

SUMMARY
Nonsense mutations that result in the expression of truncated, N-terminal, fragments of the adenomatous polyposis coli (APC) tumour suppressor protein are found in most sporadic and some hereditary colorectal cancers. These mutations can cause tumorigenesis by eliminating β-catenin-binding sites from APC, which leads to upregulation of β-catenin and thereby results in the induction of oncogenes such as MYC. Here we show that, in three distinct experimental model systems, expression of an N-terminal fragment of APC (N-APC) results in loss of directionality, but not speed, of cell motility independently of changes in β-catenin regulation. We developed a system to culture and fluorescently label live pieces of gut tissue to record high-resolution three-dimensional time-lapse movies of cells in situ. This revealed an unexpected complexity of normal gut cell migration, a key process in gut epithelial maintenance, with cells moving with spatial and temporal discontinuity. Quantitative comparison of gut tissue from wild-type mice and APC heterozygotes (APCMin/+; multiple intestinal neoplasia model) demonstrated that cells in precancerous epithelia lack directional preference when moving along the crypt-villus axis. This effect was reproduced in diverse experimental systems: in developing chicken embryos, mesoderm cells expressing N-APC failed to migrate normally; in amoeboid Dictyostelium, which lack endogenous APC, expressing an N-APC fragment maintained cell motility, but the cells failed to perform directional chemotaxis; and multicellular Dictyostelium slug aggregates similarly failed to perform phototaxis. We propose that N-terminal fragments of APC represent a gain-of-function mutation that causes cells within tissue to fail to migrate directionally in response to relevant guidance cues. Consistent with this idea, crypts in histologically normal tissues of APCMin/+ intestines are overpopulated with cells, suggesting that a lack of migration might cause cell accumulation in a precancerous state.
INTRODUCTION
A nonsense mutation in the tumour suppressor adenomatous polyposis coli (APC) is sufficient to cause colorectal cancer in humans and animal models (Nandan and Yang, 2010; Kwong and Dove, 2009). Hereditary and sporadic cancers commonly carry nonsense mutations in APC that result in the expression of N-terminal fragments of the APC protein, so that the protein lacks the more C-terminally located β-catenin-, tubulin-, actin- and EB1-binding domains (Phelps et al., 2009; Näthke, 2004). Tumorigenesis resulting from APC mutations has been attributed mainly to activation of β-catenin-regulated transcription (Morin, 1999; Barker et al., 2000). Recent studies have begun to highlight the importance of β-catenin-independent functions of APC (Okada et al., 2010; Mili et al., 2008). For instance, the phenotype produced by completely deleting the entire gene from one allele of APC seems to be more severe than that produced when this allele encodes an N-terminal APC fragment [as in APCMin/+ mice or the corresponding individuals with familial adenomatous polyposis (FAP)], despite lower levels of active β-catenin being present in mice with the gene deletion (Cheung et al., 2010). Furthermore, individuals with an APC allele that leads to production of an N-terminal APC protein fragment that only contains about 150 amino acids (compared with the 850 amino acids encoded by the APCMin allele) present with a much less severe case of the disease, called attenuated FAP (Spirio et al., 1993; Lamlum, 1999). Our understanding of the nature and impact of additional functions of APC and how direct effects of retained N-APC fragments contribute to its role in tumorigenesis remains incomplete (Phelps et al., 2009; Näthke, 2004).
Colorectal cancer usually follows the loss of the second APC allele, through loss of heterozygosity. It was recently shown that stepwise mutations of the two APC alleles resulted in dramatically faster tumorigenesis when compared with simultaneous mutation (Fischer et al., 2011). This suggests that the details of how APC heterozygosity and complete loss is achieved affect the specific phenotype of the resulting tumours and adenoma, consistent with the idea that the specific APC fragments that are expressed contribute to this process.
One function that is likely to be differentially affected by the length of N-terminal APC fragments is the ability of APC to regulate cytoskeletal proteins. Based on APC interactions with actin and microtubules, we hypothesized that heterozygosity for APC (as in APCMin/+), characterized by expression of an N-terminal fragment of APC, could affect cell migration (Phelps et al., 2009; Näthke, 2004). To test this idea, we measured cell motility in live gut tissue explants.
The small intestine is lined with a columnar epithelium organized into crypts and villi that absorb nutrients and water. These short-lived epithelial cells proliferate in the crypt base before migrating from the crypt towards intestinal villus tips or colonic crypt collars, where they are shed into the gut lumen (Barker et al., 2008). Originally, cell motility in the gut was established by recording the position of BrdU-labelled cells in thin sections at different times after BrdU injections (Wong and Wright, 1999; Potten et al., 2002). To gain more detailed insight into gut cell motility, we recorded live tissue explants using three-dimensional (3D) time-lapse movies. We were surprised to find that cells move in every direction, not just towards the villus tip. Cells in wild-type tissue migrated towards the villus more often than away, whereas N-terminal APC mutants had a sharply reduced loss in this directional preference.
Because the migratory behaviour of enterocytes in live gut has not been well described in vivo, we also examined migration in chicken embryos, which have a better-defined migratory path (Yang et al., 2002). We found that expression of N-APC in early chicken embryos produced a phenotype that is consistent with loss of directionality, similar to what we observed in mouse tissues.
To directly test whether the migration defects in mouse and chicken were due to direct effects of N-APC fragments, we expressed N-APC in Dictyostelium discoideum after carefully determining the absence of APC in its genome. We observed a strong and specific defect in the directionality of chemotaxis in response to expressing N-APC. In this case, loss of function of APC or deregulation of β-catenin can be excluded as contributing factors.
Together, these data reveal that nonsense mutations in a single allele of APC that produce an N-terminal APC protein fragment result in a directionality defect in tissue cell migration in the gut. Expression of such APC fragments results in similar phenotypes in multiple experimental systems, despite the presence of one or two wild-type copies of APC, indicating a dominant effect. Expression of N-APC results in the same phenotype in a system lacking native APC, indicating that such nonsense mutations result in a gain of function. The cell overcrowding in the crypt base we observe and which is typical of tumorigenesis might result from this directionality defect. We therefore propose that APC might act as both a tumour suppressor and a proto-oncogene.
RESULTS
Enterocytes in APCMin/+ small intestine lose directional preference in migration
The mutated APC allele in heterozygous APCMin/+ tissue contains a premature stop at codon 850 (Su et al., 1992). This germline mutation creates a genetic predisposition to colorectal cancer in mice and humans. Similar truncating mutations in this region are found in the majority of sporadic human colorectal cancers (Näthke, 2004). To determine the primary physiological consequence of this mutation in gut tissue, we developed a method to culture large pieces of mouse gut for up to 2 weeks on a microscope, based on a published protocol for culturing primary colonocytes (Booth et al., 1995).
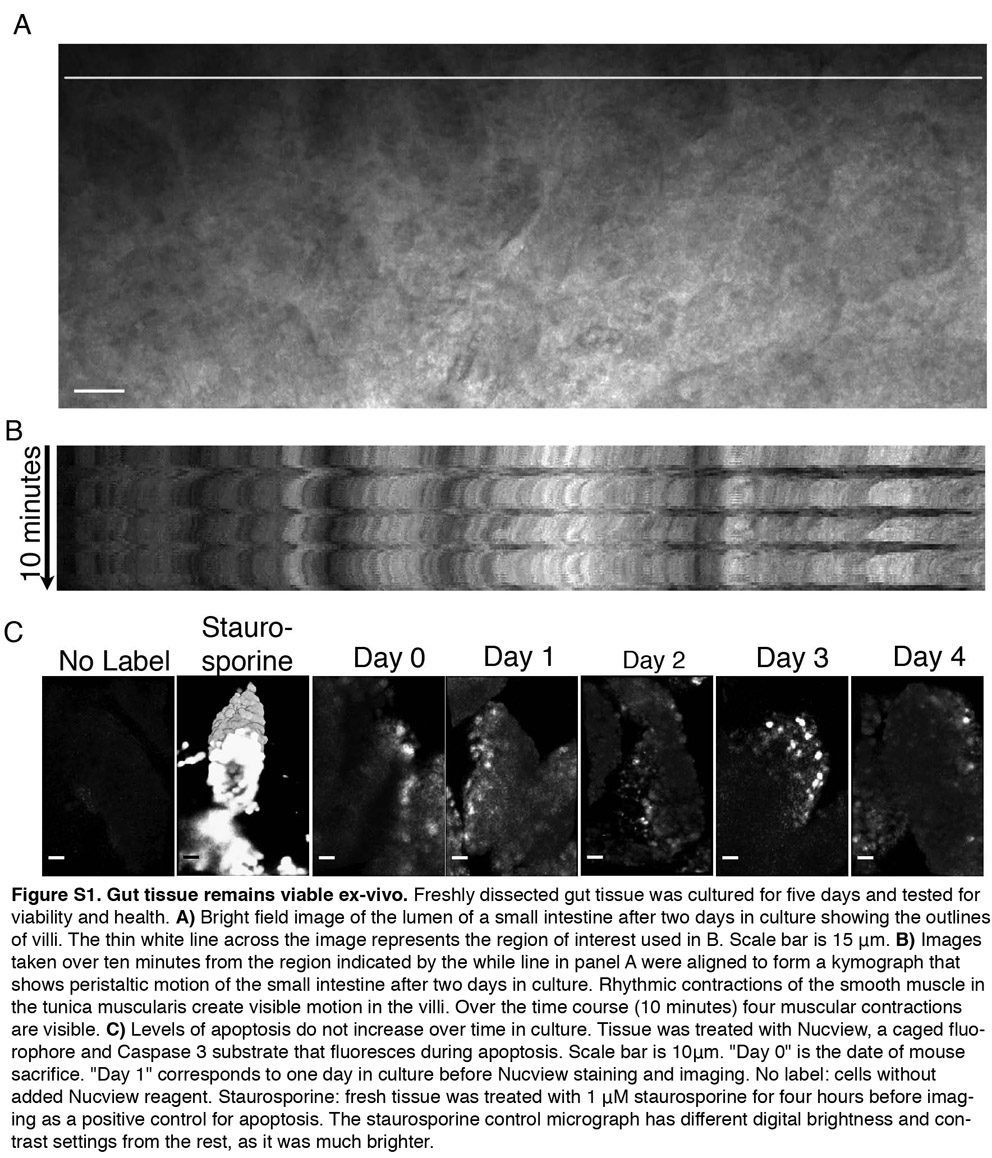
Continued health and viability of the cultured tissue during recording was confirmed by observing a similar distribution and number of apoptotic cells over time in culture, continuous rhythmic contraction of the smooth muscle (supplementary material Fig. S1, Movie 1), continuous secretion of mucus, and persistent rapid movement of organelles within cells (data not shown).
To label cells, we transfected tissue with Organelle Lights, a reagent containing a virus that allows transfection of cells with GFP that is tagged with a nuclear localization sequence (see Methods). This labelled a small proportion of epithelial cells to give a speckled pattern of fluorescence signal, which is ideal for tracking individual cells. Multiphoton fluorescence movies permitted tracking of motile enterocytes on the intestinal villus (Fig. 1; supplementary material Movies 2, 3). The distance between the villus tip and the proximal edge of the labelled nucleus was measured in every frame to yield the relative change in distance from the villus tip over time (Fig. 1A). On the basis of BrdU-labelling pulse-chase type experiments, intestinal epithelial cells in mice were previously reported to travel a distance of 50–100 μm a day from the crypt towards the villus (Wong and Wright, 1999; Potten et al., 2002; Wright and Alison, 1984). Using data from 33 cells, we measured a mean net forward migration rate of 64 μm a day in live tissue, consistent with these previous findings.
Fig. 1.

Enterocytes fail to migrate consistently towards the villus tip in live APCMin/+ tissue. (A) Time-lapse movie of an intestinal villus within a tissue explant using multiphoton fluorescence. The red box indicates the zoomed region of interest shown in the remaining panels. Ten minutes elapse between frames. Red arrowheads indicate the initial position of a moving cell within the tissue. Green arrowheads indicate the position of the moving cell in the current frame. This cell moves continuously towards the villus tip. Scale bar: 10 μm. (B,C) Relative distance of labelled epithelial cells from the villus tip over time. The black line represents the pattern of motion expected based on previous experiments, assuming constant migration towards the villus tip at a rate of 100 μm per day. Each coloured line indicates the distance from the villus tip of a single representative cell over time. Cell migration seemed to be discontinuous and multidirectional. (B) Cell migration in tissue from wild-type mice. Despite irregularity in cell migration patterns, wild-type cells exhibited a net movement towards the villus tip over time. (C) Cell migration in tissue from APCMin/+ mice. Cells in APCMin/+ tissue, although still dynamic, did not follow as clear a path towards the villus tip as those in wild-type tissue.
Surprisingly, cells moved in small clusters rather than in a continuous sheet (supplementary material Movies 2–4). Furthermore, cells moved discontinuously and showed variability in speed and direction. Cells moved both towards and away from the villus tip. In some cases, cells remained still for most of the duration of a time-lapse, but moved at high speed during short bursts of motility (Table 1).
Table 1.
Quantitation of cell migration in tissue

This surprisingly complex migration pattern of enterocyte movement nonetheless produced a net migration towards the villus tip within the previously published range (Fig. 1B) (Wong and Wright, 1999; Wright and Alison, 1984). A summary of our measurements is provided in Table 1. For the purpose of describing motion, we refer to individual cell migration events as periods of cell motility preceded and followed by pauses in migration. We observed cell migration events both towards and away from the villus tip, but migration events towards the villus tip occurred nearly twice as often as away (0.29 versus 0.17 events per hour, respectively) (Table 1). The mean total migration was 3.05 μm/hour, with a net movement towards the villus tip at 1.88 μm/hour. We calculated that 63% of motion was towards the villus tip (called the efficiency of movement) (Table 1). Taking migration speeds and frequencies into account, we calculated a ratio of forward:reverse movement of 4.8:1, and a net migration rate of 64 μm/day towards the villus tip.
APCMin/+ mice are a well-characterized model for FAP, a genetic condition characterized by the formation of up to thousands of adenomatous polyps in humans (Su et al., 1992). In APCMin/+ tissue, the frequency of forward migration events is the same as in wild type (P=0.25) and enterocytes remained motile, with 0.29 migration events per hour towards the villus tip. However, in this tissue we observed 0.25 migration events per hour in the opposite direction, away from the villus tip. Thus, there was no strong preference for migration towards the villus tip (P=0.28) (Fig. 1C, Table 1). We calculated an efficiency of movement towards the villus tip of 14%. Taking migration speeds and frequencies into account, we calculate a ratio of forward:reverse movement of 1.6:1, and a net migration rate of 16.8 μm/day towards the villus tip, confirming previously described reduced migration in fixed tissue (Mahmoud et al., 1997).
This defect in directed cell motility predicts that cells accumulate inappropriately and excessively in APCMin/+ crypts. We previously showed that crypts of APCMin/+ tissue bulge at their base when compared with wild-type crypts (Quyn et al., 2010). To further explore this, we counted the number of DAPI-stained nuclei within the bottom third and the centre third of individual crypts. We found that crypt bases in APCMin/+ tissue contain 72% more cells per unit of crypt outer surface area than do wild type (3.1±0.5 vs 1.8±0.2 per 100 mm2, respectively; P=9.5×10−13). This was less pronounced in the centre regions of crypts. Here, APCMin/+ tissue contained 55% more cells than did wild-type tissue (2.8±0.5 vs 1.8±0.3 per 100 mm2, respectively; P=4.0×10−9). These data support our finding that cells in APCMin/+ tissue fail to emigrate efficiently from the crypt.
Expression of N-APC causes changes in directional migration in cells of the primitive streak in chicken embryos
The migration defects we observed in tissue expressing N-APC fragments raised the possibility that N-APC fragments have an effect on cell behaviour that is dominant over the remaining wild-type APC allele. To investigate this possibility, we used chicken embryos, which have the advantage of a rapid, clearly defined mesoderm cell migration at a particular stage of development. This allowed us to determine whether the migration defect we observed in APCMin/+ tissue was a direct effect of an N-APC protein fragment. We expressed a YFP-tagged N-APC fragment driven by the β-actin promoter into early embryos using electroporation and investigated the migration of mesoderm cells emerging from the primitive streak of an 18- to 19-hour-old embryo (Yang et al., 2002). To monitor labelled, YFP–N-APC-expressing, cells, we grafted transfected primitive streak tissue into the corresponding position in an unlabelled host embryo and compared them with control cells expressing only YFP. The movement of fluorescent cells was recorded during the subsequent 12–15 hours of development. Tracks of individual cells (thin lines) or groups of cells, such as the graft (thick lines), are shown from the beginning of the experiment (Fig. 2; supplementary material Movies 5, 6) (Yang et al., 2002). This technique previously revealed a distinctive and reproducible pattern of cell migration that varied depending on the point of origin within the streak (Yang et al., 2002). Cells initially moved out laterally, away from the streak, and, once the fluorescent-protein-expressing node had regressed past these cells, they moved back towards the midline (Fig. 2).
Fig. 2.
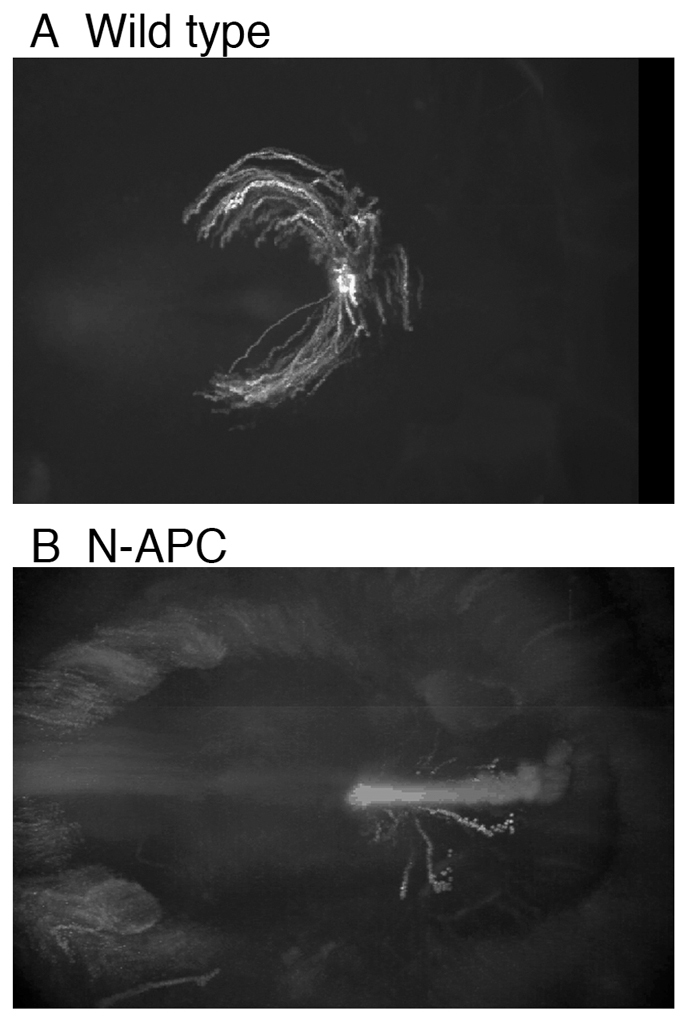
Movement trajectories of cells from the primitive streak are disrupted by expression of N-APC in chick embryos. Twelve- to thirteen-hour-old embryos were transfected with EGFP or N-APC fused to GFP (A and B, respectively) and, after 4–5 hours of incubation, the primitive streak from a transfected donor embryo was transplanted into the corresponding position in a non-transfected host embryo. Images were recorded over the next 15 hours at 3-minute intervals. Images shown are after 15 hours. The lines show the paths taken by cells over time.
Tissue that expressed N-APC remained motile, yet failed to move in the expected pattern (Fig. 2B). Fewer cells moved a significant linear distance. Those that moved a significant distance failed to move in the characteristic arc pattern observed in wild-type cells. Furthermore, the fragmentation of the tracks suggested a loss of coherence in direction in these moving cells. The migratory defect of N-APC-expressing cells was observed regardless of the point of origin of cells within the primitive streak, which affects the exact direction of the migratory path (Yang et al., 2002) (data not shown).
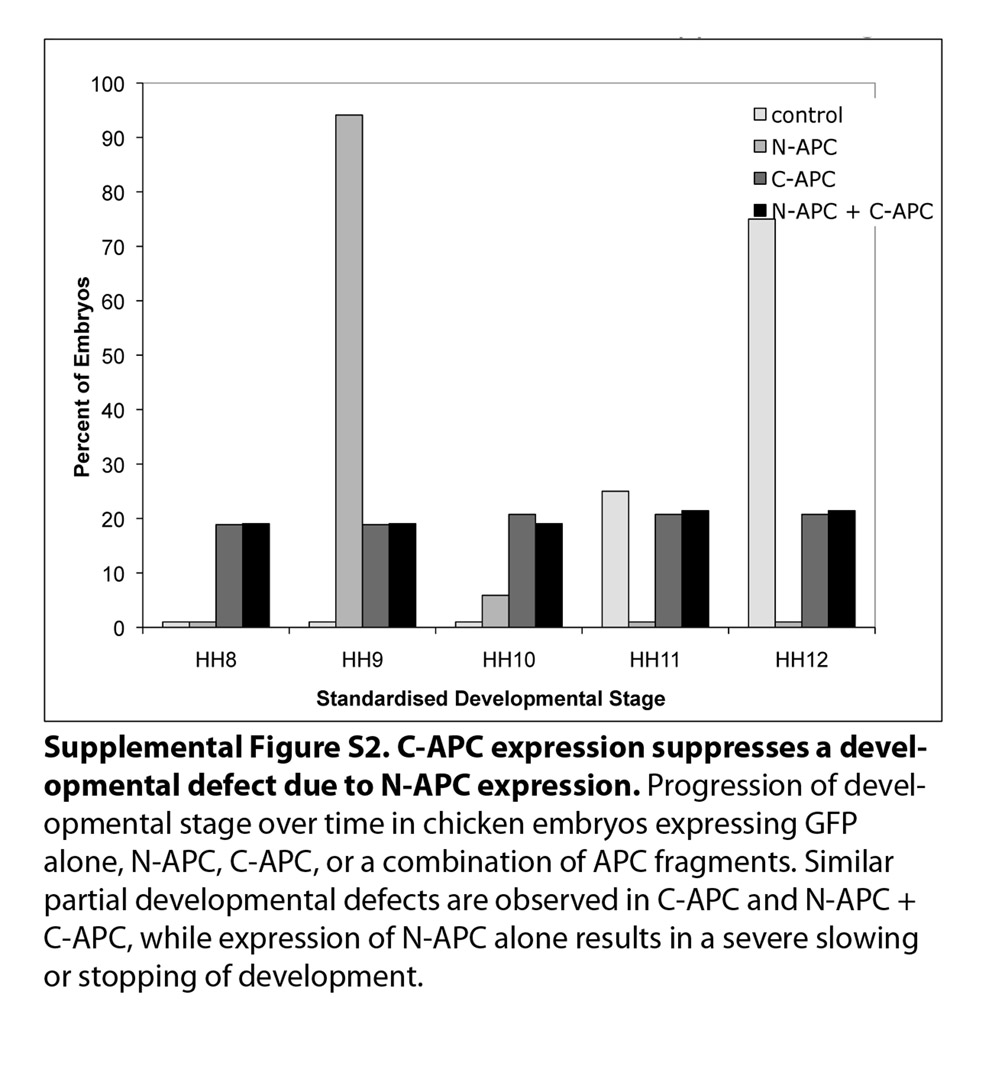
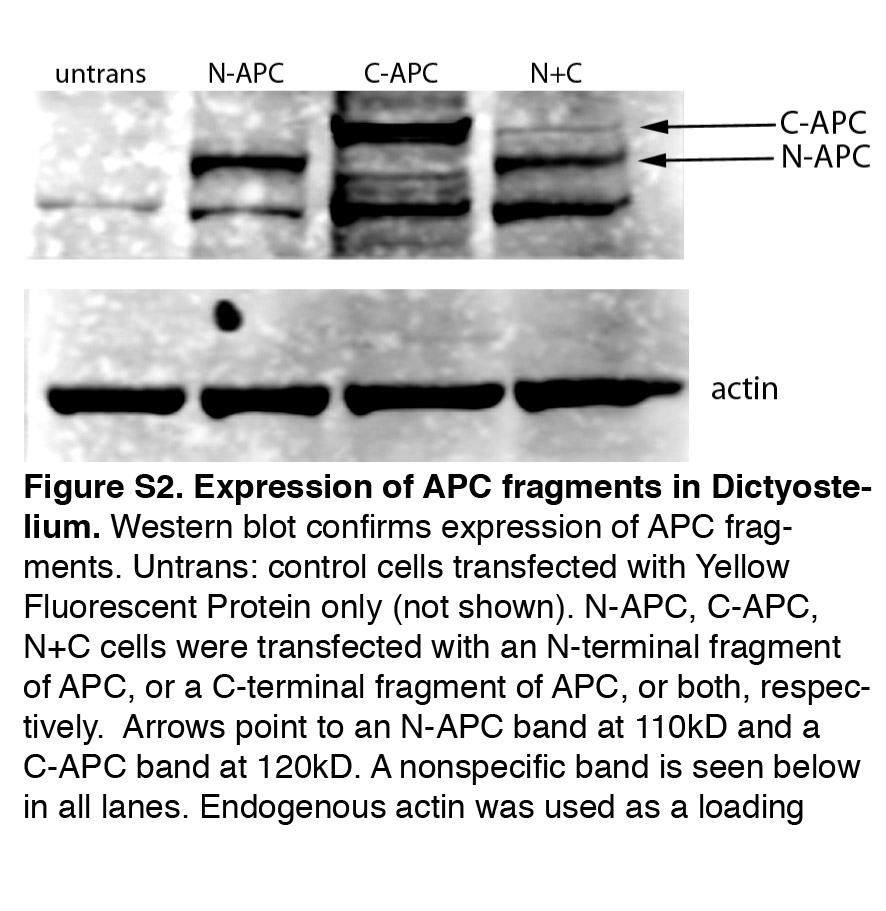
Consistent with a significant migration defect, expression of N-APC fragments in an entire embryo strongly delayed or completely abolished embryonic development (supplementary material Fig. S2). Co-expression of a C-terminal fragment of APC (residues 1014–2843; C-APC) suppressed the growth defect in embryos expressing N-APC, whereas C-APC on its own only slowed development marginally, suggesting that binding of the N-APC fragment to C-APC can inhibit its dominant effect (Li and Näthke, 2005).
Dictyostelium as a model for testing the role of N-APC in directed cell migration
Tumorigenesis due to mutations in APC is often attributed to deregulated β-catenin signalling. Changes in β-catenin are not predicted to result from exogenously expressing N-APC fragments. Nonetheless, the complexity of the transcriptional changes induced by deregulated β-catenin and the inability to generate a form of APC that is solely defective in β-catenin regulation prompted us to use another experimental system that lacks endogenous APC and thus does not provide these complications. To this end we turned to Dictyostelium discoideum to determine whether the observed defect in directional migration was due to a function of the N-APC fragment alone or caused by potential indirect effects. However, it was essential to first establish the extent of similarities in APC and β-catenin signalling pathway components in the Dictyostelium genome.
No significant matches were found in the Dictyostelium discoideum genomic sequence corresponding to either full-length APC or overlapping tiled fragments of the APC protein sequence (see Methods). On the basis of these analyses, we conclude that Dictyostelium does not contain an analogue of the APC gene nor other smaller proteins that could engage in binding interactions associated with an APC homologue.
Additionally, a high-throughput two-hybrid screen in Dictyostelium using N-APC as bait did not produce interactions with any members of the simple β-catenin signalling pathway native to Dictyostelium (Harwood, 2008) (data not shown).
N-APC fragments cause loss of directed migration in Dictyostelium
To determine whether loss of directed migration was a general effect of expressing N-terminal fragments of APC, we stably transfected Dictyostelium discoideum with N-APC (supplementary material Fig. S3). Movement of individual cells expressing the first 1018 amino acids of the APC protein towards a point source of cyclic adenosine monophosphate (cAMP) was recorded. Control cells expressing YFP migrated efficiently towards cAMP in nearly straight lines (Fig. 3A; supplementary material Movie 7). Cells expressing N-APC remained highly motile, but did not migrate towards the cAMP source (Fig. 3B; supplementary material Movie 8). We observed a mean twofold decrease in orientation towards the cAMP source [Cos (a)=0.91±0.13 for wild type, versus 0.41±0.28 for N-APC; P=2×10−9] (Fig. 3D). There was no statistically significant difference in migration speed (9.4±2.7 μm/minute for wild type versus 7.2±1.9 for N-APC; P=0.08) (Fig. 3C). Expression of the C-terminal third of APC, which can bind to N-APC regions, completely rescued the N-APC phenotype [Cos(a)=0.93±0.06; P=0.24], suggesting an autoregulatory function within the full-length APC protein that is lacking in the truncated protein (supplementary material Movie 9), which is consistent with the ability of the N-APC and C-APC fragments to form an inactive heterodimer (Li and Näthke, 2005; Li et al., 2008).
Fig. 3.
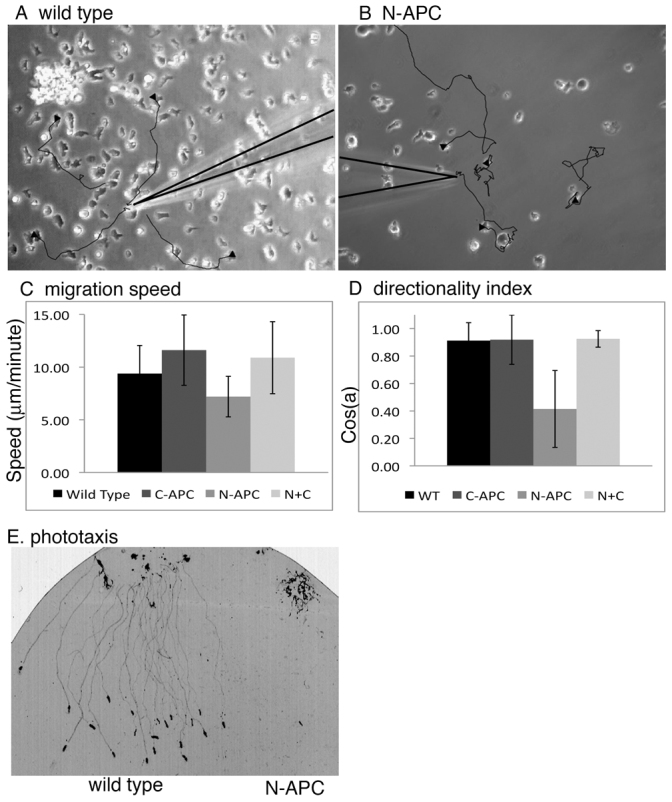
Dictyostelium expressing N-APC lack the ability to maintain direction during migration. (A,B) The first frame of a time-lapse movie of Dictyostelium amoebae chemotaxing towards a micropipette filled with cAMP is shown. Black lines indicate the migration paths taken by four individual amoebae over the course of the movie. Arrowheads indicate the cell position at the start of the movie. (A) Control amoebae expressing YFP. (B) Amoebae expressing N-APC. (C,D) xy coordinates of chemotaxing Dictyostelium cells were recorded from time-lapse movies. The median distances covered by cells over time is shown in C. (D) Values are a moving average of the cosine of the angle made by connecting the xy position of a cell at three consecutive time points. A cosine of 1 indicates movement in a straight line; zero indicates a turn of 90 degrees. The mean of 20 cells is shown. Error bars show standard deviation. (E) Coomassie Blue stain of tracks left by Dictyostelium slugs during phototaxis towards light at the bottom of the image. Wild-type slugs migrate efficiently towards the light, whereas slugs expressing N-APC do not.
To measure the effects of N-APC on multicellular structures, Dictyostelium were induced to form slugs, which phototax towards light (Bukahrova et al., 2005). When slugs travel, they synthesize an extracellular slime sheath that can be detected by staining with Coomassie Blue. Staining slug migration trails revealed that expression of N-APC completely inhibited phototaxis, although slugs still formed (Fig. 3E). This indicates that the expression of N-APC affects migration both in single and multicellular stages of development.
Because APC interacts with actin and actin-binding proteins (Moseley et al., 2007; Watanabe et al., 2004), we compared the distribution of F-actin in migrating Dictyostelium cells with or without N-APC (supplementary material Fig. S4). We found F-actin to be depolarized in N-APC-expressing cells so that it decorated the entire cell cortex rather than concentrating at the leading edge as in control cells. Overall, F-actin was reduced at the leading edge in N-APC-expressing cells, whereas non-cortical actin was increased.
DISCUSSION
Our aim was to identify changes resulting from mutation of APC in precancerous stages. To this end we developed a technique to maintain large pieces of gut tissue healthy in culture for a sufficient time to capture cell migration. We applied this technique to measure cell migration in tissue. Whereas previous studies examined overall cell migration in gut epithelium using fixed tissue samples, this technique allowed observation of individual cells over time, revealing their motility characteristics in 3D (Wong and Wright, 1999; Potten et al., 2002).
Using 3D movie analysis, we measured differences in cellular behaviour between wild-type and APCMin/+ tissue. We confirmed the hypothesis that cells heterozygous for APC are defective in migration along the crypt-villus axis. We found that cell migration still occurred in APCMin/+ tissue, but cells lacked directionality owing to an increase in non-productive migration. However, we cannot rule out the possibility that cell migration in tissue explants differs from that in a live animal.
Our initial observations of decreased migration were made in mouse gut, in which a migration pattern or mechanisms that regulate migration have not been defined. So we aimed to identify the effects of N-APC fragments on migration in other systems with well-defined migration patterns, and also take advantage of the ease of manipulation and imaging afforded by these simpler systems.
We expressed N-APC in chicken embryos. The primordial streak of embryos consists of epithelial cells that exit the streak, migrating in a characteristic arc pattern (Yang et al., 2002). Expression of N-APC caused a loss of the majority of directed cell migration, despite the presence of two wild-type endogenous APC genes. It is difficult to distinguish between loss of cell motility and loss of directionality of cell motility in this assay; however, closer inspection of the traced path of N-APC-expressing cells also revealed a lost of coherence in the migrating path of cells. The tracks deviated from a continuous line and ‘broke up’, consistent with a loss of directionality.
We also used Dictyostelium, a classic model for the study of cell motility and polarity, to test effects of APC fragments on these processes (Weijer, 2009). Expression of N-APC caused a loss in the ability of Dictyostelium cells to chemotax towards a cAMP source, although the speed of cell motility was unchanged. Co-expression of a C-terminal fragment of APC completely rescued this phenotype.
These data combine to strongly suggest that there is a dominant defect in directionality of migration that results from gain-of-function N-APC protein fragments. This effect was detected even when full-length APC protein was also expressed. The mutant mouse tissue we examined contained one wild-type copy of APC, and the chicken tissue contained two wild-type copies of APC. N-APC showed a dramatic phenotype in Dictyostelium despite the absence of a native APC gene. This is a particularly relevant result that contributes to the ever-expanding list of β-catenin-independent functions for APC (Okada et al., 2010; Mili et al., 2008). The activation of β-catenin introduced by stabilizing mutations and nonsense mutations in APC produce similar but not identical cancers in mouse models, confirming that some β-catenin-independent functions for APC can contribute to tumorigenesis (Harada et al., 1999). Our finding that expression of N-APC severely compromised directed cell migration in whole tissue is at odds with the fact that both individuals with FAP and APCMin/+ mice develop relatively normally despite the presence of an N-APC fragment in all their tissues. This becomes even more puzzling given the high penetrance of this phenotype in the other experimental multicellular and unicellular systems examined here. There are a number of possible explanations. In our experiments involving chicken embryos and Dictyostelium, N-APC was overexpressed. This might limit the ability of the full-length APC in the chicken to buffer any effect, and the absence of APC in Dictyostelium provides no such buffer. The rescue of the dominant effect of N-APC by co-expression of C-APC fragments, which can bind to N-APC and change its protein interactions, supports this idea. It is also possible that gut epithelium lacks compensatory mechanisms that can contribute to the regulation of migration and are likely to operate in other tissues, which renders them less sensitive to the effects of N-APC. We know little about how cell migration operates in gut epithelium, making it difficult to know precisely how to reconcile these results. However, the correlation that exists between the ability of agents such as circumin and non-steroidal anti-inflammatory agents such as sulindac to reduce tumour incidence, and their ability to increase migration, illustrates the importance of understanding the mechanisms that govern migration in this tissue. Our study has begun to shed some light on this process and we have also developed tools that will greatly enhance our ability to study it.
The N-terminal fragment of APC is known to associate with and activate at least one guanine exchange factor, which activates actin-polarizing GTPases, and these exchange factors are required for adenoma formation in the APCMin/+ mouse (Zhang et al., 2011; Kawasaki et al., 2009). Our findings are consistent with the idea that nonsense mutations in APC that result in expression of N-terminal fragments can cause a lack of actin polarization. This in turn could enhance the tumorigenic potential of cells in gut epithelium by compromising their directed migration to delay their exit from the proliferative crypt environment. The resulting increase in their residence time could enhance the chance of transforming mutations to occur and persist.
METHODS
Mice
Mice used for this study were wild type or APCMin/+ of the C57BL/6 genetic background (Su et al., 1992). They were males and females aged 4–6 months.
Tissue explant culture
Colon and small intestine was harvested from CO2-asphyxiated mice and placed into pre-warmed medium. The gut lumen was washed and cut into 2-mm squares, which were placed into individual 35-mm dishes. Culture medium and incubation conditions were adapted from a published protocol for culturing primary mouse colonocytes (Booth et al., 1995). Medium was based on Dulbecco’s modified Eagle medium with 2.5% fetal bovine serum, 1 unit each of penicillin and streptomycin, and 1 μg/ml insulin (Sigma). Tissue was incubated at 7.5% CO2 at 37°C. Once daily, medium was changed and excess mucus was removed by washing with a micropipette.
Fluorescent labelling
To detect apoptosis, we used the NucView reagent (Biotium, Hayward, CA) following the manufacturer’s protocol: 5 μl of NucView reagent was added to 200 μl Leibowitz medium (Invitrogen, Carlsbad, CA) containing the sample. Tissue was added and incubated in the dark for 15 minutes before detecting green fluorescence using FITC filters.
To label live cells, we transfected tissue with a commercial viral transfection system, Organelle Lights (Invitrogen, Carlsbad, CA). Two pieces of tissue were placed in 1 ml Hank’s buffered saline solution lacking Ca2+ and Mg2+, plus 0.5 ml Organelle Lights reagent. This was incubated in the dark at room temperature with gentle agitation for 1–2 hours. The pieces of tissue were returned to 35-mm dishes containing 3 ml culture medium with the addition of BacMam enhancer solution and incubated overnight.
To count nuclei and image the shape of intestinal crypts, whole-mount pieces of tissue were fixed in 4% paraformaldehyde and stained with rhodamine-phalloidin and DAPI as described (Appleton et al., 2009).
Microscopy
Brightfield imaging was carried out on a Leica DMIRB (Leica Biosystems) using a 10×/NA1.3 objective and a Hamamatsu Orca C4742-95 camera (Hamamatsu Photonics UK Ltd, Welwyn Garden City, UK).
Fluorescence imaging was carried out using a Bio-Rad Radiance 2100MP multiphoton microscope (Bio-Rad) based on a Nikon Eclipse TE2000-U inverted microscope (Nikon UK Ltd). Multiphoton excitation was provided by a Coherent Chameleon Ti-Sapphire laser at 790 nm to simultaneously excite rhodamine and DAPI for fixed specimens, and 820 nm to excite GFP in live specimens. The objective was a Nikon 40×/NA 1.3 oil-immersion lens.
Specimens were mounted onto the microscope using a Bioptechs FCS2 flow chamber.
Dictyostelium genome analysis
No significant matches were found in the Dictyostelium discoideum genomic sequence corresponding to either full-length APC or overlapping tiled fragments of the APC protein sequence (P25054) when searched with BLAST or PSI-BLAST (Altschul et al., 1990; Altschul et al., 1997). The translated genome sequence was searched with regular expression and pFam models corresponding to known APC functional sites to identify any potential divergent genes that either retain functional elements through sequence divergence or that fulfil a similar role to that of APC. In particular, the pFam models PF05924 (SAMP), PF05937 (EB1_binding), PF05923 (APC_crr), PF05972 (APC_15aa) and PF05956 (APC_basic) show no significant matches. By contrast, there are several proteins containing multiple Armadillo (Arm) repeats (PF00514), of which at least one could be a candidate for a β-catenin homologue (see below).
There are no strong matches to β-catenin (P35222); however, several proteins contain appropriate Arm domains and AarA has been reported to have many of the functions of β-catenin (Harwood, 2008). Similar results were found for IQGAP1, 2 and 3 (P46940, Q13576, Q86VI3), KAP3 (P70188), MARE1 (Q15691) and MARE2 (Q15555), with either no homologue identified, or no APC-binding motif present.
Dictyostelium migration assays
To induce chemotaxis, starving Dictyostelium cells in shaking suspension were pulsed with 40 nM cAMP every 6 minutes for 6 hours. After the cells had attached to a glass coverslip submerged in phosphate buffer (20 mM KH2PO4/K2HPO4, pH 6.8) the chemotactic response towards a micropipette containing 100 μM cAMP was recorded on a Zeiss Axiovert 135 microscope. To measure directionality, the cosine (Cos) of the angle ‘a’ between positions of cells at every time point were taken. A Cos(a) of 1 corresponds to movement in a straight line, whereas a Cos(a) of 0 corresponds to no directed movement. Phototaxis experiments were as described previously (Dormann et al., 2001).
TRANSLATIONAL IMPACT.
Clinical issue
Colorectal cancer (CRC) is one of the leading causes of cancer death worldwide. Early detection can greatly improve survival, so identifying the very first changes that occur in histologically normal gut tissue during tumorigenesis is a key priority. A prevalent feature of CRC is mutation of the adenomatous polyposis coli (APC) gene, encoding a protein that negatively regulates β-catenin and has many other cellular functions that are commonly linked to tumorigenesis. However, the mechanisms by which APC mutations affect normal tissue before transformation are unclear, in part because identifying early changes in APC-mutant tissue has been difficult using conventional methods.
Results
In this paper, the authors sought to identify new, reliable biomarkers for early detection and/or prognosis of CRC through examining live cell and tissue behaviour in real time. They recorded time-lapse movies of fluorescently labelled cells in live mouse gut to test whether a heterozygous APC truncation mutation affected cell migration, which is key for maintenance of the gut epithelium. They found that cell migration in this tissue is surprisingly complex, with cells showing variable speed and direction. Importantly, quantitative comparison of normal versus heterozygous APC-mutant tissue showed that cells in morphologically normal precancerous tissue are impaired in their directional preference when moving along the crypt-villus axis: in contrast to normal tissue, cells in APC-mutant tissue do not predominantly move forwards along the crypt-villus axis, despite the expression of one wild-type APC allele. This directionality defect seems to be dominant. De novo expression of N-terminal APC fragments similar to those in human cancers also induced cell migration defects in three other model systems. Specifically, the expression of N-terminal APC fragments induced a directionality defect in chicken embryos, and in single and multicellular Dictyostelium discoideum (an organism lacking endogenous APC).
Implications and future directions
These data suggest that mutations producing N-terminal APC fragments constitute a gain of function that causes potential tumorigenic effects that are distinct from effects on β-catenin signalling. Thus, APC might serve as both a tumour suppressor and a proto-oncogene. These results should inform the interpretation of future clinical and animal studies on tumorigenesis associated with APC mutations. Furthermore, the authors suggest that investigation of therapies for CRC that seek to restore the directionality of cell migration are warranted. Finally, these data might also help to explain why treatment of CRC with nonsteroidal anti-inflammatory agents, such as sulindac, which stimulates cell migration, help to limit the disease.
Chicken embryos
Chicken embryo transfection, primitive streak transplantation, time-lapse movie recording and tracking of paths of cell migration were performed as described previously (Yang et al., 2002).
Image analysis
Image analysis was performed using Volocity 4.4 (Improvision, Waltham, MA) and routines based on ImageJ.
To measure cell number in individual crypts, first the length of individual crypts was measured using 3D images of wild-type or APCMin/+ mice intestines stained with DAPI and phalloidin (Appleton et al., 2009). The crypt was divided from top to bottom into equally long thirds and the number of nuclei in the bottom and middle sections were counted using DAPI signals. The circumference in the middle of each segment was also measured. The outer surface area of a crypt was then calculated, assuming a crypt to take the shape of a regular cylinder so that surface area could be calculated by multiplying crypt height by circumference. This allowed us to express cell number in different segments relative to crypt surface area.
Statistical analysis
Statistical hypothesis testing was conducted using the Mann-Whitney non-parametric test for differences between two sample distributions (Sokal and Rohlf, 1995).
Supplementary Material
Acknowledgments
We thank Doreen Cantrell and Colin Henderson, University of Dundee, and their research teams, for providing mouse tissue. We thank the members of the light microscopy facility at the University of Dundee, particularly Sam Swift and Calum Thomson, for technical help. We also thank James S. Schuessler for critical evaluation of this manuscript.
Footnotes
COMPETING INTERESTS
The authors declare that they do not have any competing or financial interests.
AUTHOR CONTRIBUTIONS
S.A.N. wrote the manuscript, prepared the figures and performed the mouse gut experiments. Z.L., I.P.N. and D.D. performed the Dictyostelium experiments. D.F. and R.E.M. quantitated cell migration in mouse gut tissue. D.M.A.M. performed the Dictyostelium bioinformatics work. X.Y. and D.S. performed experiments using chicken embryos. P.L.A. supported the imaging for the Dictyostelium and chicken experiments. C.J.W. supported X.Y. and D.D. and contributed ideas and reagents. I.S.N. helped write the manuscript, conceived the ideas for this work and holds the grant that supported this work.
FUNDING
This work was supported by a program grant from Cancer Research UK [grant number: C430/11243 to I.S.N.].
SUPPLEMENTARY MATERIAL
Supplementary material for this article is available at http://dmm.biologists.org/lookup/suppl/doi:10.1242/dmm.008607/-/DC1
REFERENCES
- Altschul S. F., Gish W., Miller W., Myers E. W., Lipman D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410 [DOI] [PubMed] [Google Scholar]
- Altschul S. F., Madden T. L., Schäffer A. A., Zhang J., Zhang Z., Miller W., Lipman D. J. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Appleton P. L., Quyn A. J., Swift S., Näthke I. (2009). Preparation of wholemount mouse intestine for high-resolution three-dimensional imaging using two-photon microscopy. J. Microsc. 234, 196–204 [DOI] [PubMed] [Google Scholar]
- Barker N., Morin P. J., Clevers H. (2000). The Yin-Yang of TCF/beta-catenin signaling. Adv. Cancer Res. 77, 1–24 [DOI] [PubMed] [Google Scholar]
- Barker N., van de Wetering M., Clevers H. (2008). The intestinal stem cell. Genes Dev. 22, 1856–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C., O’Shea J. A., Potten C. S. (1995). Maintenance of functional stem cells in isolated and cultured adult intestinal epithelium. Exp. Cell Res. 249, 359–366 [DOI] [PubMed] [Google Scholar]
- Bukahrova T., Weijer G., Bosgraaf L., Dormann D., Haastert P. J., Weijer C. J. (2005). Paxillin is required for cell-substrate adhesion, cell sorting and slug migration during Dictyostelium development. J. Cell Sci. 118, 4295–4310 [DOI] [PubMed] [Google Scholar]
- Cheung A. F., Carter A. M., Kostova K. K., Woodruff J. F., Crowley D., Bronson R. T., Haigis K. M., Jacks T. (2010). Complete deletion of Apc results in severe polyposis in mice. Oncogene 29, 1857–1864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dormann D., Abe T., Weijer C. J., Williams J. (2001). Inducible nuclear translocation of a STAT protein in Dictyostelium prespore cells: implications for morphogenesis and cell-type regulation. Development 128, 1081–1088 [DOI] [PubMed] [Google Scholar]
- Fischer J. M., Miller A. J., Shibata D., Liskay R. M. (2011). Different phenotypic consequences of simultaneous versus stepwise Apc loss. Oncogene doi: 10.1038/onc.2011.385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harada N., Miyoshi H., Murai N., Oshima H., Tamai Y., Oshima M., Taketo M. M. (1999). Intestinal polyposis in mice with a dominant stable mutation of the β-catenin gene. EMBO J. 18, 5931–5942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harwood A. J. (2008). Dictyostelium development: a prototypic Wnt pathway? Methods Mol. Biol. 469, 21–32 [DOI] [PubMed] [Google Scholar]
- Kawasaki Y., Tsuji S., Muroya K., Furukawa S., Shibata Y., Okuno M., Ohwada S., Akiyama T. (2009). The adenomatous polyposis coli-associated exchange factors Asef and Asef2 are required for adenoma formation in Apc(Min/+)mice. EMBO Rep. 10, 1355–1362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwong L. N., Dove W. F. (2009). APC and its modifiers in colon cancer. Adv. Exp. Med. Biol. 656, 85–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamlum H., Ilyas M., Rowan A., Clark S., Johnson V., Bell J., Frayling I., Efstathiou J., Pack K., Payne S., et al. (1999). The type of somatic mutation at APC in familial adenomatous polyposis is determined by the site of the germline mutation: a new facet to Knudson’s ‘two-hit’ hypothesis. Nat. Med. 5, 1071–1075 [DOI] [PubMed] [Google Scholar]
- Li Z., Näthke I. S. (2005). Tumor-associated NH2-terminal fragments are the most stable part of the adenomatous polyposis coli protein and can be regulated by interactions with COOH-terminal domains. Cancer Res. 65, 5195–5204 [DOI] [PubMed] [Google Scholar]
- Li Z., Kroboth K., Newton I. P., Näthke I. S. (2008). Novel self-association of the APC molecule affects APC clusters and cell migration. J. Cell Sci. 121, 1916–1925 [DOI] [PubMed] [Google Scholar]
- Mahmoud N. N., Boolbol S. K., Bilinski R. T., Martucci C., Chadburn A., Bertagnoli M. M. (1997). Ape gene mutation is associated with a dominant-negative effect upon intestinal cell migration. Cancer Res. 57, 5045–5050 [PubMed] [Google Scholar]
- Mili S., Moissoglu K., Macara I. G. (2008). Genome-wide screen reveals APC-associated RNAs enriched in cell protrusions. Nature 453, 115–119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morin P. J. (1999). Beta-catenin signaling and cancer. BioEssays 21, 1021–1030 [DOI] [PubMed] [Google Scholar]
- Moseley J. B., Bartolini F., Okada K., Wen Y., Gundersen G. G., Goode B. L. (2007). Regulated binding of adenomatous polyposis coli protein to actin. J. Biol. Chem. 282, 12661–12668 [DOI] [PubMed] [Google Scholar]
- Nandan M. O., Yang V. W. (2010). Genetic and chemical models of colorectal cancer in mice. Curr. Colorectal Cancer Rep. 6, 51–59 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Näthke I. S. (2004). The adenomatous polyposis coli protein: the Achilles heel of the gut epithelium. Annu. Rev. Cell Dev. Biol. 20, 337–366 [DOI] [PubMed] [Google Scholar]
- Okada K., Bartolini F., Deaconescu A. M., Moseley J. B., Dogic Z., Grigorieff N., Gundersen G. G., Goode B. L. (2010). Adenomatous polyposis coli protein nucleates actin assembly and synergizes with the formin mDia1. J. Cell Biol. 189, 1087–1096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phelps R. A., Broadbent T. J., Stafforini D. M., Jones D. A. (2009). New perspectives on APC control of cell fate and proliferation in colorectal cancer. Cell Cycle 8, 2549–2556 [DOI] [PubMed] [Google Scholar]
- Potten C. S., Owen G., Booth D. (2002). Intestinal stem cells protect their genome by selective segregation of template DNA strands. J. Cell Sci. 115, 2381–2388 [DOI] [PubMed] [Google Scholar]
- Quyn A. J., Appleton P. L., Carey F. A., Steele R. J., Barker N., Clevers H., Ridgway R. A., Sansom O. J., Näthke I. S. (2010). Spindle orientation bias in gut epithelial stem cell compartments is lost in precancerous tissue. Cell Stem Cell 6, 175–181 [DOI] [PubMed] [Google Scholar]
- Sokal R. R., Rohlf F. J. (1995). Biometry. Third Edition pp. 427–431 New York: W.H. Freeman and Company [Google Scholar]
- Spirio L., Olschwang S., Groden J., Robertson M., Samowitz W., Joslyn G., Gelbert L., Thliveris A., Carlson M., Otterud B., et al. (1993). Alleles of the APC gene: an attenuated form of familial polyposis. Cell 75, 951–957 [DOI] [PubMed] [Google Scholar]
- Su L. K., Kinzler K. W., Vogelstein B., Preisinger A. C., Moser A. R., Luongo C., Gould K. A., Dove W. F. (1992). ApcMin, a mutation in the murine Apc gene, predisposes to mammary carcinomas and focal alveolar hyperplasias. Proc. Natl. Acad. Sci. USA 90, 8977–8981 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T., Wang S., Noritake J., Sato K., Fukata M., Takefuji M., Nakagawa M., Izumi N., Akiyama T., Kaibuchi K. (2004). Interaction with IQGAP1 links APC to Rac1, Cdc42, and actin filaments during cell polarization and migration. Dev. Cell 7, 871–883 [DOI] [PubMed] [Google Scholar]
- Weijer C. J. (2009). Collective cell migration in development. J. Cell Sci. 122, 3215–3223 [DOI] [PubMed] [Google Scholar]
- Wong W. M., Wright N. A. (1999). Cell proliferation in gastrointestinal mucosa. J. Clin. Pathol. 52, 321–333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright N. A., Alison M. (1984). The Biology of Epithelial Cell Populations. pp. 364–367 Oxford: Clarendon Press [Google Scholar]
- Yang X., Dormann D., Münsterberg A. E., Weijer C. J. (2002). Cell movement patterns during gastrulation in the chick are controlled by positive and negative chemotaxis mediated by FGF4 and FGF8. Dev. Cell 3, 425–437 [DOI] [PubMed] [Google Scholar]
- Zhang Z., Chen L., Gao L., Lin K., Zhu L., Lu Y., Shi X., Gao Y., Zhou J., Xu P., et al. (2011). Structural basis for the recognition of Asef by adenomatous polyposis coli. Cell Res. doi: 10.1038/cr.2011.119 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
{kind=link}
{kind=link}
{kind=link}
{kind=link}