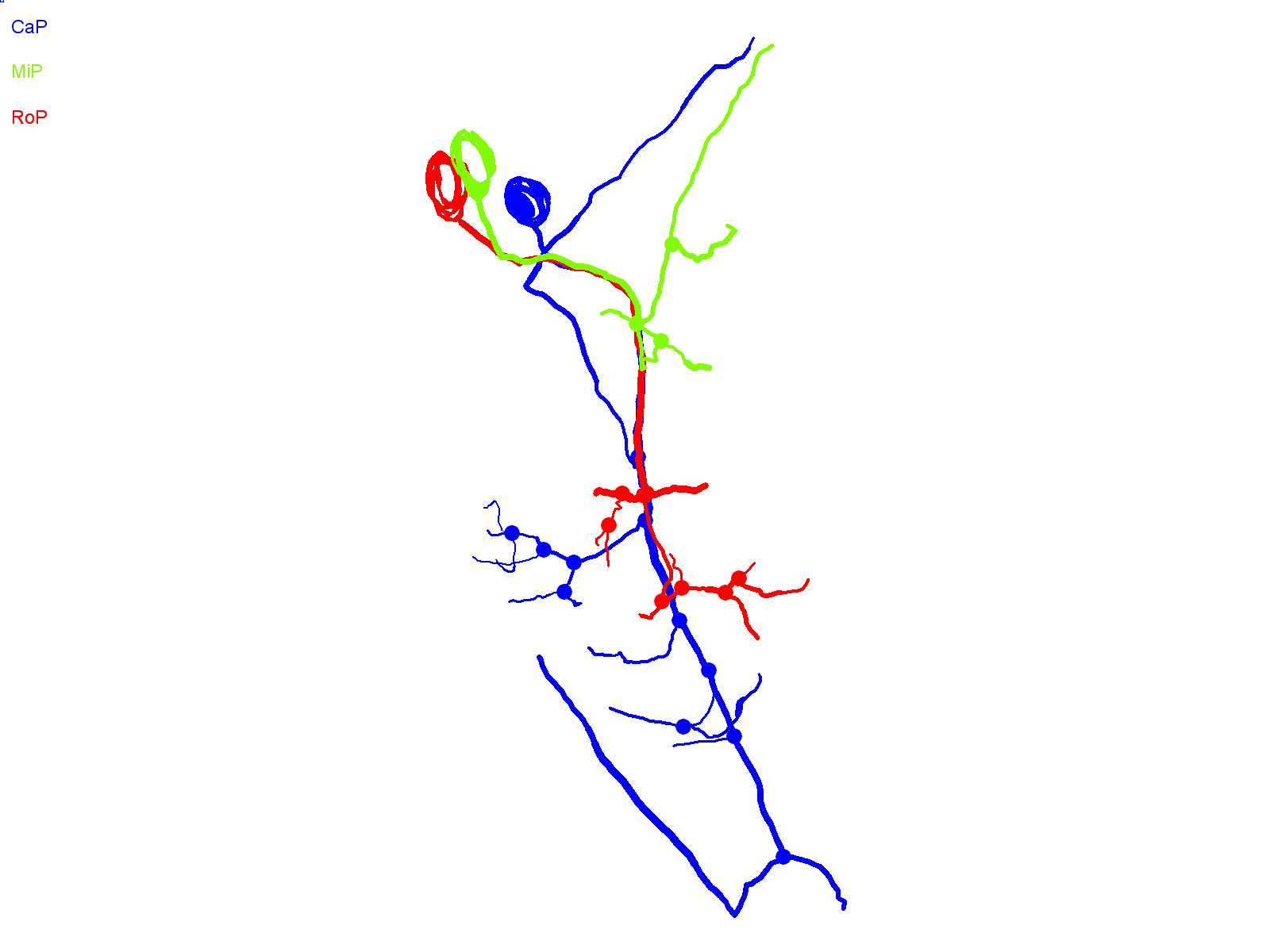
Fig. 4.

CaP neurons that express aoR3H tend to branch excessively at the distal end of the axon. (A) Projected confocal images obtained from an aoR3H-expressing CaP neuron at ∼36 and ∼48 hpf are shown. At ∼48 hpf, the distal axonal arbor is highly branched. (B) Projected Neurolucida trace of the ∼48 hpf image stack is shown from lateral (left) and cross-sectional (right) perspectives. See Fig. 1 legend for color code, etc.
FL infant-onset mutation dramatically alters CaP development
To compare axonal trajectories in CaP neurons expressing EGFP alone or the wild-type Kv3.3, ioFL or aoR3H fusion proteins, the 3D digital reconstruction of each analyzed cell was projected along the anterior-posterior axis to provide a cross-sectional view. Then, the projections of all cells in each group were aligned along the proximal axon from the cell body to the choice point and overlaid (Fig. 5A–D). The axons of EGFP-, wild-type- and aoR3H-expressing neurons followed similar, highly reproducible trajectories into the ventral myotome and around the ventral muscle (Fig. 5A–C). In contrast, axonal trajectories in ioFL-expressing CaP neurons were much less consistent, with a profusion of abnormal, long processes extending from the vicinity of the choice point (Fig. 5D). When color-coded projections of all the groups were overlaid, it was evident that the axons and collaterals of ioFL-expressing cells diverged from the normal trajectory to a significantly larger extent than the axons of CaP neurons expressing EGFP, wild-type Kv3.3 or aoR3H (Fig. 5E).
Fig. 5.

Axon collaterals of ioFL-expressing CaP neurons diverge significantly from the normal trajectory. (A–D) Cross-sectional projections of Neurolucida traces for all cells in a group have been aligned along the main axonal shaft between the cell body and the choice point and superimposed: (A) EGFP (black; n=12 cells from 7 animals); (B) wild-type Kv3.3 (green; n=13 cells from 6 animals); (C) aoR3H mutation (blue; n=19 cells from 7 animals); (D) ioFL mutation (red; n=16 cells from 7 animals). (E) All cells in all groups have been superimposed retaining the same color code used in A–D. (F) Scatter plot shows the total process length (axons + branches + collaterals) for individual cells in each group. The boxes encompass length values from the 25th to 75th percentile of the data. The horizontal line in the box shows the median value, whereas the horizontal lines above and below the box show the maximal and minimal values, respectively. Values of means ± s.e.m. are provided in Table 1. Process length in cells expressing ioFL or aoR3H mutations was significantly greater than in cells expressing EGFP (P=0.0183 by ANOVA followed by Newman-Keuls multiple comparison test: ioFL or aoR3H vs wild type, *P<0.05; EGFP, n=12; wild type, n=13; ioFL, n=16; aoR3H, n=19).
We compared the total length of the processes (axon + collaterals + branches) in EGFP-, wild-type Kv3.3-, ioFL- and aoR3H-expressing CaP neurons (Fig. 5F, Table 1). We found that processes were significantly longer in ioFL- and aoR3H-expressing CaP neurons than in EGFP-expressing cells. In contrast, the length of processes in CaP neurons expressing exogenous wild-type Kv3.3 did not differ significantly from any other group.
Table 1.
Quantitative comparison of axon parameters

Although ioFL- and aoR3H-expressing CaP neurons had significantly longer processes than EGFP-expressing cells, we suspected that the extra length came from different regions of the axon. Because CaP neurons expressing the ioFL mutation had a profusion of proximal processes, whereas those expressing the aoR3H mutation appeared to have excessive branching from the distal axon, we analyzed the numbers and lengths of proximal and distal branches separately (Fig. 6, Table 1). We defined proximal branches as those diverging from the main axon before or at the choice point, and distal branches as those diverging after the choice point.
Fig. 6.
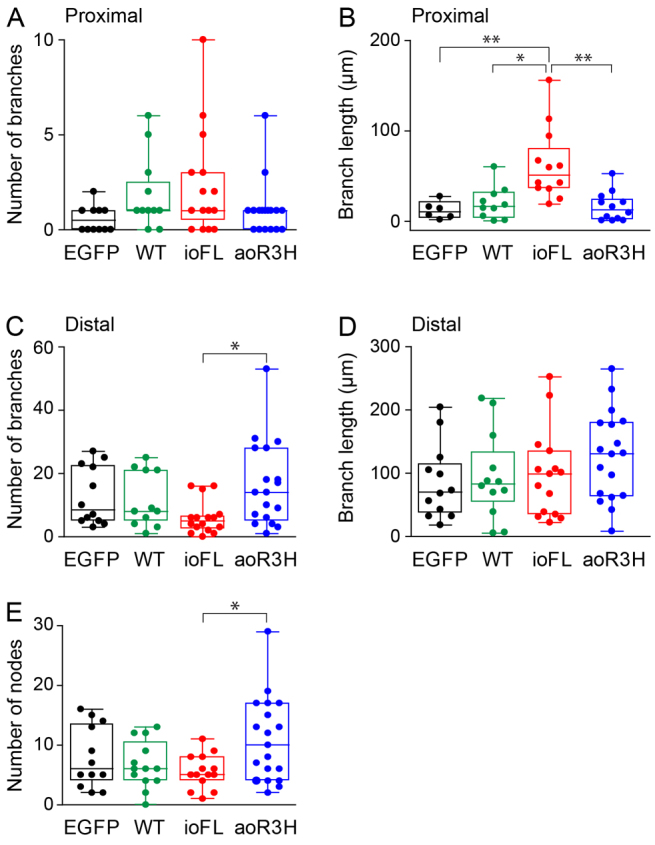
Infant- and adult-onset mutations have differential effects on proximal and distal branches in CaP neurons. Scatter plots show the numbers (A,C) and total lengths (B,D) of proximal (A,B) and distal (C,D) branches in EGFP-, wild-type Kv3.3-, ioFL- and aoR3H-expressing CaP neurons at ∼48 hpf. The boxes and lines have the same meaning as in Fig. 5F. Values of means ± s.e.m. are provided in Table 1. The color code is the same as in Fig. 5. (A) The number of proximal branches did not differ significantly among the groups (P=0.0849 by ANOVA). (B) The proximal branches in ioFL-expressing CaP neurons were significantly longer than in EGFP-, wild-type Kv3.3- or aoR3H-expressing CaP neurons (P=0.0004 by ANOVA followed by Dunn’s multiple comparison test: EGFP vs ioFL, **P<0.01; wild type vs ioFL, *P<0.05; aoR3H vs ioFL, **P<0.01). (C) The number of distal branches was significantly greater in aoR3H-expressing CaP neurons than in ioFL-expressing CaP neuons (P=0.023 by ANOVA followed by Dunn’s multiple comparison test: aoR3H vs ioFL, *P<0.05). (D) The lengths of distal branches did not differ significantly among the groups. (E) The scatter plot shows the number of nodes in EGFP-, wild-type Kv3.3-, ioFL- and aoR3H-expressing CaP neurons at ∼48 hpf. The number of nodes in aoR3H-expressing CaP neurons was significantly greater than in ioFL-expressing CaP neurons (P<0.05 by ANOVA followed by Newman-Keuls multiple comparison test: aoR3H vs ioFL, *P<0.05). For A–E: EGFP, n=12; wild type, n=13; ioFL, n=16; aoR3H, n=19.
The number of proximal branches did not differ significantly between EGFP-, wild-type Kv3.3-, ioFL- and aoR3H-expressing CaP neurons (Fig. 6A, Table 1). However, the length of proximal branches was significantly greater in ioFL-expressing cells than in all the other groups (Fig. 6B, Table 1). Fig. 6B shows that a large fraction of ioFL-expressing CaP neurons had proximal collaterals that were longer than the vast majority of those found in EGFP-, wild-type- or aoR3H-expressing cells. These abnormally long processes were responsible for most of the pathfinding errors observed in ioFL-expressing cells. Most of the aberrant processes (∼75%) in ioFL-expressing CaP neurons invaded the ventral region of the dorsal muscle or the dorsal region of the ventral muscle. The remainder (∼25%) extended into the dorsal region of the dorsal muscle. These territories are normally innervated exclusively by RoP or MiP. In control animals, the dorsal muscle is innervated by MiP. The dorsal ∼third of the ventral muscle is innervated by RoP, whereas a non-overlapping territory corresponding to the ventral ∼two-thirds of the ventral muscle is innervated by CaP (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). Unlike ioFL-expressing CaP cells, long collaterals that extended deep into RoP or MiP territory were not observed in CaP neurons that expressed EGFP alone, wild-type Kv3.3 or the adult-onset mutation, aoR3H.
A different picture emerged when distal branches were considered. The number of distal branches in aoR3H-expressing cells varied more than in the other groups (Fig. 6C, Table 1). Cells expressing aoR3H had a significantly greater number of distal branches than cells expressing ioFL. However, the number of distal branches in aoR3H cells did not differ significantly from that in EGFP-expressing or wild-type-Kv3.3-expressing cells. Interestingly, the difference between aoR3H and ioFL cells reflected the finding that ioFL-expressing cells tended to have fewer distal branches compared with the other groups, although this rose to the level of statistical significance only compared with aoR3H. The length of distal branches did not differ among the groups (Fig. 6D, Table 1).
We also analyzed the complexity of the axonal arbor. Each branch point or bifurcation was defined as a node. We found that aoR3H-expressing cells tended to have more nodes than cells in the other groups (Fig. 6E). This tendency was statistically significant when compared with ioFL-expressing cells but not with EGFP- or wild-type-expressing cells (Table 1).
DISCUSSION
Differential effects of mutations on development might underlie the age of SCA13 onset
The early-onset ioFL and late-onset aoR3H mutations have distinctly different effects on neuronal development that might determine whether SCA13 emerges during infancy or adulthood. The infant-onset mutation significantly disrupted axonal pathfinding in CaP neurons, which frequently extended axon collaterals into inappropriate postsynaptic territories in the medial and dorsal myotome that are normally innervated exclusively by RoP or MiP motor neurons (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). Importantly, the aberrant collaterals originated in the vicinity of the choice point, where CaP, MiP and RoP axons normally diverge to innervate their cell-specific territories. Our results indicate that the ioFL mutation interferes with the ability of CaP axons to choose the correct trajectory. Importantly, long proximal processes that invade RoP or MiP territory are not normally seen in CaP neurons at any stage of development (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). Therefore, the ioFL phenotype cannot be attributed to a change in the rate of development. In control animals, primary motor neurons innervate exclusive, non-overlapping regions of the myotome (Eisen et al., 1986; Myers et al., 1986; Westerfield et al., 1986). Furthermore, during normal development, CaP neurons do not extend branches into inappropriate territory that are later pruned, but instead grow unerringly to the correct target region (Eisen et al., 1986; Myers et al., 1986). Therefore, the abnormal axonal trajectories in ioFL-expressing cells do not reflect a failure to refine the terminal axonal arbor. Errors in pathfinding might be relevant to infant-onset SCA13, because interaction with incorrect synaptic partners is thought to trigger programmed cell death during brain development (Oppenheim, 1991).
Importantly, expression of exogenous wild-type Kv3.3 had no significant effect on development compared with EGFP-expressing control cells. These data indicate that the ioFL phenotype is due specifically to the SCA13 mutation and is not an artifact of exogenous Kv3.3 expression. The adult-onset mutation had little impact on CaP development apart from a trend towards increased branching in the distal axonal arbor.
From these results, we speculate that, in the infant-onset form of SCA13, the development of cerebellar neurons is severely disrupted, increasing their susceptibility to cell death early in life and giving rise to the shrunken cerebellum seen in affected children. Kv3.3 channels fulfill the same physiological role in mammalian and zebrafish neurons, which is to promote sustained high-frequency firing of action potentials (Rudy and McBain, 2001; Issa et al., 2011). It is therefore reasonable to suggest that SCA13 mutations affect the development of human neurons by mechanisms similar to those at play in zebrafish neurons. The developmental defects caused by the infant-onset mutation in cerebellar neurons might differ from those seen in motor neurons, but they are expected to be significantly more detrimental to neuronal health and viability during brain development than any phenotypes associated with the adult-onset mutation.
We do not mean to imply that developmental effects of the adult-onset mutation would have no consequences for the emergence of SCA13 later in life, however. In spinocerebellar ataxia type 1 (SCA1), insults that occur during a critical period of cerebellar development dramatically increase the severity of neurodegeneration that emerges during adulthood (Serra et al., 2006). Therefore, relatively subtle developmental changes resulting from the aoR3H mutation could play a role in triggering SCA13 in adults.
Axonal pathfinding defects might result from changes in electrical excitability
The pathfinding defects observed in ioFL-expressing CaP neurons might result from effects of the mutation on neuronal excitability. Although early phases of neuronal development occur independently of action-potential firing, spiking activity has well-established roles during the later phases of neuronal development, contributing to axonal pathfinding, the selection of post-synaptic targets and the refinement of synaptic connections (Ming et al., 2001; Hanson and Landmesser, 2004; Moody and Bosma, 2005; Hanson and Landmesser, 2006; Spitzer, 2006). Kv3.3 channels are crucial regulators of action-potential firing in fast-spiking neurons. Kv3 channels facilitate sustained high-frequency firing because they have unique gating properties, including a depolarized activation range, fast activation and extremely fast deactivation (Rudy and McBain, 2001). ioFL is a dominant gain-of-function mutation that specifically alters these gating properties, shifting activation to negative voltages, slowing opening and dramatically slowing closing (Waters et al., 2006; Minassian et al., 2012). Such changes in gating would be expected to alter electrical excitability in Kv3.3-expressing neurons (Issa et al., 2011; Minassian et al., 2012). Our identification of pathfinding defects in ioFL-expressing neurons suggests that the specialized gating properties of Kv3.3 channels play a previously unsuspected role in neuronal development.
Expression of the ioFL mutation had no apparent effect on early phases of CaP development, which occur independently of spiking activity. Our images show that axons in ioFL-expressing cells entered the periphery and traveled normally to the choice point and beyond. Importantly, the initial outgrowth of the CaP axon does not require electrical activity in the neuron. The axon follows a route marked by pre-existing clusters of acetylcholine receptors on the muscle surface (Flanagan-Steet et al., 2005; Panzer et al., 2006; Moreno and Ribera, 2009). At this early stage of development, CaP neurons are inexcitable. Rudimentary excitability does not emerge until ∼24 hpf, when CaP cells are able to fire a single action potential in response to positive-current injection (Moreno and Ribera, 2009). More mature firing properties have been established by 48 hpf, when the neurons are able to spike repetitively in response to stimulation and during spontaneous fictive swimming (Moreno and Ribera, 2009; Issa et al., 2011).
Consistent with the hypothesis that changes in action potential firing contribute to the developmental effects of the ioFL mutation, the pathfinding phenotype became apparent during a relatively advanced phase of development, with significant abnormalities emerging between ∼36 and ∼48 hpf (Eisen et al., 1986; Melançon et al., 1997). The pathfinding errors seen in ioFL-expressing cells suggest that CaP axons misinterpret or fail to respond to guidance cues. Poo and colleagues have established a direct relationship between electrical excitability and guidance decisions, showing that the pattern of action-potential firing in Xenopus neurons controls whether a diffusible guidance cue is attractive or repellent to the migrating axon (Ming et al., 2001).
Increased complexity of the axonal arbor in CaP neurons expressing the aoR3H mutation might also result from changes in excitability. Refinement of terminal arbors is controlled by neuronal activity (Moody and Bosma, 2005). The trend towards increased complexity of the distal axonal branches seen in aoR3H-expressing CaP neurons might result from failure to prune excess synapses. It is worth noting that the aoR3H and ioFL mutations have distinct effects on Kv3.3 function. The aoR3H mutation generates a dominant negative subunit that suppresses Kv3 current amplitude with no effects on channel gating (Waters et al., 2006; Mock et al., 2010; Minassian et al., 2012). Owing to their different effects on channel function, ioFL and aoR3H are expected to alter neuronal excitability in distinct ways, which could underlie their differential effects on CaP development.
We have shown that Kv3.3 channels play a crucial role in controlling CaP excitability during the developmental period analyzed in our experiments (Issa et al., 2011). Kv3.3 protein can first be detected in CaP neurons starting at ∼24 hpf. At ∼48 hpf, exogenous expression of the aoR3H mutant subunit impairs the ability of CaP neurons to fire repetitively for sustained periods of time and decreases the number of spikes fired per burst during fictive swimming. In parallel, aoR3H decreases the amplitude and precision of the startle response, an early arising, fast, large-amplitude behavior that is driven by CaP neurons. These changes in excitability and motor behavior result directly from the dominant negative effect of the aoR3H subunit on Kv3 current amplitude (Issa et al., 2011). In contrast to aoR3H, the dominant gain-of-function ioFL mutation is unlikely to affect the capacity for sustained spiking but is predicted to alter the firing rate during repetitive activity (Minassian et al., 2012).
We conclude that infant- and adult-onset SCA13 mutations have differential effects on neuronal development that could determine the age that disease emerges. Neurons that expressed the ioFL infant-onset mutation frequently interacted with inappropriate synaptic partners. If this were to occur in individuals who carry the mutation, it could result in excessive programmed cell death during cerebellar development.
METHODS
Animal maintenance
Animal procedures were approved by the Chancellor’s Animal Research Committee at the University of California at Los Angeles (UCLA). Zebrafish (Danio rerio) were housed at the UCLA Zebrafish Core Facility at 28°C under a 14 hour/10 hour light/dark cycle and bred to obtain embryos.
Molecular biology
EGFP was expressed using a previously described tol2 plasmid containing the minimal promoter from the zebrafish gata2 gene, three copies of the 125 bp motor neuron enhancer from the mouse Mnx1 (Hb9) gene, and the EGFP coding sequence (Nakano et al., 2005; Issa et al., 2011). To express Kv3.3-EGFP fusion proteins, the plasmid was modified to encode zebrafish Kv3.3a splice variant 1 (GenBank Accession #HQ118212.1) with EGFP fused in frame at the C-terminus via a four-residue linker comprising alanine-glycine-glycine-arginine before the initiation methionine in EGFP. Three fusion-protein plasmids were made that contained sequences encoding wild-type Kv3.3a, the adult-onset R335H (aoR3H) mutation, or the infant-onset F363L (ioFL) mutation (Mock et al., 2010). These mutations are equivalent to R420H and F448L in human Kv3.3 (Waters et al., 2006).
Confocal microscopy
Plasmid DNA (150–200 pg) encoding EGFP or one of the three Kv3.3-EGFP fusion proteins was injected into the single cell of wild-type (AB) embryos at the one-cell stage using a Picospritzer III (Parker Instruments). Injected animals were screened for EGFP fluorescence at ∼30 hpf using a Zeiss Discovery V12 epifluorescence microscope. At ∼36 hpf, expressing animals were anesthetized with 0.02% tricaine (MS-222) in embryo water and then embedded in low melt agarose (0.8%). The agarose was then covered with embryo water containing 0.02% tricaine.
Confocal imaging was performed on live, morphologically normal animals expressing EGFP or one of the Kv3.3-EGFP fusion proteins. Confocal imaging was also performed using a germline transgenic line [Tg(Mmu.MNX1-MNE:EGFP)] that expresses EGFP in motor neurons under the control of three copies of the 125 bp motor neuron enhancer from the mouse Mnx1 (Hb9) gene (Nakano et al., 2005; Issa et al., 2011). Images were acquired at 36–41 (∼36) hpf and again at 48–52 (∼48) hpf using an Olympus FV300 Fluoview laser scanning confocal microscope equipped with a LUMPLFL 40 × 0.8 NA water immersion lens. Fluorescence was excited using a 488 nm argon laser and detected using a 510 nm high pass emission filter. Optical sections were 1 αm in thickness. Transmitted light and fluorescent images were acquired in parallel at the maximal resolution of 1024×1024 pixels using Fluoview software.
Neurolucida analysis and 3D digital reconstruction
Confocal images and image aspect ratio parameters were imported into Neurolucida for digital tracing and 3D reconstruction (MBF Bioscience, v.7.52) (Glaser and Glaser, 1990). We restricted our analysis to neurons located in somites 10–18 so that the analyzed population would be similar in size and developmental stage. Quantified parameters of traced neurons were exported into Excel for plotting. Statistical analysis was performed using GraphPad Prism software v.3.03. Statistical tests are described in the figure legends.
Supplementary Material
Acknowledgments
We are grateful to Ms Yu Lee for excellent technical assistance and to Dr Georgeann O’Brien for advice about Neurolucida. We thank Dr Joanna Jen and members of the Papazian lab for helpful discussions and their comments on the manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
D.M.P. conceived of, designed and directed the research. F.A.I. designed and performed the research and analyzed data. A.F.M. was responsible for molecular biology. A.S. provided essential analysis tools. D.M.P. and F.A.I. wrote the paper.
FUNDING
This work was supported by the National Institutes of Health [grant R01NS058500 to D.M.P.]. F.A.I. was partially supported by a National Institutes of Health Training Grant [T32NS007101 (PI: Thomas J. O’Dell)].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.010157/-/DC1
REFERENCES
- Brooke R. E., Moores T. S., Morris N. P., Parson S. H., Deuchars J. (2004). Kv3 voltage-gated potassium channels regulate neurotransmitter release from mouse motor nerve terminals. Eur. J. Neurosci. 20, 3313–3321 [DOI] [PubMed] [Google Scholar]
- Brooke R. E., Atkinson L., Edwards I., Parson S. H., Deuchars J. (2006). Immunohistochemical localization of the voltage-gated potassium ion channel Kv3.3 in the rat medulla oblongata and thoracic spinal cord. Brain Res. 1070, 101–115 [DOI] [PubMed] [Google Scholar]
- Chang S. Y., Zagha E., Kwon E. S., Ozaita A., Bobik M., Martone M. E., Ellisman M. H., Heintz N., Rudy B. (2007). Distribution of Kv3.3 potassium channel subunits in distinct neuronal populations of mouse brain. J. Comp. Neurol. 502, 953–972 [DOI] [PubMed] [Google Scholar]
- Eisen J. S. (1994). Development of motoneuronal phenotype. Annu. Rev. Neurosci. 17, 1–30 [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Melançon E. (2001). Interactions with identified muscle cells break motoneuron equivalence in embryonic zebrafish. Nat. Neurosci. 4, 1065–1070 [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Myers P. Z., Westerfield M. (1986). Pathway selection by growth cones of identified motoneurons in live zebrafish embryos. Nature 320, 269–271 [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Pike S. H., Debu B. (1989). The growth cones of identified motoneurons in embryonic zebrafish select appropriate pathways in the absence of specific cellular interactions. Neuron 2, 1097–1104 [DOI] [PubMed] [Google Scholar]
- Eisen J. S., Pike S. H., Romancier B. (1990). An identified motoneuron with variable fates in embryonic zebrafish. J. Neurosci. 10, 34–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa K. P., Minassian N. A., Stevanin G., Waters M., Garibyan V., Forlani S., Strzelczyk A., Bürk K., Brice A., Dürr A., et al. (2010). KCNC3: Phenotype, mutations, channel biophysics – a study of 260 familial ataxia patients. Hum. Mutat. 31, 191–196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa K. P., Waters M. F., Garibyan V., Bird T. D., Gomez C. M., Ranum L. P. W., Minassian N. A., Papazian D. M., Pulst S. M. (2011). Frequency of KCNC3 DNA variants as causes of spinocerebellar ataxia 13 (SCA13). PLoS ONE 6, e17811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flanagan-Steet H., Fox M. A., Meyer D., Sanes J. R. (2005). Neuromuscular synapses can form in vivo by incorporation of initially aneural postsynaptic specializations. Development 132, 4471–4481 [DOI] [PubMed] [Google Scholar]
- Glaser J. R., Glaser E. M. (1990). Neuron imaging with Neurolucida – a PC-based system for image combining microscopy. Comput. Med. Imaging Graph. 14, 307–317 [DOI] [PubMed] [Google Scholar]
- Hanson M. G., Landmesser L. T. (2004). Normal patterns of spontaneous activity are required for correct motor axon guidance and the expression of specific guidance molecules. Neuron 43, 687–701 [DOI] [PubMed] [Google Scholar]
- Hanson M. G., Landmesser L. T. (2006). Increasing the frequency of spontaneous rhythmic activity disrupts pool-specific axon fasciculation and pathfinding of embryonic spinal motoneurons. J. Neurosci. 26, 12769–12780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herman-Bert A., Stevanin G., Netter J., Rascol O., Brassat D., Calvas P., Camuzat A., Yuan Q., Schalling M., Dürr A., et al. (2000). Mapping of spinocerebellar ataxia 13 to chromosome 19q13.3–q13.4 in a family with autosomal dominant cerebellar ataxia and mental retardation. Am. J. Hum. Genet. 67, 229–235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issa F. A., Mazzochi C., Mock A. F., Papazian D. M. (2011). Spinocerebellar ataxia type 13 mutant potassium channel alters neuronal excitability and causes locomotor deficits in zebrafish. J. Neurosci. 31, 6831–6841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D. W. C., Westerfield M. (1990). The formation of terminal fields in the absence of competitive interactions among primary motoneurons in zebrafish. J. Neurosci. 10, 3947–3959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melançon E., Liu D. W. C., Westerfield M., Eisen J. S. (1997). Pathfinding by identified zebrafish motoneurons in the absence of muscle pioneers. J. Neurosci. 17, 7796–7804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minassian N. A., Lin M. A., Papazian D. M. (2012). Altered Kv3.3 gating in early onset spinocerebellar ataxia type 13. J. Physiol. 590, 1599–1614 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ming G., Henley J., Tessier-Lavigne M., Song H., Poo M. (2001). Electrical activity modulates growth cone guidance by diffusible factors. Neuron 29, 441–452 [DOI] [PubMed] [Google Scholar]
- Mock A. F., Richardson J. L., Hsieh J.-Y., Rinetti G., Papazian D. M. (2010). Functional effects of spinocerebellar ataxia type 13 mutations are conserved in zebrafish Kv3. 3 channels. BMC Neurosci. 11, 99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody W. J., Bosma M. M. (2005). Ion channel development, spontaneous activity, and activity-dependent development in nerve and muscle cells. Physiol. Rev. 85, 883–941 [DOI] [PubMed] [Google Scholar]
- Moreno R. L., Ribera A. B. (2009). Zebrafish motor neuron subtypes differ electrically prior to axonal outgrowth. J. Neurophysiol. 102, 2477–2484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers P. Z., Eisen J. S., Westerfield M. (1986). Development and axonal outgrowth of identified motoneurons in the zebrafish. J. Neurosci. 6, 2278–2289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano T., Windrem M., Zappavigna V., Goldman S. A. (2005). Identification of a conserved 125 base-pair Hb9 enhancer that specifies gene expression to spinal motor neurons. Dev. Biol. 283, 474–485 [DOI] [PubMed] [Google Scholar]
- Oppenheim R. W. (1991). Cell death during development of the nervous system. Annu. Rev. Neurosci. 14, 453–501 [DOI] [PubMed] [Google Scholar]
- Panzer J. A., Song Y., Balice-Gordon R. J. (2006). In vivo imaging of preferential motor axon outgrowth and synaptogenesis at prepatterned acetylcholine receptor clusters in embryonic zebrafish skeletal muscle. J. Neurosci. 26, 934–947 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike S. H., Eisen J. S. (1990). Identified primary motoneurons in embryonic zebrafish select appropriate pathways in the absence of other primary motoneurons. J. Neurosci. 10, 44–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudy B., McBain C. J. (2001). Kv3 channels: Voltage-gated K+ channels designed for high-frequency repetitive firing. Trends Neurosci. 24, 517–526 [DOI] [PubMed] [Google Scholar]
- Serra H. G., Duvick L., Zu T., Carlson K., Stevens S., Jorgensen N., Lysholm A., Burright E., Zoghbi H. Y., Clark H. B., et al. (2006). RORα-mediated Purkinje cell development determines disease severity in adult SCA1 mice. Cell 127, 697–708 [DOI] [PubMed] [Google Scholar]
- Spitzer N. C. (2006). Electrical activity in early neuronal development. Nature 444, 707–712 [DOI] [PubMed] [Google Scholar]
- Waters M. F., Minassian N. A., Stevanin G., Figueroa K. P., Bannister J. P. A., Nolte D., Mock A. F., Evidente V. G., Fee D., Müller U., et al. (2006). Mutations in the voltage-gated potassium channel KCNC3 cause degenerative and developmental CNS phenotypes. Nat. Genet. 38, 447–451 [DOI] [PubMed] [Google Scholar]
- Wen H., Brehm P. (2005). Paired motor neuron-muscle recordings in zebrafish test the receptor blockade model for shaping synaptic current. J. Neurosci. 25, 8104–8111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westerfield M., McMurray J. V., Eisen J. S. (1986). Identified motoneurons and their innervation of axial muscles in the zebrafish. J. Neurosci. 6, 2267–2277 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}