

Abstract
The hallmark of pancreatic tumours, the desmoplastic reaction, provides a unique microenvironment that affects pancreatic tumour behaviour, its ability to grow and metastasize as well as resist the effects of chemotherapy. Complex molecular interactions and pathways give rise to the desmoplastic reaction. Breakdown or penetration of the desmoplastic reaction may hold the key to overcoming the limits of delivery of efficacious chemotherapy or the development of new targeted treatments. Herein we discuss such new developments to fight the desmoplastic reaction, including inhibitors of the epidermal growth factor, fibroblast growth factor, the hedgehog pathway, as well as new molecular targets like CD40 agonist and its effects on T cells, extracellular matrix modifying enzymes such as LOXL2 inhibitor and novel tumour penetrating peptides for delivery of drugs.
1. Introduction
It is well recognised that the growth of dense, collagen-rich, extracellular matrix and stroma with high interstitial pressure around pancreatic tumours, known as the desmoplastic reaction, creates a unique microenvironment that paradoxically promotes both tumour growth and metastatic spread and at the same time forms a barrier to chemotherapy penetration. Targeting components of the tumour stroma that contribute to the desmoplastic reaction is a promising new platform of investigation. Most strategies comprise of increasingly newly identified peptides that aim to enhance chemotherapeutic and even radiotherapeutic efficacy, by increasing tumour accumulation, penetration, and drug-distribution and targeting signalling pathways, which are directly implicated in the formation of desmoplastic reaction.
The hallmark of the desmoplastic reaction in tumours originating from solid epithelial glands is a dense amount of interstitial fibrillar collagen (type I and III) and accelerated proliferation of fibroblasts. Tumour-stromal interactions between pancreatic cancer cells and stromal fibroblasts lead to enhanced key gene expression promoting primary tumour incidence, tumour growth, metastasis, and angiogenesis. The tumour cells themselves are able to produce extracellular matrix (ECM) proteins and integrins [1, 2] and interact with ECM by expressing functionally active ingredients [3, 4]. The stromal production is facilitated by an abundance of growth factors including fibroblast growth factors, epidermal growth factors receptor ligands, transforming growth factor beta isoforms, and connective tissue growth factors [5]. This environment nourishes the cancer cells and facilitates invasive and metastatic potential. In this regard, any agents that target profibrotic growth factors such as small molecule tyrosine kinase inhibitors that interfere with the epidermal growth factor (EGF) receptor, FDG, platelet-derived growth factor (PDGF) receptor signalling may be useful in suppressing the proliferation of fibroblast and stellate cells (Table 1).
Table 1.
Classification of antidesmoplastic agents.
| Agent | Class |
|---|---|
| PD 98059 | MEK 1 inhibitor |
| U0126 | MEK inhibitor |
| LY294002 | ERK inhibitor |
| PP1-PP2 | TβR inhibitors |
| SB431542 and SB525334 | TβRI selective inhibitor |
| LY2109761 | TβRI/II dual inhibitor |
| SD-208 | TβRI inhibitor |
| AP 12009 | TGFβ2 mRNA phosphorothioate antisense oligodeoxynucleotide |
| 2G8 | Neutralising antibody to TβR2 neutralising antibody |
| IPI-926 | SMO Semisynthetic cyclopamine analogue inhibitor |
| GDC-0449 | 2-arylpyridine class SMO inhibitor |
| iRGD | Disulfide-based cyclic RGD tumour-penetrating peptide |
| CP870,893 | IgG2 antibody to CD40 |
| AB0023 | Allosteric inhibitor of LOX-L2 |
2. Discussion
2.1. Transforming Growth Factor Beta (TGFβ)
Many growth factors expressed by human pancreatic carcinoma cells have the ability to induce fibroblast proliferation, for example, transforming growth factor β1 (TGFβ1) and fibroblast growth factor (FGF) 2 and are associated with advanced tumour stage and decreased survival.
TGFβ is a potent cytokine that regulates mammalian development, differentiation, and homeostasis and normally exerts anticancer activities by prohibiting cell proliferation, motility, invasion, and metastases. In the process of tumourigenesis genetic and epigenetic events and aberrant alterations within the tumour confer TGFβ oncogenic activities, causing direct metastatic progression via stimulation of epithelial-mesenchymal transition (EMT). EMT also confers stem cell like properties to transitioned cells such as self renewal, tumour initiating capability, and chemoresistance [6].
TGFβ exerts its effects through TGFβ 1 and 2 receptors (TβR1 and TβR2), and Smad transcription regulators. TGFβ binding to TβR2 initiates a cascade that leads to Smad 2 and 3 activation, which in turn binds to Smad 4; the activated complex is transcriptionally active in the nucleus [7]. The growth inhibitory effect of TGFβ is thought to be mediated by Smad-dependent TGFβ signalling. In pancreatic defects in Smad proteins, especially Smad 4 or TβR2 lead to resistance to the growth inhibitory effects of TGFβ. These events in combination with activated K-Ras result in rapid tumour development. In human pancreatic cancer cells, TGFβ1, overexpression correlates with collagen I levels, suggesting that TGFβ1 is directly able to elicit the desmoplastic reaction, an observation which has been confirmed in experimental models of pancreatic cancer [8]. There is also cross-talk between collagen, TGFβ1, and MT1-MMP. MT1-MMP overexpression has been linked with fibrosis and various signalling pathways including Snail pathway, cadherins, Ras/MEK/ERK.
TGFβ also induces Snail family of transcription factors through the Smad pathway. In PDAC, collagen activates TGFβ signalling, in turn leading to increased Snail expression; whereas blocking TGF signalling with a highly specific TβRI inhibitor blocks collagen-induced Snail expression [9]. In addition, knocking down Smad 3 abrogates Snail-induced collagen fibrosis. Therefore TGFβ is a critical signalling pathway in the development and propagation of the desmoplastic reaction. The TGFβ pathway has been targeted using various strategies including small molecule inhibitors of TβRI, TGFβ-specific neutralizing antibodies, and antisense compounds [10].
As already discussed above, TGF binding to TβR2 receptor leads to activation of Smad proteins which mediate gene expression related to cell growth control. Part of this effect is mediated by the Ras/MEK/ERK signalling cascade. MEK 1 inhibitor PD 98059 reduced TGFβ1 related increase of tumour cell scattering migration and invasion [11358848] and enhances efficacy of gemcitabine. More recently, another molecule, Lefty, was identified downstream of the Ras/MEK/ERK pathway to mediate growth inhibition in pancreatic cell lines. Activation of the pathway in pancreatic cancer suppresses Lefty activation and enables cancer cells to escape growth inhibition. Inhibition of the pathway enhances TGF-mediated lefty upregulation with potential therapeutic applications [11]. The Smad pathway is also blocked by PP1 and PP2, Src family kinase inhibitors that inhibit TGFβ-Smad signalling [12].
TβR1 inhibitors have also been used in combination with gemcitabine in an attempt to improve chemopenetration. Two such molecules, SB431542 and SB525334 are able to augment the cytotoxic effects of gemcitabine [13]; SB525334 also increased apoptotic cell death and affected both the AKT pathway, and TβR1 receptor, the former crucial in gemcitabine resistance and the latter known to affect cell migration. In a similar fashion, LY2109761 suppressed both basal and TGFβ1-induced cell migration and invasion. In combination with gemcitabine, it reduced tumour burden, prolonged survival, and reduced spontaneous abdominal metastases [14]. The first human Phase I study of oral TβR1 inhibitor LY2157299 in patients with treatment-refractory malignant glioma is currently underway with promising results [15].
Another small molecule, SD-208, blocking TβR1, resulted in inhibition of expression of genes associated with tumour progression and inhibition of invasiveness in a cell-based assay. SD-208 treatment reduced proliferation and induced apoptosis in the primary tumours, and reduced fibrosis in the tumour microenvironment [16]. Similarly, Trabedersen (AP 12009) is a phosphorothioate antisense oligodeoxynucleotide specific for human TGFβ2 mRNA with antitumour activity in human pancreatic cancer, such as reduction in tumour growth, lymph node metastases, and angiogenesis [17]. The TβR2 has also been targeted by specific neutralising antibodies. 2G8 an anti-rat monoclonal antibody specifically binds and blocks TβR2, inhibiting Smad 2. As a result, reducing tumour cell migration and inhibition of tumour cell migration as well as reduced EMT transcription factors are observed, which may translate in possible delayed tumour progression. This antibody has also been shown to inhibit tumour metastases in vivo [18].
More recently, further TβR molecular pathways have been identified such as the regulation of cell adhesive properties by decreasing expression of E cadherin. These results in increased expression of invasion associated integrins and integrin binding proteins, promoting invasion and metastasis, ECM and related protein production (collagen, fibronectin, decreases collagenase, heparinize, and stromelysins) as well as plasminogen activator inhibitor 1 and tissue inhibitor of metalloprotease that inhibit ECM degradation and increase proteolytic activity of cells [19]. Furthermore, there have been reports of significant association between plasma TGFβ1 and overall survival in patients with locally advanced metastatic disease, Smad 4 loss correlation with lower survival with potential important implications in treatment decision [20, 21]. Clearly the increasing understanding of TGFβ and its functions has brought a new era in molecular therapeutics. However, acquired resistance to small molecule inhibitors is a problem that has already manifested, with resultant carcinomas more aggressive and inflammatory [22]. The recent discovery that there is transcriptional talk between TGFβ and stem cell pathways holds more promising research to come [23].
3. Fibroblast Growth Factor (FGF)
Another important function of TGFβ is that it increases production of mitogenic growth factors including fibroblast growth factor. Fibroblasts are responsible for synthesis, degradation, and remodelling of ECM and can modulate behaviour of cancer cells through cytokine secretion and modification of ECM environment. Fibroblasts are thought to be mesenchymal cells, known as stellate cells, which have differentiated into myofibroblasts that secrete collagen I, which is highly resistant to proteolysis. Stellate cells are thought to mediate the invasive potential of PDAC cells and promote EMT [24] as well as resistance to radiotherapy [25]. FGF mediates its effects through different receptor isoforms. In particular, FGFR1 IIIb isoform is associated with inhibition of cancer cell proliferation, migration, and invasion, whereas FGFR1 IIIc enhances cell proliferation. FGFR2 IIIb increases venous invasion but FGFR2 IIIc is associated with metastases, more aggressive tumours and confers PDAC cells features suggestive of cancer stem cells [26]. The FGF binding protein is dramatically upregulated in pancreatic cancer and is linked to the initiation and progression of pancreatic cancer [27]. Various preclinical studies have shown FGFR signalling inhibition may play a role in inhibiting tumour growth [28]. Neutralising monoclonal antibodies to FGF2 has been shown to suppress hepatocellular cancer growth by blocking angiogenesis and inhibiting downstream cellular signalling.
4. CD44 and Hyaluronan
Another key role of fibroblasts in the desmoplastic reaction is hyaluronan synthesis and its interaction with CD44. CD44 is another integral cell-surface glycoprotein; overexpression of its variant forms, driven by IFN gamma, has been associated with malignant transformation of pancreatic tumours [29, 30]. In fact, pretreatment levels of CD44 and its variants have been correlated with TNM staging and may well be able to serve as tumour markers in head and neck cancers [31].
CD44 is also critical in pancreatic carcinogenesis as it is the major cell surface receptor for hyaluronan, as well as matrix metalloproteinases. Hyaluronan, is a glycosaminoglycan, able to interact with extracellular matrix molecules (hyaladherins) affecting matrix structure but also cell function through its interaction with CD44, making it another key component of the stromal reaction. In addition, its breakdown products, via hyaluronidase activity, promote angiogenesis and in turn tumour neovascularisation [32]. Hyaluronan is produced by fibroblasts in response to factors released from tumour cells, such as lactate, or by direct cell-cell contact [33]. Hyaluronan-rich stroma is associated with poor prognosis in many epithelial cancers including pancreatic and together with CD44 promotes tumour cell growth, migration, and metastases [33, 34]. It is thought that hyaluronan provides increased barrier integrity and chemoresistance through CD44-dependent reorganisation of the tumour cytoskeleton [35], where as the anti-CD44 monoclonal antibody IM7 (anti-CD44 IgG2b mAb IM7) improves vascular permeability [36]. Disruption of the hyaluronan-CD44 interaction is a key therapeutic target to prevent tumour refractoriness secondary to drug resistance [37]. One such strategy implores a hyaluronan synthesis inhibitor, 4-Methylumbelliferone (4-MU), has been shown to inhibit cell migration, proliferation, and invasion [38, 39]. The ability of 4-MU to suppress hyaluronan synthesis and accumulation has recently been linked to suppression of bone metastases in breast cancer [40]. Its inhibitory effect has been shown to slow down the development of human pancreatic cancer cell lines in vitro and in mice [41, 42] but also to enhance the efficacy of gemcitabine [43]. In a similar fashion, the action of PEGylated human recombinant PH20 hyaluronidase (PEGPH20) acting as a hyaluronan depletor improved chemopermeability of doxorubicin and gemcitabine and when given in combination with the latter led to inhibition of pancreatic tumour growth and improved survival over gemcitabine alone (median survival 28.5 days versus 15) [44, 45].
5. Hedgehog Pathway
Hedgehog is a signalling pathway that is genetically altered and aberrantly activated in the majority of pancreatic cancers leading to tumour initiation, progression, and metastatic spread. In addition, it has been implicated in the initiation and maintenance of the desmoplastic reaction (Figure 1). The hallmark of the desmoplastic reaction is a dense amount of interstitial fibrillar collagen (type I and III) and accelerated proliferation of fibroblasts. The latter are thought to be mesenchymal cells, known as stellate cells, which have differentiated into myofibroblasts that secrete collagen I, which is highly resistant to proteolysis. Hedgehog (HH) signalling promotes myofibroblast differentiation and induces stroma-derived growth promoting molecules, which are in turn tumourigenic. In addition, HH ligands induce matrix metalloproteinases and TGFβ1, which are both highly active in the desmoplastic reaction formation and directly involved in fibrosis. The pathway is activated when sonic hedgehog ligands (SHH) bind to the patched receptor (PTCH) relieving the inhibitory effects of Patch (PTCH) on smoothened (SMO) and activating the GL1 family of transcription factors which turn on the Hedgehog genes such as PTCH, epidermal-derived, platelet-derived, and vascular-endothelial growth factors, cyclins B, D, and E and GLI1. Bulk cancer cells secrete hedgehog ligands to activate the pathway in stroma and cancer stem cells, promoting the formation of desmoplastic reaction and facilitating maintenance of cancer stem cells involved in metastases. Ectopic production of HH ligands has been associated with pancreatic tumourigenesis [47]. In addition, overexpression of SMO in cancer-associated stromal fibroblasts has been observed that in turn activates the HH signalling pathway [48]. Evidence also suggests that tumour cells secrete HH ligand to induce tumour-promoting HH target genes in a paracrine fashion in adjacent stroma to support tumour growth [49, 50].
Figure 1.
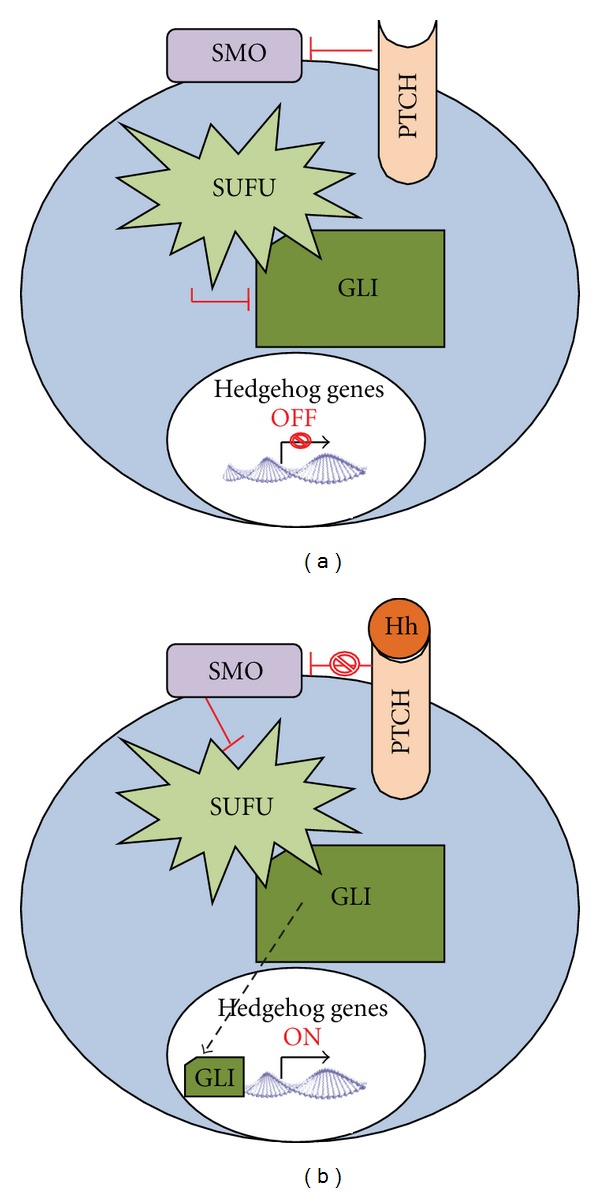
The Hedgehog pathway [46].
Blocking the hedgehog pathway in vitro studies, with the small molecule cyclopamine, a naturally occurring antagonist of the hedgehog signalling pathway component (smoothened-transmembrane receptor), leads to abrogation of pancreatic metastases and potential improvement in chemodelivery [51, 52]. IPI-926 a semisynthetic cyclopamine analogue was developed to inhibit SMO. It has been shown to reduce the desmoplastic reaction and increase tumour vascular density by blocking hedgehog signalling and hence blocking metastatic spread and tumour initiation. Inhibition of Hedgehog signalling has been shown to enhance the delivery of drugs in vitro [53] and can occur in many platforms including HH ligand inhibition, SMO antagonism, and Gli transcriptional activity inhibition.
Several studies have been designed to assess the synergistic function of Hedgehog inhibitors delivered alongside with established antineoplastic agents [54]. In one such study, Stephenson et al. tested the safety profile of IPI-926 in previously untreated metastatic pancreatic cancer in a phase Ib trial. They noted that IPI-926 facilitated the delivery of gemcitabine by diminishing tumour-associated desmoplasia with 31% of patients showing partial response and 63% showing reduction in CA 19-9. Treatment was confounded by grade 3 toxicity fatigue and transaminitis. A randomised double-blind placebo-controlled study is underway to assess survival comparison between the treatment and placebo arms, where the treatment arm will receive daily 160 mg oral IPI-926 plus gemcitabine infusion at 100 mg/m2 once weekly for 3 weeks of a 28-day cycle [NCT01130142]. Unfortunately the Phase II trial by Infinity was recently stopped because of futility of treatment [55].
Another SMO inhibitor, GDC-0449/Erivedge, also known as vismodegib, is an orally administrable molecule 2-arylpyridine class that inhibits SMO and is highly selective for SHH-Gli signalling, though to act by inhibiting SHH pathway at the level of Gli genes. Gli signalling has been implicated in the regulation of cell proliferation, cell cycle, and cell survival. GDC-0449 has been shown to inhibit pancreatic cancer cell viability, Gli-DNA binding and transcriptional activity and induces apoptosis in three pancreatic cancer cell lines and stem cells [56]. It also inhibited expression of HH receptors, such as Patched and SMO and effectors. Preclinical studies have demonstrated antitumour activity in xenograft models of pancreatic cancer [57]. LoRusso et al. presented their Phase I trial results in 2011 utilising GDC-0449 in patients with refractory, locally advanced or metastatic solid tumours, including 8 with pancreatic cancer [58] [21300762]. The molecule was able to produce tumour responses in 20 patients with BCC and medulloblastoma. The best observed response for pancreatic cancer was seen in one patient with stable disease at 2.8 months. Most promising was that Gli1 downregulation was noted and the treatment was associated with low toxicity. Recently following Phase II trials in BCC, the drug was approved by the FDA for the treatment of metastatic or locally advanced BCC that cannot be treated with surgery or radiotherapy. The trial showed partial response in 30% of patients with metastatic disease and complete or partial response in 43% of patients with locally advanced disease (ERIVANCE trial BCC/SHH4476g AACR). The theory behind GDC-0449 altering HH signalling is being tested in a Phase II study with vismodegib in the preoperative setting for patients with local, resectable disease to detect change in HH signalling in the normal tumour surrounding tissue (Proof of Mechanism Study of an Oral Hedgehog Inhibitor GDC-0449 in Patients With Resectable Pancreatic Ductal Adenocarcinoma in the Pre-operative Window Period, also known as HIPPoS by Cambridge University Hospitals NHS Foundation, NCT01096732, estimated primary completion date September 2012) looking at whether blocking the HH pathway will directly affect tumour cells or the surrounding normal tissue.
One of the main reasons for ultimate resistance to therapy is due to the existence of cancer stem cells which are resistant to chemotherapy and lead to treatment failure. The Michigan group are currently evaluating the combination of vismodegib with gemcitabine for patients with advanced disease and its effect to cancer stem cells and HH pathway (cancer stem cells and inhibition of HH pathway signalling in advanced pancreas cancer: a pilot study of GDC in combination with gemcitabine-NCT01195415), in a hope that pretreatment with GDC-0449 will inhibit the HH pathway in cancer cells and downstream tumour microenvironment enhancing treatment efficacy for gemcitabine. One of the primary endpoints is to evaluate the effect of HH signalling inhibition on pancreatic cancer stem cells by assessing the number of cancer stem cells before and after GDC-0449 treatment. Preliminary results of this trial show that three out of five patients who received pretreatment with GDC-0449 followed by gemcitabine treatment showed partial response, reduction in CA 19-9 levels, and increased vacuolated structures in tumour cells of one patient. The estimated primary completion date for this study is June 2013.
With a similar target in mind, another open label, single arm, multicentre Phase II trial is currently evaluating the progression free survival in patients with metastatic adenocarcinoma treated with vismodegib in combination with gemcitabine and nab-Paclitaxel (a Phase II Study of Gemcitabine and Nab-Paclitaxel in Combination With GDC-0449 (Hedgehog Inhibitor) in Patients With Previously Untreated Metastatic Adenocarcinoma of the Pancreas by Sidney Kimmel comprehensive Cancer Centre at John Hopkins-NCT01088815, estimated primary completion date December 2012). Abraxane is thought to weaken the stroma allowing for better chemotherapeutic efficacy of gemcitabine, using GDC-0449 to destroy the stroma but also to kill cancer stem cells. Furthermore Abraxane has shown clinical activity in patients overexpressing secreted protein acidic and rich in cysteine (SPARC), as it binds to the albumin portion of paclitaxel, potentially providing a tool to reverse gemcitabine resistance. Measurement of SPARC levels may also serve as a prognostic factor for treatment success [59, 60].
Other Phase I trials currently underway are assessing combination treatments with GDC-0449 such as in combination with Sirolimus or Erlotinib and Gemcitabine. Preliminary results are encouraging and have shown disease stabilisation and low drug-related toxicities (DLTs) for Erlotinib with Gemcitabine and GDC-0449. (Gemcitabine Hydrochloride With or Without GDC-0449 in Treating Patients With Recurrent or Metastatic Pancreatic Cancer by University of Chicago NCT01064622 to assess progression free survival; Sirolimus and Vismodegib in Treating Patients With Solid Tumours or Pancreatic Cancer That is Metastatic or Cannot Be Removed By Surgery by Mayo Clinic NCT01537107, primary completion date January 2014; GDC-0449 and Erlotinib Hydrochloride With or Without Gemcitabine Hydrochloride in Treating Patients With Metastatic Pancreatic Cancer or Solid Tumours That Cannot Be Removed by Surgery by Mayo clinic NCT00878163). Preliminary results are showing stable disease and low DLTs [61].
An important consideration is that SMO is localised in the primary cilium of the cell, which is critical in HH signalling and cancer progression. Primary cilia are required for the activation of the HH pathway in normal cells but are lost in many cancers. Some drugs may be ineffective in the absence of primary cilia [62]. Hence further research into overcoming this barrier should be considered when designing new platforms.
6. iRGD: a Tumour Penetrating Peptide for Peptide-Mediated Delivery of Drugs
One of the main reasons for treatment failure remains inability to penetrate the stromal reaction and the generation of elevated intratumour interstitial pressure. Crossing the vascular wall and penetrating into the tumour parenchyma is the main challenge for efficacious drug delivery. Recent attention has been paid to penetrating peptides for peptide-mediated drug delivery, especially peptides containing an RGD integrin recognition motif which allows them to bind to av integrins on the tumour cell surface. However to date, conventional RGD peptides have only been able to penetrate blood vessels but not the extravascular tumour parenchyma. A newly devised peptide, iRGD, a disulfide-based cyclic RGD peptide, seems to have overcome this obstacle by also targeting a downstream receptor, neuropilin-1. iRGD is a synthetic peptide containing a motif that binds to av integrins on tumour endothelium. Upon binding, the peptide is proteolytically cleaved to expose a CRGDK fragment, losing its integrin affinity but gaining affinity for neuropilin-1 instead. The new complex triggers tissue penetration, thus this peptide penetrates through the tumour vasculature into the tumour parenchyma [63].
Since the peptide is able to penetrate into the tumour parenchyma, coupling of the peptide with drugs may improve the drug delivery and efficacy, especially as iRGD seems to home to tumours but not normal tissue. av integrin and neuropilin-1 expression is largely restricted to tumours but most importantly the response is tumour specific because the peptide cleavage will only occur if there has been prior integrin activation. The hypothesis has been tested in mouse tumour models including pancreatic adenocarcinoma where various drugs including doxorubicin, nab-paclitaxel (abraxane), and doxorubicin liposomes as well as trastuzumab were coadministered with the peptide, without the need for chemical conjugation therefore preserving drug activity and improving tolerability. Tumour accumulation was increased 12-fold for abraxane, 14-fold for the doxorubicin liposomal nanoparticle, and 7-fold for the free drug and 40-fold for trastuzumab indicating that iRGD leads to enhance drug delivery to cancer cells [64]. The manufacturing company has already initiated SBIR trials with iRGD in combination with gemcitabine with preliminary data showing that iRGD enhances the anti-tumoural activity of gemcitabine in orthotopic models of pancreatic cancer [65].
7. CD40 Agonist
CD40 is a type I transmembrane glycoprotein receptor of the TNF-receptor superfamily widely expressed by immune cells such as dendritic cells, B cells, and macrophages but also endothelial cells, smooth muscle cells, fibroblasts, and epithelial cells. The CD40 ligand (CD40L) primarily expressed in the surface of activated T cells interacts with CD40+ B cells to produce multiple regulatory signals including T-cell and B-cell-dependent proliferation, immunoglobulin production and switching, and apoptosis. CD40L+T cells augment the antigen-presenting function of CD40+ B cells and other antigen-presenting cells (APCs) generating a number of interactions between CD4 and CD8 T cells [66, 67].
Interestingly, CD40 is also expressed in the membrane and cytoplasm of tumour cells but is absent from non-proliferating tissues. Its activation promotes apoptotic death and generation of tumour specific T-cell responses that contribute to tumour elimination [68]. The exact mechanism of CD40-CD40L interaction is still unclear as CD40 expression has been correlated with worse tumour prognosis, TNM stage, and lymph node metastases, perhaps because the CD40L is rarely expressed on pancreatic cancer TILs and hence unable to downregulate CD40+ cancer growth. In fact, presence of CD40L expression has been linked to improved survival [69]. In addition, epigenetic alterations of miRNA-regulated CD40 expression lead to downregulation of CD40 expression in pancreatic cancer cells promoting invasion and metastasis [70]. CD40 also engages in endothelial cells to induce in vitro tubule formation and expression of matrix metalloproteinases [71]. In a recent Phase I trial by He et al. [72], recombinant soluble human CD40L was used to block CD40 and demonstrated significant growth inhibitory effect in vitro. Specifically they showed the ligand was able to cause not only growth arrest but also cancer cell apoptosis. CD40 binding antibodies have the potential to modulate pancreatic cancer cell growth. Binding of recombinant soluble CD40L or with a CD40 reactive monoclonal antibody may produce a direct inhibitory effect on cancer cells. CD40 agonist antibody CP-870,893 can achieve substantial regression of tumours in some patients with inoperable pancreatic binding antibodies may bind to epitopes distinct from those involved in the natural CD40-CD40L interaction. Similarly CD40 monoclonal antibodies may cause collateral activation of antibody dependent cellular cytotoxicity.
CP-870,893 is a fully human IgG2 antibody that selectively interacts with CD40 at a distinct site from its ligand-binding region. Binding enhances MHCII expression as well as dendritic cell activity and is therapeutically effective against several CD40 + human tumours. In a Phase 1 dose escalation open label study CP-870,893 was combined with gemcitabine in patients with chemotherapy naive surgically incurable pancreatic cancer [73], tumour regression was observed a subsequent mouse model that tumour regression was T cell and gemcitabine independent but dependent on macrophages, that infiltrated the tumour and facilitated the depletion of the tumour stroma. Soon underway a small open label single-arm Phase I study looking at preoperative gemcitabine together with CP870,893 followed by addition of CP-870,893 to adjuvant chemoradiotherapy for patients with newly diagnosed resectable pancreatic cancer. Patients will receive standard surgery followed by chemoradiotherapy; one dose of gemcitabine/CP870,893 will be preoperatively and 3 doses postoperatively.
8. LOX-L2
Lysyl oxidase like 2 belongs to the lysyl oxidase family of extracellular matrix modifying enzymes. This group of enzymes plays an important role in connective tissue biogenesis, cellular adhesion, motility and migration, gene transcription regulation, and senescence, as well as cancer progression. Increased LOX-L2 expression has been identified in many cancers including the pancreas. In breast cancer, high levels of LOX-L2 expression appear to correlate with decreased overall survival and metastases free survival (P = 0.023 and P = 0.0367, resp.) [74]. Interestingly, LOX-L2 does not appear to be required for primary tumour growth but enables metastases in vivo.
LOXL2 serves as an extracellular matrix metalloenzyme and has been shown to catalyse the first step in the formation of crosslinks in fibrillar collagen and elastin [75, 76]. Cross-linking of collagen activates other enzymes involved in matrix remodelling such as MMPs, enhancing tumour cell invasion [77]. Therefore LOX-L2 is directly able to modify the ECM, and its overexpression leads to propagation of the desmoplastic reaction. Positive association between LOX-L2, TIMP1, and MMP9 has also been noted in human colorectal cancer [78–80]. LOXL2 inhibition has also been associated with reduction in activated fibroblasts, endothelial cells, desmoplasia, and decrease in transforming growth factor-beta signalling making LOX-L2 a potential target for fighting the desmoplastic reaction [81].
Preclinical evidence suggests that in vivo blocking LOXL2 both in vivo and in vitro is highly effective in preventing distant metastases in breast cancer through regulation of tissue inhibitor of metalloproteinase 1 (TIMP1), leading to increased TIMP1 and MMP 9 activity and facilitating ECM remodelling [82].
In pancreatic cancer cell lines, gene silencing by inhibition with small interfering RNAs has been shown to result not only in cell death but also in increased sensitivity to gemcitabine treatment [83]. In this study, LOXL2 appeared to regulate E2F5 transcription factor associated with invasion and metastases. Blocking not only LOXL2 but its effectors too, such as E2F5 or even RAMP3, a molecule downstream of LOXL2 thought to mediate some of its tumourigenic activity [81], might also prove beneficial as antitumourigenic agents.
In addition, development of specific allosteric inhibitors of LOXL2, such as AB0023, bind remote to its catalytic domain, allowing inhibition of LOXL2 regardless of substrate concentration [84]. This concept has many prospects: the ability to confer a molecule high specificity and selectivity for the cancer without affecting normal tissues, development of high affinity binders, and using different specificities of LOXL2 targeting antibodies to alter the outcome.
More excitingly, recently an intracellular function of LOXL2 has been described for the first time in relation to E-cadherin and histone H3; In normal cells, methylation of lysine 4 within histone 3 activates CDH1 transcription and E-cadherin formation, while histone deacetylation plays an important role in downregulation of E-cadherin in human pancreatic cancer promoting tumour cell migration and proliferation [85]. Loss of the cell adhesion molecule E-cadherin is critical in pancreatic tumourigenesis. LOXL2 has been found to act in the nucleus of cancer cells and deaminates the lysine 4 amino group of H3 leading to downregulation of CDH1, decreased E-cadherin expression, fewer cellular adhesions facilitating tumour growth and metastases [86].
9. Radiotherapy
As already mentioned above, there is data suggesting that pancreatic stellate cells confer protection against radiotherapy through β1-integrin and FAK signaling [25]. β1-integrin signaling and in particular integrin-mediated adhesion to extracellular matrix proteins has been implicated in mediating cell survival in response to radiation in different cancer cell lines [87]. Other PSC-specific matrix proteins such as periostin, stimulate growth, and confer resistance even under the effects of radiotherapy, continuing to enhance the desmoplastic reaction by producing excessive extracellyular matrix proteins [88]. Inhibition of the pathway enhances the efficacy of radiotherapy [30, 89]. More recently the role of caveolin-1 (Cav-1) as a critical signaling molecule within the β1-integrin and FAK pathway was described. Knockdown models of caveolin-1 increased radiosensitisation in human pancreatic cell lines [90]. Further research in this domain is required to enhance in vivo radiosensitivity.
10. Conclusion
Increasing understanding of the desmoplastic reaction and the heterogeneity of alterations of signalling pathways in pancreatic cancer is already providing us with new insights into how to fight desmoplasia. Preliminary evidence encourages the idea that attenuating the desmoplastic reaction may help limit the molecular and clinical course of pancreatic cancer, contain its progression, and enhance the response to chemotherapy. There is a long way to go until this evidence will become practice.
Conflict of Interests
The authors have no potential conflict of interests.
References
- 1.Lohr M, Trautmann B, Gottler M, et al. Human ductal adenocarcinomas of the pancreas express extracellular matrix proteins. British Journal of Cancer. 1994;69(1):144–151. doi: 10.1038/bjc.1994.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tani T, Lumme A, Linnala A, et al. Pancreatic carcinomas deposit laminin-5, preferably adhere to laminin- 5, and migrate on the newly deposited basement membrane. American Journal of Pathology. 1997;151(5):1289–1302. [PMC free article] [PubMed] [Google Scholar]
- 3.Löhr M, Trautmann B, Göttler M, et al. Expression and function of receptors for extracellular matrix proteins in human ductal adenocarcinomas of the pancreas. Pancreas. 1996;12(3):248–259. doi: 10.1097/00006676-199604000-00007. [DOI] [PubMed] [Google Scholar]
- 4.Weinel RJ, Rosendahl A, Neumann K, et al. Expression and function of VLA-α2, -α3, -α5 and -α6-integrin receptors in pancreatic carcinoma. International Journal of Cancer. 1992;52(5):827–833. doi: 10.1002/ijc.2910520526. [DOI] [PubMed] [Google Scholar]
- 5.Korc M. Pancreatic cancer—associated stroma production. American Journal of Surgery. 2007;194(4 supplement):S84–S86. doi: 10.1016/j.amjsurg.2007.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wendt MK, Tian M, Schiemann WP. Deconstructing the mechanisms and consequences of TGF-β-induced EMT during cancer progression. Cell and Tissue Research. 2012;347(1):85–101. doi: 10.1007/s00441-011-1199-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Elliott RL, Blobe GC. Role of transforming growth factor beta in human cancer. Journal of Clinical Oncology. 2005;23(9):2078–2093. doi: 10.1200/JCO.2005.02.047. [DOI] [PubMed] [Google Scholar]
- 8.Löhr M, Schmidt C, Ringel J, et al. Transforming growth factor-beta1 induces desmoplasia in an experimental model of human pancreatic carcinoma. Cancer Research. 2001;61(2):550–555. [PubMed] [Google Scholar]
- 9.Shields MA, Dangi-Garimella S, Krantz SB, Bentrem DJ, Munshi HG. Pancreatic cancer cells respond to type I collagen by inducing snail expression to promote membrane type 1 matrix metalloproteinase-dependent collagen invasion. The Journal of Biological Chemistry. 2011;286(12):10495–10504. doi: 10.1074/jbc.M110.195628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahn M, Kloeker S, Berry BS. TGF-β inhibitors for the treatment of cancer. Expert Opinion on Investigational Drugs. 2005;14(6):629–643. doi: 10.1517/13543784.14.6.629. [DOI] [PubMed] [Google Scholar]
- 11.Miyata N, Azuma A, Hozawa S, et al. Transforming growth factor beta and Ras/MEK/ERK signaling regulate the expression level of a novel tumor suppressor lefty. Pancreas. 2012;41(5):745–752. doi: 10.1097/MPA.0b013e31823b66d3. [DOI] [PubMed] [Google Scholar]
- 12.Ungefroren H, Sebens S, Groth S, Gieseler F, Fändrich F. The src family kinase inhibitors PP2 and PP1 block TGF-beta1-mediated cellular responses by direct and differential inhibition of type I and type II TGF-beta receptors. Current Cancer Drug Targets. 2011;11(4):524–535. doi: 10.2174/156800911795538075. [DOI] [PubMed] [Google Scholar]
- 13.Kim YJ, Hwang JS, Hong YB, Bae I, Seong YS. Transforming growth factor beta receptor I inhibitor sensitizes drug-resistant pancreatic cancer cells to gemcitabine. Anticancer Research. 2012;32(3):799–806. [PMC free article] [PubMed] [Google Scholar]
- 14.Melisi D, Ishiyama S, Sclabas GM, et al. LY2109761, a novel transforming growth factor β receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Molecular Cancer Therapeutics. 2008;7(4):829–840. doi: 10.1158/1535-7163.MCT-07-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ahnert JR, Baselga J, Calvo E, et al. First human dose (FHD) study of the oral transforming growth factor-beta receptor I kinase inhibitor LY2157299 in patients with treatment-refractory malignant glioma. Journal of Clinical Oncology. 2011;29(supplement, abstract 3011) ASCO Annual Meeting. [Google Scholar]
- 16.Medicherla S, Li L, Jing YM, et al. Antitumor activity of TGF-beta inhibitor is dependent on the microenvironment. Anticancer Research B. 2007;27(6):4149–4157. [PubMed] [Google Scholar]
- 17.Schlingensiepen KH, Jaschinski F, Lang SA, et al. Transforming growth factor-beta 2 gene silencing with trabedersen (AP 12009) in pancreatic cancer. Cancer Science. 2011;102(6):1193–1200. doi: 10.1111/j.1349-7006.2011.01917.x. [DOI] [PubMed] [Google Scholar]
- 18.Ostapoff K, Cenik B, Schwarz R, Brekken RA. Effect of 2G8, a TGF-beta-R2 inhibitor, on TGF-beta signaling and migration in an immunocompetent pancreatic cancer model. Journal of Clinical Oncology. 2012;30(supplement 4, abstract 230) ASCO Annual Meeting. [Google Scholar]
- 19.Hilbig A, Oettle H. Transforming growth factor beta in pancreatic cancer. Current Pharmaceutical Biotechnology. 2011;12(12):2158–2164. doi: 10.2174/138920111798808356. [DOI] [PubMed] [Google Scholar]
- 20.Schultz NA, Dehlendorff C, Werner J, et al. Diagnostic MicroRNA serum profile in pancreatic cancer. Journal of Clinical Oncology. 2012;30(supplement 4, abstract 160) ASCO Annual Meeting. [Google Scholar]
- 21.Strimpakos AS, Syrigos KN, Saif MW. Translational research. New findings and potential future applications in pancreatic adenocarcinoma. Journal of the Pancreas. 2012;13(2):177–179. [PubMed] [Google Scholar]
- 22.Connolly EC, Saunier EF, Quigley D, et al. Outgrowth of drug-resistant carcinomas expressing markers of tumor aggression after long-term TβRI/II Kinase inhibition with LY2109761. Cancer Research. 2011;71(6):2339–2349. doi: 10.1158/0008-5472.CAN-10-2941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fuxe J, Vincent T, De Herreros AG. Transcriptional crosstalk between TGFβ and stem cell pathways in tumor cell invasion: role of EMT promoting Smad complexes. Cell Cycle. 2010;9(12):2363–2374. doi: 10.4161/cc.9.12.12050. [DOI] [PubMed] [Google Scholar]
- 24.Kikuta K, Masamune A, Watanabe T, et al. Pancreatic stellate cells promote epithelial-mesenchymal transition in pancreatic cancer cells. Biochemical and Biophysical Research Communications. 2010;403(3-4):380–384. doi: 10.1016/j.bbrc.2010.11.040. [DOI] [PubMed] [Google Scholar]
- 25.Mantoni TS, Lunardi S, Al-Assar O, Masamune A, Brunner TB. Pancreatic stellate cells radioprotect pancreatic cancer cells through β1-integrin signaling. Cancer Research. 2011;71(10):3453–3458. doi: 10.1158/0008-5472.CAN-10-1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ishiwata T, Matsuda Y, Yamamoto T, Uchida E, Korc M, Naito Z. Enhanced expression of fibroblast growth factor receptor 2 IIIc promotes human pancreatic cancer cell proliferation. American Journal of Pathology. 2012;180(5):1928–1941. doi: 10.1016/j.ajpath.2012.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tassi E, Wellstein A. Tumor angiogenesis: initiation and targeting—therapeutic targeting of an FGF-binding protein, an angiogenic switch molecule, and indicator of early stages of gastrointestinal adenocarcinomas. Cancer Research and Treatment. 2006;38(4):189–197. doi: 10.4143/crt.2006.38.4.189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wagner M, Lopez ME, Cahn M, Korc M. Suppression of fibroblast growth factor receptor signaling inhibits pancreatic cancer growth in vitro and in vivo. Gastroenterology. 1998;114(4):798–807. doi: 10.1016/s0016-5085(98)70594-3. [DOI] [PubMed] [Google Scholar]
- 29.Ringel J, Jesnowski R, Schmidt C, et al. CD44 in normal human pancreas and pancreatic carcinoma cell lines. Teratog Carcinog Mutagen. 2001;21(1):97–106. [PubMed] [Google Scholar]
- 30.Nam JM, Chung Y, Hsu HC, Park CC. β1 integrin targeting to enhance radiation therapy. International Journal of Radiation Biology. 2009;85(11):923–928. doi: 10.3109/09553000903232876. [DOI] [PubMed] [Google Scholar]
- 31.Kawano T, Yanoma S, Nakamura Y, et al. Evaluation of soluble adhesion molecules CD44 (CD44st, CD44v5, CD44v6), ICAM-1, and VCAM-1 as tumor markers in head and neck cancer. American Journal of Otolaryngology. 2005;26(5):308–313. doi: 10.1016/j.amjoto.2005.02.005. [DOI] [PubMed] [Google Scholar]
- 32.Kobayashi N, Miyoshi S, Mikami T, et al. Hyaluronan deficiency in tumor stroma impairs macrophage trafficking and tumor neovascularization. Cancer Research. 2010;70(18):7073–7083. doi: 10.1158/0008-5472.CAN-09-4687. [DOI] [PubMed] [Google Scholar]
- 33.Edward M, Gillan C, Micha D, Tammi RH. Tumour regulation of fibroblast hyaluronan expression: a mechanism to facilitate tumour growth and invasion. Carcinogenesis. 2005;26(7):1215–1223. doi: 10.1093/carcin/bgi064. [DOI] [PubMed] [Google Scholar]
- 34.Toole BP, Slomiany MG. Hyaluronan: a constitutive regulator of chemoresistance and malignancy in cancer cells. Seminars in Cancer Biology. 2008;18(4):244–250. doi: 10.1016/j.semcancer.2008.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Singleton PA, Mirzapoiazova T, Guo Y, et al. High-molecular-weight hyaluronan is a novel inhibitor of pulmonary vascular leakiness. American Journal of Physiology. 2010;299(5):L639–L651. doi: 10.1152/ajplung.00405.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tanaka Y, Makiyama Y, Mitsui Y. Anti-CD44 monoclonal antibody (IM7) induces murine systemic shock mediated by platelet activating factor. Journal of Autoimmunity. 2002;18(1):9–15. doi: 10.1006/jaut.2001.0559. [DOI] [PubMed] [Google Scholar]
- 37.Toole BP, Slomiany MG. Hyaluronan, CD44 and Emmprin: partners in cancer cell chemoresistance. Drug Resistance Updates. 2008;11(3):110–121. doi: 10.1016/j.drup.2008.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Edward M, Quinn JA, Pasonen-Seppänen SM, McCann BA, Tammi RH. 4-Methylumbelliferone inhibits tumour cell growth and the activation of stromal hyaluronan synthesis by melanoma cell-derived factors. British Journal of Dermatology. 2010;162(6):1224–1232. doi: 10.1111/j.1365-2133.2010.09699.x. [DOI] [PubMed] [Google Scholar]
- 39.Kultti A, Pasonen-Seppänen S, Jauhiainen M, et al. 4-Methylumbelliferone inhibits hyaluronan synthesis by depletion of cellular UDP-glucuronic acid and downregulation of hyaluronan synthase 2 and 3. Experimental Cell Research. 2009;315(11):1914–1923. doi: 10.1016/j.yexcr.2009.03.002. [DOI] [PubMed] [Google Scholar]
- 40.Urakawa H, Nishida Y, Wasa J, et al. Inhibition of hyaluronan synthesis in breast cancer cells by 4-methylumbelliferone suppresses tumorigenicity in vitro and metastatic lesions of bone in vivo. International Journal of Cancer. 2012;130(2):454–466. doi: 10.1002/ijc.26014. [DOI] [PubMed] [Google Scholar]
- 41.Hajime M, Shuichi Y, Makoto N, et al. Inhibitory effect of 4-methylesculetin on hyaluronan synthesis slows the development of human pancreatic cancer in vitro and in nude mice. International Journal of Cancer. 2007;120(12):2704–2709. doi: 10.1002/ijc.22349. [DOI] [PubMed] [Google Scholar]
- 42.Morohashi H, Kon A, Nakai M, et al. Study of hyaluronan synthase inhibitor, 4-methylumbelliferone derivatives on human pancreatic cancer cell (KP1-NL) Biochemical and Biophysical Research Communications. 2006;345(4):1454–1459. doi: 10.1016/j.bbrc.2006.05.037. [DOI] [PubMed] [Google Scholar]
- 43.Nakazawa H, Yoshihara S, Kudo D, et al. 4-methylumbelliferone, a hyaluronan synthase suppressor, enhances the anticancer activity of gemcitabine in human pancreatic cancer cells. Cancer Chemotherapy and Pharmacology. 2006;57(2):165–170. doi: 10.1007/s00280-005-0016-5. [DOI] [PubMed] [Google Scholar]
- 44.Jacobetz MA, Chan DS, Neesse A, et al. Hyaluronan impairs vascular function and drug delivery in a mouse model of pancreatic cancer. doi: 10.1136/gutjnl-2012-302529. Gut. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Thompson CB, Shepard HM, O’Connor PM, et al. Enzymatic depletion of tumor hyaluronan induces antitumor responses in preclinical animal models. Molecular Cancer Therapeutics. 2010;9(11):3052–3064. doi: 10.1158/1535-7163.MCT-10-0470. [DOI] [PubMed] [Google Scholar]
- 46.Cheng H, Merika E, Syrigos KN, Saif MW. Novel agents for the treatment of pancreatic adenocarcinoma. Highlights from the “2011 ASCO Annual Meeting”. Chicago, IL, USA; June 3–7. Journal of Pancreas. 2011;12(4):334–338. [PubMed] [Google Scholar]
- 47.Thayer SP, Di Magliano MP, Heiser PW, et al. Hedgehog is an early and late mediator of pancreatic cancer tumorigenesis. Nature. 2003;425(6960):851–856. doi: 10.1038/nature02009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Walter K, Omura N, Hong SM, et al. Overexpression of smoothened activates the Sonic hedgehog signaling pathway in pancreatic cancer-associated fibroblasts. Clinical Cancer Research. 2010;16(6):1781–1789. doi: 10.1158/1078-0432.CCR-09-1913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian H, Callahan CA, Dupree KJ, et al. Hedgehog signaling is restricted to the stromal compartment during pancreatic carcinogenesis. Proceedings of the National Academy of Sciences of the United States of America. 2009;106(11):4254–4259. doi: 10.1073/pnas.0813203106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yauch RL, Gould SE, Scales SJ, et al. A paracrine requirement for hedgehog signalling in cancer. Nature. 2008;455(7211):406–410. doi: 10.1038/nature07275. [DOI] [PubMed] [Google Scholar]
- 51.Feldmann G, Fendrich V, McGovern K, et al. An orally bioavailable small-molecule inhibitor of Hedgehog signaling inhibits tumor initiation and metastasis in pancreatic cancer. Molecular Cancer Therapeutics. 2008;7(9):2725–2735. doi: 10.1158/1535-7163.MCT-08-0573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kelleher FC, McDermott R. Aberrations and therapeutics involving the developmental pathway Hedgehog in pancreatic cancer. Vitamins and Hormones. 2012;88:355–378. doi: 10.1016/B978-0-12-394622-5.00016-X. [DOI] [PubMed] [Google Scholar]
- 53.Olive KP, Jacobetz MA, Davidson CJ, et al. Inhibition of Hedgehog signaling enhances delivery of chemotherapy in a mouse model of pancreatic cancer. Science. 2009;324(5933):1457–1461. doi: 10.1126/science.1171362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bisht S, Brossart P, Maitra A, Feldmann G. Agents targeting the Hedgehog pathway for pancreatic cancer treatment. Current Opinion in Investigational Drugs. 2010;11(12):1387–1398. [PubMed] [Google Scholar]
- 55.Richards DA, Stephenson J, Wolpin BM, et al. A phase Ib trial of IPI-926, a hedgehog pathway inhibitor, plus gemcitabine in patients with metastatic pancreatic cancer. Journal of Clinical Oncology. 2012;30(supplement 4, abstract 213) ASCO Annual Meeting. [Google Scholar]
- 56.Singh BN, Fu J, Srivastava RK, Shankar S. Hedgehog signaling antagonist GDC-0449 (Vismodegib) inhibits pancreatic cancer stem cell characteristics: molecular mechanisms. PLOS ONE. 2011;6(11, article e27306):p. 1. doi: 10.1371/journal.pone.0027306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.De Smaele E, Ferretti E, Gulino A. Vismodegib, a small-molecule inhibitor of the hedgehog pathway for the treatment of advanced cancers. Current Opinion in Investigational Drugs. 2010;11(6):707–718. [PubMed] [Google Scholar]
- 58.LoRusso PM, Rudin CM, Reddy JC, et al. Phase I trial of hedgehog pathway inhibitor vismodegib (GDC-0449) in patients with refractory, locally advanced or metastatic solid tumors. Clinical Cancer Research. 2011;17(8):2502–2511. doi: 10.1158/1078-0432.CCR-10-2745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fukahori M. Efficacy of gemcitabine as second-line therapy after S-1 therapy failure in advanced pancreatic carcinoma. Journal of Clinical Oncology. 2012;30(supplement 4, abstract 248) ASCO Annual Meeting. [Google Scholar]
- 60.Choi M, Kim R, Saif MW. What options are available for refractory pancreatic cancer? Journal of Pancreas. 2012;13(2):163–165. [PubMed] [Google Scholar]
- 61.Palmer SR, Erlichman C, Fernandez-Zapico M, et al. Phase I trial erlotinib, gemcitabine, and the hedgehog inhibitor, GDC-0449. Journal of Clinical Oncology. 2011;29(supplement, abstract 3092) ASCO Annual Meeting. [Google Scholar]
- 62.Hassounah NB, Bunch TA, McDermott KM. Molecular pathways: the role of primary cilia in cancer progression and therapeutics with a focus on hedgehog signaling. Clinical Cancer Research. 2012;18(9):2429–2435. doi: 10.1158/1078-0432.CCR-11-0755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sugahara KN, Teesalu T, Karmali PP, et al. Tissue-penetrating delivery of compounds and nanoparticles into tumors. Cancer Cell. 2009;16(6):510–520. doi: 10.1016/j.ccr.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sugahara KN, Teesalu T, Prakash Karmali P, et al. Coadministration of a tumor-penetrating peptide enhances the efficacy of cancer drugs. Science. 2010;328(5981):1031–1035. doi: 10.1126/science.1183057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Guzman M. Preclinical Development of iRGD for pancreatic cancer Grant 1R43CA162766-01. Grant 1R43CA162766-01 from National Cancer Institute, 2011.
- 66.Banchereau J, Bazon F, Blanchard D, et al. The CD40 antigen and its ligand. Annual Review of Immunology. 1994;12:881–922. doi: 10.1146/annurev.iy.12.040194.004313. [DOI] [PubMed] [Google Scholar]
- 67.Biancone L, Cantaluppi V, Camussi G. CD40-CD154 interaction in experimental and human disease (review) International Journal of Molecular Medicine. 1999;3(4):343–353. doi: 10.3892/ijmm.3.4.343. [DOI] [PubMed] [Google Scholar]
- 68.Fonsatti E, Maio M, Altomonte M, Hersey P. Biology and clinical applications of CD40 in cancer treatment. Seminars in Oncology. 2010;37(5):517–523. doi: 10.1053/j.seminoncol.2010.09.002. [DOI] [PubMed] [Google Scholar]
- 69.Shoji Y, Miyamoto M, Ishikawa K, et al. The CD40-CD154 interaction would correlate with proliferation and immune escape in pancreatic ductal adenocarcinoma. Journal of Surgical Oncology. 2011;103(3):230–238. doi: 10.1002/jso.21812. [DOI] [PubMed] [Google Scholar]
- 70.Mees ST, Mardin WA, Sielker S, et al. Involvement of CD40 targeting miR-224 and miR-486 on the progression of pancreatic ductal adenocarcinomas. Annals of Surgical Oncology. 2009;16(8):2339–2350. doi: 10.1245/s10434-009-0531-4. [DOI] [PubMed] [Google Scholar]
- 71.Ottaiano A, Pisano C, De Chiara A, et al. CD40 activation as potential tool in malignant neoplasms. Tumori. 2002;88(5):361–366. doi: 10.1177/030089160208800502. [DOI] [PubMed] [Google Scholar]
- 72.He S, Zhao H, Fei M, et al. Expression of the co-signaling molecules CD40-CD40L and their growth inhibitory effect on pancreatic cancer in vitro. Oncology Reports. 2012;28(1):262–268. doi: 10.3892/or.2012.1790. [DOI] [PubMed] [Google Scholar]
- 73.Beatty GL, Chiorean EG, Fishman MP, et al. CD40 agonists alter tumor stroma and show efficacy against pancreatic carcinoma in mice and humans. Science. 2011;331(6024):1612–1616. doi: 10.1126/science.1198443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Gennari P, Raffi GB, Baldi E. Evaluation of a few indices of hepatic and renal function in a group of workers chronically exposed to anti parasitic agents. Giornale di Clinica Medica. 1975;56(11-12):423–430. [PubMed] [Google Scholar]
- 75.Vadasz Z, Kessler O, Akiri G, et al. Abnormal deposition of collagen around hepatocytes in Wilson’s disease is associated with hepatocyte specific expression of lysyl oxidase and lysyl oxidase like protein-2. Journal of Hepatology. 2005;43(3):499–507. doi: 10.1016/j.jhep.2005.02.052. [DOI] [PubMed] [Google Scholar]
- 76.Reiser K, McCormick RJ, Rucker RB. Enzymatic and nonenzymatic cross-linking of collagen and elastin. FASEB Journal. 1992;6(7):2439–2449. doi: 10.1096/fasebj.6.7.1348714. [DOI] [PubMed] [Google Scholar]
- 77.Erler JT, Bennewith KL, Cox TR, et al. Hypoxia-induced lysyl oxidase is a critical mediator of bone marrow cell recruitment to form the premetastatic niche. Cancer Cell. 2009;15(1):35–44. doi: 10.1016/j.ccr.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hinton CV, Avraham S, Avraham HK. Role of the CXCR4/CXCL12 signaling axis in breast cancer metastasis to the brain. Clinical and Experimental Metastasis. 2010;27(2):97–105. doi: 10.1007/s10585-008-9210-2. [DOI] [PubMed] [Google Scholar]
- 79.Friedl P, Wolf K. Tube travel: the role of proteases in individual and collective cancer cell invasion. Cancer Research. 2008;68(18):7247–7249. doi: 10.1158/0008-5472.CAN-08-0784. [DOI] [PubMed] [Google Scholar]
- 80.Offenberg H, Brünner N, Mansilla F, Ørntoft Torben F, Birkenkamp-Demtroder K. TIMP-1 expression in human colorectal cancer is associated with TGF-B1, LOXL2, INHBA1, TNF-AIP6 and TIMP-2 transcript profiles. Molecular Oncology. 2008;2(3):233–240. doi: 10.1016/j.molonc.2008.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Barry-Hamilton V, Spangler R, Marshall D, et al. Allosteric inhibition of lysyl oxidase—like-2 impedes the development of a pathologic microenvironment. Nature Medicine. 2010;16(9):1009–1017. doi: 10.1038/nm.2208. [DOI] [PubMed] [Google Scholar]
- 82.Barker HE, Chang J, Cox TR, et al. LOXL2-mediated matrix remodeling in metastasis and mammary gland involution. Cancer Research. 2011;71(5):1561–1572. doi: 10.1158/0008-5472.CAN-10-2868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rückert F, Joensson P, Saeger HD, Grützmann R, Pilarsky C. Functional analysis of LOXL2 in pancreatic carcinoma. International Journal of Colorectal Disease. 2010;25(3):303–311. doi: 10.1007/s00384-009-0853-5. [DOI] [PubMed] [Google Scholar]
- 84.Rodriguez HM, Vaysberg M, Mikels A, et al. Modulation of lysyl oxidase-like 2 enzymatic activity by an allosteric antibody inhibitor. The Journal of Biological Chemistry. 2010;285(27):20964–20974. doi: 10.1074/jbc.M109.094136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Aghdassi A, Sendler M, Guenther A, et al. Recruitment of histone deacetylases HDAC1 and HDAC2 by the transcriptional repressor ZEB1 downregulates E-cadherin expression in pancreatic cancer. Gut. 2012;61(3):439–448. doi: 10.1136/gutjnl-2011-300060. [DOI] [PubMed] [Google Scholar]
- 86.Herranz N, Dave N, Millanes-Romero A, et al. Lysyl Oxidase-like 2 Deaminates Lysine 4 in Histone H3. Molecular Cell. 2012;46(3):369–376. doi: 10.1016/j.molcel.2012.03.002. [DOI] [PubMed] [Google Scholar]
- 87.Cordes N, Seidler J, Durzok R, Geinitz H, Brakebusch C. β1-integrin-mediated signaling essentially contributes to cell survival after radiation-induced genotoxic injury. Oncogene. 2006;25(9):1378–1390. doi: 10.1038/sj.onc.1209164. [DOI] [PubMed] [Google Scholar]
- 88.Erkan M, Kleeff J, Gorbachevski A, et al. Periostin creates a tumor-supportive microenvironment in the pancreas by sustaining fibrogenic stellate cell activity. Gastroenterology. 2007;132(4):1447–1464. doi: 10.1053/j.gastro.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 89.Park CC, Zhang HJ, Yao ES, Park CJ, Bissell MJ. β 1 integrin inhibition dramatically enhances radiotherapy efficacy in human breast cancer xenografts. Cancer Research. 2008;68(11):4398–4405. doi: 10.1158/0008-5472.CAN-07-6390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Cordes N, Frick S, Brunner TB, et al. Human pancreatic tumor cells are sensitized to ionizing radiation by knockdown of caveolin-1. Oncogene. 2007;26(48):6851–6862. doi: 10.1038/sj.onc.1210498. [DOI] [PubMed] [Google Scholar]