

Abstract
Astrocytes participate in brain functions through Ca2+ signals, including Ca2+ waves and Ca2+ oscillations. Currently the mechanisms of Ca2+ signals in astrocytes are not fully clear. Here, we present a computational model to specify the relative contributions of different Ca2+ flows between the extracellular space, the cytoplasm and the endoplasmic reticulum of astrocytes to the generation of spontaneous Ca2+ oscillations (CASs) and cortical spreading depression (CSD)-triggered Ca2+ waves (CSDCWs) in a one-dimensional astrocyte network. This model shows that CASs depend primarily on Ca2+ released from internal stores of astrocytes, and CSDCWs depend mainly on voltage-gated Ca2+ influx. It predicts that voltage-gated Ca2+ influx is able to generate Ca2+ waves during the process of CSD even after depleting internal Ca2+ stores. Furthermore, the model investigates the interactions between CASs and CSDCWs and shows that the pass of CSDCWs suppresses CASs, whereas CASs do not prevent the generation of CSDCWs. This work quantitatively analyzes the generation of astrocytic Ca2+ signals and indicates different mechanisms underlying CSDCWs and non-CSDCWs. Research on the different types of Ca2+ signals might help to understand the ways by which astrocytes participate in information processing in brain functions.
Introduction
For the past few decades, the role of astrocytes has been thought to be restricted to passive, histological support elements in the central nervous system [1]. However, new functions of astrocytes have recently been identified [2], [3]. Astrocytes can release chemical transmitters that regulate synaptic transmission, activate neurons, and influence cerebral microcirculation [4], [5], and their dysfunction is implicated with neurological conditions such as epilepsy and Alzheimer’s disease [6], [7]. Ca2+-mediated signals are the predominant model of communication between astrocytes [8]. Two main types of Ca2+ responses are observed in astrocytes, including Ca2+ oscillations and Ca2+ waves [9]. Ca2+ oscillations are characterized as transient Ca2+ increases that are restricted to the single cells [10], whereas Ca2+ waves are characterized as Ca2+ elevations propagating within and between neighboring astrocytes [11]. Ca2+ waves in the astrocyte networks are considered to represent an effective form of intercellular signaling in the central nervous system [12].
Many experiments have suggested that Ca2+ oscillations in astrocytes are based on inositol 1,4,5-trisphosphate (IP3) receptor/Ca2+ channels (IP3R) [13], [14]. The opening of these channels can release Ca2+ from internal stores of the endoplasmic reticulum (ER), in a process known as calcium-induced calcium release (CICR). In addition to the Ca2+ released from internal stores, Ca2+ influx from the extracellular fluid is also reported to be needed to generate Ca2+ oscillations [15] and voltage-gated calcium channels (VGCCs) have been found to contribute to this Ca2+ influx [13], [16]. However, other works show that extracellular Ca2+ is not required for the occurrence of Ca2+ oscillations [17]. Ca2+ released from the ER is usually considered to be the key factor in the generation of Ca2+ waves which are induced by ATP or IP3 [18], [19], but this is not necessary when Ca2+ waves are triggered by cortical spreading depression (CSD) [20], [21]. CSD refers to a pathophysiological phenomenon and manifests as a self-propagation wave of electrical silence, resulting in the depolarization of neurons and astrocytes and a redistribution of ions [22], [23], which is thought to underlie the migraine aura and develop after cerebral ischemia and trauma [24], [25]. The contradictive results about astrocytic Ca2+ signals suggest that the underlying mechanisms in the generation of Ca2+ oscillations and Ca2+ waves are still unclear.
Interestingly, spontaneous Ca2+ oscillations (CASs) and CSD-triggered Ca2+ waves (CSDCWs) have been reported in the same experiments [21]. Different models have been used to investigate the mechanisms of astrocytic Ca2+ signals. However, these models either focused just on CASs [26], [27], or on Ca2+ waves induced by ATP or IP3 but not by CSD (non-CSDCWs) [28]–[30]. Bennett et al. simulated the CSDCWs, but Ca2+ flows within the astrocytes, for example, the Ca2+ released from CICR and the Ca2+ uptaken into the ER, were neglected in their model [31]. In addition, the role of Ca2+ from the ER for the generation of different types of Ca2+ waves (CSDCWs and non-CSDCWs) could not be fully explained by these models.
In the present study, we investigated CASs and CSDCWs in a one-dimensional astrocyte network by an expanded version of our previous model, which simulated the VGCCs-mediated CASs [32], to account for the contributions of different Ca2+ flows between the extracellular space, the cytoplasm and the ER of astrocytes to the generation of these Ca2+ signals. We first explored the mechanisms for the generation of CASs and CSDCWs, and then investigated the interactions between CASs and CSDCWs, and finally addressed the transition from CASs to CSDCWs. Our results quantitatively analyze the generation of astrocytic Ca2+ signals and indicate different mechanisms underlying CSDCWs and non-CSDCWs.
Methods
The model consisted of a single lane of astrocytes which were assumed as spherical somas with a radius of 5 µm. In a single-cell context, three compartments were considered, including the extracellular space (ECS), the intracellular space (ICS), and the ER internal space, as seen in Fig. 1A. As to the astrocyte networks, astrocytes were coupled to the adjacent ones by the transfer of IP3 from cytosol to cytosol through gap junctions (Fig. 1B).
Figure 1. A schematic diagram of the model.

(A) As to the single astrocyte, Ca2+ influx through voltage-gated calcium channels (VGCCs) triggers the fluctuation of Ca2+ in the intracellular space (ICS), enhancing the production of inositol 1,4,5-triphosphate (IP3), which is catalyzed by phospholipase C (PLC). Ca2+ and IP3 bind to IP3 receptors (IP3R), activating the process of calcium-induced calcium release (CICR). The endoplasmic reticulum (ER) is filled with Ca2+ by the sarcoplasmic/endoplasmic reticulum calcium ATPase (SERCA). A Ca2+ pump discharges Ca2+ from the ICS into the extracellular space (ECS). K+ in the ECS is partly uptaken into the ICS during cortical spreading depression (CSD). JVGCC, JCICR and JSERCA represent the Ca2+ flow through VGCCs, CICR and SERCA, respectively. Jpump represents the Ca2+ flow through the Ca2+ pump. Jleak represents the leak Ca2+ flow. Jupt represents the K+ flow untaken into ICS, and Jdis represents the K+ flow discharged into ECS. (B) Single astrocytes are coupled to the adjacent ones by the transfer of IP3 from cytosol to cytosol through gap junctions to form a one-dimensional astrocyte network.
Ca2+ Flows through the Astrocytic Membrane
VGCCs include high-voltage-activated channels and low-voltage-activated channels. Low-voltage-activated channels have been demonstrated to have little effect on CASs and CSDCWs [16], [31]–[33]. In the present model, only high-voltage-activated channels (as a group) were considered. Similar operations were also applied by other groups [31], [33] in the study of CSDCWs. The Hodgkin-Huxley equation was used to model VGCCs:
| (1) |
where, I VGCC is the Ca2+ current that flows into astrocytes via VGCCs, and g VGCC is the membrane conductance. As shown in [31], m ∞ and h ∞ are gated parameters that regulate the activation and inactivation of the VGCCs, respectively.
| (2a) |
where
| (2b) |
| (3a) |
where
| (3b) |
Vm is the astrocytic membrane potential, and its calculation is defined in the following text. ECa is the Nernst potential of Ca2+.
| (4) |
where, R is the ideal gas constant, T is the absolute temperature, zCa is the valence of Ca2+, and F is the Faraday constant. Ca o and Ca i represent the Ca2+ concentration in the ECS and in the ICS, respectively. According to [32], the Ca2+ current of VGCCs described in Eq. (1) was converted into flux to calculate its contribution to the increase of Ca2+ in astrocytes:
| (5) |
where, V astro is the volume of an astrocyte.
Ca2+ in astrocytes is partly discharged into the ECS by the Ca2+ pump, and the calculation is adapted from [31]:
| (6) |
where g pump is the membrane conductance of the pump and additionally, the current in Eq. (6) was also converted into flux:
| (7) |
The leak flux into the ECS was calculated following [26], [32]:
| (8) |
where, L ext is the rate of Ca2+ efflux from astrocytes.
Ca2+ Flows through the Membrane of ER
As shown in Fig. 1A and in our previous model [32], Ca2+ flows through the membrane of ER included Ca2+ released from CICR, Ca2+ uptaken into the ER via the sarcoplasmic/endoplasmic reticulum Ca2+ ATPase (SERCA) and leak flux through the membrane of ER, which are described as J CICR, J SERCA and J leakics in Eqs. (S1)-(S3) of Materials S1, respectively. Combining Eqs. (5), (7), (8) and Eqs. (S1–S3), the dynamics of Ca2+ in the astrocytic cytosol were expressed as
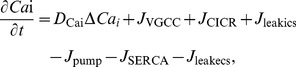 |
(9) |
where, D Cai is the diffusion coefficient for Ca2+ in the cytosol and Δ is the Laplace operator. The first term on the right of Eq. (9) represents the diffusion of Ca2+ in the ICS. Accordingly, Ca2+ in the ER (Ca ER) was determined by
| (10) |
The dynamics of Ca o were neglected owing to its slight changes during Ca2+ oscillations in previous models. However, Ca o changes dramatically during CSD [23], [34]. Combining Eqs. (5), (7), (8) and the influence from CSD, Ca2+ in the ECS was modeled by
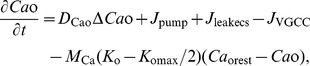 |
(11) |
where, D Cao is the diffusion coefficient for Ca2+ in the ECS. K o represents the K+ concentration in the ECS. M ca is a constant, K omax is the maximal K o during CSD, and Ca orest is Ca o at resting state. The first term on the right of Eq. (11) represents the diffusion of Ca2+ in the ECS. The last term on the right of Eq. (11) describes the decrease of Ca o during CSD, and its precise mechanism needs to be further explored.
IP3 in the ICS
IP3 is a known intracellular messenger, which can bind to the IP3R to cause Ca2+ to flow out of the ER. In a single-cell context, IP3 in the ICS (IP 3) was catalyzed by phospholipase C (PLC) as defined in Eq. (S4) of Materials S1. Experimental evidences show that Ca2+ waves in astrocytes are mediated following the transfer of IP3 [35], [36]. In the present astrocyte networks, the astrocyte was coupled to its nearest neighbors by the transfer of IP3 through gap junctions. Following [37], the change of IP 3 in astrocyte i due to the gap junction with astrocyte j is
| (12) |
where i, j are indices of adjacent astrocytes, and γ is the coupling strength. Combining the diffusion of IP3 inside the cells [38] and Eq. (S4), IP3 in astrocyte i was calculated as
| (13) |
where, DIP3 is the diffusion coefficient for IP3 in the ICS. IPpro represents the production of IP3 and IP deg represents its degradation, as seen in Eq. (S4) of Materials S1.
Cortical Spreading Depression
CSD causes neurons and astrocytes to depolarize and greatly changes the ion concentration [23], [34]. We incorporated CSD in this model to initiate astrocytic Ca2+ waves. Because high extracellular K+ is required for the propagation of CSD [39], K o was described as the classical diffusion-reaction equation:
| (14a) |
where, D Ko is the diffusion coefficient for K+ in the ECS, and the first term on the right of Eq. (14a) represents the diffusion of K+ in the ECS. f(K o) describes the reaction process of K o, which is adapted from [40]:
| (14b) |
where, M KK is a rate constant, K orest is K o at the resting level, and K θ is the threshold for the triggering of CSD. The first term on the right of Eq. (14b) meets the following requirements: maintaining homeostasis at the resting level, triggering explosive subsequent growth in K o when K o is higher than the threshold K θ, and preventing K o rising when K o is beyond the ceiling Komax [40]. The second term represents the recovery of K o. R k, which restores K o to the normal level, was modeled by:
| (15) |
where, M KR and M R are constants.
Astrocytes are reported to be fast buffers and play an important role in the clearance of excess K o [41], [42]. During the process of CSD, K o would partly be untaken by astrocytes. The K+ concentration in the astrocytes (K i) was calculated as.
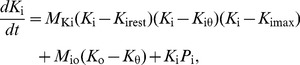 |
(16) |
where, M Ki and M io are rate constants. K irest is K i at the resting level, K iθ is the threshold for the fast elevation of K i, and K imax is the ceiling K i during CSD. The first term on the right of Eq. (16) follows the formalism of K o to maintain cytosol K+ homeostasis, the senond term is used to detect the change in K o and is assumed to be effective only when K o is beyond the threshold for the triggering of CSD, and the third term represents the discharge of K i.
| (17) |
where, A Ki and A rr are constants. The first term on the right of Eq. (17) represents the discharge of K i, and the second is a decay term.
The astrocytic membrane potential is a complicated parameter to calculate in computational models. To avoid these complex calculations, a simplified method was adopted. It is well known that the K+ Nernst potential is close to V m; therefore, in this study, the K+ Nernst potential was used to approximate V m by adding a modulation factor, which was chosen based on previously experiments [43].
| (18) |
where, z K is the valence of K+ and ε is the modulation factor.
Implementation
The chain model consisted of 3N astrocytes, where N ranged from 1 to 100. All of the computations and visualizations of this model were implemented in the Matlab environment (Matlab 7.0, MathWorks Inc., USA). The Crank-Nicholson algorithm was used to solve the differential equations [44], with a zero-flux boundary condition and a time step of 15 ms. The parameter values used in the model are shown in Table S1, and initial values of the variables are list in Table S2.
Results
The Contributions of Different Ca2+ Flows to CASs
In this model, CASs occurred in single astrocytes without any stimulus. Fig. 2A shows that in the ICS, cytosol Ca2+ oscillated with the amplitude of 0.63×10−3 mM and the frequency of 0.0044 Hz, which were consistent with the experimental results [17], [45]. The duration of cytosol Ca2+ oscillations, measured at the half-amplitude level, was 21 s. In the ER (Fig. 2B), the oscillations of Ca2+ showed a larger amplitude and a longer duration than those in the ICS. As been reported in the experiments [46], [47], the peak time point for oscillations came earlier in the ER than in the ICS (Fig. 2A, B). The duration of IP3 fluctuation in the ICS was longer than that of Ca2+ oscillations in the ICS but shorter than that of Ca2+ oscillations in the ER (Fig. 2D). In the ECS, a slight decline in Ca2+ was noticed before each Ca2+ oscillation in the ICS (Fig. 2C). The change of Ca2+ concentration in the ECS indicated that CASs in the ICS are related to the Ca2+ in the ECS [15], [16].
Figure 2. Characteristics of spontaneous Ca2+ oscillations (CASs) in the computerized astrocytic model.

(A) Ca2+ oscillations in the ICS occur without any stimulus. (B) Ca2+ oscillations in the ER occur earlier and last longer than those in the ICS. (C) The concentration of Ca2+ in the ECS decreases slightly before Ca2+ oscillations in the ICS and then increases after the oscillations. (D) IP3 oscillates in the ICS.
CASs in the ICS depended on Ca2+ influx from the ECS and Ca2+ released from the ER [13], [15], [16], but the precise contributions of these Ca2+ flows are not clear. Here, we investigated the influence of different Ca2+ flows on the generation of CASs. In this model, CICR that released Ca2+ to the ICS and SERCA that extracted Ca2+ to the ER were considered to be the main processes that modulated the exchange of Ca2+ between the ICS and the ER. VGCCs were thought to regulate extracellular Ca2+ that flowed into the ICS. To understand the contributions of these different processes, we inhibited CICR, SERCA, and VGCCs separately to compare with the control condition, which had no inhibition of CICR, SERCA, and VGCCs.
The CICR inhibition was achieved by reducing the release of Ca2+ from the ER (reducing M CICR in Eq. (S1) of Materials S1). Fig. 3A shows that the frequency of CASs progressively decreased with the inhibition of CICR, consistent with the reported experimental results that CICR plays an important role in the generation of Ca2+ oscillations, and oscillations cannot occur without Ca2+ released from the ER [13], [48], [49]. In addition, CICR inhibition decreased the amplitude of CASs, whereas the duration was increased, as seen in Fig. 3B, C and in Fig. S1. The SERCA inhibition was achieved by reducing the uptake of Ca2+ into the ER (reducing M SERCA in Eq. (S2) of Materials S1). SERCA inhibition decreased the ability to form large-amplitude oscillations (Fig. 3B) [13], [17], [48], as well as the duration (Fig. 3C). Previous results indicate a decline in the frequency of CASs with SERCA inhibition [16], [45], whereas our simulation results suggest that the frequency of CASs first increases (Fig. 3A) and then Ca2+ dynamics evolve into small oscillations before finally disappearing, as seen in Fig. S2. The membrane conductance, g VGCC, was reduced to simulate the VGCCs inhibition. After the reduction, the frequency of CASs decreased (Fig. 3A), confirming that VGCCs can mediate CASs [16], [32]. The amplitude and duration of CASs also decreased as a result of VGCCs inhibition (Fig. 3B, C). In the condition that CASs were not fully blocked, CICR or SERCA inhibition had a significant effect on the frequency, amplitude and duration of CASs, whereas VGCCs inhibition had little effect on the amplitude and duration but a great effect on the frequency, as seen in Fig. 3 and in Fig. S3. These results indicate that the elevation of Ca2+ in the CASs mainly came from the Ca2+ released from internal stores.
Figure 3. The influence of different Ca2+ flows on CASs.

By inhibiting CICR (dashed triangle), the frequency (A) and amplitude (B) of CASs decrease, but the duration (C) increases. CASs do not occur when CICR is inhibited more than 95%. By inhibiting SERCA (dashed circle), the frequency (A) of CASs increases, but both the amplitude (B) and the duration (C) decrease. CASs do not occur when SERCA is inhibited more than 70%. Inhibiting VGCCs (solid star) has little effect on the amplitude (B) and duration (C) but great on frequency (A) before CASs disappear. CASs do not occur when VGCCs are inhibited more than 45%.
The Contributions of Different Ca2+ Flows to CSDCWs
According to Kager et al. and Chapuisat et al. [50], [51], local extracellular K+ concentration (around the central astrocytes in the astrocyte networks in this model) was elevated to 12 mM to evoke a CSD. CSD would cause the local extracellular K+ to increase further and spread to the surrounding [50], [51]. Usually, the propagation of CSD is indicated as the spread of significantly increased K o [40]. As shown in Fig. S4A, K o was significantly increased and spread in this model after locally increasing extracellular K+ concentration, suggesting the emergence of CSD, and then K i was also increased to partly clear the excess K+ in the ECS (Fig. S4B). Owing to the depolarization of astrocytes caused by CSD (Fig. S4C), Ca2+ influx via VGCCs increased. A Ca2+ wave was induced by CSD and propagated in the astrocyte networks at a speed of 58 µm/s, with the amplitude of 6.4×10−3 mM and the duration of 56.7 s, as seen in Fig. 4A. The amplitude and duration of CSDCWs were significantly larger than those of CASs in the ICS.
Figure 4. The influence of different Ca2+ flows on CSD-triggered Ca2+ waves (CSDCWs).

A typical CSDCW is characterized as the significant elevation of Ca2+ in the ICS at successive astrocytes in the network (A), associated with the increase of Ca2+ in the ER (B), the increase of IP3 in the ICS (D) and the decrease of Ca2+ in the ECS (C). CICR inhibition shortens the duration of increased Ca2+ in the ICS (E), slows the recovery of Ca2+ in the ER (F), decreases Ca2+ in the ECS more than in the control condition (G) and increases the duration of increased IP3 in the ICS (H). SERCA inhibition increases the amplitude of Ca2+ in the ICS (I) and decreases that in the ER (J). Ca2+ in the ECS is increased compared to the control (K). Because high Ca2+ in the ICS would inhibit the process of CICR, the increase of IP3 in the ICS is shortened (L). After VGCCs inhibition, Ca2+ is largely weakened in the ICS (M), in the ER (N) and in the ECS (O). The changes of IP3 are also shortened because of the low Ca2+ in the ICS (P).
It was readily apparent that Ca2+ levels in the ER and in the ECS changed as the CSDCWs spread (Fig. 4B, C). Here, we also took into account the processes of CICR, SERCA and VGCCs to study the contributions of different Ca2+ flows to the generation of CSDCWs.
Compared to the control condition, the CICR inhibition shortened the duration of the increased Ca i (Fig. 4E). Due to that Ca2+ could not be rapidly released from the ER through the process of CICR, the rate of recovery of Ca ER was reduced (Fig. 4F). To maintain the amplitude of Ca2+ in the ICS, more Ca2+ in the ECS was needed to flow into the ICS to make up for the reduction of Ca2+ released from the ER, causing Ca o to decrease further (Fig. 4G) and the duration of IP3 elevation to be increased (Fig. 4H). After SERCA inhibition, Ca2+ in the ICS could not be extracted into the ER promptly by SERCA, and this led to a decrease in Ca ER (Fig. 4J) and an accumulation of Ca i (Fig. 4I). Owing to this accumulation, Ca2+ influx was reduced, and Ca o was increased compared to the control (Fig. 4K). Because high Ca2+ in the ICS inhibits the process of CICR [29], the duration of IP3 elevation in the ICS was shortened (Fig. 4L). After VGCCs inhibition, the changes of Ca2+ were strongly weakened in the ICS, in the ER and in the ECS (Fig. 4M, N and O). Compared to the control (Fig. 4D), the duration of IP3 elevation was also shortened owing to low Ca2+ in the ICS (Fig. 4P). These results suggest that CSDCWs are primarily triggered by Ca2+ influx via VGCCs, and the Ca2+ efflux from the ER contributes to the generation of Ca2+ waves to a lesser degree.
Interactions between CASs and CSDCWs
Experiments showed that the appearance of CSDCWs depressed CASs, and then CASs reappeared after the pass of CSDCWs in single astrocytes [21]. To investigate the interactions between CASs and CSDCWs in this study, we focused on Ca2+ signals in single astrocytes, and the results were similar to other astrocytes which underwent CASs and CSDCWs in the astrocyte networks. Comparing Fig. 5A with Fig. 5B, it shows that the CAS (marked with an asterisk in Fig. 5A, B) was absent after the appearance of CSDCW, and reappeared a few minutes later after the peak of the CSDCW. Moreover, CSDCWs could be induced immediately after the occurrence of the CAS (marked with an arrow in Fig. 5B), which indicated that CASs did not prevent the generation of CSDCWs. To further understand the effect of CSDCWs on CASs, the appearance time of CSDCWs was manipulated, by changing the time points of locally elevating extracellular K+ concentration (marked with red bars in Fig. 5B), to investigate the changes in peak-to-peak interval between the CSDCW and the following CAS (see t2 in Fig. 5B). Fig. 5B shows that by regulating the appearance time of CSDCWs, the peak-to-peak interval between the CSDCW and the following CAS remained almost constant. This suggests that CSDCWs had a similar effect on the latency to the onset of the following CAS, and the latency was not affected by the appearance time of CSDCWs relative to the previous CAS (see t1 in Fig. 5B). By depleting the Ca2+ store, CASs were completely abolished, but CSDCWs still propagated with shorter duration (Fig. 5C), suggesting that there were different mechanisms underlying the generation of CASs and CSDCWs. Furthermore, CSDCWs could spread without Ca2+ flow from ER, which also indicates different mechanisms underlying CSDCWs and non-CSDCWs [21].
Figure 5. Interactions between CASs and CSDCWs.

(A) A series of CASs occur without any stimulus. (B) The appearance of CSDCWs depresses CASs, and CASs reappear a few minutes after the pass of CSDCWs (the affected CASs are marked with asterisks in A and B). CSDCWs can appear immediately after CASs (marked with an arrow in B). By regulating the appearance time of CSDCWs through changing the time points of locally elevating K o (marked with red bars in B), the peak-to-peak interval between the CSDCW and the following CAS is similar, and it is not related to the peak-to-peak interval between the CSDCW and the previous CAS. t1, the peak-to-peak interval between the CSDCW and the previous CAS. t2, the peak-to-peak interval between the CSDCW and the following CAS. (C) Depletion of Ca2+ stores in the ER abolishes CASs, but CSDCWs still spread. The bar illustrates the time of locally elevating K o.
Transition from CASs to CSDCWs
Local K o (around the central astrocytes in the astrocyte networks in this model) was increased gradually to explore the transition from CASs to CSDCWs in single astrocytes. It shows that increasing K o would induce the elevation of Ca i and the generation of CASs in the astrocytes near to the stimulation site (Fig. 6A), and higher K o caused an earlier onset time for oscillations compared to the control (the onset of CAS under the control condition is marked with an blue arrow). By increasing K o in the present of CASs facilitated the further elevation of Ca i and the elevated Ca i deferred the occurrence of successive oscillations (Fig. 6B and its illustration). When K o was increased to 12 mM, a CSDCW was evoked and propagated to neighboring astrocytes.
Figure 6. Transition from CASs to CSDCWs.

(A) By locally increasing K o to 5 or 9 mM, Ca2+ in ICS is increased, which facilitates the occurrence of CASs. When K o is increased to 12 mM, a CSDCW is induced. (B) Increasing K o in the present of CASs elevates Ca2+ in ICS and then the elevated Ca2+ will postpone the occurrence of the following CASs. Following oscillations are shown in the illustration. The bar illustrates the time of locally elevating K o, the concentrations of which are shown in the legend. Arrows indicate the appearance time of CASs under the control condition.
Discussion
In this model, we incorporated CASs and CSDCWs in a one-dimensional astrocyte network, and investigated the contributions of different Ca2+ flows, including Ca2+ flows between the extracellular space, the cytoplasm and the ER of astrocytes, to the generation of these Ca2+ signals. The results show that CASs depended primarily on Ca2+ released from internal stores in astrocytes, whereas CSDCWs depended mainly on voltage-gated Ca2+ influx. The appearance of CSDCWs would suppress CASs, whereas CASs did not prevent the generation of CSDCWs. Furthermore, our results suggest that after Ca2+ stores have been depleted, CSDCWs could still propagate due to voltage-gated Ca2+ influx, different from the non-CSDCWs.
The predominant model of communication between astrocytes is Ca2+-mediated signals, which are determined by an intricate interplay between Ca2+ influx, buffering and extrusion pathways [8]. Experimental results show that CASs require extracellular Ca2+ and operating VGCCs [16], whereas others suggest that single CASs are observed many minutes later after the elimination of extracellular Ca2+ [17]. Our simulations support the former and furthermore, we show that in the present of CASs, both CICR inhibition and SERCA inhibition had a significant effect on the amplitude and duration of CASs, whereas VGCCs inhibition had little effect on the amplitude and duration but a great effect on the frequency. It indicated that the elevation of Ca2+ in the CASs mainly came from the Ca2+ released from the ER, and that Ca2+ influx from the ECS might play a role in triggering the process of CICR to generate CASs and replenishing the Ca2+ load during CASs [27].
In situ and in vivo experiments show that CASs occur 0.15 to 1 time per minute (0.0025 to 0.017 Hz) [17], [52]–[54]. The frequency of CASs in our model is 0.0044 Hz, which is within the reported frequency range and suggests that the modeled CASs here recapitulate the physiological astrocytic Ca2+ responses. Local Ca2+ transients in the astrocytic processes have mean frequency of 0.028 Hz, which is significantly higher than the frequency of Ca2+ transients in the astrocyte cell body [55]. Although Ca2+ transients in the processes of astrocytes are not considered in the present model, the generation of Ca2+ transients in the processes of astrocytes is reported to depend mainly on the Ca2+ released from internal stores [55], which is similar as the CASs in the astrocyte cell body in our model.
Ca2+ waves in astrocytes are usually thought to be induced by ATP or IP3, and they depend on Ca2+ released from internal stores [18], [19]. This kind of Ca2+ waves was represented as non-CSDCWs in this study. There are at least three differences between the CSDCWs and non-CSDCWs. First, after depletion of internal stores, non-CSDCWs cannot be generated [18], [19], while CSDCWs could still propagate [21]. Second, theoretical and experimental results suggest that CSDCWs have a larger amplitude than that of non-CSDCWs [21], [28], [31]. Third, CSDCWs spread faster than non-CSDCWs [11], [21]. All of these suggest that there may be different mechanisms underlying CSDCWs and non-CSDCWs. In this model, the amplitude and speed of CSDCWs were similar to reported results [31], significantly larger than those of non-CSDCWs [11], [28]. Moreover, by depleting the internal store in this study, CSDCWs could still propagate. This is because that even without Ca2+ efflux from the internal stores, CSD can cause astrocytes to depolarize, which opens the VGCCs, and Ca2+ influx via VGCCs is able to generate Ca2+ waves. It suggests that the generation of CSDCWs depends primarily on Ca2+ influx via VGCCs. VGCCs have been reported to be not physiologically relevant for intracellular Ca2+ signals [56], while CSDCWs pertain not to this case, because CSD would cause astrocytes to depolarize and Ca2+ influx coupled to depolarization was recorded during CSDCWs [21]. It is reported that after CSD has stopped, which means that astrocytes do not depolarize and no membrane current are detectable, Ca2+ waves still spread but with a significantly reduced amplitude and speed [21], suggesting that non-CSDCWs might depend mainly on Ca2+ flows uncoupled to depolarization. In this study, we only simulated CSDCWs, focusing on the voltage-gated Ca2+ influx pathway, and this might explain why the amplitude of CSDCWs was not strongly attenuated after depletion of internal stores compared to the experiment data [21], though the duration was reduced.
We investigated the interactions between CASs and CSDCWs, and showed that CSDCWs depressed CASs and CASs reappeared after the pass of CSDCWs, which was consistent with previous experiments [21]. Furthermore, CSDCWs postponed the onset of the following CASs with a similar time lag, and this might be related to the process of CSD. As an “all or none” process [57], CSD would elevate Ca2+ in the cytosol to a similar level. As a result, it took astrocytes equivalent time to recover from the effect of CSD, which might determine the latency to the onset of following CASs. Locally increasing K o would cause astrocytes to depolarize and increase Ca2+ influx via VGCCs, which facilitated the occurrence of CASs and the elevation of Ca i. Meanwhile, the onset of subsequent Ca2+ oscillation was postponed as the elevation of Ca i. This is because that in addition to the increased amplitude of Ca i caused by increasing K o, the duration of Ca i was also increased. As a result, astrocytes needed more time to recover from the elevation of Ca i, and the subsequent oscillations came later.
Ca2+ signals in astrocytes are quite variable in the spatiotemporal organization. CASs in cortical layer 2/3 show infrequent synchronous pattern, whereas in layer 1 CASs are frequent asynchronous [58]. CSDCWs in gray matter of the neocortex are reported to propagate with a higher speed than non-CSDCWs in white matter [21], [59]. The complexity may be related to the mechanisms that control Ca2+ entry from the extracellular space as well as Ca2+ release from internal stores. VGCCs, which are expected to play significant functional roles in Ca2+ influx in astrocytes [60], are key transducers of membrane potential changes into intracellular Ca2+ transients. According to our study, voltage-gated Ca2+ influx had a great contribution to CSDCWs, and might play a role in triggering CASs. However, the distribution of VGCCs expression in the brain tissue is not uniform. Experiments show apparent lack of VGCCs in rat hippocampus and visual cortex [56], while different types of VGCCs are present in mouse hippocampus [61]. Astrocytes differ in membrane currents and are heterogeneous with respect to VGCCs expression [62], but several types of astrocytes can coexist within the same brain region [63], which might lead to the heterogeneity of Ca2+ signals among astrocytes.
Experiments show that CASs are based on the process of CICR [13], [14], but Ca2+ entry via the external medium has also been found to contribute to CASs [13], [16]. In our model, we quantitatively analyzed the contributions of different Ca2+ flows to the generation of astrocytic Ca2+ signals, and showed that voltage-gated Ca2+ influx played a role in regulating the frequency of CASs and might be important for initiating CASs [64]. Depleting internal Ca2+ stores is reported to block non-CSDCWs [18], [19], but not CSDCWs [20], [21]. Consistent with the experimental results, our model showed that after Ca2+ stores have been depleted CSDCWs could still propagate. Furthermore, we showed that the propagation of CSDCWs after depleting internal Ca2+ stores was due to voltage-gated Ca2+ influx, indicating different mechanisms underlying CSDCWs and non-CSDCWs. Future research should focus on the different mechanisms underlying astrocytic Ca2+ signals.
A recent study has simulated the transition from single CASs to non-CSDCWs, and the results suggested that long-distance non-CSDCWs are favored when the internal Ca2+ dynamics implements the frequency modulation-encoding oscillations [28]. We explored the transition from CASs to CSDCWs and showed that CSDCWs would occur when there was enough Ca2+ influx caused by the depolarizing stimulus with high extracellular K+. Frequency modulation-encoding oscillations are mediated by the process of CICR [65]. Because CSDCWs depended mainly on voltage-gated Ca2+ influx rather than the process of CICR, frequency modulation-encoding oscillations would not determine the transition from CASs to CSDCWs. It will be interesting to study the interactions between non-CSDCWs and CSDCWs, because that non-CSDCWs compromise a large number of synchronized astrocytes, which might affect the incidence of CSDCWs. However, we failed to elicit non-CSDCWs, which might be due to that in our model the internal Ca2+ dynamics does not implement the frequency modulation-encoding oscillations and that ATP diffusion in the ECS is not considered. Ca2+ in non-CSDCWs originates mainly from internal stores of astrocytes [18], [19], while Ca2+ in CSDCWs, according to our study, originates mainly from Ca2+ influx, and Ca2+ in internal stores also contributes to CSDCWs. Base on these findings, non-CSDCWs seem not to suppress the incidence of CSDCWs but to change their properties. This might be supported by the experiments that after depletion of the internal Ca2+ stores, CSDCWs could still be recorded but the amplitude of Ca2+ signal was reduced [21]. In contrast, CSDCWs might affect the occurrence of non-CSDCWs by taking up the process of CICR. Future experiments are needed to elucidate the interactions between non-CSDCWs and CSDCWs.
Due to changes in the membrane potential are difficult to model because of the many and complex processes involved, we and others [66] use a simplified model of using the K+ Nernst potential to approximate the membrane potential, but add a modulation factor. The value of the modulation factor was chosen based on the considerations: after adding the modulation factor, astrocyte membrane potential is within the range where CASs would occur [32]; according to experimental observations [43], [67], the frequency of CASs is significantly decreased when the temperature in this model increases from 20 to 37°C (Fig. S5). Ca2+ recording from astrocytes in vivo shows that astrocytes were either quiescent or responded with a few Ca2+ transients [12], [68], while Ca2+ transients occurred more frequently in slices prepared at 28°C [17]. When the slices were prepared at 37°C, no statistical difference was found in the percentage of active astrocytes and the frequency of Ca2+ events between the in vivo and in situ results [52]. Moreover, the frequency of CASs was showed to be temperature-dependent: from 20 to 37°C, CASs occurred frequently at low temperature and became less frequent at higher temperature [43], [67]. The mechanism underlying this would be the decreased activity of IP3R channels or the increased activity of SERCA at higher temperature [69], [70], which reduces the Ca2+ released from internal stores and the frequency of CASs. Another limitation is that we did not consider the volume of the extracellular space. Although the volume of astrocytes is not altered during CSD [71], the volume of the extracellular space is decreased. To model these Ca2+ signals more accurately, the changes of volume of the extracellular space need be taken into account.
This model could be improved at least in two aspects. First, Ca2+ waves in astrocytes are thought to be transmitted by gap junction or by extracellular diffusion of ATP [72], [73]. The former seems predominant in the neocortex [21], [74], and the latter in the archicortex and spinal cord [75], [76]. In the present model, the astrocytes are coupled by the transfer of IP3 through gap junction, and the ATP diffusion is not considered. Hence, the results in the present study are expected to be relevant to the brain structures in neocortex. To compare the properties of Ca2+ waves in different brain areas, the gap junction and ATP diffusion should be considered together. ATP-mediated Ca2+ waves have been modeled in the astrocyte network [31], [77]. However, because of the complex details in the ATP production and propagation, it’s not very easy to integrate these models into our model. Nevertheless, this part of the improvement will be the goal of our future work. Second, astrocytes communicate not only with themselves, but also with neurons, and Ca2+ signals in astrocytes could be affected by the neuronal activity [11], [16]. Transmitters released from neurons, for example, glutamate, should be considered to understand the Ca2+ signals, especially the CSDCWs [31].
In summary, we analyze the contributions of different Ca2+ flows to the generation of CASs and CSDCWs, and indicate different mechanisms underlying CSDCWs and non-CSDCWs. An experiment test could be done is to example the effects of Ca2+ influx, especially Ca2+ influx via VGCCs, on the generation and propagation of CSDCWs. Research on the different types of Ca2+ signals might help to understand the different ways by which astrocytes participate in the brain functions.
Supporting Information
The influence of CICR on CASs. From (A) to (E), the inhibition of CICR is 0%, 30%, 70%, 90% and 100%, respectively. By inhibiting CICR gradually, the frequency and amplitude of CASs decrease, while the duration increases.
(TIF)
The influence of SERCA on CASs. From (A) to (E), the inhibition of SERCA is 0%, 20%, 50%, 70% and 90%, respectively. By inhibiting SERCA gradually, the amplitude and duration of CASs decrease, while the frequency increases. Ca2+ dynamics evolve into small oscillations before disappearing.
(TIF)
The influence of VGCCs on CASs. From (A) to (E), the inhibition of VGCCs is 0%, 20%, 30%, 40% and 50%, respectively. In the present of CASs, inhibiting VGCCs has little influence on the amplitude and duration of CASs, but great on frequency.
(TIF)
Dynamics of CSD. During CSD, K+ in the ECS (A) and in the ICS (B) is significantly increased and astrocytes are depolarized at successive astrocytes in the network (C). The bar illustrates the time of locally elevating K o to evoke a CSD.
(TIF)
The influence of temperature on the frequency of CASs. From (A) to (D), the value of temperature used in the model is 20, 25, 31, and 36°C, respectively. As the temperature increases, the frequency of CASs decreases. (E) CASs occur frequently at low temperature and become less frequent at higher temperature.
(TIF)
The parameter values used in the model.
(PDF)
The initial values of the variables.
(PDF)
Supporting materials that briefly describe the Ca2+ flows through the membrane of ER and the dynamics of IP3 in single astrocytes in the previous model of CASs.
(DOC)
Acknowledgments
We thank Sherri Tran for helpful comments and critical reading of the manuscript.
Funding Statement
This work is supported by Science Fund for Creative Research Group of China (Grant No. 61121004), the National High Technology Research and Development Program of China (Grant No. 2012AA011602), the Program for New Century Excellent Talents in University (Grant No. NCET-08-0213), and the National Natural Science Foundation of China (Grant Nos. 30970964, 30800339). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Temburni MK, Jacob MH (2001) New functions for glia in the brain. Proc Natl Acad Sci U S A 98: 3631–3632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Volterra A, Meldolesi J (2005) Astrocytes, from brain glue to communication elements: the revolution continues. Nat Rev Neurosci 6: 626–640. [DOI] [PubMed] [Google Scholar]
- 3. Nedergaard M, Ransom B, Goldman SA (2003) New roles for astrocytes: redefining the functional architecture of the brain. Trends Neurosci 26: 523–530. [DOI] [PubMed] [Google Scholar]
- 4. Haydon PG, Carmignoto G (2006) Astrocyte control of synaptic transmission and neurovascular coupling. Physiol Rev 86: 1009–1031. [DOI] [PubMed] [Google Scholar]
- 5. Simard M, Arcuino G, Takano T, Liu QS, Nedergaard M (2003) Signaling at the gliovascular interface. J Neurosci 23: 9254–9262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Tian GF, Azmi H, Takano T, Xu Q, Peng W, et al. (2005) An astrocytic basis of epilepsy. Nat Med 11: 973–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Barres BA (2008) The mystery and magic of glia: a perspective on their roles in health and disease. Neuron 60: 430–440. [DOI] [PubMed] [Google Scholar]
- 8. Nimmerjahn A (2009) Astrocytes going live: advances and challenges. J Physiol 587: 1639–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Codazzi F, Teruel MN, Meyer T (2001) Control of astrocyte Ca(2+) oscillations and waves by oscillating translocation and activation of protein kinase C. Curr Biol. 11: 1089–1097. [DOI] [PubMed] [Google Scholar]
- 10. Pasti L, Volterra A, Pozzan T, Carmignoto G (1997) Intracellular calcium oscillations in astrocytes: a highly plastic, bidirectional form of communication between neurons and astrocytes in situ. J Neurosci 17: 7817–7830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Dani JW, Chernjavsky A, Smith SJ (1992) Neuronal activity triggers calcium waves in hippocampal astrocyte networks. Neuron 8: 429–440. [DOI] [PubMed] [Google Scholar]
- 12. Hirase H, Qian L, Bartho P, Buzsaki G (2004) Calcium dynamics of cortical astrocytic networks in vivo. PLoS Biol 2: E96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Parri HR, Crunelli V (2003) The role of Ca2+ in the generation of spontaneous astrocytic Ca2+ oscillations. Neuroscience 120: 979–992. [DOI] [PubMed] [Google Scholar]
- 14. Saheki Y, Li ST, Matsushita M, Wu YM, Cai WH, et al. (2005) A new approach to inhibiting astrocytic IP3-induced intracellular calcium increase in an astrocyte-neuron co-culture system. Brain Res 1055: 196–201. [DOI] [PubMed] [Google Scholar]
- 15. Manning TJ Jr, Sontheimer H (1997) Spontaneous intracellular calcium oscillations in cortical astrocytes from a patient with intractable childhood epilepsy (Rasmussen’s encephalitis). Glia 21: 332–337. [PubMed] [Google Scholar]
- 16. Aguado F, Espinosa-Parrilla JF, Carmona MA, Soriano E (2002) Neuronal activity regulates correlated network properties of spontaneous calcium transients in astrocytes in situ. J Neurosci 22: 9430–9444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Parri HR, Gould TM, Crunelli V (2001) Spontaneous astrocytic Ca2+ oscillations in situ drive NMDAR-mediated neuronal excitation. Nat Neurosci 4: 803–812. [DOI] [PubMed] [Google Scholar]
- 18. Fam SR, Gallagher CJ, Salter MW (2000) P2Y(1) purinoceptor-mediated Ca(2+) signaling and Ca(2+) wave propagation in dorsal spinal cord astrocytes. J Neurosci 20: 2800–2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Syyong HT, Yang HH, Trinh G, Cheung C, Kuo KH, et al. (2009) Mechanism of asynchronous Ca(2+) waves underlying agonist-induced contraction in the rat basilar artery. Br J Pharmacol 156: 587–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chuquet J, Hollender L, Nimchinsky EA (2007) High-resolution in vivo imaging of the neurovascular unit during spreading depression. J Neurosci 27: 4036–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Peters O, Schipke CG, Hashimoto Y, Kettenmann H (2003) Different mechanisms promote astrocyte Ca2+ waves and spreading depression in the mouse neocortex. J Neurosci 23: 9888–9896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Leao AAP (1944) Spreading depression of activity in the cerebral cortex. J Neurophysiol 7: 359–390. [DOI] [PubMed] [Google Scholar]
- 23. Smith JM, Bradley DP, James MF, Huang CL (2006) Physiological studies of cortical spreading depression. Biol Rev Camb Philos Soc 81: 457–481. [DOI] [PubMed] [Google Scholar]
- 24. Lauritzen M, Dreier JP, Fabricius M, Hartings JA, Graf R, et al. (2011) Clinical relevance of cortical spreading depression in neurological disorders: migraine, malignant stroke, subarachnoid and intracranial hemorrhage, and traumatic brain injury. J Cereb Blood Flow Metab 31: 17–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gorji A (2001) Spreading depression: a review of the clinical relevance. Brain Res Brain Res Rev 38: 33–60. [DOI] [PubMed] [Google Scholar]
- 26. Lavrentovich M, Hemkin S (2008) A mathematical model of spontaneous calcium(II) oscillations in astrocytes. J Theor Biol 251: 553–560. [DOI] [PubMed] [Google Scholar]
- 27. Sneyd J, Tsaneva-Atanasova K, Yule DI, Thompson JL, Shuttleworth TJ (2004) Control of calcium oscillations by membrane fluxes. Proc Natl Acad Sci U S A 101: 1392–1396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Goldberg M, De Pitta M, Volman V, Berry H, Ben-Jacob E (2010) Nonlinear gap junctions enable long-distance propagation of pulsating calcium waves in astrocyte networks. PLoS Comput Biol 6. [DOI] [PMC free article] [PubMed]
- 29. Atri A, Amundson J, Clapham D, Sneyd J (1993) A single-pool model for intracellular calcium oscillations and waves in the Xenopus laevis oocyte. Biophys J 65: 1727–1739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Edwards JR, Gibson WG (2010) A model for Ca2+ waves in networks of glial cells incorporating both intercellular and extracellular communication pathways. J Theor Biol 263: 45–58. [DOI] [PubMed] [Google Scholar]
- 31. Bennett MR, Farnell L, Gibson WG (2008) A quantitative model of cortical spreading depression due to purinergic and gap-junction transmission in astrocyte networks. Biophys J 95: 5648–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zeng S, Li B, Zeng S, Chen S (2009) Simulation of spontaneous Ca2+ oscillations in astrocytes mediated by voltage-gated calcium channels. Biophys J 97: 2429–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Dronne MA, Boissel JP, Grenier E (2006) A mathematical model of ion movements in grey matter during a stroke. J Theor Biol 240: 599–615. [DOI] [PubMed] [Google Scholar]
- 34. Martins-Ferreira H, Nedergaard M, Nicholson C (2000) Perspectives on spreading depression. Brain Res Brain Res Rev 32: 215–234. [DOI] [PubMed] [Google Scholar]
- 35. Charles A (1998) Intercellular calcium waves in glia. Glia 24: 39–49. [DOI] [PubMed] [Google Scholar]
- 36. Boitano S, Dirksen ER, Sanderson MJ (1992) Intercellular propagation of calcium waves mediated by inositol trisphosphate. Science 258: 292–295. [DOI] [PubMed] [Google Scholar]
- 37. Hofer T, Venance L, Giaume C (2002) Control and plasticity of intercellular calcium waves in astrocytes: a modeling approach. J Neurosci 22: 4850–4859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bennett MR, Farnell L, Gibson WG (2005) A quantitative model of purinergic junctional transmission of calcium waves in astrocyte networks. Biophys J 89: 2235–2250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Obrenovitch TP, Zilkha E (1995) High extracellular potassium, and not extracellular glutamate, is required for the propagation of spreading depression. J Neurophysiol 73: 2107–2114. [DOI] [PubMed] [Google Scholar]
- 40.Reggia JA, Montgomery D (1994) Modeling cortical spreading depression. Proc Annu Symp Comput Appl Med Care: 873–877. [PMC free article] [PubMed]
- 41. Walz W (2000) Role of astrocytes in the clearance of excess extracellular potassium. Neurochem Int 36: 291–300. [DOI] [PubMed] [Google Scholar]
- 42. Newman EA (1986) High potassium conductance in astrocyte endfeet. Science 233: 453–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schipke CG, Heidemann A, Skupin A, Peters O, Falcke M, et al. (2008) Temperature and nitric oxide control spontaneous calcium transients in astrocytes. Cell Calcium 43: 285–295. [DOI] [PubMed] [Google Scholar]
- 44.Ding H, Chen S, Zeng S, Zeng S, Liu Q, et al. Computation and visualization of spreading depression based on reaction-diffusion equation with recovery. In: Qingming L, Lihong VW, Valery VT, editors; 2008. SPIE. 72801F.
- 45. Tashiro A, Goldberg J, Yuste R (2002) Calcium oscillations in neocortical astrocytes under epileptiform conditions. J Neurobiol 50: 45–55. [DOI] [PubMed] [Google Scholar]
- 46. De Young GW, Keizer J (1992) A single-pool inositol 1,4,5-trisphosphate-receptor-based model for agonist-stimulated oscillations in Ca2+ concentration. Proc Natl Acad Sci U S A 89: 9895–9899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Ullah G, Jung P, Cornell-Bell AH (2006) Anti-phase calcium oscillations in astrocytes via inositol (1, 4, 5)-trisphosphate regeneration. Cell Calcium 39: 197–208. [DOI] [PubMed] [Google Scholar]
- 48. Nett WJ, Oloff SH, McCarthy KD (2002) Hippocampal astrocytes in situ exhibit calcium oscillations that occur independent of neuronal activity. J Neurophysiol 87: 528–537. [DOI] [PubMed] [Google Scholar]
- 49. Hisatsune C, Nakamura K, Kuroda Y, Nakamura T, Mikoshiba K (2005) Amplification of Ca2+ signaling by diacylglycerol-mediated inositol 1,4,5-trisphosphate production. J Biol Chem 280: 11723–11730. [DOI] [PubMed] [Google Scholar]
- 50. Chapuisat G, Dronne MA, Grenier E, Hommel M, Gilquin H, et al. (2008) A global phenomenological model of ischemic stroke with stress on spreading depressions. Prog Biophys Mol Biol 97: 4–27. [DOI] [PubMed] [Google Scholar]
- 51. Kager H, Wadman WJ, Somjen GG (2002) Conditions for the triggering of spreading depression studied with computer simulations. J Neurophysiol 88: 2700–2712. [DOI] [PubMed] [Google Scholar]
- 52. Sasaki T, Kuga N, Namiki S, Matsuki N, Ikegaya Y (2011) Locally synchronized astrocytes. Cereb Cortex 21: 1889–1900. [DOI] [PubMed] [Google Scholar]
- 54. Kuga N, Sasaki T, Takahara Y, Matsuki N, Ikegaya Y (2011) Large-scale calcium waves traveling through astrocytic networks in vivo. J Neurosci 31: 2607–2614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Di Castro MA, Chuquet J, Liaudet N, Bhaukaurally K, Santello M, et al. (2011) Local Ca2+ detection and modulation of synaptic release by astrocytes. Nat Neurosci 14: 1276–1284. [DOI] [PubMed] [Google Scholar]
- 56. Carmignoto G, Pasti L, Pozzan T (1998) On the role of voltage-dependent calcium channels in calcium signaling of astrocytes in situ. J Neurosci 18: 4637–4645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Somjen GG (2001) Mechanisms of spreading depression and hypoxic spreading depression-like depolarization. Physiol Rev 81: 1065–1096. [DOI] [PubMed] [Google Scholar]
- 58. Takata N, Hirase H (2008) Cortical layer 1 and layer 2/3 astrocytes exhibit distinct calcium dynamics in vivo. PLoS One 3: e2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Schipke CG, Boucsein C, Ohlemeyer C, Kirchhoff F, Kettenmann H (2002) Astrocyte Ca2+ waves trigger responses in microglial cells in brain slices. FASEB J 16: 255–257. [DOI] [PubMed] [Google Scholar]
- 60. D’Ascenzo M, Vairano M, Andreassi C, Navarra P, Azzena GB, et al. (2004) Electrophysiological and molecular evidence of L-(Cav1), N- (Cav2.2), and R- (Cav2.3) type Ca2+ channels in rat cortical astrocytes. Glia 45: 354–363. [DOI] [PubMed] [Google Scholar]
- 61. Akopian G, Kressin K, Derouiche A, Steinhauser C (1996) Identified glial cells in the early postnatal mouse hippocampus display different types of Ca2+ currents. Glia 17: 181–194. [DOI] [PubMed] [Google Scholar]
- 62. Verkhratsky A, Steinhauser C (2000) Ion channels in glial cells. Brain Res Brain Res Rev 32: 380–412. [DOI] [PubMed] [Google Scholar]
- 63. Matyash V, Kettenmann H (2010) Heterogeneity in astrocyte morphology and physiology. Brain Res Rev 63: 2–10. [DOI] [PubMed] [Google Scholar]
- 64. Fiacco TA, McCarthy KD (2006) Astrocyte calcium elevations: properties, propagation, and effects on brain signaling. Glia 54: 676–690. [DOI] [PubMed] [Google Scholar]
- 65. De Pitta M, Volman V, Levine H, Pioggia G, De Rossi D, et al. (2008) Coexistence of amplitude and frequency modulations in intracellular calcium dynamics. Phys Rev E Stat Nonlin Soft Matter Phys 77: 030903. [DOI] [PubMed] [Google Scholar]
- 66. Despa SI (1996) Membrane potential changes in activated cells: connection with cytosolic calcium oscillator. Biosystems 39: 233–240. [DOI] [PubMed] [Google Scholar]
- 67. Kawamura M Jr, Kawamura M (2011) Long-term facilitation of spontaneous calcium oscillations in astrocytes with endogenous adenosine in hippocampal slice cultures. Cell Calcium 49: 249–258. [DOI] [PubMed] [Google Scholar]
- 68. Wang X, Lou N, Xu Q, Tian GF, Peng WG, et al. (2006) Astrocytic Ca2+ signaling evoked by sensory stimulation in vivo. Nat Neurosci 9: 816–823. [DOI] [PubMed] [Google Scholar]
- 69. Landeira-Fernandez AM, Morrissette JM, Blank JM, Block BA (2004) Temperature dependence of the Ca2+-ATPase (SERCA2) in the ventricles of tuna and mackerel. Am J Physiol Regul Integr Comp Physiol 286: R398–404. [DOI] [PubMed] [Google Scholar]
- 70. Dode L, Van Baelen K, Wuytack F, Dean WL (2001) Low temperature molecular adaptation of the skeletal muscle sarco(endo)plasmic reticulum Ca2+-ATPase 1 (SERCA 1) in the wood frog (Rana sylvatica). J Biol Chem 276: 3911–3919. [DOI] [PubMed] [Google Scholar]
- 71. Zhou N, Gordon GR, Feighan D, MacVicar BA (2010) Transient swelling, acidification, and mitochondrial depolarization occurs in neurons but not astrocytes during spreading depression. Cereb Cortex 20: 2614–2624. [DOI] [PubMed] [Google Scholar]
- 72. Falcke M (2004) Reading the patterns in living cells - the physics of Ca(2+) signaling. Advances in Physics 53: 255–440. [Google Scholar]
- 73. Scemes E, Giaume C (2006) Astrocyte calcium waves: what they are and what they do. Glia 54: 716–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Haas B, Schipke CG, Peters O, Sohl G, Willecke K, et al. (2006) Activity-dependent ATP-waves in the mouse neocortex are independent from astrocytic calcium waves. Cereb Cortex 16: 237–246. [DOI] [PubMed] [Google Scholar]
- 75. Bowser DN, Khakh BS (2007) Vesicular ATP is the predominant cause of intercellular calcium waves in astrocytes. J Gen Physiol 129: 485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gallagher CJ, Salter MW (2003) Differential properties of astrocyte calcium waves mediated by P2Y1 and P2Y2 receptors. J Neurosci 23: 6728–6739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Stamatakis M, Mantzaris NV (2006) Modeling of ATP-mediated signal transduction and wave propagation in astrocytic cellular networks. J Theor Biol 241: 649–668. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
The influence of CICR on CASs. From (A) to (E), the inhibition of CICR is 0%, 30%, 70%, 90% and 100%, respectively. By inhibiting CICR gradually, the frequency and amplitude of CASs decrease, while the duration increases.
(TIF)
The influence of SERCA on CASs. From (A) to (E), the inhibition of SERCA is 0%, 20%, 50%, 70% and 90%, respectively. By inhibiting SERCA gradually, the amplitude and duration of CASs decrease, while the frequency increases. Ca2+ dynamics evolve into small oscillations before disappearing.
(TIF)
The influence of VGCCs on CASs. From (A) to (E), the inhibition of VGCCs is 0%, 20%, 30%, 40% and 50%, respectively. In the present of CASs, inhibiting VGCCs has little influence on the amplitude and duration of CASs, but great on frequency.
(TIF)
Dynamics of CSD. During CSD, K+ in the ECS (A) and in the ICS (B) is significantly increased and astrocytes are depolarized at successive astrocytes in the network (C). The bar illustrates the time of locally elevating K o to evoke a CSD.
(TIF)
The influence of temperature on the frequency of CASs. From (A) to (D), the value of temperature used in the model is 20, 25, 31, and 36°C, respectively. As the temperature increases, the frequency of CASs decreases. (E) CASs occur frequently at low temperature and become less frequent at higher temperature.
(TIF)
The parameter values used in the model.
(PDF)
The initial values of the variables.
(PDF)
Supporting materials that briefly describe the Ca2+ flows through the membrane of ER and the dynamics of IP3 in single astrocytes in the previous model of CASs.
(DOC)