

Abstract
Protein clearance is critical for the maintenance of the integrity of neuronal cells, and there is accumulating evidence that in most—if not all—neurodegenerative disorders, impaired protein clearance fundamentally contributes to functional and structural alterations eventually leading to clinical symptoms. Dysfunction of protein clearance leads to intra- and extraneuronal accumulation of misfolded proteins and aggregates. The pathological hallmark of Lewy body disorders (LBDs) is the abnormal accumulation of misfolded proteins such as alpha-synuclein (Asyn) and amyloid-beta (Abeta) in a specific subset of neurons, which in turn has been related to deficits in protein clearance. In this paper we will highlight common intraneuronal (including autophagy and unfolded protein stress response) and extraneuronal (including interaction of neurons with astrocytes and microglia, phagocytic clearance, autoimmunity, cerebrospinal fluid transport, and transport across the blood-brain barrier) protein clearance mechanisms, which may be altered across the spectrum of LBDs. A better understanding of the pathways underlying protein clearance—in particular of Asyn and Abeta—in LBDs may result in the identification of novel biomarkers for disease onset and progression and of new therapeutic targets.
1. Introduction
Lewy body disorders (LBDs) is an umbrella term that includes diseases with alpha-synuclein (Asyn) aggregates as fibrils in Lewy bodies (LBs) and Lewy neurites. Several lines of evidence support a pathogenic role of misfolded Asyn in LBDs [1–3]. Parkinson's disease (PD) without dementia (PDND) is the most common subtype of LBDs, followed by PD with dementia (PDD) and dementia with Lewy bodies (DLBs) [4, 5]. Like in PD, the core feature of PDD is a diagnosis of PD according to the UK Brain Bank Society criteria [6] but also includes cognitive symptoms severe enough to fulfil dementia criteria at least one year after PD diagnosis with insidious onset, slow progression, and impairment in more than one cognitive domain [7]. DLB is the second most prevalent neurodegenerative dementia after Alzheimer's disease (AD). Clinical diagnosis is based on the presence of a dementia syndrome, accompanied by at least two out of the three following symptoms: fluctuating cognition with pronounced variations in attention and alertness, visual hallucinations, and Parkinsonism [8]. In contrast to PDD, the onset of dementia in DLB is before or within one year of any Parkinsonism.
About one-fourth of LBDs patients show cortical amyloid-beta (Abeta) deposition, with the highest proportion in DLB subjects, followed by PDD subjects [9–14]. According to clinical, neuroimaging, and neuropathological data, co-occurrence of Asyn and Abeta is regularly associated with dementia in LBDs [13–15].
Based on the central role that Asyn and Abeta play in the pathogenesis of LBDs and the increasing body of literature pointing to defective clearance of misfolded proteins as a key mechanism to the pathogenesis of LBDs, this paper aims at providing a condensed review of this latter topic. Although not exhaustive, it may provide a basic understanding of such mechanisms eventually contributing to the development of novel disease biomarkers (which are currently not available [16, 17]) and neuromodulatory treatment strategies for these still incurable and chronic diseases.
2. General Mechanisms of Protein Clearance
The main (intraneuronal) pathways for the degradation and recycling of proteins are the ubiquitin/proteasome system (UPS) and the autophagy-lysosomal pathway (macroautophagy, microautophagy, and chaperone-mediated autophagy (CMA)).
The UPS degrades short-lived nuclear and cytosolic proteins or misfolded proteins in the endoplasmic reticulum [18]. It plays a key role in signal transduction, cell cycle progression, apoptosis, and cellular differentiation and has been implicated in several human diseases, including neurodegenerative diseases, cancer, inflammation, and autoimmunity [18–20].
Autophagy is a process involving the degradation of components inside lysosomes [21, 22]. It has a variety of physiological and pathophysiological roles in protein and organelle clearance, development, defence against microorganisms, cell death, and antigen presentation [23]. In macroautophagy, organelles and macromolecular components are first surrounded by a double membrane, defined as the autophagosome or autophagic vacuole (AV), which then fuses with lysosomes to form autolysosomes. In microautophagy, the transfer of cytosolic components the lysosomal compartment happens by direct invagination of the lysosomal membrane without prior sequestration into the autophagosome. Finally, in CMA, individual proteins are targeted to lysosomes by the binding of a hsc70-containing chaperone/cochaperone complex.
All these pathways are observable in neurons, and both impairment and excessive activation of these pathways are linked to neurodegenerative processes [24].
(Extraneuronal) clearance pathways include interaction of neurons with astro- and microglia and with infiltrating macrophages. Microglial cells are considered the professional phagocytes in the brain. However, other populations of cells may also act as phagocytes, including astrocytes, neural stem cells, and neurons [25–28]. There is no clear evidence for a defective protein clearance by the brain innate immune system as a primary pathogenetic event in neurodegenerative diseases. However, phagocytosis of misfolded proteins by astro- and microglia triggers the release of proinflammatory cytokines and chemokines and reactive oxygen/nitrogen species, which may, under pathological conditions, further promote neuronal dysfunction and degeneration [29, 30].
The adaptive immune system is also involved in the clearance of misfolded proteins in the brain. Naturally occurring autoantibodies are detectable in body fluids of healthy controls and in patients with neurodegenerative disorders [31–33]. Finally, protein transport from the parenchyma to the cerebrospinal fluid (CSF) and across the blood-brain barrier are relevant clearance mechanisms and have been shown to be affected in LBDs.
Intra- and extraneuronal clearance mechanisms closely interact: as an example, protein aggregates can stimulate the cell surface innate immune receptors, initiating intracellular signaling cascades that, in turn, stimulate phagocytosis. An overview of how these mechanisms may interact is schematically illustrated in Figure 1.
Figure 1.
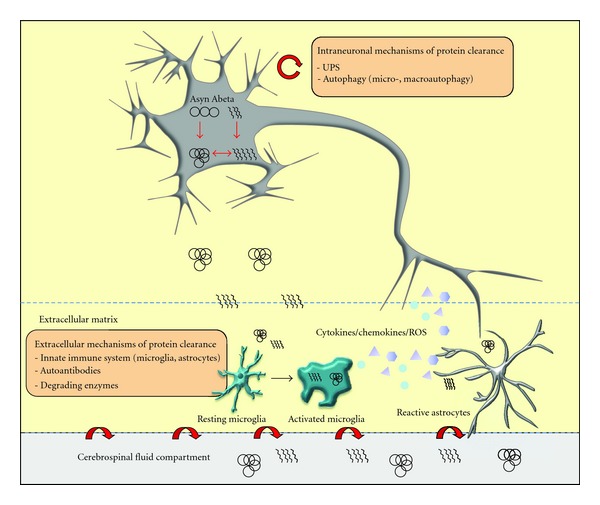
Intra- and extraneuronal mechanisms of protein clearance. The main intracellular pathways for the degradation and recycling of proteins are the ubiquitin/proteasome system (UPS) and the autophagy-lysosomal pathway (microautophagy, macroautophagy, and chaperone-mediated autophagy (CMA)). Extracellular clearance pathways include interaction of neurons with astro- and microglia, and with infiltrating macrophages, autoantibodies, and protein transport from the parenchyma to the cerebrospinal fluid and across the blood-brain barrier. The engulfment of misfolded proteins by astro- and microglia triggers the release of proinflammatory cytokines and chemokines as well as reactive oxygen/nitrogen species, which may, under pathological conditions, further promote neuronal dysfunction and degeneration.
In the following, all these mechanisms are discussed in more detail with special emphasis on Asyn and Abeta clearance.
3. Asyn Clearance
3.1. Intraneuronal Mechanisms
Abnormal deposition of Asyn occurs early in the disease process of LBDs and may follow an ascending pattern in most of the cases, starting from lower brainstem areas and then affecting limbic and cortical areas [34]. Deposition may start even earlier in the autonomic peripheral nervous system [35, 36]. The mechanisms responsible for Asyn degradation have been controversial, but it appears that, under normal conditions, Asyn is degraded by both the UPS and the autophagy-lysosomal pathway [37–39], whereas the autophagy-lysosomal pathway mediates clearance of accumulated and aggregated Asyn [38]. In agreement with this observation, activation of autophagy leads to increased wild-type Asyn clearance and neuroprotection [37, 40]. Normal Asyn binds to the CMA-specific receptor LAMP-2A on the lysosomal membrane and is subsequently degraded by CMA. However, mutant forms of Asyn (A53T and A30P) and Asyn modified by dopamine tightly bind to the CMA receptors on the lysosomal membrane and inhibit both their own degradation and that of other CMA substrates [38, 41]. The dysfunction of CMA triggers neuronal dysfunction and increases vulnerability to stress. Interestingly, both mutant and wild-type Asyn can decrease proteasomal activity and increase vulnerability to neurodegeneration, leading to a vicious cycle where an increased amount of intraneuronal Asyn can block its clearance by itself [38, 42–46].
Several studies provide evidence for impaired autophagy-mediated clearance mechanisms in PD. The main proteins involved in CMA (Lamp2a and Hsc70) are decreased in the SN and amygdala from PD patients [46]. Microtubule-associated protein 1A/1B-light chain 3 (LC3, a marker for autophagic vacuoles) colocalizes with Asyn in most LBs and Lewy neurites [46].
Genetic mutations leading to Parkinsonism support the hypothesis that defective clearance mechanisms are centrally involved also in idiopathic forms of LBDs. Interaction between PINK1 and Parkin can modulate mitophagy [47, 48]. PINK1 itself directly activates autophagy and interacts with autophagic proteins such as Beclin1 [49]. A reduced clearance of mitochondria was also demonstrated in cells lacking DJ-1 which is another protein associated with recessive forms of Parkinsonism [50]. Mutations in the LRRK2 gene, the most common form of late onset autosomal dominant Parkinsonism, may also cause neuronal cell death via impairment of protein degradation pathways as they influence the autophagy-lysosomal pathway, leading to Asyn accumulation and aggregation [51, 52]. Heterozygous mutations in the gene encoding the lysosomal enzyme glucocerebrosidase (GBA) convey an approximately 5-fold risk for PD. These mutations are associated with lysosomal dysfunction and influence binding of Asyn to its specific receptor at the lysosome membrane: a recent study demonstrated that the accumulation of the GBA substrate glucosylceramide stabilizes Asyn soluble oligomers which, in turn, can inhibit normal GBA lysosomal activity. This creates a positive feedback loop that could directly contribute to the neurodegenerative process [53]. The heterozygous GBA mutation also seems to directly influence fatty acid metabolism: PD patients with these mutations have lower CSF levels of palmitoleic, oleic, linoleic, arachidonic, eicosapentaenoic, and docosahexaenoic acids compared with both idiopathic PD patients and controls [54].
3.2. Extraneuronal Mechanisms (Mainly)
Although Asyn is a cytosolic protein, a low quantity of the protein is secreted via vesicle exocytosis and is then present in biological fluids including plasma and CSF [55–57]. The mechanisms responsible for the extraneuronal clearance of Asyn are not entirely clear; however, there is increasing evidence that adjacent cells such as astrocytes and microglia as well as the adaptive immune system and local protein transport mechanisms are crucially involved.
Loss of midbrain dopaminergic neurons and striatal degeneration can be preceded by neuroinflammation marked by activated microglia and an increase in proinflammatory cytokines and chemokines [29, 30, 58–62]. Astrocytes are also involved in the initiation and progression of the disease [63]. A recent study showed that astrocytic Asyn initiates noncell autonomous killing of neurons [64]. Indeed, extraneuronal forms of Asyn can activate glial cells and trigger inflammation and subsequent release of proinflammatory molecules, a common pathological hallmark of LBDs [60, 65]. Several studies have reported Asyn-containing inclusions in astroglia of PD and DLB patients [66], and phagocytic microglial cells are very efficient scavengers of extraneuronal Asyn aggregates [63, 67].
Results of recent GWAS studies argue for the relevance of the immune system, in the pathophysiology of LBDs, as variations in the human leukocyte antigen (HLA) region were associated with occurrence of LBDs [68, 69]. This may also be mediated through microglia as these cells are capable of presenting antigens to lymphocytes [70] via the HLA domain. Activation of HLA-positive microglia is observable in affected brain regions of PD patients [71]. Asyn autoantibodies are more prevalent in sera of PD patients than in controls [31]. These results were basically confirmed by a recent study where serum Asyn autoantibody levels were higher in demented LBDs patients than in controls [72].
An overload of local Asyn can also occur due to defective transport mechanisms of Asyn from the neuron to the CSF. Most studies investigating CSF total Asyn levels in PD showed that these levels are reduced in the CSF of PD patients compared to controls, whereas oligomeric Asyn levels seem to be higher in PD compared to control CSF [57, 73–77]. A recent study showed that CSF levels of phosphorylated Asyn correlate weakly with PD severity and, if corrected for total Asyn, contributed to the differential diagnosis between PD, multiple system atrophy (MSA), and progressive supranuclear palsy (PSP) [78]. However, it is neither clear to date how Asyn is transported from the parenchyma to the CSF, nor why total Asyn is reduced and pathological forms of Asyn are elevated in the CSF of PD patients.
4. Abeta Clearance: Lessons from Alzheimer's Disease Research
Comparable to LBDs pathophysiology, in AD, only some rare genetic mutations lead to increased APP expression or to changes in Abeta stability or aggregation [79]. The common late onset sporadic AD seems to be far better explained by impaired Abeta clearance mechanisms [80]. Thus, assuming that similar mechanisms may occur in (late onset) AD and LBDs and that Abeta pathology is a pathophysiologically relevant feature of LBDs, a discussion of (ineffective) Abeta clearance mechanisms as they occur in AD may substantially contribute to our understanding of how Abeta may (or may not) be cleared in LBDs.
4.1. Intraneuronal Mechanisms
There is convincing evidence from electron microscopy studies [81] that the autophagy-lysosomal pathway is involved in the pathogenesis of AD. AVs which are the major reservoir of intraneuronal Abeta are abundant in affected neurons, especially in those with neurofibrillary tangles [82]. These studies argue for an impaired maturation of autophagolysosomes and impaired intraneuronal retrograde transport in AD. Defective clearance of Abeta-generating AVs may result in Abeta accumulation [83, 84]. Several further findings corroborate the hypothesis that impaired autophagy plays a key role in the pathogenesis of neuronal degeneration in AD. Beclin1 is decreased in AD brains, and decreased neuronal autophagy and subsequent lysosomal dysfunction and neurodegeneration are observed in mice carrying a heterozygous deletion of Beclin1 [85]. Presenilin 1 (PS1) is essential for maturation of the lysosomal proton pump and affects autophagocytosis and protein turnover [86].
4.2. Extraneuronal Clearance Mechanisms (Mainly)
Abeta concentration is tightly regulated by amyloid-degrading proteolytic enzymes and perivascular drainage [87–89]. Neprilysin (NEP) is an Abeta-degrading protein found at presynaptic terminals and in body fluids [90, 91]. It is a preferentially membrane-bound, presynaptically located protein with an extracellular catalytic site which can degrade Abeta [90, 91]. A soluble form of NEP is detectable in body fluids such as blood and CSF, emanating from a slow release from the membranes [92]. Most interestingly, reduced CSF NEP activity levels have been shown to occur in early AD [93, 94].
Another protein involved in defective clearance mechanisms in AD is cystatin C. Neurons, among other cells, are able to produce and secrete this protein [95]. Fourfold higher levels of cystatin C in the CSF than in blood [96] indicate a relevant role of the protein in CNS pathways. Cystatin C binds monomeric Abeta and carries soluble Abeta [97, 98]. There is evidence that AD patients have reduced CSF cystatin C levels [99, 100]. This makes it intriguing to hypothesize that a deficient Abeta-binding capacity, as induced by a lack of (functional) cystatin C, may contribute to the amyloidogenic process in AD [99]. Indeed, increased expression of this protein has been shown to reduce parenchymal Abeta load in mouse models of AD [101, 102]. Of note, the BB genotype of the cystatin C-encoding gene—which leads to reduced cystatin C secretion from the neuron to the extracellular space [103, 104]—conveys susceptibility to AD [105].
Inflammatory reactions, characterized by activated microglia and astrocytes surrounding amyloid deposits, are intimately associated with the onset and progress of AD [106–108]. Reactive astrocytes with Abeta-positive granules are found in close proximity to amyloid plaques. Human astrocytes express scavenger receptors and several Abeta-degrading enzymes such as NEP, insulin-degrading enzyme (IDE), endothelin-converting enzyme (ECE), angiotensin-converting enzyme (ACE), plasminogen activators, and the matrix metalloproteinases-9 and -2 (MMP-9, MMP-2) [109–112]. These findings suggest an important role of astrocytes in Abeta clearance.
Clinicopathological studies suggest that microglial activation is an early event in AD pathology [113, 114]. Activated microglia surround amyloid fibril deposits, and postmortem studies have shown significant amounts of Abeta in microglial cells of AD patients treated with immunization therapy [115, 116]. Microglia express toll-like receptors (TLRs), a family of highly conserved molecules that recognize pathogen-associated molecular patterns, including both exogenous and endogenous ligands [117]. TLR2 and TLR4 have been associated with the removal of Abeta, indicating that the innate immune system plays a key role in preventing the brain from Abeta deposits [118–120].
In addition, the adaptive immune system is obviously involved in CNS parenchyma clearance mechanisms. Naturally occurring antibodies directed against Abeta have been detected in the CSF and plasma of patients with AD and healthy control subjects. Some studies have shown reduced CSF levels of anti-Abeta antibodies in patients with AD compared with healthy control subjects [32, 121] and in individuals at increased risk for AD [122]. Another study reported that a subset of conformation-specific, cross-reactive antibodies that may protect against amyloidogenic toxic peptides are reduced in AD patients [123]. As a consequence of these findings, a number of phase II and III clinical trials are currently under way to test the effect of such autoantibodies in AD patients [124]. First results are promising: a recent study using carbon 11-labeled Pittsburgh Compound B ([11C]PiB) positron emission tomography (PET) has shown that passive immunization can reduce the level of brain amyloid in vivo after 18 months of antibody treatment [125].
5. Abeta Clearance in Lewy Body Disorders
5.1. Intraneuronal Mechanisms
To the best of our knowledge there is no study available that investigated intraneuronal mechanisms of Abeta clearance in LBDs. There is however indirect evidence that deficits of (intraneuronal) defence mechanisms against Abeta toxicity may exist, at least in demented LBDs patients. We recently showed that CSF levels of uric acid, an antioxidant detectable in neurons and associated with PD progression, were significantly lower in demented than in nondemented LBDs patients. In addition, these levels correlated positively with CSF Abeta42 levels, with highest correlation values in controls and lowest in demented LBDs patients [126]. In the light of the recent finding that CSF Abeta levels increase within hours after trauma and thus reflect a sufficient and fast response to neuronal stress [127], a weak correlation of CSF Abeta42 with uric acid may indicate deficits in this repair mechanism.
5.2. Extraneuronal Mechanisms (Mainly)
Lowered CSF NEP activity levels have been found in demented LBDs patients, compared to nondemented LBDs patients and controls [128]. In addition, CSF NEP activity levels correlated positively with CSF Abeta42. These data argue for a role of NEP in the pathophysiology of cognitive decline in LBDs.
Also cystatin C seems to be relevantly involved in Abeta-associated cognitive decline in LBDs. Our group investigated CSF and serum levels in LBDs patients [129] and found lower CSF cystatin C levels in demented LBDs patients compared to PDND and controls. In additions, these levels correlated positively with age at onset of dementia but not with parameters associated with Parkinsonism. Notably, the correlation between CSF cystatin C and CSF Abeta42 levels was highly significant in nondemented individuals, but not significant in demented patients. This indicates that cystatin C-related Abeta transport from the neuron to the CSF is impaired in demented LBDs patients. This hypothesis is corroborated by genetic results [129]. The risk genotype of the CST3 gene, BB, was detectable only in demented LBDs patients and was associated with low CSF cystatin C levels.
The role of the innate immune system in Abeta clearance in LBDs is not well understood although neuroinflammatory reactions are a common finding in LBDs and are considered to play a key role in the neurodegenerative process [63]. There is a tight association of microglia with degenerating LB-containing neurons [130]. Activated microglia is associated with Asyn-positive oligodendrocytes in MSA patients and in an animal model of this disease [131, 132]. Astrocytic abnormalities also occur [133, 134].
The adaptive immune system is also involved in Abeta-associated mechanisms in LBDs. Autoantibodies against Abeta were elevated in serum and CSF of demented LBDs patients, compared to controls and were even higher than in other forms of dementia such vascular dementia [72]. Still, many questions remain about the contribution of the immune system, such as microglia, macrophages, and T cells but also other immune cells, to clearance of misfolded proteins in the CNS.
6. Conclusion
Although many questions remain open, recent literature suggests that impairment of protein clearance is one of the key factors mediating the degeneration of vulnerable neuronal populations in LBDs. Both intra- and extraneuronal clearance mechanisms are impaired in LBDs. An improved understanding of such pathways can provide the basis for new developments in the biomarker era and, ultimately, contribute to the development of neuromodulatory or even causal treatment strategies.
References
- 1.Kahle PJ. α-Synucleinopathy models and human neuropathology: similarities and differences. Acta Neuropathologica. 2008;115(1):87–95. doi: 10.1007/s00401-007-0302-x. [DOI] [PubMed] [Google Scholar]
- 2.Gasser T. Molecular pathogenesis of Parkinson disease: insights from genetic studies. Expert Reviews in Molecular Medicine. 2009;11, article e22 doi: 10.1017/S1462399409001148. [DOI] [PubMed] [Google Scholar]
- 3.Scholz SW, Houlden H, Schulte C, et al. SNCA variants are associated with increased risk for multiple system atrophy. Annals of Neurology. 2009;65(5):610–614. doi: 10.1002/ana.21685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lippa CF, Duda JE, Grossman M, et al. DLB and PDD boundary issues: diagnosis, treatment, molecular pathology, and biomarkers. Neurology. 2007;68(11):812–819. doi: 10.1212/01.wnl.0000256715.13907.d3. [DOI] [PubMed] [Google Scholar]
- 5.Baba M, Nakajo S, Tu PH, et al. Aggregation of α-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. American Journal of Pathology. 1998;152(4):879–884. [PMC free article] [PubMed] [Google Scholar]
- 6.Gelb DJ, Oliver E, Gilman S. Diagnostic criteria for Parkinson disease. Archives of Neurology. 1999;56(1):33–39. doi: 10.1001/archneur.56.1.33. [DOI] [PubMed] [Google Scholar]
- 7.Emre M, Aarsland D, Brown R, et al. Clinical diagnostic criteria for dementia associated with Parkinson’s disease. Movement Disorders. 2007;22(12):1689–1707. doi: 10.1002/mds.21507. [DOI] [PubMed] [Google Scholar]
- 8.McKeith IG, Dickson DW, Lowe J, et al. Diagnosis and management of dementia with Lewy bodies: third report of the DLB consortium. Neurology. 2005;65(12):1863–1872. doi: 10.1212/01.wnl.0000187889.17253.b1. [DOI] [PubMed] [Google Scholar]
- 9.Jellinger KA, Seppi K, Wenning GK. Clinical and neuropathological correlates of Lewy body disease (multiple letters) Acta Neuropathologica. 2003;106(2):188–189. doi: 10.1007/s00401-003-0721-2. [DOI] [PubMed] [Google Scholar]
- 10.Boller F, Mizutani T, Roessmann U, Gambetti P. Parkinson disease, dementia, and Alzheimer disease: clinicopathological correlations. Annals of Neurology. 1980;7(4):329–335. doi: 10.1002/ana.410070408. [DOI] [PubMed] [Google Scholar]
- 11.Braak H, Rüb U, Jansen Steur ENH, Del Tredici K, de Vos RAI. Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology. 2005;64(8):1404–1410. doi: 10.1212/01.WNL.0000158422.41380.82. [DOI] [PubMed] [Google Scholar]
- 12.Hughes AJ, Daniel SE, Blankson S, Lees AJ. A clinicopathologic study of 100 cases of Parkinson’s disease. Archives of Neurology. 1993;50(2):140–148. doi: 10.1001/archneur.1993.00540020018011. [DOI] [PubMed] [Google Scholar]
- 13.Maetzler W, Liepelt I, Berg D. Progression of Parkinson’s disease in the clinical phase: potential markers. The Lancet Neurology. 2009;8(12):1158–1171. doi: 10.1016/S1474-4422(09)70291-1. [DOI] [PubMed] [Google Scholar]
- 14.Ballard C, Ziabreva I, Perry R, et al. Differences in neuropathologic characteristics across the Lewy body dementia spectrum. Neurology. 2006;67(11):1931–1934. doi: 10.1212/01.wnl.0000249130.63615.cc. [DOI] [PubMed] [Google Scholar]
- 15.Brooks DJ. Imaging amyloid in Parkinson’s disease dementia and dementia with Lewy bodies with positron emission tomography. Movement Disorders. 2009;24(supplement 2):S742–S747. doi: 10.1002/mds.22581. [DOI] [PubMed] [Google Scholar]
- 16.Maetzler W, Liepelt I, Reimold M, et al. Cortical PIB binding in Lewy body disease is associated with Alzheimer-like characteristics. Neurobiology of Disease. 2009;34(1):107–112. doi: 10.1016/j.nbd.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 17.Gerlach M, Maetzler W, Broich K, et al. Biomarker candidates of neurodegeneration in Parkinson’s disease for the evaluation of disease-modifying therapeutics. Journal of Neural Transmission. 2012;119(1):39–52. doi: 10.1007/s00702-011-0682-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hershko A, Ciechanover A. The ubiquitin system. Annual Review of Biochemistry. 1998;67:425–479. doi: 10.1146/annurev.biochem.67.1.425. [DOI] [PubMed] [Google Scholar]
- 19.Schwartz AL, Ciechanover A. The ubiquitin-proteasome pathway and pathogenesis of human diseases. Annual Review of Medicine. 1999;50:57–74. doi: 10.1146/annurev.med.50.1.57. [DOI] [PubMed] [Google Scholar]
- 20.Ciechanover A, Brundin P. The ubiquitin proteasome system in neurodegenerative diseases: sometimes the chicken, sometimes the egg. Neuron. 2003;40(2):427–446. doi: 10.1016/s0896-6273(03)00606-8. [DOI] [PubMed] [Google Scholar]
- 21.Cuervo AM. Autophagy: in sickness and in health. Trends in Cell Biology. 2004;14(2):70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- 22.Klionsky DJ. Autophagy. Current Biology. 2005;15(8):R282–R283. doi: 10.1016/j.cub.2005.04.013. [DOI] [PubMed] [Google Scholar]
- 23.Mizushima N. Autophagy: process and function. Genes and Development. 2007;21(22):2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- 24.Martinez-Vicente M, Cuervo AM. Autophagy and neurodegeneration: when the cleaning crew goes on strike. The Lancet Neurology. 2007;6(4):352–361. doi: 10.1016/S1474-4422(07)70076-5. [DOI] [PubMed] [Google Scholar]
- 25.Dewitt DA, Perry G, Cohen M, Doller C, Silver J. Astrocytes regulate microglial phagocytosis of senile plaque cores of Alzheimer’s disease. Experimental Neurology. 1998;149(2):329–340. doi: 10.1006/exnr.1997.6738. [DOI] [PubMed] [Google Scholar]
- 26.Lee CYD, Landreth GE. The role of microglia in amyloid clearance from the AD brain. Journal of Neural Transmission. 2010;117(8):949–960. doi: 10.1007/s00702-010-0433-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takuma K, Fang F, Zhang W, et al. RAGE-mediated signaling contributes to intraneuronal transport of amyloid-β and neuronal dysfunction. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(47):20021–20026. doi: 10.1073/pnas.0905686106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Fuentealba RA, Liu Q, Zhang J, et al. Low-density lipoprotein receptor-related protein 1 (LRP1) mediates neuronal Aβ42 uptake and lysosomal trafficking. PLoS ONE. 2010;5(7) doi: 10.1371/journal.pone.0011884.e11884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koprich JB, Reske-Nielsen C, Mithal P, Isacson O. Neuroinflammation mediated by IL-1β increases susceptibility of dopamine neurons to degeneration in an animal model of Parkinson’s disease. Journal of Neuroinflammation. 2008;5, article 8 doi: 10.1186/1742-2094-5-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Deleidi M, Hallett PJ, Koprich JB, Chung CY, Isacson O. The toll-like receptor-3 agonist polyinosinic:polycytidylic acid triggers nigrostriatal dopaminergic degeneration. Journal of Neuroscience. 2010;30(48):16091–16101. doi: 10.1523/JNEUROSCI.2400-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Papachroni KK, Ninkina N, Papapanagiotou A, et al. Autoantibodies to α-synuclein in inherited Parkinson’s disease. Journal of Neurochemistry. 2007;101(3):749–756. doi: 10.1111/j.1471-4159.2006.04365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weksler ME, Relkin N, Turkenich R, LaRusse S, Zhou L, Szabo P. Patients with Alzheimer disease have lower levels of serum anti-amyloid peptide antibodies than healthy elderly individuals. Experimental Gerontology. 2002;37(7):943–948. doi: 10.1016/s0531-5565(02)00029-3. [DOI] [PubMed] [Google Scholar]
- 33.Brettschneider S, Morgenthaler NG, Teipel SJ, et al. Decreased serum amyloid β1–42 autoantibody levels in Alzheimer’s disease, determined by a newly developed immuno-precipitation assay with radiolabeled amyloid β1–42 peptide. Biological Psychiatry. 2005;57(7):813–816. doi: 10.1016/j.biopsych.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 34.Braak H, Del Tredici K, Rüb U, de Vos RAI, Jansen Steur ENH, Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiology of Aging. 2003;24(2):197–211. doi: 10.1016/s0197-4580(02)00065-9. [DOI] [PubMed] [Google Scholar]
- 35.Hallett PJ, McLean JR, Kartunen A, Langston JW, Isacson O. α-Synuclein overexpressing transgenic mice show internal organ pathology and autonomic deficits. Neurobiology of Disease. 2012;47(2):258–267. doi: 10.1016/j.nbd.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaufmann H, Nahm K, Purohit D, Wolfe D. Autonomic failure as the initial presentation of Parkinson disease and dementia with Lewy bodies. Neurology. 2004;63(6):1093–1095. doi: 10.1212/01.wnl.0000138500.73671.dc. [DOI] [PubMed] [Google Scholar]
- 37.Webb JL, Ravikumar B, Atkins J, Skepper JN, Rubinsztein DC. α-Synuclein is degraded by both autophagy and the proteasome. The Journal of Biological Chemistry. 2003;278(27):25009–25013. doi: 10.1074/jbc.M300227200. [DOI] [PubMed] [Google Scholar]
- 38.Cuervo AM, Stafanis L, Fredenburg R, Lansbury PT, Sulzer D. Impaired degradation of mutant α-synuclein by chaperone-mediated autophagy. Science. 2004;305(5688):1292–1295. doi: 10.1126/science.1101738. [DOI] [PubMed] [Google Scholar]
- 39.Tofaris GK, Layfield R, Spillantini MG. α-Synuclein metabolism and aggregation is linked to ubiquitin-independent degradation by the proteasome. FEBS Letters. 2001;509(1):22–26. doi: 10.1016/s0014-5793(01)03115-5. [DOI] [PubMed] [Google Scholar]
- 40.Spencer B, Potkar R, Trejo M, et al. Beclin 1 gene transfer activates autophagy and ameliorates the neurodegenerative pathology in α-synuclein models of Parkinson’s and Lewy body diseases. Journal of Neuroscience. 2009;29(43):13578–13588. doi: 10.1523/JNEUROSCI.4390-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Martinez-Vicente M, Talloczy Z, Kaushik S, et al. Dopamine-modified α-synuclein blocks chaperone-mediated autophagy. Journal of Clinical Investigation. 2008;118(2):777–778. doi: 10.1172/JCI32806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Stefanis L, Larsen KE, Rideout HJ, Sulzer D, Greene LA. Expression of A53T mutant but not wild-type α-synuclein in PC12 cells induces alterations of the ubiquitin-dependent degradation system, loss of dopamine release, and autophagic cell death. Journal of Neuroscience. 2001;21(24):9549–9560. doi: 10.1523/JNEUROSCI.21-24-09549.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tanaka Y, Engelender S, Igarashi S, et al. Inducible expression of mutant α-synuclein decreases proteasome activity and increases sensitivity to mitochondria-dependent apoptosis. Human Molecular Genetics. 2001;10(9):919–926. doi: 10.1093/hmg/10.9.919. [DOI] [PubMed] [Google Scholar]
- 44.Petrucelli L, O’Farrell C, Lockhart PJ, et al. Parkin protects against the toxicity associated with mutant α-synuclein: proteasome dysfunction selectively affects catecholaminergic neurons. Neuron. 2002;36(6):1007–1019. doi: 10.1016/s0896-6273(02)01125-x. [DOI] [PubMed] [Google Scholar]
- 45.Snyder H, Mensah K, Theisler C, Lee J, Matouschek A, Wolozin B. Aggregated and monomeric α-synuclein bind to the S6′ proteasomal protein and inhibit proteasomal function. The Journal of Biological Chemistry. 2003;278(14):11753–11759. doi: 10.1074/jbc.M208641200. [DOI] [PubMed] [Google Scholar]
- 46.Alvarez-Erviti L, Rodriguez-Oroz MC, Cooper JM, et al. Chaperone-mediated autophagy markers in Parkinson disease brains. Archives of Neurology. 2010;67(12):1464–1472. doi: 10.1001/archneurol.2010.198. [DOI] [PubMed] [Google Scholar]
- 47.Vives-Bauza C, Przedborski S. Mitophagy: the latest problem for Parkinson’s disease. Trends in Molecular Medicine. 2011;17(3):158–165. doi: 10.1016/j.molmed.2010.11.002. [DOI] [PubMed] [Google Scholar]
- 48.Geisler S, Holmström KM, Skujat D, et al. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nature Cell Biology. 2010;12(2):119–131. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 49.Michiorri S, Gelmetti V, Giarda E, et al. The Parkinson-associated protein PINK1 interacts with Beclin1 and promotes autophagy. Cell Death and Differentiation. 2010;17(6):962–974. doi: 10.1038/cdd.2009.200. [DOI] [PubMed] [Google Scholar]
- 50.McCoy MK, Cookson MR. DJ-1 regulation of mitochondrial function and autophagy through oxidative stress. Autophagy. 2011;7(5):531–532. doi: 10.4161/auto.7.5.14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tong Y, Yamaguchi H, Giaime E, et al. Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of α-synuclein, and apoptotic cell death in aged mice. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(21):9879–9884. doi: 10.1073/pnas.1004676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gómez-Suaga P, Luzón-Toro B, Churamani D, et al. Leucine-rich repeat kinase 2 regulates autophagy through a calcium-dependent pathway involving NAADP. Human Molecular Genetics. 2012;21(3):511–525. doi: 10.1093/hmg/ddr481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Mazzulli JR, Xu YH, Sun Y, et al. Gaucher disease glucocerebrosidase and α-synuclein form a bidirectional pathogenic loop in synucleinopathies. Cell. 2011;146(1):37–52. doi: 10.1016/j.cell.2011.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Schmid SP, Schleicher ED, Cegan A, et al. Cerebrospinal fluid fatty acids in glucocerebrosidase-associated Parkinson’s disease. Movement Disorders. 2012;27(2):288–293. doi: 10.1002/mds.23984. [DOI] [PubMed] [Google Scholar]
- 55.Lee HJ, Patel S, Lee SJ. Intravesicular localization and exocytosis of α-synuclein and its aggregates. Journal of Neuroscience. 2005;25(25):6016–6024. doi: 10.1523/JNEUROSCI.0692-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.El-Agnaf OM, Salem SA, Paleologou KE, Cooper LJ, Fullwood NJ, et al. α-Synuclein implicated in Parkinson’s disease is present in extracellular biological fluids, including human plasma. The FASEB Journal. 2003;17(13):1945–1947. doi: 10.1096/fj.03-0098fje. [DOI] [PubMed] [Google Scholar]
- 57.Mollenhauer B, Locascio JJ, Schulz-Schaeffer W, Sixel-Döring F, Trenkwalder C, Schlossmacher MG. α-Synuclein and tau concentrations in cerebrospinal fluid of patients presenting with parkinsonism: a cohort study. The Lancet Neurology. 2011;10(3):230–240. doi: 10.1016/S1474-4422(11)70014-X. [DOI] [PubMed] [Google Scholar]
- 58.Cicchetti F, Brownell AL, Williams K, Chen YI, Livni E, Isacson O. Neuroinflammation of the nigrostriatal pathway during progressive 6-OHDA dopamine degeneration in rats monitored by immunohistochemistry and PET imaging. European Journal of Neuroscience. 2002;15(6):991–998. doi: 10.1046/j.1460-9568.2002.01938.x. [DOI] [PubMed] [Google Scholar]
- 59.Sánchez-Pernaute R, Ferree A, Cooper O, Yu M, Brownell AL, Isacson O. Selective COX-2 inhibition prevents progressive dopamine neuron degeneration in a rat model of Parkinson’s disease. Journal of Neuroinflammation. 2004;1, article 6 doi: 10.1186/1742-2094-1-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Deleidi M, Isacson O. Viral and inflammatory triggers of neurodegenerative diseases. Science Translational Medicine. 2012;4(121) doi: 10.1126/scitranslmed.3003492.121ps3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Teismann P, Sathe K, Bierhaus A, et al. Receptor for advanced glycation endproducts (RAGE) deficiency protects against MPTP toxicity. Neurobiology of Aging. 2012;33(10):2478–2490. doi: 10.1016/j.neurobiolaging.2011.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Maetzler W, Berg D, Schalamberidze N, et al. Osteopontin is elevated in Parkinson’s disease and its absence leads to reduced neurodegeneration in the MPTP model. Neurobiology of Disease. 2007;25(3):473–482. doi: 10.1016/j.nbd.2006.10.020. [DOI] [PubMed] [Google Scholar]
- 63.Halliday GM, Stevens CH. Glia: initiators and progressors of pathology in Parkinson’s disease. Movement Disorders. 2011;26(1):6–17. doi: 10.1002/mds.23455. [DOI] [PubMed] [Google Scholar]
- 64.Gu XL, Long CX, Sun L, Xie C, Lin X, Cai H. Astrocytic expression of Parkinson’s disease-related A53T α-synuclein causes neurodegeneration in mice. Molecular Brain. 2010;3(1, article 12) doi: 10.1186/1756-6606-3-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mackenzie IRA. Activated microglia in dementia with Lewy bodies. Neurology. 2000;55(1):132–134. doi: 10.1212/wnl.55.1.132. [DOI] [PubMed] [Google Scholar]
- 66.Braak H, Sastre M, Del Tredici K. Development of α-synuclein immunoreactive astrocytes in the forebrain parallels stages of intraneuronal pathology in sporadic Parkinson’s disease. Acta Neuropathologica. 2007;114(3):231–241. doi: 10.1007/s00401-007-0244-3. [DOI] [PubMed] [Google Scholar]
- 67.Lee HJ, Suk JE, Bae EJ, Lee SJ. Clearance and deposition of extracellular α-synuclein aggregates in microglia. Biochemical and Biophysical Research Communications. 2008;372(3):423–428. doi: 10.1016/j.bbrc.2008.05.045. [DOI] [PubMed] [Google Scholar]
- 68.Hamza TH, Zabetian CP, Tenesa A, et al. Common genetic variation in the HLA region is associated with late-onset sporadic Parkinson’s disease. Nature Genetics. 2010;42(9):781–785. doi: 10.1038/ng.642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Saiki M, Baker A, Williams-Gray CH, et al. Association of the human leucocyte antigen region with susceptibility to Parkinson’s disease. Journal of Neurology, Neurosurgery and Psychiatry. 2010;81(8):890–891. doi: 10.1136/jnnp.2008.162883. [DOI] [PubMed] [Google Scholar]
- 70.Kreutzberg GW. Microglia: a sensor for pathological events in the CNS. Trends in Neurosciences. 1996;19(8):312–318. doi: 10.1016/0166-2236(96)10049-7. [DOI] [PubMed] [Google Scholar]
- 71.Orr CF, Rowe DB, Mizuno Y, Mori H, Halliday GM. A possible role for humoral immunity in the pathogenesis of Parkinson’s disease. Brain. 2005;128(11):2665–2674. doi: 10.1093/brain/awh625. [DOI] [PubMed] [Google Scholar]
- 72.Maetzler W, Berg D, Synofzik M, et al. Autoantibodies against amyloid and glial-derived antigens are increased in serum and cerebrospinal fluid of lewy body-associated dementias. Journal of Alzheimer’s Disease. 2011;26(1):171–179. doi: 10.3233/JAD-2011-110221. [DOI] [PubMed] [Google Scholar]
- 73.Hong Z, Shi M, Chung KA, et al. DJ-1 and α-synuclein in human cerebrospinal fluid as biomarkers of Parkinson’s disease. Brain. 2010;133(3):713–726. doi: 10.1093/brain/awq008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Shi M, Bradner J, Hancock AM, et al. Cerebrospinal fluid biomarkers for Parkinson disease diagnosis and progression. Annals of Neurology. 2011;69(3):570–580. doi: 10.1002/ana.22311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Mollenhauer B, Cullen V, Kahn I, et al. Direct quantification of CSF α-synuclein by ELISA and first cross-sectional study in patients with neurodegeneration. Experimental Neurology. 2008;213(2):315–325. doi: 10.1016/j.expneurol.2008.06.004. [DOI] [PubMed] [Google Scholar]
- 76.Tokuda T, Salem SA, Allsop D, et al. Decreased α-synuclein in cerebrospinal fluid of aged individuals and subjects with Parkinson’s disease. Biochemical and Biophysical Research Communications. 2006;349(1):162–166. doi: 10.1016/j.bbrc.2006.08.024. [DOI] [PubMed] [Google Scholar]
- 77.Tokuda T, Qureshi MM, Ardah MT, et al. Detection of elevated levels of α-synuclein oligomers in CSF from patients with Parkinson disease. Neurology. 2010;75(20):1766–1772. doi: 10.1212/WNL.0b013e3181fd613b. [DOI] [PubMed] [Google Scholar]
- 78.Wang Y, Shi M, Chung KA, et al. Phosphorylated α-synuclein in Parkinson’s disease. Science Translational Medicine. 2012;4(121) doi: 10.1126/scitranslmed.3002566.ra120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Scheuner D, Eckman C, Jensen M, et al. Secreted amyloid β-protein similar to that in the senile plaques of Alzheimer’s disease is increased in vivo by the presenilin 1 and 2 and APP mutations linked to familial Alzheimer’s disease. Nature Medicine. 1996;2(8):864–870. doi: 10.1038/nm0896-864. [DOI] [PubMed] [Google Scholar]
- 80.Mawuenyega KG, Sigurdson W, Ovod V, et al. Decreased clearance of CNS β-amyloid in Alzheimer’s disease. Science. 2010;330(6012, article 1774) doi: 10.1126/science.1197623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nixon RA, Wegiel J, Kumar A, et al. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. Journal of Neuropathology and Experimental Neurology. 2005;64(2):113–122. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 82.Yu WH, Cuervo AM, Kumar A, et al. Macroautophagy—a novel β-amyloid peptide-generating pathway activated in Alzheimer’s disease. Journal of Cell Biology. 2005;171(1):87–98. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Nixon RA. Autophagy, amyloidogenesis and Alzheimer disease. Journal of Cell Science. 2007;120(23):4081–4091. doi: 10.1242/jcs.019265. [DOI] [PubMed] [Google Scholar]
- 84.Boland B, Kumar A, Lee S, et al. Autophagy induction and autophagosome clearance in neurons: relationship to autophagic pathology in Alzheimer’s disease. Journal of Neuroscience. 2008;28(27):6926–6937. doi: 10.1523/JNEUROSCI.0800-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pickford F, Masliah E, Britschgi M, et al. The autophagy-related protein beclin 1 shows reduced expression in early Alzheimer disease and regulates amyloid β accumulation in mice. Journal of Clinical Investigation. 2008;118(6):2190–2199. doi: 10.1172/JCI33585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Lee JH, Yu WH, Kumar A, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141(7):1146–1158. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Weller RO, Yow HY, Preston SD, Mazanti I, Nicoll JAR. Cerebrovascular disease is a major factor in the failure of elimination of Aβ from the aging human brain: implications for therapy of Alzheimer’s disease. Annals of the New York Academy of Sciences. 2002;977:162–168. doi: 10.1111/j.1749-6632.2002.tb04812.x. [DOI] [PubMed] [Google Scholar]
- 88.Nalivaeva NN, Fisk LR, Belyaev ND, Turner AJ. Amyloid-degrading enzymes as therapeutic targets in Alzheimer’s disease. Current Alzheimer Research. 2008;5(2):212–224. doi: 10.2174/156720508783954785. [DOI] [PubMed] [Google Scholar]
- 89.Hawkes CA, Härtig W, Kacza J, et al. Perivascular drainage of solutes is impaired in the ageing mouse brain and in the presence of cerebral amyloid angiopathy. Acta Neuropathologica. 2011;121(4):431–443. doi: 10.1007/s00401-011-0801-7. [DOI] [PubMed] [Google Scholar]
- 90.Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze β-amyloid(1–40) but shows no effect on β-amyloid precursor protein metabolism. Peptides. 1995;16(4):647–652. doi: 10.1016/0196-9781(95)00021-b. [DOI] [PubMed] [Google Scholar]
- 91.Iwata N, Tsubuki S, Takaki Y, et al. Metabolic regulation of brain Aβ by neprilysin. Science. 2001;292(5521):1550–1552. doi: 10.1126/science.1059946. [DOI] [PubMed] [Google Scholar]
- 92.Spillantini MG, Sicuteri F, Salmon S, Malfroy B. Characterization of endopeptidase 3.4.24.11 (“enkephalinase”) activity in human plasma and cerebrospinal fluid. Biochemical Pharmacology. 1990;39(8):1353–1356. doi: 10.1016/0006-2952(90)90012-a. [DOI] [PubMed] [Google Scholar]
- 93.Nalivaeva NN, Fisk L, Kochkina EG, et al. Effect of hypoxia/ischemia and hypoxic preconditioning/reperfusion on expression of some amyloid-degrading enzymes. Annals of the New York Academy of Sciences. 2004;1035:21–33. doi: 10.1196/annals.1332.002. [DOI] [PubMed] [Google Scholar]
- 94.Iwata N, Takaki Y, Fukami S, Tsubuki S, Saido TC. Region-specific reduction of Aβ-degrading endopeptidase, neprilysin, in mouse hippocampus upon aging. Journal of Neuroscience Research. 2002;70(3):493–500. doi: 10.1002/jnr.10390. [DOI] [PubMed] [Google Scholar]
- 95.Nishio C, Yoshida K, Nishiyama K, Hatanaka H, Yamada M. Involvement of cystatin C in oxidative stress-induced apoptosis of cultured rat CNS neurons. Brain Research. 2000;873(2):252–262. doi: 10.1016/s0006-8993(00)02540-3. [DOI] [PubMed] [Google Scholar]
- 96.Grubb A. Diagnostic value of analysis of cystatin C and protein HC in biological fluids. Clinical Nephrology. 1992;38(supplement 1):S20–S27. [PubMed] [Google Scholar]
- 97.Selenica ML, Wang X, Ostergaard-Pedersen L, Westlind-Danielsson A, Grubb A. Cystatin C reduces the in vitro formation of soluble Aβ1–42 oligomers and protofibrils. Scandinavian Journal of Clinical and Laboratory Investigation. 2007;67(2):179–190. doi: 10.1080/00365510601009738. [DOI] [PubMed] [Google Scholar]
- 98.Sastre M, Calero M, Pawlik M, et al. Binding of cystatin C to Alzheimer’s amyloid β inhibits in vitro amyloid fibril formation. Neurobiology of Aging. 2004;25(8):1033–1043. doi: 10.1016/j.neurobiolaging.2003.11.006. [DOI] [PubMed] [Google Scholar]
- 99.Hansson SF, Andréasson U, Wall M, et al. Reduced levels of amyloid-β-binding proteins in cerebrospinal fluid from Alzheimer’s disease patients. Journal of Alzheimer’s Disease. 2009;16(2):389–397. doi: 10.3233/JAD-2009-0966. [DOI] [PubMed] [Google Scholar]
- 100.Simonsen AH, McGuire J, Podust VN, et al. A novel panel of cerebrospinal fluid biomarkers for the differential diagnosis of Alzheimer’s disease versus normal aging and frontotemporal dementia. Dementia and Geriatric Cognitive Disorders. 2007;24(6):434–440. doi: 10.1159/000110576. [DOI] [PubMed] [Google Scholar]
- 101.Kaeser SA, Herzig MC, Coomaraswamy J, et al. Cystatin C modulates cerebral β-amyloidosis. Nature Genetics. 2007;39(12):1437–1439. doi: 10.1038/ng.2007.23. [DOI] [PubMed] [Google Scholar]
- 102.Mi W, Pawlik M, Sastre M, et al. Cystatin C inhibits amyloid-β deposition in Alzheimer’s disease mouse models. Nature Genetics. 2007;39(12):1440–1442. doi: 10.1038/ng.2007.29. [DOI] [PubMed] [Google Scholar]
- 103.Benussi L, Ghidoni R, Steinhoff T, et al. Alzheimer disease-associated cystatin C variant undergoes impaired secretion. Neurobiology of Disease. 2003;13(1):15–21. doi: 10.1016/s0969-9961(03)00012-3. [DOI] [PubMed] [Google Scholar]
- 104.Paraoan L, Ratnayaka A, Spiller DG, Hiscott P, White MRH, Grierson I. Unexpected intracellular localization of the AMD-associated cystatin C variant. Traffic. 2004;5(11):884–895. doi: 10.1111/j.1600-0854.2004.00230.x. [DOI] [PubMed] [Google Scholar]
- 105.Dehghan A, Köttgen A, Yang Q, et al. Association of three genetic loci with uric acid concentration and risk of gout: a genome-wide association study. The Lancet. 2008;372(9654):1953–1961. doi: 10.1016/S0140-6736(08)61343-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.McGeer EG, McGeer PL. Brain inflammation in Alzheimer disease and the therapeutic implications. Current Pharmaceutical Design. 1999;5(10):821–836. [PubMed] [Google Scholar]
- 107.Akiyama H, Barger S, Barnum S, et al. Inflammation and Alzheimer’s disease. Neurobiology of Aging. 2000;21(3):383–421. doi: 10.1016/s0197-4580(00)00124-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Alexander JJ, Anderson AJ, Barnum SR, Stevens B, Tenner AJ. The complement cascade: Yin-Yang in neuroinflammation—neuro-protection and -degeneration. Journal of Neurochemistry. 2008;107(5):1169–1187. doi: 10.1111/j.1471-4159.2008.05668.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Husemann J, Silverstein SC. Expression of scavenger receptor class B, type I, by astrocytes and vascular smooth muscle cells in normal adult mouse and human brain and in Alzheimer’s disease brain. American Journal of Pathology. 2001;158(3):825–832. doi: 10.1016/S0002-9440(10)64030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Allaman I, Gavillet M, Bélanger M, et al. Amyloid-β aggregates cause alterations of astrocytic metabolic phenotype: impact on neuronal viability. Journal of Neuroscience. 2010;30(9):3326–3338. doi: 10.1523/JNEUROSCI.5098-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Leissring MA. The AβCs of Aβ-cleaving proteases. The Journal of Biological Chemistry. 2008;283(44):29645–29649. doi: 10.1074/jbc.R800022200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Turner AJ, Nalivaeva NN. New insights into the roles of metalloproteinases in neurodegeneration and neuroprotection. International Review of Neurobiology. 2007;82:113–135. doi: 10.1016/S0074-7742(07)82006-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Hoozemans JJM, van Haastert ES, Veerhuis R, et al. Maximal COX-2 and ppRb expression in neurons occurs during early Braak stages prior to the maximal activation of astrocytes and microglia in Alzheimer’s disease. Journal of Neuroinflammation. 2005;2, article 27 doi: 10.1186/1742-2094-2-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Cagnin A, Brooks DJ, Kennedy AM, et al. In-vivo measurement of activated microglia in dementia. The Lancet. 2001;358(9280):461–467. doi: 10.1016/S0140-6736(01)05625-2. [DOI] [PubMed] [Google Scholar]
- 115.Itagaki S, McGeer PL, Akiyama H, Zhu S, Selkoe D. Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. Journal of Neuroimmunology. 1989;24(3):173–182. doi: 10.1016/0165-5728(89)90115-x. [DOI] [PubMed] [Google Scholar]
- 116.Nicoll JA, Barton E, Boche D, Neal JW, Ferrer I, et al. Aβ species removal after Aβ42 immunization. Journal of Neuropathology and Experimental Neurology. 2006;65(11):1040–1048. doi: 10.1097/01.jnen.0000240466.10758.ce. [DOI] [PubMed] [Google Scholar]
- 117.Glezer I, Simard AR, Rivest S. Neuroprotective role of the innate immune system by microglia. Neuroscience. 2007;147(4):867–883. doi: 10.1016/j.neuroscience.2007.02.055. [DOI] [PubMed] [Google Scholar]
- 118.Liu Y, Walter S, Stagi M, et al. LPS receptor (CD14): a receptor for phagocytosis of Alzheimer’s amyloid peptide. Brain. 2005;128(8):1778–1789. doi: 10.1093/brain/awh531. [DOI] [PubMed] [Google Scholar]
- 119.Tahara K, Kim HD, Jin JJ, Maxwell JA, Li L, Fukuchi KI. Role of toll-like receptor signalling in Aβ uptake and clearance. Brain. 2006;129(11):3006–3019. doi: 10.1093/brain/awl249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Richard KL, Filali M, Préfontaine P, Rivest S. Toll-like receptor 2 acts as a natural innate immune receptor to clear amyloid β1–42 and delay the cognitive decline in a mouse model of Alzheimer’s disease. Journal of Neuroscience. 2008;28(22):5784–5793. doi: 10.1523/JNEUROSCI.1146-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Du Y, Dodel R, Hampel H, et al. Reduced levels of amyloid β-peptide antibody in Alzheimer disease. Neurology. 2001;57(5):801–805. doi: 10.1212/wnl.57.5.801. [DOI] [PubMed] [Google Scholar]
- 122.Maetzler W, Langkamp M, Lerche S, Godau J, Brockmann K, et al. Lowered serum amyloid-beta1–42 autoantibodies in individuals with lifetime depression. Journal of Alzheimer’s Disease. 2012;32(1):95–100. doi: 10.3233/JAD-2012-120625. [DOI] [PubMed] [Google Scholar]
- 123.Britschgi M, Olin CE, Johns HT, et al. Neuroprotective natural antibodies to assemblies of amyloidogenic peptides decrease with normal aging and advancing Alzheimer’s disease. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(29):12145–12150. doi: 10.1073/pnas.0904866106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hampel H, Prvulovic D, Teipel S, et al. The future of Alzheimer’s disease: the next 10 years. Progress in Neurobiology. 2011;95(4):718–728. doi: 10.1016/j.pneurobio.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 125.Rinne JO, Brooks DJ, Rossor MN, et al. 11C-PiB PET assessment of change in fibrillar amyloid-β load in patients with Alzheimer’s disease treated with bapineuzumab: a phase 2, double-blind, placebo-controlled, ascending-dose study. The Lancet Neurology. 2010;9(4):363–372. doi: 10.1016/S1474-4422(10)70043-0. [DOI] [PubMed] [Google Scholar]
- 126.Maetzler W, Stapf AK, Schulte C, et al. Serum and cerebrospinal fluid uric acid levels in lewy body disorders: associations with disease occurrence and amyloid-β pathway. Journal of Alzheimer’s Disease. 2011;27(1):119–126. doi: 10.3233/JAD-2011-110587. [DOI] [PubMed] [Google Scholar]
- 127.Brody DL, Magnoni S, Schwetye KE, et al. Amyloid-β dynamics correlate with neurological status in the injured human brain. Science. 2008;321(5893):1221–1224. doi: 10.1126/science.1161591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Maetzler W, Stoycheva V, Schmid B, et al. Neprilysin activity in cerebrospinal fluid is associated with dementia and amyloid-β42 levels in lewy body disease. Journal of Alzheimer’s Disease. 2010;22(3):933–938. doi: 10.3233/JAD-2010-101197. [DOI] [PubMed] [Google Scholar]
- 129.Maetzler W, Schmid B, Synofzik M, et al. The CST3 BB genotype and low cystatin C cerebrospinal fluid levels are associated with dementia in lewy body disease. Journal of Alzheimer’s Disease. 2010;19(3):937–942. doi: 10.3233/JAD-2010-1289. [DOI] [PubMed] [Google Scholar]
- 130.Iseki E, Marui W, Akiyama H, Uéda K, Kosaka K. Degeneration process of Lewy bodies in the brains of patients with dementia with Lewy bodies using α-synuclein-immunohistochemistry. Neuroscience Letters. 2000;286(1):69–73. doi: 10.1016/s0304-3940(00)01090-9. [DOI] [PubMed] [Google Scholar]
- 131.Ishizawa K, Komori T, Sasaki S, Arai N, Mizutani T, Hirose T. Microglial activation parallels system degeneration in multiple system atrophy. Journal of Neuropathology and Experimental Neurology. 2004;63(1):43–52. doi: 10.1093/jnen/63.1.43. [DOI] [PubMed] [Google Scholar]
- 132.Stefanova N, Reindl M, Neumann M, et al. Oxidative stress in transgenic mice with oligodendroglial α-synuclein overexpression replicates the characteristic neuropathology of multiple system atrophy. American Journal of Pathology. 2005;166(3):869–876. doi: 10.1016/s0002-9440(10)62307-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Hishikawa N, Hashizume Y, Yoshida M, Niwa JI, Tanaka F, Sobue G. Tuft-shaped astrocytes in Lewy body disease. Acta Neuropathologica. 2005;109(4):373–380. doi: 10.1007/s00401-004-0967-3. [DOI] [PubMed] [Google Scholar]
- 134.Terada S, Ishizu H, Yokota O, et al. Glial involvement in diffuse Lewy body disease. Acta Neuropathologica. 2003;105(2):163–169. doi: 10.1007/s00401-002-0622-9. [DOI] [PubMed] [Google Scholar]