

Abstract
Objective:
To assess the frequency and clinical characteristics of patients with mutations of major amyotrophic lateral sclerosis (ALS) genes in a prospectively ascertained, population-based epidemiologic series of cases.
Methods:
The study population includes all ALS cases diagnosed in Piemonte, Italy, from January 2007 to June 2011. Mutations of SOD1, TARDBP, ANG, FUS, OPTN, and C9ORF72 have been assessed.
Results:
Out of the 475 patients included in the study, 51 (10.7%) carried a mutation of an ALS-related gene (C9ORF72, 32; SOD1, 10; TARDBP, 7; FUS, 1; OPTN, 1; ANG, none). A positive family history for ALS or frontotemporal dementia (FTD) was found in 46 (9.7%) patients. Thirty-one (67.4%) of the 46 familial cases and 20 (4.7%) of the 429 sporadic cases had a genetic mutation. According to logistic regression modeling, besides a positive family history for ALS or FTD, the chance to carry a genetic mutation was related to the presence of comorbid FTD (odds ratio 3.5; p = 0.001), and age at onset ≤54 years (odds ratio 1.79; p = 0.012).
Conclusions:
We have found that ∼11% of patients with ALS carry a genetic mutation, with C9ORF72 being the commonest genetic alteration. Comorbid FTD or a young age at onset are strong indicators of a possible genetic origin of the disease.
Amyotrophic lateral sclerosis (ALS) is a degenerative disorder of adult age involving the motor system, with a progressive and invariably fatal course. In 5% of cases it is considered to be genetically transmitted (familial ALS [fALS])1,2 while in the remaining cases it occurs sporadically in the population (sporadic ALS [sALS]). Mutations in several genes have been found to cause ALS, namely superoxide dismutase 1 (SOD1),3 TAR DNA binding protein (TARDBP),4 angiogenin (ANG),5 fused in sarcoma (FUS),6,7 optineurin (OPTN),8 and the recently described chromosome 9 open reading frame 72 (C9ORF72).9,10
Several studies have assessed the frequency of mutations of single genes,11 but few studies have compared relative frequencies of SOD1, TARDBP, ANG, and FUS11–15; all these studies were based on tertiary center series of cases, and were consequently skewed toward a younger population of patients with a larger number of fALS16; moreover, only one of them reported data on C9ORF72, which is now recognized as the major contributor to this form of neurodegeneration. Therefore there are no data on the frequency of mutations of ALS-related genes in a population-based setting that would be representative of the general ALS population, with the exception of our previous article on SOD1 mutation in a province of Torino, Italy, in the period 2000–2005.16
The aim of this study is to assess the frequency of mutations of major ALS genes in a prospectively ascertained, population-based epidemiologic series of cases identified through the Piemonte and Valle d'Aosta register for ALS (PARALS) and to compare the clinical characteristics of patients with and without genetic mutations.
METHODS
The study population includes all ALS cases diagnosed in Piemonte and Valle d'Aosta, Italy, during the 4.5-year period January 1, 2007, to June 30, 2011. The cases were recruited through the PARALS, a prospective epidemiologic register involving all the neurologic departments of the 2 regions of Northern Italy. The register was established in 1995 and is still in operation. Epidemiologic data regarding the 1995–2004 period have been published elsewhere.17
Case collection.
The main sources of cases were the neurology departments of the 2 regions. Investigators used an ad hoc questionnaire to collect patients' demographic data, disease history, neurologic and laboratory findings, and treatments. Diagnostic EMG examination was performed in all patients according to standard procedures. The secondary sources for case collection were the Piemonte and Valle d'Aosta Central Regional Archives and mortality coding from the Italian Bureau of Statistics. Clinical records of cases found through secondary sources were obtained, and relevant clinical information for each case was analyzed in order to verify whether the patient met the eligibility criteria for the diagnosis of ALS; all living patients were contacted by phone and visited by one of the neurologists involved in the study.
Diagnostic criteria.
The diagnosis of ALS was based on El Escorial revised criteria.18 Patients with definite, probable, and probable laboratory-supported ALS were included in the register.
Definition of fALS.
Currently there is not a consensus on the diagnostic criteria for fALS,19 and the recent identification of C9ORF72 gene hexanucleotide expansions as a major cause of both ALS and frontotemporal dementia (FTD) co-occurring in the same pedigrees20–22 has further complicated the analysis of ALS pedigrees. For the present article, we used 2 different definitions of fALS: first, a narrower definition requiring the presence of at least 1 first- or second-degree relative affected by ALS in the pedigree of the index case23; second, a broader definition, considering as fALS also the patients with 1 first-degree relative with FTD, when the index case carried a GGGGCC hexanucleotide expansion in the first intron of C9ORF72 gene.
Genetic analysis.
All the coding exons and 50 bp of the flanking intron-exon boundaries of SOD1, of exon 6 of TARDBP, and of exons 14 and 15 of FUS and exons 5, 9, 12, and 14 of OPTN and the only exon of ANG have been PCR amplified, sequenced using the Big-Dye Terminator v3.1 sequencing kit (Applied Biosystems Inc.), and run on an ABIPrism 3130 genetic analyzer. These exons were selected as the vast majority of known pathogenic variants are known to lie within these mutational hotspots. A repeat-primed PCR assay was used to screen for the presence of the GGGGCC hexanucleotide expansion in the first intron of C9ORF72.10
Cognitive assessment.
From January 2007 patients' neurobehavioral dysfunction was assessed with the Frontal Systems Behavior Scale.24 From June 2009, >95% of patients also underwent a complete cognitive battery according to the consensus criteria for the diagnosis of frontotemporal cognitive and behavioral syndromes in ALS.25 Patients were classified in 2 categories based on neurobehavioral and cognitive testing: subjects with normal cognition and subjects with FTD. Patients with FTD were further subclassified as having behavioral variant frontotemporal dementia (bvFTD), progressive aphasia, or semantic dementia, according to the proposed criteria for the classification of dementia in ALS.25
Controls.
Controls were neurologically healthy subjects, resident in the area of the study, and age- and gender-matched to cases. SOD1 was sequenced in 130 controls, C9ORF72 in 245, TARDBP in 196, ANG in 140, FUS in 280, and OPTN in 96.
Statistical methods.
Comparisons between means were made with analysis of variance. Cochran-Armitage test for trend was used to test the frequency of mutated cases in different age groups. Logistic regression (stepwise forward) was used to identify factors independently related to presence of a genetic mutation. Variables included in the logistic regression were age (≤54, 55–69, ≥70 years), gender, type of onset (bulbar vs spinal), comorbid FTD, and family history positive for 1 first- or second-degree relative with ALS or FTD. A p value <0.05 was considered significant. Data were processed using SPSS statistical package version 18 (IBM Corporation, Chicago, IL). No patients were lost to follow-up. The follow-up included a total of 1,148.5 person/years.
Ethical approval.
The study has been approved by the ethical committees of the involved centers. All patients and controls signed a written informed consent. Databases were treated according to the Italian regulations for privacy.
RESULTS
During the period of the study a total of 601 patients were diagnosed with ALS in the study area. Of these, 40 did not give their consent for genetic analysis and 42 died before blood sampling. Forty-four patients were found only through secondary sources and were therefore not tested for DNA (figure). Therefore, a total of 475 patients (79.0%) were included in the mutational analysis study. The patients not included in the genetic analysis had an older mean age at onset than those included in the study (68.7 [SD 10.7] vs 65.5 [SD 10.6] years, p = 0.02), but were similar in terms of site of onset and clinical phenotype (data not shown).
Figure. Flow chart showing capture rate and the sequence of participant selection.

ALS = amyotrophic lateral sclerosis.
Frequency of familial ALS.
Thirty-six patients (7.6%) belonging to 34 families met the narrower criteria for fALS. A total of 46 (9.7%) patients, belonging to 44 apparently unrelated families, met the broader criteria for fALS.
Frequency of genetic mutations.
Thirty-one (67.4%) out of the 46 patients with fALS carried a mutation in one of the tested ALS genes. In contrast, only 20 (4.7%) out of the 429 apparently sALS cases had a genetic mutation. Overall, 51 patients (10.7% of the whole series) carried a mutation of an ALS-related gene. Younger patients were nearly 3 times more likely to carry a mutation in one of the known ALS genes compared to older patients (Cochran-Armitage test for trend, p = 0.03, table 1).
Table 1.
Frequency of mutated patients (all tested genes) according to agea
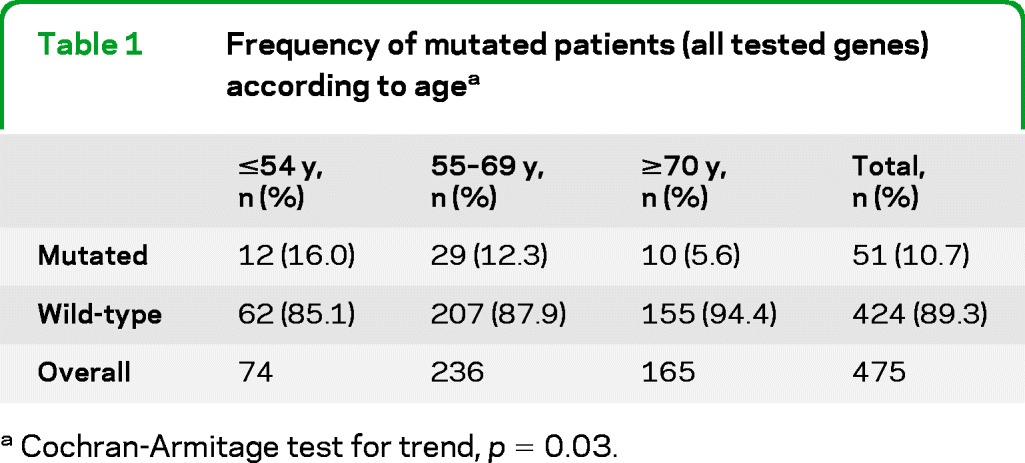
Cochran-Armitage test for trend, p = 0.03.
Clinical factors related to the risk of carrying a pathogenic mutation.
According to logistic regression modeling, the chance of a patient having a genetic mutation was most strongly related to a positive family history for ALS or FTD in a first- or second-degree relative (odds ratio [OR] 22.2, 95% confidence interval [CI] 9.6–52.6; p = 0.0001). The presence of comorbid FTD (OR 3.5, 95% CI 1.7–7.4; p = 0.001) and age at onset ≤54 years (OR 1.79; 95% CI 1.1–3.6; p = 0.012) were also significant independent factors influencing the likelihood of carrying a genetic mutation.
The clinical characteristics of patients carrying or not carrying gene mutations are reported in table 2. The different genetic mutations are characterized by rather different phenotypes. No pathogenetic mutations of ANG were found, and none of the patients in this series carried mutations of 2 different genes.
Table 2.
Main clinical characteristics of patients with different mutations and without known mutations
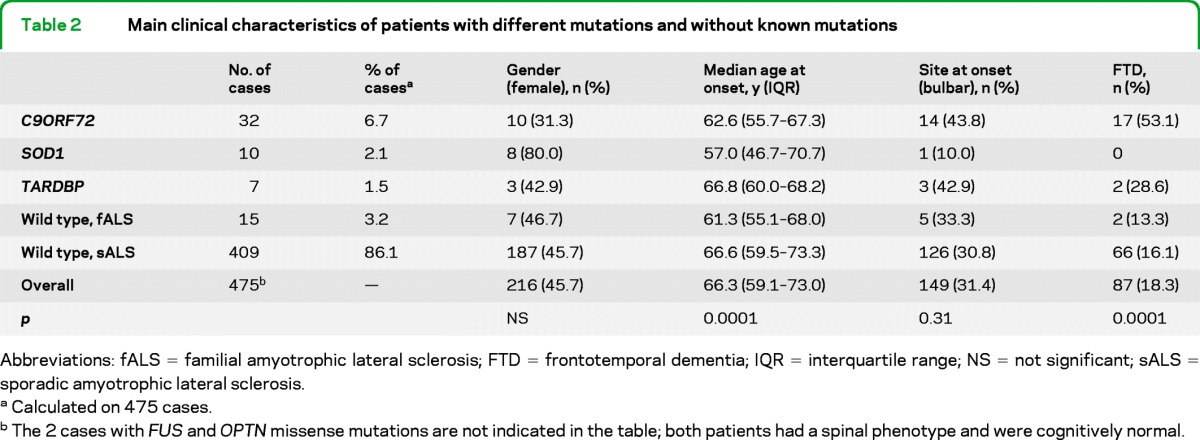
Abbreviations: fALS = familial amyotrophic lateral sclerosis; FTD = frontotemporal dementia; IQR = interquartile range; NS = not significant; sALS = sporadic amyotrophic lateral sclerosis.
Calculated on 475 cases.
The 2 cases with FUS and OPTN missense mutations are not indicated in the table; both patients had a spinal phenotype and were cognitively normal.
C9ORF72 hexanucleotide repeat expansions.
Thirty-two (6.7%) ALS cases carried a hexanucleotide repeat expansion of C9ORF72 gene, making it the commonest mutation found in our series, representing 62.7% of all mutated cases. Among these cases, 24 (75.0%) had a family history for ALS, FTD, or both. The mean age at onset of ALS in patients with C9ORF72 hexanucleotide repeat expansion was 61.6 years (SD 8.5) (range 43–71; median 62.6, interquartile range 55.7–67.3). Fourteen patients (45.2%) had a bulbar onset and 10 (31.3%) were female. More than half of the patients with C9ORF72 hexanucleotide repeat expansions had a florid FTD, significantly more than patients with other mutations and those without mutation. Symptomatology was consistent with bvFTD in all our cases carrying the pathogenic expansion. Three patients had psychotic-like symptoms. Comorbid FTD was significantly more frequent among patients with a family history positive for ALS, FTD, or both (p = 0.008) (table 3).
Table 3.
Frequency of FTD among patients with C9ORF72 hexanucleotide repeat expansion according to the presence of a family history for ALS, FTD, or both (p = 0.008)
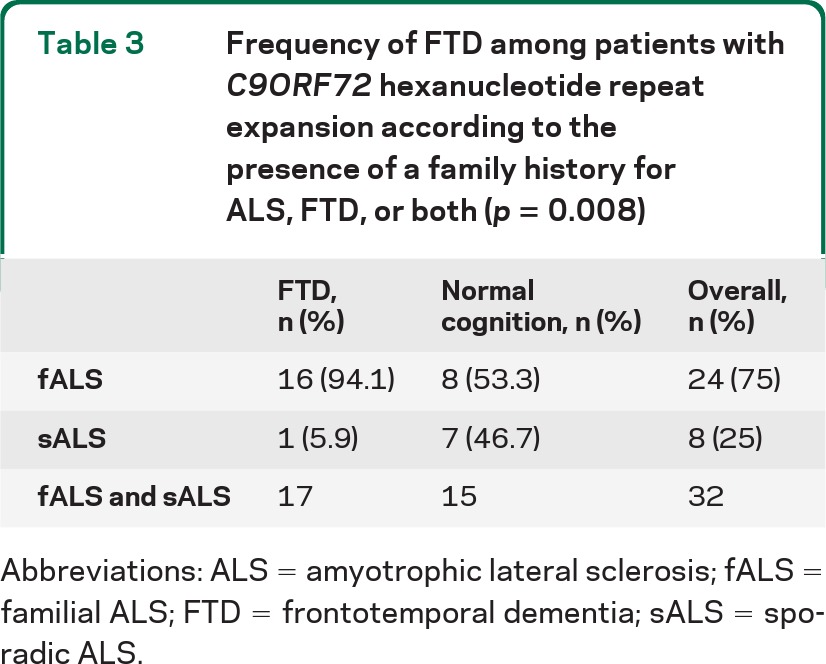
Abbreviations: ALS = amyotrophic lateral sclerosis; fALS = familial ALS; FTD = frontotemporal dementia; sALS = sporadic ALS.
SOD1 mutations.
SOD1 mutations were the second most common mutations, representing 2.1% of all ALS cases and 19.6% of all mutations. The following missense mutations were identified: c59A>G (p.N19S) (1 sALS), c.115C>G (p.L38V) (1 fALS), c.142G>T (p.V47F) (1 fALS), c.271G>A (p.D90N) (1 sALS, in heterozygosis), c.272A>C (p.D90A, in heterozygosis) (1 sALS), c.281G>A (p.G93D) (2 fALS), c.328 G>T (p.D109Y) (2 unrelated sALS cases), and c.435G>C (p.L144F) (1 sALS). Patients with SOD1 mutations had a mean age at onset of 54.4 years (SD 20.3) (range 16–85; median 57.0, interquartile range 44.7–70.7). Only 1 patient had a bulbar onset. No patients with SOD1 mutations had cognitive impairment.
TARDBP mutations.
Seven patients carried a missense mutation of the TARDBP gene, accounting for 1.5% of all ALS cases and 13.7% of all mutations. The most common TARDBP mutation was the c.1144G>A (p.A382T) missense mutation (1 fALS and 3 sALS). Other identified mutations were c.1009A>G (p.M337V) (1 fALS), c.1102G>A (p.G368S) (1 sALS), and c.1169 A>G (p.N390S) (1 sALS). Patients with TARDBP mutations had a mean age at onset of 64.4 years (SD 6.1) (range 52–70, interquartile range, 60.0–68.2). Three out of the 7 patients (42.9%) had a bulbar onset and 3 (42.9%) presented with bvFTD.
FUS mutations.
Only 1 patient had the c.1542G>C (p.R514S) missense mutation of the FUS gene. A detailed description of this patient's clinical picture and pedigree has been reported elsewhere.26
OPTN mutations.
We detected 1 patient with fALS carrying the never previously described c.1499T>C (p.L500P) missense mutation in the exon 14 of the OPTN gene. This mutation was predicted to be pathogenic by 2 different programs, Polyphen2 (http://genetics.bwh.harvard.edu/pph2) and PMut (http://mmb.pcb.ub.es/PMut/).
fALS with no identified mutations.
Fifteen patients had a positive family history for ALS, but did not carry mutations of one of the major ALS genes. These patients had a mean age of 62.1 years (SD 9.2; range 47–80; median 61.3, interquartile range 55.1–68.0). Only 2 patients (13.3%) had bvFTD, and 3 (33.3%) had a bulbar onset.
Pedigrees of apparently sporadic patients carrying genetic mutations.
In the pedigrees of the 20 apparently sporadic patients with C9ORF72, SOD1, and TARDBP mutations, no relatives with dementia, Parkinson disease, or other neurodegenerative disorders were detected.
DISCUSSION
In our study, we analyzed the genetics etiology of a large epidemiologic series of patients with ALS. The patients were prospectively identified through the Piemonte and Valle d'Aosta register for ALS during a period of 4.5 years and ∼80% of all identified cases underwent genetic testing. More than 10% of the tested patients carried a mutation of one of the major ALS genes, with C9ORF72 hexanucleotide expansion being the most commonly identified genetic mutation.
Only one population-based study on ALS genetics has been performed to date in one of the provinces of Piemonte in the period 2000–2005.16 In that article, 5.7% of the 386 patients with ALS had a positive family history for ALS and 5 patients (1.3%) (2 of whom were apparently sporadic) carried a SOD1 mutation. In the clinical-based series that have compared the frequencies of various ALS genes,11–15 the most frequent mutations have been found in the SOD1 gene (5% to 12% of cases) followed by TARDBP and FUS (2% to 4% of cases). In the only study having also assessed C9ORF72, this was the commonest mutated gene.15 All but one of these studies included only patients with fALS. The higher mutational rates observed in these studies compared to ours most likely arose from their use of patients referred to ALS clinics. Such referral cohorts are known to be biased toward the inclusion of younger patients who are more likely to carry a genetic mutation.16
The frequency of fALS in the Piemonte population is unexpectedly higher than that reported by previous epidemiologic studies,1,2 with the exception of certain genetically conserved populations, such as Finnish,10,27 Northern Swedish and Northern Norwegians,28 and Sardinians.29 Using the restrictive criteria for fALS (i.e., the presence of a first- or second-degree relative with ALS),23 the frequency of fALS in our series was 7.6%, higher than the 5.1% reported in a meta-analysis of published studies.2 This difference is probably related to the systematic collection of patients' pedigrees in the Piemonte register after 2007 as well as to the existence of a historical archive of ALS cases diagnosed in the Piemonte region going back to the 1950s which allows us to search for surnames recurring in patients' pedigrees and to identify cases according to the communities of origin.
The recent identification of C9ORF72 as a major cause of both ALS and FTD and the frequent identification of apparently sALS carrying genetic mutations challenges the classic definition of fALS.2,20 Based on this, we broadened the criteria for fALS to include patients with C9ORF72 hexanucleotide repeat expansions and one first- or second-degree relative with FTD. With these broader criteria, the frequency of fALS increased to 9.7% of the whole series. We believe that this rate represents a more realistic estimation of familial disease within the ALS population. The fact that two-thirds of patients meeting these criteria for fALS had a gene detected strongly supports our use of more inclusive criteria than those previously proposed.23
C9ORF72 hexanucleotide repeat expansions have been found in 6.7% of all ALS cases and represent the commonest mutations in this epidemiologic population of Italian ancestry. About two-thirds of these cases have a positive family history for ALS or FTD. In keeping with the first clinical descriptions of C9ORF72,21,22,30–32 patients carrying this mutation are younger than those without genetic mutations and about half of them have a bvFTD. The high frequency of positive family history for FTD in these cases agrees with the findings of 2 recent articles.20,30 It is still unclear whether PCR is sufficient for diagnosis of C9ORF72 mutations in the clinical setting. Moreover, accurate measurements of the repeat expansion size by Southern blot analysis are necessary to making genotype-phenotype correlations in patients with this mutation.22
The frequency of SOD1 mutations, 19.6% of all mutations and 2.1% of all fALS cases, is in keeping with the frequency observed in the same area in the 2000–2005 period.16 About half of the patients carrying SOD1 mutations were apparently sporadic and none of the cases with SOD1 mutations had FTD.33 The pathogenicity of some SOD1 missense mutations has been questioned.34 However, most mutations in our series are recognized pathogenic mutations and the others are likely to be mutations with a reduced penetrance.35
The third most common gene was TARDBP, identified in 1.5% of all ALS cases. The commonest TARDBP mutation was p.A382T, identified in 3 cases of Sardinian ancestry and 1 case of Sicilian ancestry. We have previously published that this particular founder mutation is highly prevalent on Sardinia.29 About half of the cases carrying TARDBP mutations had a bvFTD, a relatively common feature in association with TARDBP mutations.29
Not surprisingly, we found no putatively pathogenetic mutations of ANG. In fact, a recent meta-analysis reported that ANG mutations have been detected in only ∼0.5% of patients with ALS of European ancestry36 and the frequency was even lower in Italian patients.37–40 Although it remains possible that our cohort was not sufficiently powered to detect ANG mutations, our data confirm that mutations in this gene are not a frequent cause of motor neuron degeneration.
Overall, ALS cases carrying genetic mutations had some clinical peculiarities. First, the median age at onset of patients with genetic mutations was significantly lower than that of patients without genetic mutation. As a consequence, the probability of carrying a genetic mutation is inversely related with the age at onset of patients: the younger the age at onset, the higher the likelihood of having a genetic mutation. Second, 19 (21.8%) out of the 87 of patients with a comorbid FTD carried a genetic mutation (either C9ORF72 or TARDBP) compared to only 31 (8.0%) out of the 357 patients with normal cognition. Logistic regression analysis also demonstrated that comorbid FTD increased the chance of having a mutation by 3.5 and an age at onset <55 by 1.6. Therefore, our data, if confirmed with other studies, might suggest to offer the testing also to patients with comorbid FTD and those younger than 55, in addition to patients with a clear positive family history for ALS or FTD, who are obvious candidates for genetic testing.
A limitation of this study is that ∼20% of cases did not undergo genetic analysis. However, cases who were not captured did not differ on any relevant clinical or demographic characteristic from those who were included in the study.
This epidemiologic study has found that at least 10% of patients with ALS carry a genetic mutation of one of the major ALS genes, with C9ORF72 being the commonest genetic alteration in this Italian mainland population. We found that the presence of comorbid FTD or a young age at onset are strong indicators of a possible genetic origin of the disease, with possible implication both for future genetic studies and for genetic counseling in ALS.
Supplementary Material
ACKNOWLEDGMENT
The authors thank the patients and their families for collaborating in this study.
GLOSSARY
- ALS
amyotrophic lateral sclerosis
- bvFTD
behavioral variant frontotemporal dementia
- CI
confidence interval
- fALS
familial ALS
- FTD
frontotemporal dementia
- OR
odds ratio
- PARALS
Piemonte and Valle d'Aosta register for ALS
- sALS
sporadic ALS
Footnotes
Supplemental data at www.neurology.org
AUTHOR CONTRIBUTIONS
Study concept and design: Dr. Chiò, Dr. Calvo, Dr. Mora, Dr. Mazzini, Dr. Restagno, Dr. D'Alfonso, Dr. Traynor. Acquisition of data: Dr. Calvo, Dr. Moglia, Dr. Pisano, Dr. Mazzini, Dr. Corrado, Dr. Majounie, Dr. Renton, Dr. Ossola, Dr. Brunetti. Analysis and interpretation of data: Dr. Chiò, Dr. Calvo, Dr. Mora, Dr. Mazzini, Dr. Restagno, Dr. D'Alfonso. Drafting of the manuscript: Dr. Chiò, Dr. Mora, Dr. Traynor. Critical revision of the manuscript for important intellectual content: Dr. Chiò, Dr. Calvo, Dr. Mazzini, Dr. Cantello, Dr. Mora, Dr. Moglia, Dr. Corrado, Dr. D'Alfonso, Dr. Majounie, Dr. Renton, Dr. Pisano, Dr. Ossola, Dr. Brunetti, Dr. Traynor, Dr. Restagno. Obtained funding: Dr. Chiò, Dr. Restagno, Dr. D'Alfonso. Administrative, technical, and material support: Dr. Corrado, Dr. Majounie, Dr. Renton, Dr. Ossola, Dr. Brunetti. Study supervision: Dr. Chiò, Dr. Mazzini, Dr. Mora. Dr. Chiò had full access to all of the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis. All authors have approved the submitted version of the paper.
DISCLOSURE
A. Chiò has received research support from Italian Ministry of Health (Ricerca Finalizzata), Regione Piemonte (Ricerca Finalizzata), University of Torino, Federazione Italiana Giuoco Calcio, Fondazione Vialli e Mauro onlus, and European Commission (Health Seventh Framework Programme), and serves on a scientific advisory board for Biogen Idec and Cytokinetics. A. Calvo has received research support from Regione Piemonte (Ricerca Finalizzata) and Compagnia di San Paolo. L. Mazzini has received research support from Fondazione Borgonovo. R. Cantello reports no disclosures. G. Mora has received research support from Italian Ministry of Health (Ricerca Finalizzata). C. Moglia and L. Corrado report no disclosures. S. D'Alfonso has received research support from Fondazione Cariplo. E. Majunie, A. Renton, F. Pisano, I. Ossola, M. Brunetti, and B. Traynor report no disclosures. G. Restagno reports has received research support from Italian Ministry of Health (Ricerca Finalizzata) and Regione Piemonte (Ricerca Finalizzata). Go to Neurology.org for full disclosures.
REFERENCES
- 1.Logroscino G, Traynor BJ, Hardiman O, et al. Descriptive epidemiology of amyotrophic lateral sclerosis: new evidence and unsolved issues. J Neurol Neurosurg Psychiatry 2008;79:6–11. [DOI] [PubMed] [Google Scholar]
- 2.Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry 2011;82:623–627. [DOI] [PubMed] [Google Scholar]
- 3.Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993;362:59–62. [DOI] [PubMed] [Google Scholar]
- 4.Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science 2008;319:1668–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic' amyotrophic lateral sclerosis. Nature Genet 2006;38:411–413. [DOI] [PubMed] [Google Scholar]
- 6.Kwiatkowski TJ, Jr, Bosco DA, Leclerc AL, et al. Mutations in the FUS/TLS gene on chromosome 16 cause familial amyotrophic lateral sclerosis. Science 2009;323:1205–1208. [DOI] [PubMed] [Google Scholar]
- 7.Vance C, Rogelj B, Hortobágyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science 2009;323:1208–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 2010;465:223–226. [DOI] [PubMed] [Google Scholar]
- 9.Dejesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-Linked ALS-FTD. Neuron 2011;72:257–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown JA, Min J, Staropoli JF, et al. SOD1, ANG, TARDBP and FUS mutations in amyotrophic lateral sclerosis: a United States clinical testing lab experience. Amyotroph Lateral Scler 2012;13:217–222. [DOI] [PubMed] [Google Scholar]
- 12.Millecamps S, Salachas F, Cazeneuve C, et al. SOD1, ANG, VAPB, TARDBP, and FUS mutations in familial amyotrophic lateral sclerosis: genotype-phenotype correlations. J Med Genet 2010;47:554–560. [DOI] [PubMed] [Google Scholar]
- 13.Kwon MJ, Baek W, Ki SC, et al. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol Aging 2012;33:e17–e23. [DOI] [PubMed] [Google Scholar]
- 14.Tsai CP, Soong BW, Lin KP, et al. FUS, TARDBP, and SOD1 in a Taiwanese cohort of familial ALS. Neurobiol Aging 2011;32:553e13–553e21. [DOI] [PubMed] [Google Scholar]
- 15.Millecamps S, Boillée S, Le Ber I, et al. Phenotype difference between ALS patients with expanded repeats in C9ORF72 and patients with mutations in other ALS-related genes. J Med Genet 2012;49:258–263. [DOI] [PubMed] [Google Scholar]
- 16.Chiò A, Traynor BJ, Lombardo F, Fimognari M, et al. Prevalence of SOD1 mutations in the Italian ALS population. Neurology 2008;70:533–537. [DOI] [PubMed] [Google Scholar]
- 17.Chiò A, Mora G, Calvo A, et al. Epidemiology of ALS in Italy: a 10-year prospective population-based study. Neurology 2009;72:725–731. [DOI] [PubMed] [Google Scholar]
- 18.Brooks BR, Miller RG, Swash M, Munsat TL, World Federation of Neurology Research Group on Motor Neuron Diseases. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 19.Byrne S, Elamin M, Bede P, Hardiman O. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J Neurol Neurosurg Psychiatry 2012;83:36–367. [DOI] [PubMed] [Google Scholar]
- 20.Byrne S, Elamin M, Bede P, et al. Cognitive and clinical characteristics of patients with amyotrophic lateral sclerosis carrying a C9ORF72 repeat expansion: a population-based cohort study. Lancet Neurol 2012;11:232–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiò A, Borghero G, Restagno G, et al. Clinical characteristics of patients with familial amyotrophic lateral sclerosis carrying the pathogenic GGGGCC hexanucleotide repeat expansion of C9ORF72. Brain 2012;135:784–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Majounie E, Renton AE, Mok K, et al. Frequency of the C9ORF72 hexanucleotide repeat expansion in patients with amyotrophic lateral sclerosis and frontotemporal dementia: a cross-sectional study. Lancet Neurol 2012;11:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Byrne S, Bede P, Elamin M, Kenna K, Lynch C, McLaughlin R, Hardiman O. Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2011;12:157–159. [DOI] [PubMed] [Google Scholar]
- 24.Grace J, Malloy P. Frontal Systems Behavior Scale (FrSBe): Professional Manual. Lutz, FL: Psychological Assessment Resources; 2001. [Google Scholar]
- 25.Strong MJ, Grace GM, Freedman M, et al. Consensus criteria for the diagnosis of frontotemporal cognitive and behavioral syndromes in amyotrophic lateral sclerosis. Amyotroph Lateral Scler 2009;10:131–146. [DOI] [PubMed] [Google Scholar]
- 26.Chiò A, Restagno G, Brunetti M, et al. Two Italian kindreds with familial amyotrophic lateral sclerosis due to FUS mutation. Neurobiol Aging 2009;30:1272–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Laaksovirta H, Peuralinna T, Schymick JC, et al. Chromosome 9p21 in amyotrophic lateral sclerosis in Finland: a genome-wide association study. Lancet Neurol 2010;9:978–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Andersen PM, Forsgren L, Binzer M, et al. Autosomal recessive adult-onset ALS associated with homozygosity for Asp90Ala CuZn-superoxide dismutase mutation: a clinical and genealogical study of 36 patients. Brain 1996;119:1153–1172. [DOI] [PubMed] [Google Scholar]
- 29.Chiò A, Borghero G, Pugliatti M, et al. Large proportion of amyotrophic lateral sclerosis cases in Sardinia due to a single founder mutation of the TARDBP gene. Arch Neurol 2011;68:594–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boeve BF, Boylan KB, Graff-Radford RM, et al. Characterization of frontotemporal dementia and/or amyotrophic lateral sclerosis associated with the GGGGCC repeat expansion in C9ORF72. Brain 2012;135:765–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cooper-Knock J, Shaw P, Hewitt C, et al. Clinico-pathological features in amyotrophic lateral sclerosis with expansions in C9ORF72. Brain 2012;135:751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sabatelli M, Conforti FL, Zollino M, et al. C9ORF72 hexanucleotide repeat expansion in the Italian sporadic ALS population. Neurobiol Aging 2012;33:1848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wicks P, Abrahams S, Papps B, et al. SOD1 and cognitive dysfunction in familial amyotrophic lateral sclerosis. J Neurol 2009;256:234–341. [DOI] [PubMed] [Google Scholar]
- 34.Felbecker A, Camu W, Valdmanis PN, et al. Four familial ALS pedigrees discordant for two SOD1 mutations: are all SOD1 mutations pathogenic? J Neurol Neurosurg Psychiatry 2010;81:572–577. [DOI] [PubMed] [Google Scholar]
- 35.Andersen PM, Al-Chalabi A. Clinical genetics of amyotrophic lateral sclerosis: what do we really know? Nat Rev Neurol 2011;7:603–615. [DOI] [PubMed] [Google Scholar]
- 36.Van Es MA, Scheelhas HJ, van Vugh PWJ, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol 2011;70:964–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Del Bo R, Scarlato M, Ghezzi S, et al. Absence of angiogenin genes modifications in Italian ALS patients. Neurobiol Aging 2008;29:314–316. [DOI] [PubMed] [Google Scholar]
- 38.Corrado L, Battistini S, Penco S, et al. Variations in the coding and regulatory sequences of angiogenin (ANG) gene are not associated to ALS (amyotrophic lateral sclerosis) in the Italian population. J Neurol Sci 2007;258:123–127. [DOI] [PubMed] [Google Scholar]
- 39.Gellera C, Colombrita C, Ticozzi N, et al. Identification of new ANG gene mutations in a large cohort of Italian patients with amyotrophic lateral sclerosis. Neurogenetics 2008;9:33–40. [DOI] [PubMed] [Google Scholar]
- 40.Conforti FL, Sprovieri T, Mazzei R, et al. A novel angiogenin gene mutation in a sporadic patient with amyotrophic lateral sclerosis from southern Italy. Neuromuscul Dis 2008;18:68–70. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.