

Abstract
The vitamin D receptor (VDR) mediates the pleiotropic biologic effects of 1α,25 dihydroxy-vitamin D3. Recent in vitro studies suggested that curcumin and poly-unsaturated fatty acids (PUFAs) also bind to VDR with low affinity. As potential ligands for the VDR, we hypothesized that curcumin and PUFAs would induce expression of known VDR target genes in cells. In this study, we tested whether these compounds regulated two important VDR target genes - human cathelicidin antimicrobial peptide (CAMP) and 1,25-dihydroxyvitamin D3 24-hydroxylase (CYP24A1)- in human monocytic cell line U937, colon cancer cell line HT-29 and keratinocyte cell line HaCaT. We demonstrated that PUFAs failed to induce CAMP or CYP24A1 mRNA expression in all three cell lines, but curcumin up-regulated CAMP mRNA and protein levels in U937 cells. Curcumin treatment induced CAMP promoter activity from a luciferase reporter construct lacking the VDR binding site and did not increase binding of the VDR to the CAMP promoter as determined by chromatin immunoprecipitation assays. These findings indicate that induction of CAMP by curcumin occurs through a vitamin D receptor-independent manner. We conclude that PUFAs and curcumin do not function as ligands for the VDR.
Keywords: innate immunity, vitamin D receptor, curcumin, poly-unsaturated fatty acid, cathelicidin
1. Introduction
The nuclear receptor superfamily is divided into four groups based on whether the receptor forms a homo- or heterodimer complex and what class of ligand is bound. The endocrine receptors form homodimers and bind steroid hormones produced by endocrine tissues. The xenobiotic receptors function as heterodimers with retinoid-X-receptor (RXR) and bind to xenobiotic compounds, dietary lipids and cholesterol metabolites. The third group forms heterodimers with RXR and binds to thyroid hormone and vitamins A and D while the orphan receptor group lacks known ligands. The vitamin D receptor (VDR, NR1I1) is widely expressed in most, if not all, human tissues and possesses characteristics of both the second and third groups. It serves as the receptor for 1α, 25-dihydroxyvitamin D3 [1,25(OH)2D3] which binds with high affinity and for the secondary bile acid lithocholic acid (LCA) that binds with low affinity. Vitamin D is obtained either from food, supplementation or synthesized in the skin by UVB irradiation of 7-dehydrocholesterol. LCA is a secondary bile acid converted from primary bile acids by gut microbiota. Upon engagement of a ligand, VDR forms a heterodimer with RXR and binds to vitamin D response elements (VDREs) present in about 2000 genomic locations and directly regulates approximately 200 genes. Target genes of the VDR contribute to bone mineral homeostasis, detoxification of exogenous and endogenous compounds, cancer prevention, mammalian hair cycling and immune function.
The ability of the VDR to bind LCA suggests that it may interact with other novel ligands. To identify additional VDR ligands, Jurutka and colleagues used a mammalian two-hybrid system to test high concentrations of curcumin (CM) and the polyunsaturated fats (PUFAs) docosahexaenoic acid (DHA), eicosapentaenoic acid (EPA), arachidonic acid (AA), and linolenic acid (LA). These compounds promoted the dimerization of VDR and RXR suggesting that they may function as novel low-affinity ligands for the VDR. More recently, curcumin was shown to induce expression of the VDR target genes CYP24A1, CYP3A4, TRPV6 and CDKN1A in the human colon cancer cell line Caco-2.
The human cathelicidin antimicrobial peptide (CAMP) gene encodes the hCAP18 pro-protein that is cleaved to release the active peptide LL-37. The CAMP gene is directly regulated by binding of the VDR to a VDRE located in its promoter region. Expression of the human CAMP mRNA and hCAP18 is strongly induced by both 1,25(OH)2D3 and LCA in keratinocytes and myeloid leukemia cell lines. Induction of the CAMP gene by LCA requires a 1000-fold higher concentration of LCA than 1,25(OH)2D3 (1 × 10−5 versus 1 × 10−9 M, respectively) as it is a low-affinity ligand for the VDR. We hypothesized that if CM and PUFAs are low-affinity ligands for the VDR then at high concentrations they may induce the human CAMP gene in cells via activation of the VDR. In this study, we showed that PUFAs did not act as VDR ligands and were unable to increase expression of the CAMP gene in keratinocyte, colon and myeloid cell lines, but CM acted through a VDR-independent pathway to increase CAMP expression.
2. Material and methods
2.1. Compounds
Curcumin (C7727-500MG), cis-4,7,10,13,16,19-docosahexaenoic acid (D2534), cis-5,8,11,14,17-eicosapentaenoic acid (E2011), arachidonic acid (A9673), linolenic acid (L2376), were purchased from Sigma Aldrich (St. Louis, MO, USA).
2.2. Cell culture
Colonic epithelial cell line HT-29 was kindly provided by Dr. Rod Dashwood (Oregon State University, Corvallis, OR). The human monocytic U937 and the keratinocyte HaCaT cell lines were a generous gift from Dr. H. Phillip Koeffler (Cedars-Sinai Medical Center, Los Angeles, CA). U937 cells were maintained in RPMI 1640 medium and HT-29 and HaCaT cells were maintained in DMEM medium (Mediatech Inc., Manassas, VA, USA). All media were supplemented with 10% (v/v) heat-inactivated FBS, 2 mM L-glutamine, and 1% Pen/Strep (Invitrogen Corporation, Carlsbad, CA, USA). Cell cultures were incubated at 37°C in a humidified 5% CO2 incubator.
2.3. Quantitative real-time PCR (qRT-PCR)
U937, HaCaT and HT-29 cells were treated with compounds as described in the figure legends. Total RNA was isolated using the SV Total RNA Isolation System according to the manufacturer’s protocol (Promega Corporation, Madison, WI, USA). RNA (1–2 μg) was converted to cDNA using SuperScript III reverse transcriptase and random hexamer primers (Invitrogen Corporation) according to the manufacturer’s recommendations. PCR reactions were set up as described previously. PCR was performed on a Bio-Rad iCycler iQ5 or CFX-96 QPCR system (Bio-Rad Laboratories, Hercules, CA, USA). All the threshold cycle (Ct) numbers were normalized to 18S rRNA. The probes and primers for the human CAMP, CYP24A1, FABP4 and RN18S1 genes used for qRT-PCR are described in Table 1.
Table 1.
Primers and probes used for qRT-PCR
| Gene | Primer Sequence | Probe Sequence |
|---|---|---|
| CAMP | F 5′-GCTAACCTCTACCGCCTCCT -3′ R 5′-GGTCACTGTCCCCATACACC -3′ |
5′-FAM-ACCCCAGGCCCACGATGGAT-BHQ1-3′ |
| CYP24A1 | F 5′-GAACGTTGGCTTCAGGAGAA -3′ R 5′-TATTTGCGGACAATCCAACA -3′ |
5′-FAM-TGCGCATCTTCCATTTGGCG-BHQ1-3′ |
| FABP4 | F 5′-AGCACCATAACCTTAGATGGGG -3′ F 5′-CGTGGAAGTGACGCCTTTCA -3′ |
5′-FAM-ATTCCACCACCAGTTTATCATCCTCTCGT-BHQ1-3′ |
|
CAMP CHIP |
F 5′-GGGCAACTTGTCCCTTGCAAGAG-3′ F 5′-TGAAAATTAGCCACGCATGA-3′ |
5′-FAM-CTCTAGGTTGGGGGTGGCTACTGTCTTCAT-BHQ1-3′ |
| RN18S1 | F 5′-AAACGGCTACCACATCCAAG -3′ R 5′-CCTCCAATGGATCCTCGTTA -3′ |
5′-FAM-AGCAGGCGCGCAAATTACCC-BHQ1-3′ |
2.4. Intracellular staining, fluorescence activated cell sorting and enzyme-linked immunosorbent assay
U937 cells were treated as indicated in the figure legends. Cells were fixed, permeabilized and blocked using the eBioscience™ Fixation and Permeabilization Kit as described by the manufacturer (eBioscience, Inc., San Diego, CA, USA). Cells were incubated with a rabbit, anti-hCAP18 polyclonal antibody and a Dylight 649 Fab’ 2 donkey anti-rabbit antibody (Jackson Immunoresearch, Pike West Grove, PA, USA). Fluorescence activated cell sorting (FACS) was performed on a BD FACSCalibur flow cytometer (BD Biosciences, San Jose, CA, USA) and the results were analyzed by BD CellQuest™ Pro software (BD Biosciences). The enzyme-linked immunosorbent assay (ELISA) was performed as described previously.
2.5 CAMP promoter luciferase reporter assay
U937 cells were electroporated using a NEON™ transfection system (Life Technologies, Grand Island, NY, USA) in Tip100 tips at 5×107 cells/mL. Electroporation conditions were 1400 mV, 30ms, 1 pulse. A total of 10 μg plasmid was used per electroporation. After transfection, cells were treated with CM or 1,25(OH)2D3 or vehicle as indicated in the figure legends. Cells were lysed and dual luciferase assays were performed as described by the manufacturer (Promega, Madison, WI, USA). The human CAMP promoter (nucleotides –693 to 14) containing the VDRE and a 5′ deletion of the promoter (ΔHindIII, nucleotides –497 to 14) lacking the VDRE were subcloned into a pXP2 firefly luciferase reporter plasmid previously. A renilla luciferase reporter (phTKRL, Promega) was co-transfected to normalize firefly luciferase activities in all experiments.
2.6. Chromatin-Immunoprecipitation Assay
Chromatin-immunoprecipitation (ChIP) experiments were performed as described previously. Briefly, U937 cells (107 cells/IP) were treated with compounds as specified in the figure legend for 24 hours. Cells then were fixed with 1% (v/v) formaldehyde for 10 minutes at room temperature and quenched by 0.1 M glycine for 5 minutes. Fixed chromatin was sheared to 200–1000 bp fragments by a bath sonicator (Bioruptor™ XL, Diagenode Inc. Denville, NJ) following the manufacturer’s recommendations. Anti-VDR antibodies (2 μg C-20 VDR antibody, sc-1008; 2 μg N-20 VDR antibody, sc-1009, Santa Cruz Biotechnology, Santa Cruz, CA) were incubated with sheared chromatin for 16 hours at 4°C. Immunocomplexes were pulled down by Protein A/G Plus Agrose beads (sc-2003, Santa Cruz Biotechnology, Santa Cruz, CA) and DNA was recovered using Chelex® 100 resin (Bio-Rad, Hercules, CA). To evaluate the VDR occupancy at the human CAMP gene promoter, quantitative PCR was performed as described in section 2.3. Occupancy by VDR was normalized with respect to chromatin input used for immunoprecipitation. Primers and probe are listed in Table 1.
2.7. Data analysis
All qRT-PCR and ELISA experiments were performed in triplicate or duplicate and results were represented as mean value with SD. Student’s t-test was performed using Sigma Plot (Systat Software, San Jose, CA) and Microsoft Excel (Microsoft Corporation, Redmond, WA).
3. Results
3.1. CAMP gene expression is induced by curcumin but not PUFAs
The human CAMP and CYP24A1 are known target genes of the VDR and induced by 1,25(OH)2D3 and LCA. We predicted that compounds that function as low-affinity ligands for the VDR would induce expression of these two genes. To test this, we treated U937 (Fig. 1A & B), HT-29 (Fig. 1C & D) and HaCaT (Fig. 1E & F) cells with CM, DHA, EPA, AA and LA for 24 hours. 1,25(OH)2D3 and LCA were included as positive controls and vehicle (ethanol or DMSO) was used for the untreated control. Because 1,25(OH)2D3 does not induce CAMP strongly in HT-29 cells, sodium butyrate (NaB), a known inducer of CAMP in colon cancer cell lines, was included for experiments with HT-29 (Fig. 1C & D). 1,25(OH)2D3 and LCA strongly induced expression of both the CAMP (Fig. 1A & E) and CYP24A1 (Fig. 1B & F) genes in U937 and HaCaT cells. In HT-29 cells CAMP was not strongly induced by 1,25(OH)2D3 or LCA, but was induced about four-fold by NaB (Fig. 1C). CYP24A1 expression was induced by LCA and 1,25(OH)2D3 in all cells tested (Fig. 1B, D & F). The PUFAs (DHA, EPA, AA and LA) did not induce human CAMP or CYP24A1 expression in U937, HaCaT or HT-29 cells (Fig. 1A–F). CM consistently induced expression of human CAMP by about 3-fold (n=3, P<0.05) in U937 and HT-29 cells (Fig. 1A & C) but not in HaCaT cells (Fig. 1E). In all three cell lines, CM did not induce CYP24A1 (Fig. 1B, D & F). To demonstrate that the PUFAs used in this study were active, we examined expression of FABP4, a gene induced by PUFAs binding the PPARγ receptor in monocytes. FABP4 expression was induced in U937 cells demonstrating that the compounds were functional (Fig. 2). To ensure that induction of CAMP or CYP24A1 did not peak prior to 24 hours, we tested CM and DHA in a time course experiment (0, 3, 6, 12 and 24 hours) and observed maximal induction of CAMP by CM at 24 hours and no induction by DHA (data not shown). Collectively, these data indicate that PUFAs do not act as low-affinity agonists for the VDR and that CM induces CAMP, but not CYP24A1.
Figure 1. Alternative VDR ligands fail to activate the human CAMP gene.

U937 cells (A, B), HT-29 cells (C, D) and HaCaT cells (E, F) were treated with 10 μM curcumin (CM), 100μM docosahexaenoic acid (DHA), 100 μM eicosapentaenoic acid (EPA), 100 μM arachidonic acid (AA), 100 μM linolenic Acid (LA), 100 μM lithocholic acid (LCA) and 1 nM 1,25(OH)2D3 for 24 hours. For HT-29 cells, 2 mM sodium butyrate (NaB) was used as positive control since 1,25(OH)2D3 is not a potent inducer of CAMP in these cells. qRT-PCR analysis of human CAMP (A, C, and E) and CYP24A1 (B, D and F) mRNA levels were normalized to 18S rRNA. Each panel is from one experiment, but is representative of three independent experiments. *Significant (P < 0.05) difference compared with untreated control.
Figure 2. PUFAs and CM induced FABP4 expression in U937 cells.

U937 cells were treated with 10 μM CM, 100 μM DHA, 100 μM EPA, 100 μM AA and 100 μM LA for 24 hours. FABP4 mRNA levels were measured by qRT-PCR using primers and probe as described in Table 1. *Significant (P < 0.05) difference compared with untreated control.
3.2. CM elevates hCAP18 levels
Treatment of U937 cells with 10 nM 1,25(OH)2D3 increases levels of hCAP18 (the protein encoded by the human CAMP gene) secreted into the medium. We monitored secreted levels of hCAP18 in the medium by ELISA. As expected, 10 nM 1,25(OH)2D3 increased secretion of hCAP18 into the medium; however, treatment with 100 μM LCA and 1 nM 1,25(OH)2D3, which induce CAMP mRNA expression to similar levels, did not enhance hCAP18 secretion and neither did CM nor the PUFAs (Fig. 3). These results suggest that modest increases in CAMP mRNA levels may not lead to secretion of hCAP18 proteins in U937 cells.
Figure 3. Curcumin and PUFAs do not increase levels of secreted hCAP18.

U937 cells were treated with 10 μM CM, 100 μM DHA, 100 μM EPA, 100 μM AA, 100 μM LA, 100 μM LCA or 1,25(OH)2D3 (1 nM and 10 nM) for 24 hours. Culture medium was collected and subjected to ELISA to measure extracellular hCAP18 protein levels. *Significant (P < 0.01) difference compared with untreated control.
To determine if induction of CAMP mRNA by CM would increase intracellular hCAP18 expression, U937 cells were treated with either 15 μM CM, 100 μM DHA, 100 μM EPA, 100 μM AA, 100 μM LA, 100 μM LCA or 10 nM 1,25(OH)2D3 for 24 hours. The hCAP18 levels were measured by intracellular staining and FACS. The PUFAs did not increase hCAP18 levels (data not shown). CM increased the intracellular hCAP18 levels, however, they were lower than those induced by LCA and 1,25(OH)2D3 (Fig. 4).
Figure 4. CM increases intracellular levels of hCAP18.

U937 cells were treated with 10 μM CM, 100 μM LCA and 1 nM 1,25(OH)2D3 for 24 hours. Intracellular hCAP18 levels were assessed by flow cytometry. This panel is from one experiment, but is representative of four independent experiments.
3.3. CM does not enhance 1,25(OH)2D3 induction of CAMP expression
It was shown previously that treatment of Caco-2 cells with CM and 1,25(OH)2D3 resulted in a combinatorial activation of a transfected VDRE-Luc reporter construct. To determine if CM plus 1,25(OH)2D3 would activate the CAMP gene better than either compound alone, we treated U937 cells with 15 μM CM and increasing doses of 1,25(OH)2D3. The CAMP mRNA levels were evaluated by qRT-PCR (Fig. 5). CM increased CAMP mRNA levels by 2.6-fold while 0.1 nM vitamin D induced CAMP by 6.5-fold. The combination induced CAMP by 5.5-fold indicating no combinatorial activation of the gene. This lack of combinatorial activation was observed with 1 nM and 10 nM 1,25(OH)2D3, as well (Fig. 5).
Figure 5. CM does not cooperatively increase CAMP expression by 1,25(OH)2D3.

U937 cells were treated with 15 μM CM in the absence or presence of increasing concentrations of 1,25(OH)2D3 for 24 hours. This data are representative of two independent experiments. Levels of CAMP expression were measured by qRT-PCR using primers and probe as described in Table 1.
3.4 Induction of the CAMP gene by CM does not require the VDRE in the CAMP promoter
We predicted that if CM induced CAMP through the VDR, then deletion of the VDRE in the CAMP promoter should abrogate the induction. We transfected CAMP promoter firefly luciferase reporters with or without the presence of the VDRE (pXP2-CAMP-luc and pXP2-CAMP-ΔHindIII-luc, respectively, Fig. 6A) into U937 cells. Consistent with our previous report, deletion of the VDRE in the CAMP promoter almost completely abolished induction of luciferase activity by 10 nM 1,25(OH)2D3 (Fig. 6B). On the other hand, CM was still capable of increasing CAMP promoter activity in the absence of the VDRE in the promoter (Fig. 6B). 10 μM CM induced the luciferase activities by about two-fold regardless of the presence or absence of the VDRE. From these experiments, we concluded that induction of the CAMP gene by CM does require the VDRE.
Figure 6. CM induces CAMP promoter activity in absence of VDRE.

A) Schematic diagrams show the structures of the two CAMP promoter-luciferase reporter constructs used in this study. Solid filled black box indicates the location of the VDRE in the CAMP promoter. B) U937 cells were electroporated with pXP2-CAMP-luc or pXP2-CAMP-ΔHindIII-luc plasmid and then treated with 10 μM CM, 10 nM 1,25(OH)2D3 or vehicle for 20 hours. Data were presented as fold changes over the corresponding untreated control. *Significant (P < 0.05, n=3) difference compared with untreated control. This bar chart summarizes three independent experiments.
3.5 CM does not increase VDR binding to the CAMP gene promoter
CM does not appear to function as a ligand for the VDR, thus we predicted that it would not increase VDR binding to the human CAMP gene promoter. To test this, we performed ChIP for VDR in U937 cells treated with CM, LCA and1,25(OH)2D3 (Fig. 7). We found that VDR binding to the CAMP promoter was increased with 1,25(OH)2D3, and LCA treatment and not by CM (Fig. 7), strongly suggesting that CM-induced human CAMP expression occurs through a VDR- independent mechanism.
Figure 7. CM does not enhance VDR binding to the human CAMP promoter.
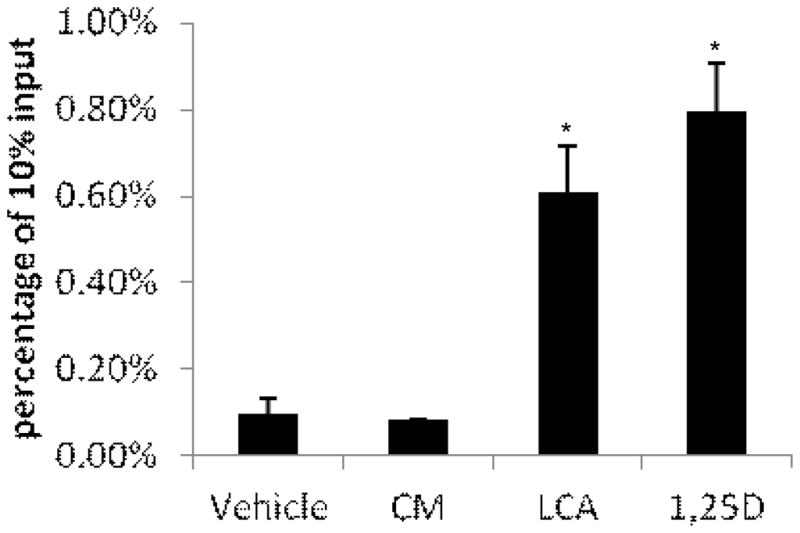
U937 cells were treated with 10 μM CM, 100 μM LCA and 10 nM 1,25(OH)2D3 for 24 hours. Chromatin-IP was performed as described in section 2.5. The panel represents two independent experiments. *Significant (P < 0.05) difference compared with untreated control.
4. Discussion
VDR agonists are of great interest because of their potential therapeutic benefits in treating cancer, psoriasis and other diseases. Thousands of analogs have been synthesized around the vitamin D backbone to reduce or eliminate its hypercalcemic side effects. Another class of VDR agonists is secondary bile acid LCA and its analogs that activate VDR target genes without inducing hypercalcemia. The identification of new agonists increases the toolbox of backbones upon which additional analogs can be developed. To this end, we tested a group of potential VDR ligands identified by a mammalian two hybrid system. We showed that CM modestly induced CAMP, but not CYP24A1 expression and that PUFAs did not induce the mRNA levels these two VDR target genes in human monocyte (U937), keratinocyte (HaCaT) or colon cancer (HT-29) cell lines. These results suggest these compounds are not functional VDR agonists. On the other hand, the known ligands, LCA and 1,25(OH)2D3 strongly induced both genes. Of the putative ligands tested, only CM increased intracellular levels of hCAP18. This induction was observed in three of four experiments and was less than either LCA or 1,25(OH)2D3. The modest induction of CAMP by CM did not appear to occur through the VDR. ChIP experiments showed that VDR binding to the CAMP promoter was not increased by CM as it is by both LCA and 1,25(OH)2D3. Furthermore, we demonstrated by reporter assays that CM activated the CAMP promoter in the absence of the VDRE.
CM at the concentration we used can elicit ER stress and a recent study showed ER stress induces human CAMP expression in keratinocytes. We tested whether ER stress induced CAMP in our cell lines and were unable to demonstrate a role for this mechanism (data not shown); therefore, ER stress elicited by CM is not a likely mechanism for induction of human CAMP gene expression in our study. Collectively, these data argue that CM and PUFAs are not low affinity ligands for the VDR and CM activates CAMP expression by a currently unknown mechanism(s).
The discrepancy between our work and the previous study could be attributed to several factors. First, recent molecular docking studies proposed that two ligand binding pockets exist in the VDR ligand binding domain: the genomic and alternative pockets. Vitamin D and its metabolites are ligands of the genomic pocket while CM is proposed to mainly bind to the alternative pocket. Therefore, in the mammalian two hybrid system, the possible binding of CM to the alternative pocket may have increased VDR/RXR dimerization; however, since CM was a weak ligand of the genomic pocket, it did not activate transcription of VDR target genes in our cell culture experiments. Second, prior studies demonstrated that CM regulated the VDR target gene CYP24A1 in Caco-2 cells. We did not observe this in U937, HaCaT or HT-29 cells, suggesting that modulation of VDR target genes by CM could be specific to the type of cell used.
Future experiments determining the crystal structure of the VDR/CM complex may further define the role of CM as a VDR alternative pocket ligand. Also, additional studies in other cell lines may be required to comprehensively understand the possible function of CM and PUFAs as VDR ligands.
Acknowledgments
This study was supported by NIH grant 5R01AI65604 (A.F.G.).
We thank Mary Fantacone for critically reviewing the manuscript and Charlotte Horn for technical assistance. In addition, we are grateful to Brian Sinnott, Yan Campbell and Jesse Rushen for their help in completing this project.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chawla A, Repa JJ, Evans RM, Mangelsdorf DJ. Nuclear receptors and lipid physiology: opening the X-files. Science. 2001;294:1866–70. doi: 10.1126/science.294.5548.1866. [DOI] [PubMed] [Google Scholar]
- 2.Pike JW, Meyer MB. The vitamin D receptor: new paradigms for the regulation of gene expression by 1,25-dihydroxyvitamin D(3) Endocrinol Metab Clin North Am. 2011;39:255–69. doi: 10.1016/j.ecl.2010.02.007. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Makishima M, Lu TT, Xie W, Whitfield GK, Domoto H, Evans RM, et al. Vitamin D receptor as an intestinal bile acid sensor. Science. 2002;296:1313–6. doi: 10.1126/science.1070477. [DOI] [PubMed] [Google Scholar]
- 4.Holick MF, MacLaughlin JA, Clark MB, Holick SA, Potts JT, Jr, Anderson RR, et al. Photosynthesis of previtamin D3 in human skin and the physiologic consequences. Science. 1980;210:203–5. doi: 10.1126/science.6251551. [DOI] [PubMed] [Google Scholar]
- 5.Fedorowski T, Salen G, Tint GS, Mosbach E. Transformation of chenodeoxycholic acid and ursodeoxycholic acid by human intestinal bacteria. Gastroenterology. 1979;77:1068–73. [PubMed] [Google Scholar]
- 6.Szeles L, Poliska S, Nagy G, Szatmari I, Szanto A, Pap A, et al. Research resource: transcriptome profiling of genes regulated by RXR and its permissive and nonpermissive partners in differentiating monocyte-derived dendritic cells. Mol Endocrinol. 2011;24:2218–31. doi: 10.1210/me.2010-0215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pike JW, Meyer MB, Martowicz ML, Bishop KA, Lee SM, Nerenz RD, et al. Emerging regulatory paradigms for control of gene expression by 1,25-dihydroxyvitamin D3. J Steroid Biochem Mol Biol. 2010;121:130–5. doi: 10.1016/j.jsbmb.2010.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Haussler MR, Haussler CA, Bartik L, Whitfield GK, Hsieh JC, Slater S, et al. Vitamin D receptor: molecular signaling and actions of nutritional ligands in disease prevention. Nutr Rev. 2008;66:S98–112. doi: 10.1111/j.1753-4887.2008.00093.x. [DOI] [PubMed] [Google Scholar]
- 9.Jurutka PW, Bartik L, Whitfield GK, Mathern DR, Barthel TK, Gurevich M, et al. Vitamin D receptor: key roles in bone mineral pathophysiology, molecular mechanism of action, and novel nutritional ligands. J Bone Miner Res. 2007;22 (Suppl 2):V2–10. doi: 10.1359/jbmr.07s216. [DOI] [PubMed] [Google Scholar]
- 10.Bartik L, Whitfield GK, Kaczmarska M, Lowmiller CL, Moffet EW, Furmick JK, et al. Curcumin: a novel nutritionally derived ligand of the vitamin D receptor with implications for colon cancer chemoprevention. J Nutr Biochem. 2011;21:1153–61. doi: 10.1016/j.jnutbio.2009.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gombart AF, Borregaard N, Koeffler HP. Human cathelicidin antimicrobial peptide (CAMP) gene is a direct target of the vitamin D receptor and is strongly up-regulated in myeloid cells by 1,25-dihydroxyvitamin D3. FASEB J. 2005;19:1067–77. doi: 10.1096/fj.04-3284com. [DOI] [PubMed] [Google Scholar]
- 12.Peric M, Koglin S, Dombrowski Y, Gross K, Bradac E, Ruzicka T, et al. VDR and MEK-ERK dependent induction of the antimicrobial peptide cathelicidin in keratinocytes by lithocholic acid. Mol Immunol. 2009;46:3183–7. doi: 10.1016/j.molimm.2009.08.010. [DOI] [PubMed] [Google Scholar]
- 13.Herdick M, Steinmeyer A, Carlberg C. Antagonistic action of a 25-carboxylic ester analogue of 1alpha, 25-dihydroxyvitamin D3 is mediated by a lack of ligand-induced vitamin D receptor interaction with coactivators. J Biol Chem. 2000;275:16506–12. doi: 10.1074/jbc.M910000199. [DOI] [PubMed] [Google Scholar]
- 14.Sorensen O, Cowland JB, Askaa J, Borregaard N. An ELISA for hCAP-18, the cathelicidin present in human neutrophils and plasma. J Immunol Methods. 1997;206:53–9. doi: 10.1016/s0022-1759(97)00084-7. [DOI] [PubMed] [Google Scholar]
- 15.Nelson JD, Denisenko O, Bomsztyk K. Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nat Protoc. 2006;1:179–85. doi: 10.1038/nprot.2006.27. [DOI] [PubMed] [Google Scholar]
- 16.Gombart AF, O’Kelly J, Saito T, Koeffler HP. Regulation of the CAMP gene by 1,25(OH)2D3 in various tissues. J Steroid Biochem Mol Biol. 2007;103:552–7. doi: 10.1016/j.jsbmb.2006.12.095. [DOI] [PubMed] [Google Scholar]
- 17.Ishizawa M, Matsunawa M, Adachi R, Uno S, Ikeda K, Masuno H, et al. Lithocholic acid derivatives act as selective vitamin D receptor modulators without inducing hypercalcemia. J Lipid Res. 2008;49:763–72. doi: 10.1194/jlr.M700293-JLR200. [DOI] [PubMed] [Google Scholar]
- 18.Schauber J, Dorschner RA, Yamasaki K, Brouha B, Gallo RL. Control of the innate epithelial antimicrobial response is cell-type specific and dependent on relevant microenvironmental stimuli. Immunology. 2006;118:509–19. doi: 10.1111/j.1365-2567.2006.02399.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pelton PD, Zhou L, Demarest KT, Burris TP. PPARgamma activation induces the expression of the adipocyte fatty acid binding protein gene in human monocytes. Biochem Biophys Res Commun. 1999;261:456–8. doi: 10.1006/bbrc.1999.1071. [DOI] [PubMed] [Google Scholar]
- 20.Peterlik M, Grant WB, Cross HS. Calcium, vitamin D and cancer. Anticancer Res. 2009;29:3687–98. [PubMed] [Google Scholar]
- 21.Takiishi T, Gysemans C, Bouillon R, Mathieu C. Vitamin D and diabetes. Endocrinol Metab Clin North Am. 2011;39:419–46. doi: 10.1016/j.ecl.2010.02.013. table of contents. [DOI] [PubMed] [Google Scholar]
- 22.Sun J. Vitamin D and mucosal immune function. Curr Opin Gastroenterol. 2011;26:591–5. doi: 10.1097/MOG.0b013e32833d4b9f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de Borst MH, de Boer RA, Stolk RP, Slaets JP, Wolffenbuttel BH, Navis G. Vitamin D deficiency: universal risk factor for multifactorial diseases? Curr Drug Targets. 2011;12:97–106. doi: 10.2174/138945011793591590. [DOI] [PubMed] [Google Scholar]
- 24.Rucevic I, Barisic-Drusko V, Glavas-Obrovac L, Stefanic M. Vitamin D endocrine system and psoriasis vulgaris--review of the literature. Acta Dermatovenerol Croat. 2009;17:187–92. [PubMed] [Google Scholar]
- 25.Eduardo-Canosa S, Fraga R, Sigueiro R, Marco M, Rochel N, Moras D, et al. Design and synthesis of active vitamin D analogs. J Steroid Biochem Mol Biol. 2010;121:7–12. doi: 10.1016/j.jsbmb.2010.03.036. [DOI] [PubMed] [Google Scholar]
- 26.Nehring JA, Zierold C, DeLuca HF. Lithocholic acid can carry out in vivo functions of vitamin D. Proc Natl Acad Sci U S A. 2007;104:10006–9. doi: 10.1073/pnas.0703512104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pae HO, Jeong SO, Jeong GS, Kim KM, Kim HS, Kim SA, et al. Curcumin induces pro-apoptotic endoplasmic reticulum stress in human leukemia HL-60 cells. Biochem Biophys Res Commun. 2007;353:1040–5. doi: 10.1016/j.bbrc.2006.12.133. [DOI] [PubMed] [Google Scholar]
- 28.Park K, Elias PM, Oda Y, Mackenzie D, Mauro T, Holleran WM, et al. Regulation of Cathelicidin Antimicrobial Peptide Expression by an Endoplasmic Reticulum (ER) Stress Signaling, Vitamin D Receptor-independent Pathway. J Biol Chem. 2011;286:34121–30. doi: 10.1074/jbc.M111.250431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menegaz D, Mizwicki MT, Barrientos-Duran A, Chen N, Henry HL, Norman AW. Vitamin D Receptor (VDR) Regulation of Voltage-Gated Chloride Channels by Ligands Preferring a VDR-Alternative Pocket (VDR-AP) Mol Endocrinol. 2011 doi: 10.1210/me.2010-0442. [DOI] [PMC free article] [PubMed] [Google Scholar]