

Abstract
Objectives:
Mutations in the GBA gene occur in 7% of patients with Parkinson disease (PD) and are a well-established susceptibility factor for PD, which is characterized by Lewy body disease (LBD) neuropathologic changes (LBDNCs). We sought to determine whether GBA influences risk of dementia with LBDNCs, Alzheimer disease (AD) neuropathologic changes (ADNCs), or both.
Methods:
We screened the entire GBA coding region for mutations in controls and in subjects with dementia and LBDNCs and no or low levels of ADNCs (pure dementia with Lewy bodies [pDLB]), LBDNCs and high-level ADNCs (LBD-AD), and high-level ADNCs but without LBDNCs (AD).
Results:
Among white subjects, pathogenic GBA mutations were identified in 6 of 79 pDLB cases (7.6%), 8 of 222 LBD-AD cases (3.6%), 2 of 243 AD cases (0.8%), and 3 of 381 controls (0.8%). Subjects with pDLB and LBD-AD were more likely to carry mutations than controls (pDLB: odds ratio [OR] = 7.6; 95% confidence interval [CI] = 1.8–31.9; p = 0.006; LBD-AD: OR = 4.6; CI = 1.2–17.6; p = 0.025), but there was no significant difference in frequencies between the AD and control groups (OR = 1.1; CI = 0.2–6.6; p = 0.92). There was a highly significant trend test across groups (χ2(1) = 19.3; p = 1.1 × 10−5), with the likelihood of carrying a GBA mutation increasing in the following direction: control/AD < LBD-AD < pDLB.
Conclusions:
GBA is a susceptibility gene across the LBD spectrum, but not in AD, and appears to convey a higher risk for PD and pDLB than for LBD-AD. PD and pDLB might be more similar to one another in genetic determinants and pathophysiology than either disease is to LBD-AD.
Lewy body disease (LBD) represents a continuum of clinicopathologic entities that includes Parkinson disease (PD), PD with dementia (PDD), and dementia with Lewy bodies (DLB). PDD is diagnosed when parkinsonism precedes dementia by at least 12 months, whereas in DLB, dementia occurs before or concurrently with parkinsonism.1 LBD neuropathologic changes (LBDNCs) include classic histologic inclusions (Lewy bodies) and α-synuclein immunopositive neuronal inclusions and processes (Lewy neurites). However, the pathologic classification of DLB is complex because some cases show LBDNCs with no or low levels of Alzheimer disease (AD) neuropathologic changes (ADNCs), which we refer to as “pure” DLB (pDLB), whereas many other cases show LBDNCs with coexistent high levels of ADNCs (LBD-AD). The pathophysiologic relationship among LBD-AD, pDLB, and PDD has not been delineated, and whether there is overlap of genetic risk factors among these disorders remains unclear.
Gaucher disease, the most common lysosomal storage disorder, results from a recessively inherited deficiency of glucocerebrosidase, which is encoded by the GBA gene. Heterozygous GBA mutation carriers have a 5- to 6-fold increased risk of developing PD.2 The relationship between GBA mutations and DLB is less clear. Several small studies have observed a higher mutation frequency in patients with DLB than in controls,3,4 but interpretation of these results is limited by methodologic issues. Furthermore, one study reported an increased mutation frequency in patients with AD but without LBDNCs.5
To address these issues, we screened for GBA in a large sample of subjects with neuropathologically determined AD, LBD-AD, and pDLB, and in controls without dementia.
METHODS
Subjects and clinical evaluation.
The study population was composed of 562 autopsied cases with dementia and 391 controls. Subjects with dementia were enrolled in 1 of 7 Alzheimer disease centers (ADCs) (Oregon Health and Science University; Rush University Medical Center; University of California, San Diego; University of Kentucky; University of Pennsylvania; University of Pittsburgh; and University of Washington), in the Rush Memory and Aging Project, or in the Group Health/University of Washington Alzheimer's Disease Patient Registry/Adult Changes in Thought Study. The Group Health/University of Washington Alzheimer's Disease Patient Registry/Adult Changes in Thought Study is a community-based longitudinal study that enrolled individuals aged 65 years or older with dementia6 or with no dementia7 from a health maintenance organization in the Seattle area. Expert diagnosticians at the ADCs, Rush Memory and Aging Project, and Group Health/University of Washington Alzheimer's Disease Patient Registry/Adult Changes in Thought Study reviewed subject clinical history, physical examinations, and neuropsychological tests at a consensus diagnosis conference. All individuals included in the dementia group received a clinical diagnosis of “probable AD,” “possible AD,” or “dementia, type unknown” according to the National Institute of Neurological and Communicative Disorders and Stroke and the Alzheimer's Disease and Related Disorders Association criteria.8 None had a clinical diagnosis of PD.
Controls were elderly adults who were enrolled in longitudinal studies of normal aging as part of the Group Health/University of Washington Alzheimer's Disease Patient Registry/Adult Changes in Thought Study, the Rush Memory and Aging Project, or 1 of 6 ADCs (Oregon Health and Science University; Rush University Medical Center; University of California, San Diego; University of Kentucky; University of Pittsburgh; and University of Washington). All controls were cognitively normal at study entry, as determined by a clinical consensus that considered both clinical history and neuropsychological testing. Controls remained free of cognitive impairment at their last evaluation, as indicated by a detailed clinical assessment (Rush ADC and Rush Memory and Aging Project),9 a Cognitive Abilities Screening Instrument10 score >85 (Group Health/University of Washington Alzheimer's Disease Patient Registry/Adult Changes in Thought Study), or a Clinical Dementia Rating score <1 and Mini-Mental State Examination score >26 (all other centers). None of the controls had a clinical diagnosis of a neurodegenerative disease, including PD. The controls consisted of 2 groups, one that underwent autopsy (n = 267) and one in which the subjects were alive at the time the dataset was closed (n = 124). All controls in the autopsied group underwent a clinical evaluation within 3 years of death.
Standard protocol approvals, registrations, and patient consents were obtained. All study procedures were approved by the institutional review boards at each participating site.
Neuropathologic classification.
All autopsied cases underwent a standard histologic evaluation using hematoxylin & eosin, modified Bielschowsky, and thioflavine S methods. Immunostaining for LBDNCs was also performed on all autopsied cases using α-synuclein immunohistochemistry with antibody LB509 (1:50 to 1:400; Zymed, San Francisco, CA), as previously described.11 Cases with questionable LB509 immunoreactivity were evaluated with a second antibody to nitrated α-synuclein (syn 303, 1:1,000).12 A section from each of the following 5 regions was assessed for LBDNCs: medulla, substantia nigra, amygdala, cingulate gyrus, and frontal cortex. The only exception was that 3 regions (substantia nigra, cingulate gyrus, and frontal cortex) rather than 5 regions were assessed for LBDNCs in cases and controls from Rush University Medical Center. Braak staging13 for neurofibrillary tangles and Consortium to Establish a Registry for Alzheimer's Disease plaque score14 were determined using modified Bielschowsky-stained sections, tau immunohistochemistry (AT8, Endogen, 1:250), or both.
All subjects with dementia underwent autopsy and were neuropathologically classified as pDLB (n = 80), LBD-AD (n = 231), or AD (n = 251) based on ADNCs and LBDNCs. AD was defined by the presence of high-level ADNCs (Braak stage IV, V, or VI and a Consortium to Establish a Registry for Alzheimer's Disease plaque score of “moderate” or “frequent”) but no LBDNCs. LBD-AD was defined by the presence of both high-level ADNCs and limbic or neocortical stage LBDNCs.11 pDLB was defined by the presence of limbic or neocortical stage LBDNCs and no or low levels of ADNCs. Note that all cases with ADNCs that did not meet criteria for high level were classified as low level, and we did not include an “intermediate level” in order to maximize statistical power. The autopsied control group showed no evidence of LBDNCs or high-level ADNCs.
Mutational analysis.
DNA was extracted from blood or brain tissue using standard methods. Because nearly 300 pathogenic mutations have been reported in patients with Gaucher disease,15 our screening approach was designed to cover the entire GBA coding region. GBA was amplified with PCR in 3 fragments of 1.7 to 3.0 kilobases in length using primers that specifically amplify the functional gene but not the nearby pseudogene. All 11 GBA exons and intron-exon boundaries were then sequenced using the Applied Biosystems Big-Dye Terminator v3.1 Cycle Sequencing Kit on an ABI PRISM 3130 genetic analyzer (Applied Biosystems, Foster City, CA). Sequence data were base-called, aligned, and scanned for variation using Mutation Surveyor (SoftGenetics, State College, PA). A mutation was considered “pathogenic” if it was previously reported in at least one patient with Gaucher disease.15,16 Fifty-five of the pDLB/LBD-AD cases were previously screened for the 2 most common GBA mutations (N370S and L444P); these results have been published elsewhere.4
Statistical analysis.
Demographic and descriptive information were summarized as means, standard deviations, and ranges for continuous measures and as proportions for discrete variables. Age and age at onset of dementia were compared among groups using analysis of variance. A Pearson χ2 test was used to evaluate group differences in sex proportions.
The Fisher exact test was used to examine differences in GBA mutation frequency among groups. Multinomial logistic regression was used to test for association between GBA mutations and each of the 3 disease groups with controls as the reference. A test for a trend in the odds of carrying a mutation by disease group was performed using the score trend test. In this analysis, controls and AD cases were coded as 0, LBD-AD cases were coded as 1, and pDLB cases were coded as 2. All statistical analyses were conducted using STATA version 10 and a p value <0.05 was considered significant.
RESULTS
The demographic and clinical characteristics of the sample are shown in table 1. A substantially greater proportion of subjects with pDLB were male in comparison to the AD, LBD-AD, and control groups (overall χ2(3) = 41.6, p < 0.001). There were no differences in age or age at onset among groups.
Table 1.
Clinical characteristics of the study population
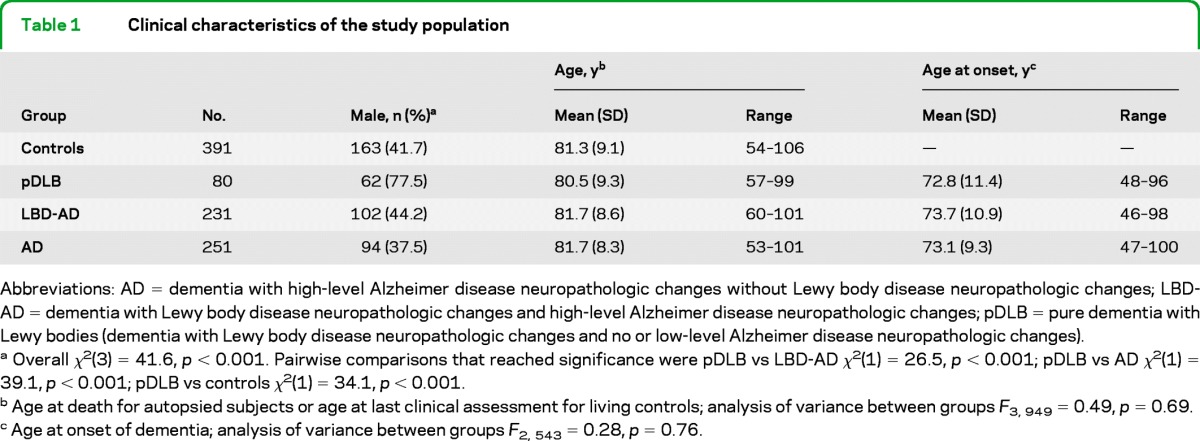
Abbreviations: AD = dementia with high-level Alzheimer disease neuropathologic changes without Lewy body disease neuropathologic changes; LBD-AD = dementia with Lewy body disease neuropathologic changes and high-level Alzheimer disease neuropathologic changes; pDLB = pure dementia with Lewy bodies (dementia with Lewy body disease neuropathologic changes and no or low-level Alzheimer disease neuropathologic changes).
Overall χ2(3) = 41.6, p < 0.001. Pairwise comparisons that reached significance were pDLB vs LBD-AD χ2(1) = 26.5, p < 0.001; pDLB vs AD χ2(1) = 39.1, p < 0.001; pDLB vs controls χ2(1) = 34.1, p < 0.001.
Age at death for autopsied subjects or age at last clinical assessment for living controls; analysis of variance between groups F3, 949 = 0.49, p = 0.69.
Age at onset of dementia; analysis of variance between groups F2, 543 = 0.28, p = 0.76.
Table 2 lists all GBA variants that were observed in the sample within exons or intronic splice sites, including 7 pathogenic mutations, 4 mutations of unknown significance, and 4 nonsynonymous polymorphisms. Note that 2 rare variants, K(−27)R and I(−20)V, were classified as polymorphisms because they have been observed to occur at a frequency of >5% in controls of African [K(−27)R; unpublished data] and Asian [I(−20)V]17 ancestry. Twenty-five subjects were found to carry GBA mutations (pathogenic, n = 19; unknown significance, n = 6), and their neuropathologic findings are summarized in table e-1 on the Neurology® Web site at www.neurology.org. Eighteen of these mutation carriers displayed LBDNCs (pDLB, n = 7; LBD-AD, n = 11), and 16 of the 18 cases had “neocortical” stage LBDNCs (figure). The remaining 2 cases had “limbic” stage LBDNCs11 with involvement of the brainstem and cingulate gyrus but sparing of the neocortex. All cases were specifically examined for the pathologic changes previously described in 4 cases of Gaucher disease with parkinsonism and LBDNCs.18 We observed neither “brainstem-type” Lewy bodies in the hippocampus nor severe neuronal loss and gliosis in the cornu ammonis 2–4 hippocampal subfields or neocortex. However, frequent α-synuclein immunopositive neuronal inclusions were present in the cornu ammonis subregions of all 16 cases with neocortical stage LBDNCs.
Table 2.
GBA variants observed
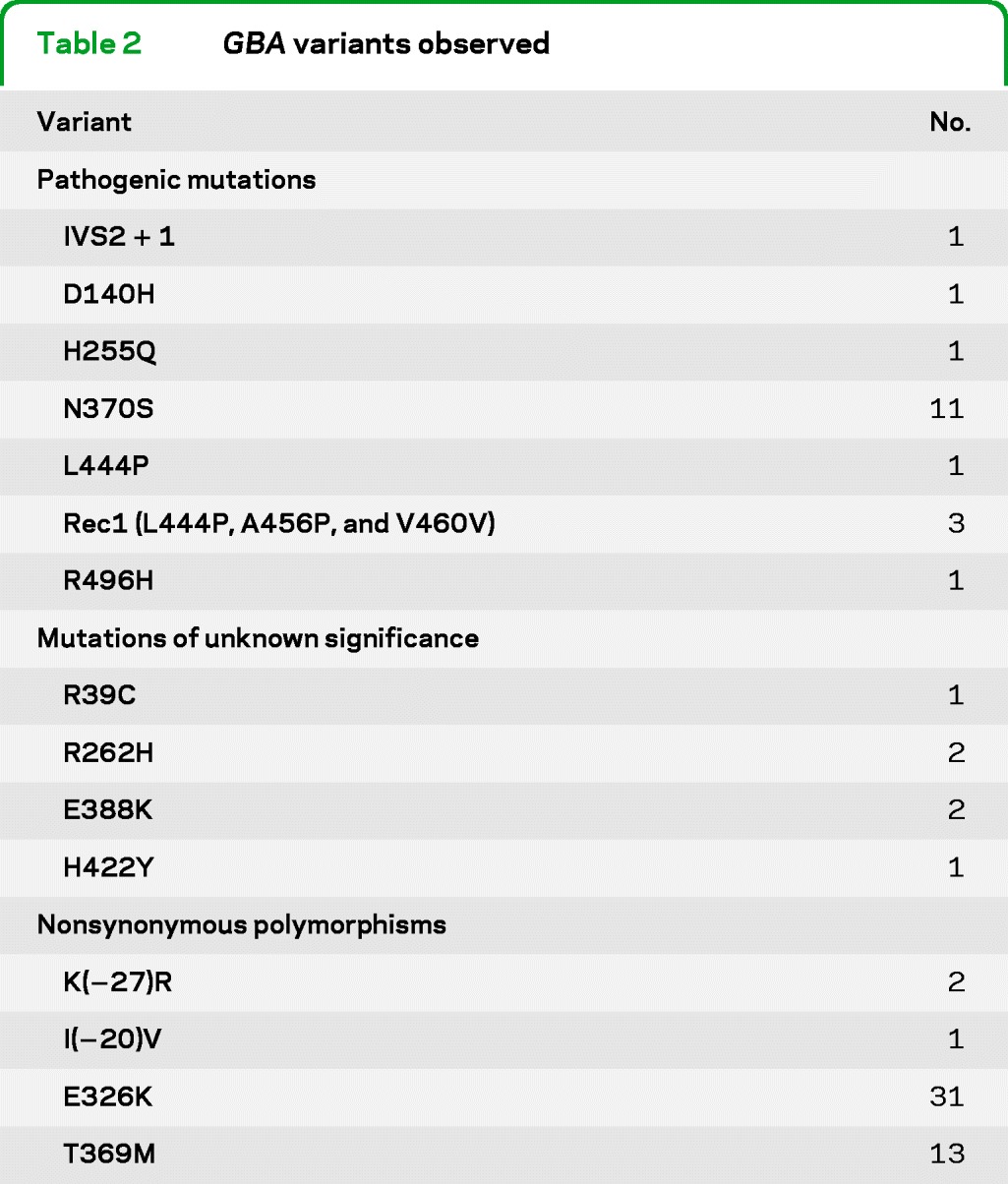
Figure. Lewy body disease neuropathologic changes in subject 18 with an N370S GBA mutation.

Multiple Lewy bodies by hematoxylin & eosin staining (A, arrowheads) and by α-synuclein immunostaining (B, arrowheads) in the substantia nigra. α-Synuclein immunopositive inclusions (arrowheads) and frequent neurites in the frontal cortex (C). Original magnifications, ×126.
The GBA mutation frequencies in each group are presented in table 3. Among white subjects, 6 of the 79 cases with pDLB (7.6%) were heterozygous for a pathogenic GBA mutation, as were 8 of the 222 cases with LBD-AD (3.6%), 2 of the 243 cases with AD (0.8%), and 3 of the 381 controls (0.8%). There was an overall difference in the frequency of pathogenic GBA mutation carriers across groups (Fisher exact test, p = 0.001). In comparison to controls, the carrier frequency was elevated in the pDLB (p = 0.001) and LBD-AD (p = 0.022) groups, but not in the AD group (p = 1.0).
Table 3.
GBA mutation frequencies by groupa
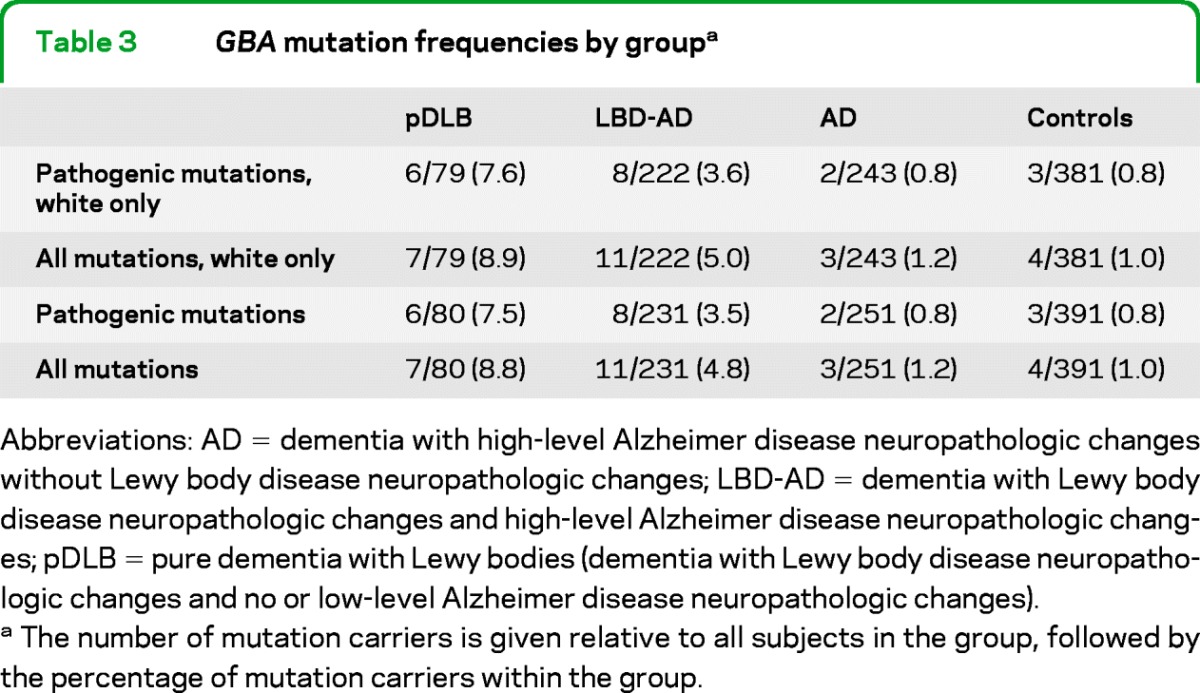
Abbreviations: AD = dementia with high-level Alzheimer disease neuropathologic changes without Lewy body disease neuropathologic changes; LBD-AD = dementia with Lewy body disease neuropathologic changes and high-level Alzheimer disease neuropathologic changes; pDLB = pure dementia with Lewy bodies (dementia with Lewy body disease neuropathologic changes and no or low-level Alzheimer disease neuropathologic changes).
The number of mutation carriers is given relative to all subjects in the group, followed by the percentage of mutation carriers within the group.
In a logistic regression model adjusted for age and sex and limited to white subjects (table 4), pathogenic GBA mutations were associated with both pDLB (odds ratio [OR] = 7.6; 95% confidence interval [CI] = 1.8–31.9; p = 0.006) and LBD-AD (OR = 4.6; 95% CI = 1.2–17.6; p = 0.025), but not AD (OR = 1.1; 95% CI = 0.2–6.6; p = 0.92). Inspection of the ORs suggested that individuals with LBD-AD might have a mutation carrier risk intermediate between the pDLB and AD/control groups. We assessed this hypothesis using a trend test. Given that the frequency of GBA mutations in controls and AD cases was nearly identical, for the purposes of this test, the controls and AD cases were collapsed into a single group. The results indicated an increased likelihood of carrying a GBA mutation across groups in the following direction: control/AD < LBD-AD < pDLB (χ2[1] = 19.3; p = 1.1 × 10−5).
Table 4.
Association of GBA mutations with dementia by group
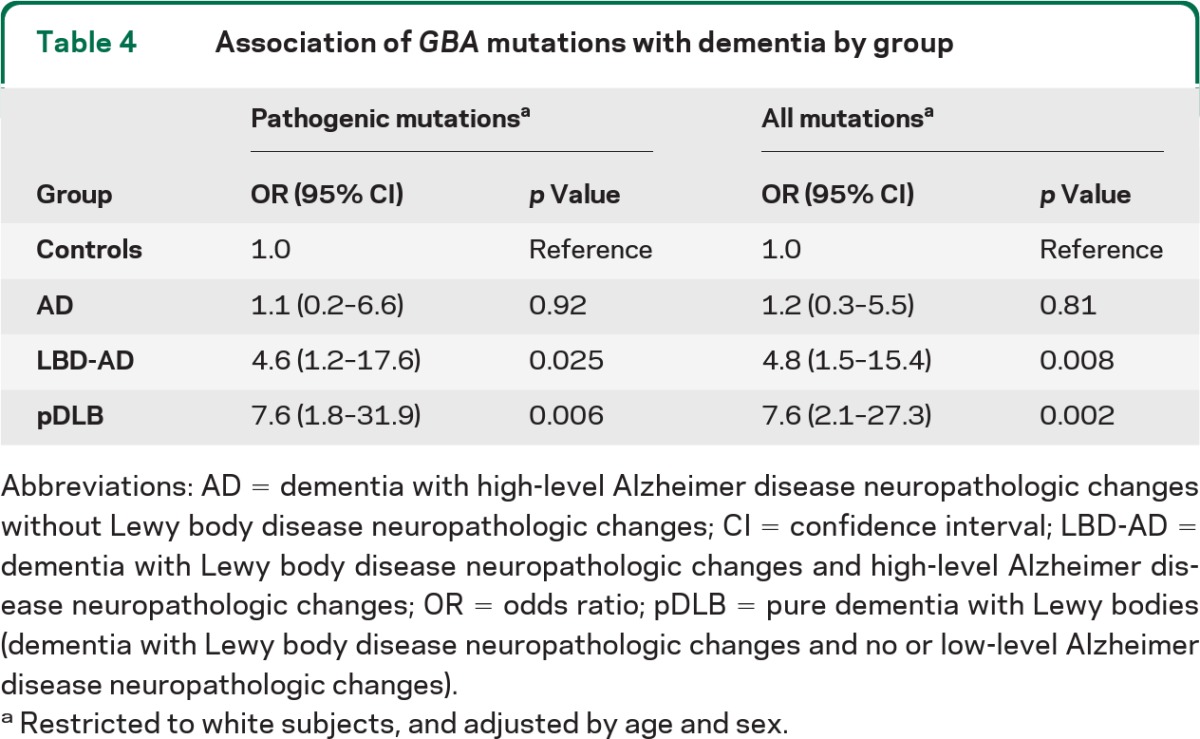
Abbreviations: AD = dementia with high-level Alzheimer disease neuropathologic changes without Lewy body disease neuropathologic changes; CI = confidence interval; LBD-AD = dementia with Lewy body disease neuropathologic changes and high-level Alzheimer disease neuropathologic changes; OR = odds ratio; pDLB = pure dementia with Lewy bodies (dementia with Lewy body disease neuropathologic changes and no or low-level Alzheimer disease neuropathologic changes).
Restricted to white subjects, and adjusted by age and sex.
DISCUSSION
Our results are novel in that they suggest that GBA conveys a higher risk for pDLB than for LBD-AD. The observed frequency of pathogenic mutations in the pDLB group (7.6%) was very similar to the 7% frequency reported in a multicenter analysis of 1,883 patients with PD in whom GBA was fully sequenced.2 In contrast to another report,5 we did not find differences in the proportion of GBA mutation carriers between controls and patients with AD.
pDLB and PDD are neuropathologically indistinguishable from one another19 whereas clinically, pDLB more closely resembles LBD-AD. Also, in contrast to LBD-AD, both pDLB and PDD exhibit a strong male predominance.20,21 The degree to which genetic factors have a role in shaping the similarities and differences among these 3 clinicopathologic entities is not well understood, in part because very few genetic studies have made a distinction between pDLB and LBD-AD. Our findings address this gap in knowledge and begin to differentiate the “genetic profiles” of PDD, pDLB, and LBD-AD. Taken together with previous work,2 our data suggest that in terms of the risk conveyed by GBA mutations, pDLB and PDD overlap more closely with one another than with LBD-AD. Whether this same pattern will prove to be true for other susceptibility genes remains to be determined.
Five studies3–5,22,23 have previously investigated the association between GBA mutations and DLB, and 2 of these studies have reported a significantly higher mutation frequency in the DLB group than in the matched controls.4,5 The observed DLB mutation carrier frequency varied several-fold across studies, from 27 of 95 cases (28%)5 to 3 of 57 cases (3.5%).4 Furthermore, one study5 reported an increased GBA mutation frequency of 10% in 60 cases with a neuropathologic diagnosis of AD and without LBDNCs. There are several factors that might explain these seemingly discrepant findings. Two of the studies lacked sufficient clinical information to exclude all cases with a clinical diagnosis of PD.5,23 Without making this distinction, one cannot accurately estimate separate mutation frequencies for PD and DLB.1 Two of the studies did not include a matched control group and instead referenced previously published mutation frequencies in controls.3,23 All previous studies had more modest sample sizes than the current study and thus only had sufficient power to detect large effects at the expected allele frequencies. One study assessed only the 2 most common pathogenic GBA mutations (N370S and L444P) rather than screening the entire coding region.4 Thus, some other pathogenic mutations were likely missed. In one study, the reported mutation frequency included rare variants of unknown functional significance and low-frequency polymorphisms.5 Finally, GBA mutation frequencies are substantially higher in Ashkenazi Jews,24 but none of the prior studies reported the proportion of subjects with Ashkenazi Jewish ancestry in the sample.
Our study had several strengths. The sample was large and was well characterized both clinically and neuropathologically. Importantly, all subjects with pDLB and LBD-AD had clinically documented dementia at onset rather than parkinsonism, allowing for exclusion of patients with PDD. We included a matched control group in which all subjects were screened for cognitive impairment and more than half underwent autopsy. All pathologic examinations for LBD were made using the same methods (by J.L., R.H., T.M., J.S., and P.N.). Finally, we comprehensively screened all GBA exons rather than selectively genotyping a subset of mutations. Our study also had limitations. Because the majority of subjects included in this clinicopathologic study died before the publication of the clinical criteria for DLB, we were unable to fully apply these criteria retrospectively, in particular the “fluctuating cognition” and “REM sleep behavior disorder” features.1 Also, as in previous studies, we did not collect information on the presence of Ashkenazi Jewish ancestry in all subjects and thus we cannot entirely exclude the possibility of unrecognized population structure in our dataset.
In a neuropathologic study of 14 patients with Gaucher disease,18 only the 4 cases that displayed clinical parkinsonism and dementia had LBDNCs. In all cases, the authors noted hippocampal and neocortical gliosis, and they also observed marked neuronal loss in a subset of cases with phenotypically severe type II and III disease. The LBDNCs were unusual in that “brainstem-type” Lewy bodies were seen in the hippocampus. We specifically evaluated the GBA mutation carriers identified in our study for these pathologic changes and they were not observed. However, we did observe severe and anatomically diffuse LBDNCs in the majority of GBA mutation carriers, including α-synuclein immunopositive inclusions in hippocampal neurons. We suspect that the pathologic differences observed between the Gaucher disease cases with parkinsonism and our patients with pDLB and LBD-AD might be attributable to a gene dosage effect in that the Gaucher disease patients had 2 alleles with reduced or absent function whereas our patients had only 1.
Although findings from this and other case-control studies indicate an association between GBA mutations and LBD, the molecular mechanism by which dysfunction of the gene leads to LBDNCs and neurodegeneration has not been fully elucidated. However, recent in vivo and in vitro experiments indicate that GBA mutations promote aggregation of α-synuclein25 and enhance α-synuclein–mediated neurotoxicity,26 thus providing the first evidence directly linking alteration of glucocerebrosidase function with synucleinopathy.
Despite recent progress on the role of GBA in LBD, several critical questions remain to be answered. For example, although GBA mutations are a strong risk factor for LBD, most GBA carriers remain asymptomatic. Also, for those mutation carriers who do become symptomatic, it is not clear why some present with dementia at onset (pDLB or LBD-AD) whereas others present with parkinsonism (PD). It is likely that genetic and environmental factors modify the expression of GBA-related neurodegeneration. This hypothesis should be tested with large-scale genetic association studies, which incorporate careful clinicopathologic characterization and collection of detailed environmental exposure data. Family-based studies might be particularly fruitful. Although such endeavors will be challenging, the identification of major modifying factors for GBA-induced synucleinopathies could substantially accelerate progress in developing improved treatment strategies for LBD.
Supplementary Material
ACKNOWLEDGMENT
The authors thank all subjects for participating in this work, Lynne Greenup for technical assistance, and Andrew David for editorial assistance.
GLOSSARY
- AD
Alzheimer disease
- ADC
Alzheimer disease center
- ADNCs
Alzheimer disease neuropathologic changes
- CI
confidence interval
- DLB
dementia with Lewy bodies
- LBD
Lewy body disease
- LBDNCs
Lewy body disease neuropathologic changes
- OR
odds ratio
- PD
Parkinson disease
- PDD
Parkinson disease with dementia
- pDLB
pure dementia with Lewy bodies
Footnotes
Editorial. page 1938
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Dr. Tsuang: study concept and design, acquisition of data, interpretation of data, drafting of the manuscript, and obtaining funding. Dr. Leverenz: study concept and design, acquisition of data, interpretation of data, and drafting of the manuscript. Dr. Lopez: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Hamilton: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Bennett: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Schneider: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Buchman: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Larson: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Crane: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Kaye: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Kramer: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Woltjer: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Kukull: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Nelson: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Jicha: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Neltner: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Galasko: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Masliah: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Dr. Trojanowski: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Schellenberg: critical revision of the manuscript for important intellectual content, interpretation of data, and obtaining funding. Ms. Yearout: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and technical support. Ms. Huston: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and technical support. Ms. Fritts-Penniman: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and technical support. Dr. Mata: critical revision of the manuscript for important intellectual content, acquisition of data, and interpretation of data. Ms. Wan: statistical analysis, interpretation of data, and critical revision of the manuscript for important intellectual content. Dr. Edwards: statistical analysis, interpretation of data, and critical revision of the manuscript for important intellectual content. Dr. Montine: critical revision of the manuscript for important intellectual content, acquisition of data, interpretation of data, and obtaining funding. Dr. Zabetian: study concept and design, acquisition of data, interpretation of data, drafting of the manuscript, and obtaining funding.
DISCLOSURE
D. Tsuang is funded by grants from Department of Veterans Affairs, NARSAD, and NIH. J. Leverenz has served as a consultant for Bayer and Teva Pharmaceuticals and is funded by grants from the American Parkinson Disease Association, Michael J. Fox Foundation, NIH, and Northwest Collaborative Care. O. Lopez is funded by the NIH. R. Hamilton performs diagnostic neuropathology (40% effort) and bills for these procedures. He is partly supported by grants from the NIH. D. Bennett serves on the editorial board of Neurology®; serves on the scientific advisory board for Vigorous Minds; serves/has served as a consultant to Danone Research B.V., Willmar Schwabe GmbH, Eli Lilly, and Gerson Lehrman Group; and receives research support from Danone Research B.V., the NIH, the Illinois Department of Public Health, and the Robert C. Borwell Endowment Fund. J. Schneider served on a scientific advisory board for GE Healthcare; serves as a consultant for Avid Radiopharmaceuticals, Inc.; and receives research support from Avid Radiopharmaceuticals, Inc. and the NIH. A. Buchman is funded by the NIH. E. Larson receives research support from the NIH. P. Crane is funded by grants from the Alzheimer's Association and NIH. J. Kaye receives research support from the Department of Veterans Affairs and the NIH. Individuals who work in the research centers he directs receive research support from Johnson & Johnson, Roche, and Bristol Myers Squibb. Dr. Kaye is compensated for serving on a data monitoring committee for Eli Lilly; he serves as a paid advisor for Janssen Pharmaceutical. Dr. Kaye receives reimbursement through Medicare or commercial insurance plans for providing clinical assessment and care for patients; is salaried to see patients at the Portland VA Medical Center; and serves as an unpaid Vice-Chair for the International Professional Interest Area Work Group of the International Society to Advance Alzheimer's Research and Treatments (ISTAART) and as an unpaid Commissioner for the Center for Aging Services and Technologies. P. Kramer reports no disclosures. R. Woltjer is funded by the NIH. W. Kukull is funded by grants from the Alzheimer's Association and NIH. P. Nelson is funded by grants from the NIH. G. Jicha serves as a consultant for Pfizer and Eli Lilly, and receives research support from Pfizer, Elan Corporation, Janssen, Medivation, Inc., Danone, and the NIH/NIA. J. Neltner reports no disclosures. D. Galasko serves on safety monitoring committees for clinical trials for Janssen Pharmaceuticals, Elan Pharmaceuticals, and Balance Pharmaceuticals, and has served as a paid advisor to Elan Pharmaceuticals, Phloronol, Inc., and United BioSource. He has received research funding from the National Institute on Aging. E. Masliah is funded by grants from the NIH. J. Trojanowski has received funding for travel and honoraria from Takeda Pharmaceutical Company, Ltd.; has received speaker honoraria from Pfizer; may accrue revenue on patents regarding modified avidin-biotin technique, method of stabilizing microtubules to treat Alzheimer disease, method of detecting abnormally phosphorylated tau, method of screening for Alzheimer disease or disease associated with the accumulation of paired helical filaments, compositions and methods for producing and using homogeneous neuronal cell transplants, rat comprising straight filaments in its brain, compositions and methods for producing and using homogeneous neuronal cell transplants to treat neurodegenerative disorders and brain and spinal cord injuries, diagnostic methods for Alzheimer disease by detection of multiple MRNAs, methods and compositions for determining lipid peroxidation levels in oxidant stress syndromes and diseases, compositions and methods for producing and using homogeneous neuronal cell transplants, method of identifying, diagnosing and treating α-synuclein positive neurodegenerative disorders, mutation-specific functional impairments in distinct tau isoforms of hereditary frontotemporal dementia and parkinsonism linked to chromosome-17: genotype predicts phenotype, microtubule stabilizing therapies for neurodegenerative disorders, and treatment of Alzheimer and related diseases with an antibody; and receives research support from the NIH (NIA, NINDS) and from the Marian S. Ware Alzheimer Program. G. Schellenberg serves on a scientific advisory board and receives honoraria from the American Health Assistance Foundation; has served as a consultant for and received funding for travel from Integra-Gen; holds/has filed patents regarding chromosome 14 and familial Alzheimer disease genetic markers and assays, chromosome 1 gene and gene products related to Alzheimer disease, and genetic basis of Alzheimer disease and diagnosis and treatment thereof; and receives/has received research support from the NIH (NIA, NIMH), the Autism Genome Project, Autism Speaks, CurePSP, the Rainwater Foundation, and Peebler PSP Research Foundation. He is an uncompensated member (other than travel funds to meetings of these boards) of the Alzheimer's Association Medical and Scientific Advisory Council, CSP #546 Executive Committee, Veterans Affairs Dementia Prevention Study of Vitamin E and Memantine, the Board of the Peebler PSP Research Foundation, and the Medical Advisory Board, Society of Progressive Supranuclear Palsy. D. Yearout and H. Huston receive salary support from the Department of Veterans Affairs and NIH. A. Fritts-Penniman received salary support from the Department of Veterans Affairs and NIH. I. Mata is funded by grants from the Department of Veterans Affairs, NIH, and Parkinson's Disease Foundation. J. Wan receives salary support from the Department of Veterans Affairs and NIH. K. Edwards and T. Montine are funded by grants from the NIH. C. Zabetian is funded by grants from the American Parkinson Disease Association, Department of Veterans Affairs, NIH, Northwest Collaborative Care, and Parkinson's Disease Foundation. Go to Neurology.org for full disclosures.
REFERENCES
- 1.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 2005; 65: 1863– 1872 [DOI] [PubMed] [Google Scholar]
- 2.Sidransky E, Nalls MA, Aasly JO, et al. Multicenter analysis of glucocerebrosidase mutations in Parkinson's disease. N Engl J Med 2009; 361: 1651– 1661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goker-Alpan O, Giasson BI, Eblan MJ, et al. Glucocerebrosidase mutations are an important risk factor for Lewy body disorders. Neurology 2006; 67: 908– 910 [DOI] [PubMed] [Google Scholar]
- 4.Mata IF, Samii A, Schneer SH, et al. Glucocerebrosidase gene mutations: a risk factor for Lewy body disorders. Arch Neurol 2008; 65: 379– 382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Clark LN, Kartsaklis LA, Wolf Gilbert R, et al. Association of glucocerebrosidase mutations with dementia with Lewy bodies. Arch Neurol 2009; 66: 578– 583 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Larson EB, Kukull WA, Teri L, et al. University of Washington Alzheimer's Disease Patient Registry (ADPR): 1987–1988. Aging 1990; 2: 404– 408 [PubMed] [Google Scholar]
- 7.Kukull WA, Higdon R, Bowen JD, et al. Dementia and Alzheimer disease incidence: a prospective cohort study. Arch Neurol 2002; 59: 1737– 1746 [DOI] [PubMed] [Google Scholar]
- 8.McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's Disease. Neurology 1984; 34: 939– 944 [DOI] [PubMed] [Google Scholar]
- 9.Schneider JA, Arvanitakis Z, Leurgans SE, Bennett DA. The neuropathology of probable Alzheimer disease and mild cognitive impairment. Ann Neurol 2009; 66: 200– 208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Teng EL, Hasegawa K, Homma A, et al. The Cognitive Abilities Screening Instrument (CASI): a practical test for cross-cultural epidemiological studies of dementia. Int Psychogeriatr 1994; 6: 45– 58 [DOI] [PubMed] [Google Scholar]
- 11.Leverenz JB, Hamilton R, Tsuang DW, et al. Empiric refinement of the pathologic assessment of Lewy-related pathology in the dementia patient. Brain Pathol 2008; 18: 220– 224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Duda JE, Giasson BI, Mabon ME, Lee VM, Trojanowski JQ. Novel antibodies to synuclein show abundant striatal pathology in Lewy body diseases. Ann Neurol 2002; 52: 205– 210 [DOI] [PubMed] [Google Scholar]
- 13.Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82: 239– 259 [DOI] [PubMed] [Google Scholar]
- 14.Mirra SS, Heyman A, McKeel D, et al. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD): part II: standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 1991; 41: 479– 486 [DOI] [PubMed] [Google Scholar]
- 15.Hruska KS, LaMarca ME, Scott CR, Sidransky E. Gaucher disease: mutation and polymorphism spectrum in the glucocerebrosidase gene (GBA). Hum Mutat 2008; 29: 567– 583 [DOI] [PubMed] [Google Scholar]
- 16.Beutler E, Gelbart T, Scott CR. Hematologically important mutations: Gaucher disease. Blood Cells Mol Dis 2005; 35: 355– 364 [DOI] [PubMed] [Google Scholar]
- 17.Mitsui J, Mizuta I, Toyoda A, et al. Mutations for Gaucher disease confer high susceptibility to Parkinson disease. Arch Neurol 2009; 66: 571– 576 [DOI] [PubMed] [Google Scholar]
- 18.Wong K, Sidransky E, Verma A, et al. Neuropathology provides clues to the pathophysiology of Gaucher disease. Mol Genet Metab 2004; 82: 192– 207 [DOI] [PubMed] [Google Scholar]
- 19.Dickson DW, Braak H, Duda JE, et al. Neuropathological assessment of Parkinson's disease: refining the diagnostic criteria. Lancet Neurol 2009; 8: 1150– 1157 [DOI] [PubMed] [Google Scholar]
- 20.de Lau LM, Breteler MM. Epidemiology of Parkinson's disease. Lancet Neurol 2006; 5: 525– 535 [DOI] [PubMed] [Google Scholar]
- 21.Nelson PT, Schmitt FA, Jicha GA, et al. Association between male gender and cortical Lewy body pathology in large autopsy series. J Neurol 2010; 257: 1875– 1881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Farrer MJ, Williams LN, Algom AA, et al. Glucosidase-beta variations and Lewy body disorders. Parkinsonism Relat Disord 2009; 15: 414– 416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nishioka K, Ross OA, Vilarino-Guell C, et al. Glucocerebrosidase mutations in diffuse Lewy body disease. Parkinsonism Relat Disord 2010; 17: 55– 57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gan-Or Z, Giladi N, Rozovski U, et al. Genotype-phenotype correlations between GBA mutations and Parkinson disease risk and onset. Neurology 2008; 70: 2277– 2283 [DOI] [PubMed] [Google Scholar]
- 25.Cullen V, Sardi SP, Ng J, et al. Acid beta-glucosidase mutants linked to Gaucher disease, Parkinson disease, and Lewy body dementia alter alpha-synuclein processing. Ann Neurol 2011; 69: 940– 953 [DOI] [PubMed] [Google Scholar]
- 26.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and alpha-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell 2011; 146: 37– 52 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.