

Abstract
Context:
Germline mutations in PTEN are associated with phosphatase and tensin homolog deleted on chromosome 10 (PTEN) hamartoma tumor syndrome including Cowden syndrome (CS) and Cowden-like syndrome (CSL) that predisposes to high risks of benign and malignant tumors of thyroid and breast.
Objective:
The objective of the study was to analyze the subcellular pattern of phosphorylated (P)-AKT expression in nonmedullary thyroid cancers from PTEN hamartoma tumor syndrome patients and to investigate whether the lack of PTEN in the nucleus and/or lack of proper PTEN function in the nucleus affect(s) nuclear AKT activity in CS patients.
Design:
In all, 664 patients with CS/CSL were screened for PTEN germline mutations and nonmedullary thyroid cancers. Twenty-two patients who have both pathogenic PTEN germline mutations and nonmedullary thyroid cancers were selected. Thyroid samples from these patients were stained for PTEN and P-AKT. In our in vitro study, PTEN was knocked down or overexpressed in both thyroid cancer cells and breast cancer cells, and nuclear P-AKT was compared with the control.
Results:
Loss of PTEN protein was found in thyroid adenomas and carcinomas from all 22 (100%) PTENMut+ CS/CSL patients. AKT activation was identified in 17 of 22 (77.3%) thyroid adenoma/carcinoma specimens, and most patients (63.7%) have activated nuclear AKT. Knockdown of PTEN in cells containing wild-type PTEN enhanced nuclear P-AKT, whereas expression of wild-type PTEN, but not phosphatase-dead mutants (C124S or G129E), markedly reduced nuclear P-AKT in PTEN null cells. We also showed that in breast cancer but not thyroid cancer cells, PTEN suppresses nuclear P-AKT mainly through decreasing P-AKT nuclear translocation by reducing the PIP3/P-AKT reservoir in the cytoplasm. In thyroid cancer cells, PTEN suppresses phosphorylation of AKT already resident in the nucleus.
Conclusions:
PTEN is necessary and sufficient for inhibiting AKT activation in the nucleus through its intact lipid phosphatase activity and proper subcellular localization.
Phosphatase and tensin homolog deleted on chromosome 10 (PTEN) is a phosphatase that mainly localizes in the cytoplasm and the plasma membrane and antagonizes phosphoinositol-3-kinase (PI3K) by dephosphorylating phosphatidylinositol-3,4,5-triphosphate (PIP3) and negatively regulates its downstream target, AKT. Germline mutations of the PTEN gene, localized to 10q23, cause the PTEN hamartoma tumor syndrome (PHTS), which umbrellas PTEN mutation-positive subsets of Cowden syndrome (CS), Bannayan-Riley-Ruvalcaba syndrome, Proteus syndrome, and Proteus-like syndrome. Lifetime risks for thyroid cancer, breast cancer, endometrial cancer, colorectal cancer, kidney cancer, and melanoma are increased in patients with PTEN mutations (1, 2). Furthermore, loss of PTEN protein expression due to either loss of heterozygosity of the PTEN locus, or to epigenetic silencing, is frequent in sporadic breast carcinomas and thyroid carcinomas (3).
Negative regulation of the PI3K/AKT pathway in the cytoplasm has been considered as one of the most important tumor suppressor activities of PTEN. Although PTEN, PIP3, and AKT predominantly localize to the cytoplasm, all of them have been reported to be localized in the nucleus (4–8). Our group first reported that PTEN exists in the nucleus in normal breast and in thyroid tissues (9, 10). We also revealed that, in sporadic thyroid carcinomas, nuclear PTEN immunostaining was weak in comparison with normal thyroid follicular cells and thyroid follicular adenomas (9). Tanaka et al. (8) reported that, under H2O2 stimulation, PI3K was translocated and activated in the nucleus. This suggests that PI3K can be activated in the nucleus as well as in the membrane after appropriate stimulation of the cells. Another report showed that PTEN was required for robust depletion of nuclear phosphorylated (P)-AKT (11). Finally, nuclear localized P-AKT has been reported to occur in cells in the invasive fronts of thyroid cancer and has been shown capable of regulating migration in fibroblasts (12, 13).
The data to date therefore point to nuclear PTEN potentially being required for its tumor suppressor function. We have shown that PTEN has bipartite nuclear localization sequence (NLS)-like sequences that are required for major vault protein-mediated nuclear import (14). Double NLS mutant PTEN (PTENM3M4) cannot interact with major vault protein and leads to diminished nuclear PTEN (15). Recently we have successfully established a knock-in mouse model to analyze the role of Pten in the nucleus. The PtenM3M4 homozygous mutant knock-in mice have obvious macrocephaly, which also occurs in more than 95% of PTEN mutation positive Cowden syndrome patients (16). In light of all these overall findings, we sought to address the hypothesis that a lack of PTEN in the nucleus and/or a lack of proper PTEN function in the nucleus affect(s) nuclear AKT activity.
Materials and Methods
Patients and tumors
Six hundred sixty-four individuals clinically diagnosed for CS and Cowden-like syndrome (CSL) with documented thyroid tumors had been previously analyzed for PTEN mutations (17). Among these CS/CSL individuals, 36 have PTEN pathogenic germline mutations, and specimens of thyroid tumor tissues were collected from 22 patients. All tissue specimens were collected after written informed consent and with approval of the Institutional Review Board at the Cleveland Clinic.
Reagents and constructs
The pCMV-FLAG-PTENWT construct was made by cloning PTENWT into the pCMV-FLAG vector using NotI and XbaI sites. The pCMV-FLAG-PTENC124S and pCMV-FLAG-PTENG129E constructs were made by inducing the site-specific point mutation into the pCMV-FLAG-PTENWT construct (QuikChange Site-Directed Mutagenesis Kit; Stratagene Cloning Systems, La Jolla, CA). For nuclear targeting, three tandem repeats of the Simian virus 40 large T antigen NLS peptide (DPKKKRKV) were fused to the N terminus of PTEN to make the pCMV-FLAG-NLS-PTEN construct.
Cell lines and cell culture
FTC-236 and BCPAP thyroid cancer cells and BT-549 and MCF-7 breast cancer cells were purchased from the American Type Culture Collection (Manassas, VA). MCF-7 Tet-off cells were obtained from Clontech Laboratories (Mountain View, CA) and were stably transfected with plasmids encoding pTre2hyg-vector only (vector control) or pTre2hyg-FLAG-PTENWT as previous described (18). For the PTEN short hairpin RNA (shRNA) and control shRNA stable cell lines, MCF-7 cells were plated on 35-mm culture dishes and transfected with PTEN shRNA construct (SHCLNG-NM_000314) or the pLKO.1 vector control (Sigma Aldrich, St. Louis, MO), and cells were selected with puromycin (0.5 μg/ml).
Transfection
Cells were transfected with lipofectamine 2000 in Opti-MEM (without serum) (Lerner Research Institute, Cleveland, OH). After 4 h, Opti-MEM was replaced with complete medium and cells were cultured for another 20 h until harvest.
Subcellular fractionation
We carefully optimized our fractionation protocol to preserve cellular AKT phosphorylation status. Cells were quickly washed with ice-cold PBS twice and then scraped in 1 ml (for p100 dishes) ice-cold hypotonic buffer [buffer A (10 mm 3[N-morholino]propanesulfonic acid, 1.5 mm MgCl2, 10 mm KCl, and 1% Triton X-100)] with protease inhibitors and phosphatase inhibitors. Cells were then left on ice for 15 min, vortexing every 5 min. After centrifugation at 13,000 rpm for 5 min at 4 C, the supernatant was carefully collected as the cytoplasmic fraction. The pellets were then incubated twice in buffer A for 15 min and separated by centrifugation at 13,000 rpm for 5 min at 4 C. The pellets were dissolved in 2× sodium dodecyl sulfate sample buffer (126 mm Tris HCl, 20% glycerol, and 4% sodium dodecyl sulfate). The purity of each fraction was analyzed by immunoblotting using antibodies against α-tubulin for the cytoplasm and poly(ADP-ribose) polymerase-1 for the nucleus.
SDS-PAGE and Western blot analyses
Proteins were run on SDS-PAGE gels and transferred to nitrocellulose. Blots were probed with primary antibodies and followed by incubation with secondary antibody and then visualized using enhanced chemiluminescence detection. Antibodies for total AKT and P-AKTSer473 were from Cell Signaling Technology, Inc. (Danvers, MA). Antibodies for FLAG and α-tubulin were from Sigma-Aldrich. The PTEN antibody was from Cascade Biosience (Winchester, MA).
Indirect immunofluorescence and confocal microscopy
Cells were seeded in six-well plates with coverslips. The medium was aspirated and cells were washed with PBS followed by fixing in 100% methanol for 1 min. After 5 min incubation with 0.3% Triton X-100 in PBS, the coverslips were washed three times in PBS, blocked in PBS with 10% goat serum for 30 min, incubated with primary antibodies for 1 h, washed three times in PBS, and finally incubated with Alexa Fluor dye-labeled secondary antibodies for 1 h at the concentration of 1:1500 (Invitrogen, Carlsbad, CA). The immunofluorescence antibodies were purchased and used as follows: FLAG M2 mouse monoclonal antibody from Sigma-Aldrich (catalog no. F1804, 1:100 dilution); P-AKTSer473 XP (Cell Signaling; clone D9E, 1:100 dilution); and PTEN rabbit polyclonal antibody from Cell Signaling (catalog no. 9552, 1:100 dilution). PIP2 antibody was purchased from Echelon (catalog no. Z-A045, 1:50 dilution). Cells were mounted on glass slides with Pro-Long Gold antifade reagent with 4′,6′-diamino-2-phenylindole (Invitrogen) and visualized on a Leica TCS-SP spectral laser scanning confocal microscope (Leica Microsystems Inc., Buffalo Grove, IL). Quantification of fluorescence intensity was performed by ImagePro software (Media Cybernetics, Inc., Bethesda, MD). For each cell line (WT, C124S, or G129E), 20 positively stained cells were measured and the average nuclear P-AKT staining intensities were compared with those obtained from 20 adjacent nontransfected control cells (set as 100%), and the results were presented as percentages of control cells.
Immunohistochemistry (IHC) and quantifications
For IHC, tissues were fixed in 10% formalin and embedded in paraffin according to standard procedures. Sections were processed with hematoxylin and eosin reagents or stained for P-AKT1/2/3Ser473 (catalog no. SC-7985-R; Santa Cruz Biotechnology, Santa Cruz, CA; 1:50 dilution), PTEN (clone 6H2.1; Cascade Bioscience; 1:50 dilution), and total AKT (catalog no. 9272; Cell signaling; 1:50 dilution). All studies involving human tissues were in accordance with institutional guidelines and were approved by the Institutional Review Board for Human Subjects Protection.
Results
Robust AKT phosphorylation can be detected in the nucleus of thyroid tumor tissues from CS patients with germline PTEN mutations
Analysis of the coding sequence of PTEN in genomic DNA from 22 PTENMut+ CS/CSL patients with thyroid tumors revealed a spectrum of genotypes (Table 1). More than half of the mutations (12 of 22) are truncations and frame shift mutations that result in significant decreases in PTEN protein levels because of the rapid degradation of the truncated PTEN protein.
Table 1.
Type of PTEN pathogenic mutations in PTEN mutation-positive CS/CSL patients with thyroid tumors
| Mutation type | n (%) |
|---|---|
| n | 22 |
| Missense | 6 (27.3) |
| Truncation | 7 (31.8) |
| Frameshift | 5 (22.7) |
| Interfere with splice site | 3 (13.6) |
| Synonymous | 1 (4.6) |
We then investigated whether in PTENMut+ CS/CSL patients, loss of PTEN in thyroid tumors are accompanied by elevated P-AKT levels in the nucleus. We immunostained for P-AKT and PTEN in thyroid adenomas and carcinomas from 22 PTENMut+ CS patients. Loss of PTEN protein was found in thyroid tumors from all 22 patients. On the other hand, activated AKT can be detected in atypical adenomas, follicular carcinomas, papillary carcinomas, and anaplastic carcinomas (Fig. 1, A–D). Compared with tumor tissues, adjacent normal tissues showed much weaker levels of P-AKT, although a few normal thyrocytes stained positively for nuclear P-AKT (Fig. 1E). Not surprisingly, the adjacent normal thyroid tissue from a PTEN wild-type CS patient showed robust PTEN staining and absent P-AKT expression (Fig. 1F).
Fig. 1.

Expression of P-AKT, total AKT, and PTEN by immunohistochemistry in thyroid tissues from PTENMut+ CS patients. Note nuclear-predominant (A), cytoplasmic-predominant (B), nuclear-cytoplasmic equal (C), and negative (D) P-AKT staining in thyroid tumor tissues from different CS patients. Adjacent normal tissues from CS patients with (E) or without (F) PTEN gene mutation were included as controls. Case numbers and PTEN genotypes were labeled on the left. PTEN staining was negative in thyroid tumor cells of all 22 PTENMut+ CS patients that we tested. Note the strong staining intensity of the stromal cells that serve as internal positive controls for PTEN, and also note the loss of stromal PTEN in PTC (A) and anaplastic thyroid cancer (B) but not adenomas (C and D). In the adjacent thyroid tissue (E), PTEN can be detected in a few follicular epithelial cells, whereas the expression of P-AKT is weaker than that in the tumor tissues from the same patient (A). Magnification, ×40.
In total, 17 of 22 PTENMut+ CS patients (77.3%) showed increased P-AKT in thyroid tumor tissues. Strikingly, P-AKT staining was most frequently observed in the nucleus (45.5% nuclear only; 18.2% both nuclear and cytoplasmic) (Table 2). In contrast to P-AKT, total AKT was found in both cytoplasm and nucleus of thyroid tumor tissues, regardless of P-AKT staining intensity or subcellular localization (Fig. 1). These results support our hypothesis that PTEN is important in the down-regulation of nuclear P-AKT. Loss of PTEN resulted in activation of AKT in the nucleus of thyroid.
Table 2.
Subcellular expression of P-AKT, by immunohistochemistry, in thyroid tumors from PTEN mutation-positive CS/CSL patients
| P-AKT staining |
||||
|---|---|---|---|---|
| Negative | Nuclear | Cytoplasmic | Nuclear and cytoplasmic | |
| n (%) | 5 (22.7) | 10 (45.5) | 3 (13.6) | 4 (18.2) |
| Atypical FA (W/ thyroiditis) (n = 5) | 0 | 2 | 0 | 3 |
| Atypical FA (W/O thyroiditis) (n = 7) | 3 | 4 | 0 | 0 |
| Follicular Ca. (n = 3) | 0 | 1 | 1 | 1 |
| Papillary Ca. (n = 5) | 2 | 2 | 1 | 0 |
| Anaplastic Ca. (n = 2) | 0 | 1 | 1 | 0 |
FA, Follicular adenoma; Ca., carcinoma; W, with; W/O, without.
Increased AKT phosphorylation can be detected in the nucleus of normal tissues from PHTS patients
We then asked whether CS patients harboring naturally occurring germline PTEN mutations would present with increased nuclear AKT activity, even in their nonneoplastic cells because we noticed that in several CS patients we studied in Fig. 1E, positive P-AKT staining can also be detected in the nucleus of the normal thyrocytes. In this regard, we isolated lymphoblast cells from two CS patients who harbor heterozygous germline nonsense mutations (c.388C>T, PTENWT/R130X) in PTEN and from two controls that are PTENWT/WT. The R130X nonsense mutation leads to PTEN haploinsufficiency in the patients and is associated with robust P-AKT expression in the whole-cell lysates (Fig. 2A). After subcellular fractionation, we found AKT was activated in both the cytoplasm and nucleus of lymphoblast cells in CS patients when compared with the PTENWT/WT controls (Fig. 2B). This result, together with the IHC result in Fig. 1E, suggests that even in normal cells from CS patients with PTEN mutations, nuclear P-AKT can also be elevated.
Fig. 2.
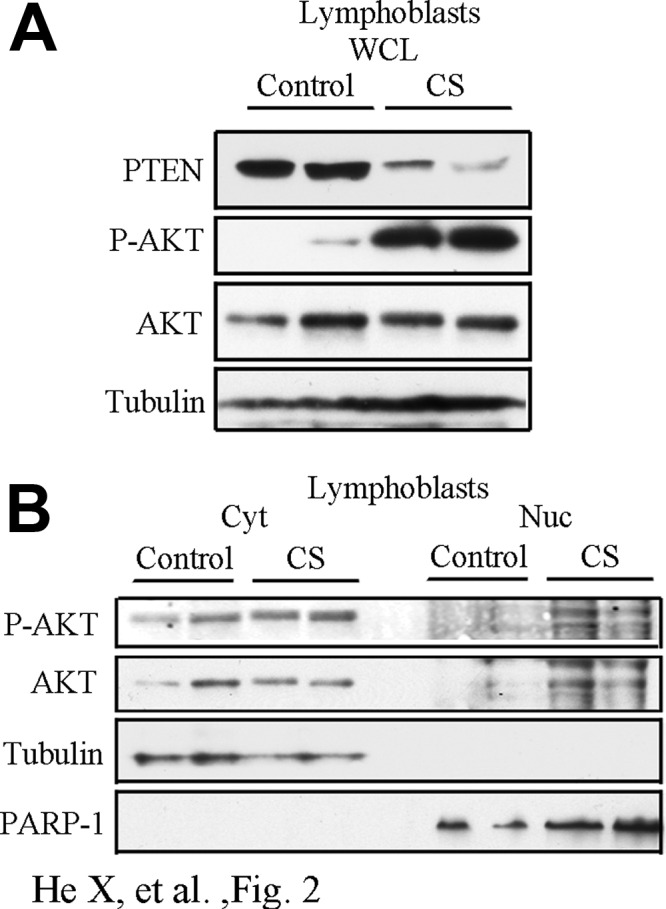
Loss of PTEN leads to hyperactivation of AKT in the nucleus of lymphoblast cells of CS patients. A, Whole-cell lysates were collected from lymphoblast cells isolated from two PTEN WT controls (control) and two Cowden syndrome patients harboring germline PTEN heterozygous R130X mutation (CS). Cell lysates were immunoblotted for PTEN and P-AKTSer473. B, Western blots of P-AKTSer473 expression in cytoplasmic/nuclear fractions of the lymphoblast cells.
Loss of PTEN resulted in activation of AKT in the nucleus of thyroid and breast cancer cell lines
Because both thyroid cancer and breast cancer represent the most common cancers of CS, we next investigate whether knockdown of PTEN in these two cancer cell lines will mimic the condition of PTEN loss in CS/CSL patients in vitro. The BCPAP thyroid cell line and MCF-7 breast cancer cell line were chosen based on the fact that they express wild-type PTEN. In both cell lines, knockdown of PTEN resulted in an up-regulation of P-AKT in both cytoplasmic and nuclear compartments (Fig. 3, A and B). Notably, an increase of total AKT can be detected in the nuclear fraction of the PTEN-knocked down MCF-7 cells but not BCPAP cells, indicating a possible increase of AKT nuclear translocation in PTEN knockdown breast cancer cells or a possible decrease of AKT nuclear export/degradation. Taken together, these data indicate that PTEN is necessary for the down-regulation of both cytoplasmic and nuclear P-AKT levels in the cell. Loss of PTEN leads to elevated P-AKT in both compartments.
Fig. 3.

Loss of PTEN leads to hyperactivation of AKT in the nucleus of breast and thyroid cancer cells. A, Left, Whole-cell lysates (WCL) were collected from MCF-7 cells stably expressing PTEN-shRNA or shRNA vector control. Western blots showed that PTEN-shRNA knocked down PTEN expression accompanied by increased P-AKT. Right, Cytoplasmic (Cyt) and nuclear (Nuc) fractions of MCF-7 cells stably expressing PTEN-shRNA or shRNA vector control were isolated and immunoblotted for P-AKT. B, Left, Whole cell lysates (WCL) were collected from BCPAP cells transfected with PTEN-shRNA or shRNA vector control. Western blots showed that PTEN-shRNA knocked down PTEN expression accompanied by increased P-AKT. Right, Cytoplasmic (Cyt) and nuclear (Nuc) fractions of BCPAP cells expressing PTEN-shRNA or shRNA vector control were isolated and immunoblotted for P-AKT. C and D, Charts show quantification of Western blot densitometry in A and B, separately (n = 2 experiments for each cell line).
PTEN decreases phosphorylation of nuclear AKT through its lipid phosphatase activity
Having demonstrated that PTEN loss is associated with an increase in both cytosolic and nuclear P-AKT levels in the cell, we then sought to determine whether PTEN is sufficient to inhibit nuclear P-AKT. BT-549 PTEN null cells were selected to investigate the activation process of nuclear P-AKT. Under serum starvation conditions, only a very small amount of activated AKT can be detected in the nucleus. Ten minutes after serum stimulation, P-AKT levels were enhanced in the nucleus (Supplemental Fig. 1, A and B, published on The Endocrine Society's Journals Online web site at http://jcem.endojournals.org). Notably, the nuclear total AKT levels slightly increased after serum stimulation, suggesting the elevation of P-AKT after serum stimulation was at least partially due to increased nuclear translocation. To determine whether nuclear PTEN is involved in AKT deactivation, we transfected PTEN-null BT549 cells with the green fluorescent protein-tagged PTEN. PTEN overexpression in BT-549 cells significantly decreased nuclear P-AKT and reduced nuclear total AKT to a lesser extent (Supplemental Fig. 1, C and D). These data indicate that PTEN inhibits nuclear P-AKT in the cell.
Because PTEN protein has both lipid phosphatase and protein phosphatase activities (19), we next investigated which of the specific phosphatase activities of PTEN were involved in down-regulating nuclear AKT. Two PTEN-null cell lines, BT-549 (breast cancer) and FTC-236 (thyroid cancer), were transfected with FLAG-tagged PTENWT or different mutant PTEN constructs. PTENC124S is a pan-phosphatase dead PTEN, which abolishes both lipid and protein phosphatase activities, whereas PTENG129E loses only lipid phosphatase activity but still retains protein phosphatase activity (Fig. 4C). One day after transfection, only cells expressing PTENWT, but neither mutant PTEN constructs, showed significant reductions in nuclear AKT phosphorylation (Fig. 4 for BT-549 and Supplemental Fig. 1E for FTC-236). Hence, our results suggest involvement of PTEN′s lipid phosphatase activity in the deactivation of nuclear AKT.
Fig. 4.

PTEN suppresses nuclear AKT, depending on its lipid phosphatase activity. A, BT-549 cells were transfected for 24 h with various FLAG-tagged PTEN constructs, as noted. Cells were fixed and stained with FLAG (green) and P-AKTSer473 (red) antibodies and were visualized by confocal microscopy. Dashed lines demarcate BT-549 cells expressing FLAG-PTEN. DAPI, 4′,6′-diamino-2-phenylindole. Scale bar, 50 μm. B, Average intensities of nuclear P-AKT of transfected cells were normalized to that of nontransfected cells in the same field. For each group, 20 positively stained cells were quantified for immunofluorescent intensities. Average intensities of nuclear P-AKTSer473 were plotted on a graph, and the statistical analysis was performed using one-way ANOVA. The error bars represent the sd. N.S., Not significant. C, BT-549 cells were transiently transfected with FLAG-tagged PTEN constructs, as noted. Western blots showed that only wild-type PTEN suppresses P-AKTSer473.
Expression of the nuclear compartment-specific PTEN can decrease nuclear P-AKT
To further evaluate the effect of PTEN in suppressing AKT phosphorylation in the nucleus, we used NLS-PTEN, which is a nuclear compartment-specific PTEN by linking PTENWT to a Simian virus 40 NLS peptide. After transfection, we observed decreased nuclear P-AKT only in cells expressing the NLS-PTEN (Supplemental Fig. 2). This suggests that nuclear PTEN is able to deplete P-AKT.
PTEN increases PIP2 in the cytoplasmic, especially in the perinuclear regions
Because we saw that PTEN decreases phosphorylation of nuclear AKT through its lipid phosphatase activity and we know PIP2 is a major product of PIP3 by PTEN′s action, we therefore set out to analyze whether PTEN expression could increase nuclear PIP2. PTEN was transfected into BT-549 cells, and then the cells were costained with PTEN and PIP2 antibodies. We found that expression of PTEN significantly increased PIP2 in the cytoplasm, especially in the perinuclear area, in which intracellular targets of PI3K activation are already known to exist (Supplemental Fig. 3) (20, 21).
Discussion
Our report represents the first systematic study in PHTS thyroid tumors showing loss of PTEN and elevation of nuclear P-AKT as two common and relevant events. We have demonstrated that CS patients who harbor germline PTEN mutations have not only increased cytoplasmic P-AKT but also elevated nuclear P-AKT. In parallel, we have also shown that proper nuclear PTEN expression is equally important for suppressing activated AKT in the nucleus. Our data provide evidence that both cytoplasmic and nuclear PTEN work together and act as a crucial negative regulator of P-AKT.
Studies from sporadic thyroid tumors suggest that AKT activation is a common event in thyroid cancers and adenomas (22). Vasko et al. (13) reported that AKT activation could be detected in six of 10 sporadic atypical follicular adenomas (60%) but not in any typical follicular adenomas. Similarly, in our study, we identified nine of 12 atypical follicular adenomas from PTENMut+ CS patients (75%) that showed activated AKT (Fig. 1A). In the study by Vasko et al., nuclear activated AKT was observed primarily in follicular thyroid cancers and in atypical follicular adenomas, whereas cytoplasmic localization of AKT was observed primarily in papillary thyroid cancers, unless the papillary thyroid cancers were invasive. In our study, although limited by the small sample size (because thyroid cancer tissues from PTENMut+ CS patients are very rare), we still observed a predominantly nuclear localization of P-AKT in all four different tumor types, with a total of 63.7% positive rate for nuclear P-AKT staining. Therefore, thyroid tumors from PTENMut+ CS patients have higher positive rates for both P-AKT and nuclear P-AKT when compared with sporadic thyroid tumors. This might be one of the reasons patients with germline PTEN pathogenic mutations have thyroid tumors younger than sporadic patients.
The activation and subcellular trafficking of AKT is a dynamic process. Components of the PI3K signaling pathway, including PI3K, PIP3, 3-phosphoinositide-dependent kinase-1, and its downstream kinase AKT, have been identified at both the cytoplasmic and the nuclear levels (23). The nuclear translocation of both P-AKT and unphosphorylated AKT had been reported in different cell models (6, 24, 25). For example, in human ambryonic kidney-293 cells, T-cell leukemia/lymphoma protein 1A promotes AKT1 nuclear translocation (25). Besides nuclear translocation, AKT can be directly activated in the nucleus upon growth factor stimulation (6) or through nuclear pyruvate dehydrogenase kinase-1, which can also be translocated from the cytoplasm after phosphorylation (26). In addition, AKT1 has been shown to have a functional nuclear export sequence that binds chromosome region maintenance protein-1 (12); thus, its localization can also be regulated via nuclear export.
We have focused our studies on the role of nuclear PTEN on AKT activation because loss of PTEN, especially nuclear PTEN expression, is not only a hallmark of PHTS but is also commonly observed in sporadic invasive cancers. We have previously shown that, in thyroid carcinomas as a group, nuclear PTEN immunostaining was mostly weak in comparison with normal thyroid follicular cells and follicular adenomas (9). We also previously showed a correlation between enhanced nuclear-predominant P-AKT with tumor invasion in thyroid follicular and papillary carcinomas (13). Because a role for PTEN in the nucleus, especially on AKT activation, largely remains unknown, we undertook this current study. By using different PTEN mutants that bifurcate the phosphatase activities, we confirmed that the lipid phosphatase activity is required for the inhibitory effect on cytoplasmic AKT and show that this activity is also important in regulating nuclear AKT. Some studies reported that nuclear compartment-specific PTEN (NLS-PTEN) was able to antagonize nuclear AKT activation (27, 28). We therefore studied whether diminished nuclear PTEN will lead to elevated nuclear P-AKT. In breast cancer cells, after serum or growth factor stimulation, AKT can be phosphorylated and subsequently translocated from plasma membrane or cytoplasm into the nucleus. In the cytoplasm, PTEN decreases PIP3/P-AKT, and so this will deplete the major origin and reservoir of nuclear P-AKT, which normally translocates from plasma membrane and cytoplasm into the nucleus. In the nucleus, the translocated P-AKT and any nuclear-activated P-AKT that escapes from cytoplasmic PTEN can still be deactivated by nuclear PTEN through a yet-unknown mechanism. Importantly, the effect of nuclear PTEN on nuclear AKT may need other components and may also depend on its phosphatase activity (11).
Different from breast cancer cells, thyroid cancer cells already have increase expression of nuclear total AKT, as demonstrated by both Western blot (Fig. 3) and IHC (Fig. 1). The abundant nuclear AKT may show a different activation mechanism because PTEN knockdown enhanced only nuclear P-AKT but did not significantly increase nuclear total AKT. In addition, our unpublished data verified that, unlike BT-549 breast cancer cells, in FTC-236 thyroid cancer cells, serum stimulation did not significantly induce AKT nuclear translocation. Therefore, it is very possible that, in thyroid cancer cells, nuclear P-AKT is mainly derived from the activation of nuclear AKT instead of translocation from cytoplasm.
Notably, we demonstrated that the nuclear compartment-specific PTEN (NLS-PTEN) was able to antagonize nuclear AKT activation. This is in accordance with previous reports by others. Hence, it is reasonable to postulate that the suppressive effect of PTEN on nuclear P-AKT is dependent on both intact lipid phosphatase activity and proper subcellular localization. In the cytoplasm, PTEN decreases PIP3/P-AKT, so this will deplete the major origin and reservoir of nuclear P-AKT, which normally translocates from the plasma membrane and cytoplasm into the nucleus of breast cancer cells. In the nucleus, P-AKT can still be deactivated by nuclear PTEN through a yet-unknown mechanism, and this is important for PTEN′s antagonizing effect on nuclear P-AKT in thyroid cancer cells. Importantly, the effect of nuclear PTEN on nuclear AKT may need other components and may also depend on its phosphatase activity (11).
Liu et al. (29) reported that nuclear PTEN-mediated growth suppression is independent of AKT down-regulation. In their study, they found nuclear PTEN leads to p70S6K inactivation without down-regulating AKT. This can be explained by the following two reasons: 1) cell line-specific difference (U251MG instead of BT-549); or 2) they interrogated P-AKT only in whole-cell lysates, whereas we used both fractionation and confocal immunofluorescence staining to examine nucleus vs. cytoplasm. Because NLS-PTEN expresses only in the nucleus, it may not change cytoplasmic P-AKT. Therefore, in the whole-cell lysates, small decreases of nuclear P-AKT will be masked in comparison with the large volume of the cytoplasmic compartment. Liu et al. (29) also reported that nuclear PTEN increased nuclear PIP2. In contrast, we found that PTEN increased PIP2 in the perinuclear area but not in the nucleus. Similar to our results, Lindsay et al. (28) reported a nuclear PIP3 pool that is insensitive to PTEN expression. By using live-cell imaging, another group also failed to detect the change of PIP3 and PIP2 in NIH3T3 cells after growth factor stimulation (30). Although the effects of PIP3 in the nucleus remain largely unknown, PIP3 can act as an adaptor to connect AKT and B23 (31). Importantly, PTEN or SH2 containing inositol-5-phosphatase abrogates the association between AKT and B23 of PIP3 in the nucleus (31). Therefore, one possible mechanism of nuclear PTEN is to abolish the association between AKT and other proteins to destabilize its phosphorylation status.
In summary, we have established that PTEN is both necessary and sufficient to inhibit AKT activation in the nucleus. Both proper subcellular localization and intact phosphatase activity are key factors determining PTEN′s suppressive effects on nuclear AKT. Characterization of nuclear PTEN in the nuclear AKT signaling pathway will provide insight into the molecular mechanism of how PTEN functions as a tumor suppressor in PHTS or in cancers with PTEN somatic mutations. Understanding the role of PTEN in the nuclear and cytoplasmic compartments should provide us with untapped pathways and mechanisms for diagnosis and prognosis and perhaps guide treatment.
Acknowledgments
This work was supported by Grant 5R01CA118980-05 (to C.E.) and Grant P01CA124570 (to M.D.R. and C.E.) from the National Cancer Institute (Bethesda, MD).
Disclosure Summary: No conflict of interest is declared.
Footnotes
- CS
- Cowden syndrome
- CSL
- Cowden-like syndrome
- IHC
- immunohistochemistry
- NLS
- nuclear localization sequence
- P
- phosphorylated
- PHTS
- PTEN hamartoma tumor syndrome
- PI3K
- phosphoinositol-3-kinase
- PIP3
- phosphatidylinositol-3,4,5-triphosphate
- PTEN
- phosphatase and tensin homolog deleted on chromosome 10
- shRNA
- short hairpin RNA.
References
- 1. Liaw D, Marsh DJ, Li J, Dahia PL, Wang SI, Zheng Z, Bose S, Call KM, Tsou HC, Peacocke M, Eng C, Parsons R. 1997. Germline mutations of the PTEN gene in Cowden disease, an inherited breast and thyroid cancer syndrome. Nat Genet 16:64–67 [DOI] [PubMed] [Google Scholar]
- 2. Tan MH, Mester JL, Ngeow J, Rybicki LA, Orloff MS, Eng C. 2012. Lifetime cancer risks in individuals with germline PTEN mutations. Clin Cancer Res 18:400–407 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Feilotter HE, Coulon V, McVeigh JL, Boag AH, Dorion-Bonnet F, Duboué B, Latham WC, Eng C, Mulligan LM, Longy M. 1999. Analysis of the 10q23 chromosomal region and the PTEN gene in human sporadic breast carcinoma. Br J Cancer 79:718–723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Andjelković M, Alessi DR, Meier R, Fernandez A, Lamb NJ, Frech M, Cron P, Cohen P, Lucocq JM, Hemmings BA. 1997. Role of translocation in the activation and function of protein kinase B. J Biol Chem 272:31515–31524 [DOI] [PubMed] [Google Scholar]
- 5. Planchon SM, Waite KA, Eng C. 2008. The nuclear affairs of PTEN. J Cell Sci 121:249–253 [DOI] [PubMed] [Google Scholar]
- 6. Wang R, Brattain MG. 2006. AKT can be activated in the nucleus. Cell Signal 18:1722–1731 [DOI] [PubMed] [Google Scholar]
- 7. Gil A, Andrés-Pons A, Pulido R. 2007. Nuclear PTEN: a tale of many tails. Cell Death Differ 14:395–399 [DOI] [PubMed] [Google Scholar]
- 8. Tanaka K, Horiguchi K, Yoshida T, Takeda M, Fujisawa H, Takeuchi K, Umeda M, Kato S, Ihara S, Nagata S, Fukui Y. 1999. Evidence that a phosphatidylinositol 3,4,5-trisphosphate-binding protein can function in nucleus. J Biol Chem 274:3919–3922 [DOI] [PubMed] [Google Scholar]
- 9. Gimm O, Perren A, Weng LP, Marsh DJ, Yeh JJ, Ziebold U, Gil E, Hinze R, Delbridge L, Lees JA, Mutter GL, Robinson BG, Komminoth P, Dralle H, Eng C. 2000. Differential nuclear and cytoplasmic expression of PTEN in normal thyroid tissue, and benign and malignant epithelial thyroid tumors. Am J Pathol 156:1693–1700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Perren A, Weng LP, Boag AH, Ziebold U, Thakore K, Dahia PL, Komminoth P, Lees JA, Mulligan LM, Mutter GL, Eng C. 1999. Immunohistochemical evidence of loss of PTEN expression in primary ductal adenocarcinomas of the breast. Am J Pathol 155:1253–1260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mistafa O, Ghalali A, Kadekar S, Högberg J, Stenius U. 2010. Purinergic receptor-mediated rapid depletion of nuclear phosphorylated Akt depends on pleckstrin homology domain leucine-rich repeat phosphatase, calcineurin, protein phosphatase 2A, and PTEN phosphatases. J Biol Chem 285:27900–27910 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Saji M, Vasko V, Kada F, Allbritton EH, Burman KD, Ringel MD. 2005. Akt1 contains a functional leucine-rich nuclear export sequence. Biochem Biophys Res Commun 332:167–173 [DOI] [PubMed] [Google Scholar]
- 13. Vasko V, Saji M, Hardy E, Kruhlak M, Larin A, Savchenko V, Miyakawa M, Isozaki O, Murakami H, Tsushima T, Burman KD, De Micco C, Ringel MD. 2004. Akt activation and localisation correlate with tumour invasion and oncogene expression in thyroid cancer. J Med Genet 41:161–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chung JH, Eng C. 2005. Nuclear-cytoplasmic partitioning of phosphatase and tensin homologue deleted on chromosome 10 (PTEN) differentially regulates the cell cycle and apoptosis. Cancer Res 65:8096–8100 [DOI] [PubMed] [Google Scholar]
- 15. Chung JH, Ginn-Pease ME, Eng C. 2005. Phosphatase and tensin homologue deleted on chromosome 10 (PTEN) has nuclear localization signal-like sequences for nuclear import mediated by major vault protein. Cancer Res 65:4108–4116 [DOI] [PubMed] [Google Scholar]
- 16. Mester JL, Tilot AK, Rybicki LA, Frazier TW, 2nd, Eng C. 2011. Analysis of prevalence and degree of macrocephaly in patients with germline PTEN mutations and of brain weight in Pten knock-in murine model. Eur J Hum Genet 19:763–768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ngeow J, Mester J, Rybicki LA, Ni Y, Milas M, Eng C. 2011. Incidence and clinical characteristics of thyroid cancer in prospective series of individuals with Cowden and Cowden-like syndrome characterized by germline PTEN, SDH, or KLLN alterations. J Clin Endocrinol Metab 96:E2063–E2071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lobo GP, Waite KA, Planchon SM, Romigh T, Nassif NT, Eng C. 2009. Germline and somatic cancer-associated mutations in the ATP-binding motifs of PTEN influence its subcellular localization and tumor suppressive function. Hum Mol Genet 18:2851–2862 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Eng C. 2003. PTEN: one gene, many syndromes. Hum Mutat 22:183–198 [DOI] [PubMed] [Google Scholar]
- 20. Niswender KD, Gallis B, Blevins JE, Corson MA, Schwartz MW, Baskin DG. 2003. Immunocytochemical detection of phosphatidylinositol 3-kinase activation by insulin and leptin. J Histochem Cytochem 51:275–283 [DOI] [PubMed] [Google Scholar]
- 21. Várnai P, Rother KI, Balla T. 1999. Phosphatidylinositol 3-kinase-dependent membrane association of the Bruton's tyrosine kinase pleckstrin homology domain visualized in single living cells. J Biol Chem 274:10983–10989 [DOI] [PubMed] [Google Scholar]
- 22. Shinohara M, Chung YJ, Saji M, Ringel MD. 2007. AKT in thyroid tumorigenesis and progression. Endocrinology 148:942–947 [DOI] [PubMed] [Google Scholar]
- 23. Lian Z, Di Cristofano A. 2005. Class reunion: PTEN joins the nuclear crew. Oncogene 24:7394–7400 [DOI] [PubMed] [Google Scholar]
- 24. Abe N, Watanabe J, Tsunoda S, Kuramoto H, Okayasu I. 2011. Significance of nuclear p-Akt in endometrial carcinogenesis: rapid translocation of p-Akt into the nucleus by estrogen, possibly resulting in inhibition of apoptosis. Int J Gynecol Cancer 21:194–202 [DOI] [PubMed] [Google Scholar]
- 25. Pekarsky Y, Koval A, Hallas C, Bichi R, Tresini M, Malstrom S, Russo G, Tsichlis P, Croce CM. 2000. Tcl1 enhances Akt kinase activity and mediates its nuclear translocation. Proc Natl Acad Sci USA 97:3028–3033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Scheid MP, Parsons M, Woodgett JR. 2005. Phosphoinositide-dependent phosphorylation of PDK1 regulates nuclear translocation. Mol Cell Biol 25:2347–2363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Trotman LC, Wang X, Alimonti A, Chen Z, Teruya-Feldstein J, Yang H, Pavletich NP, Carver BS, Cordon-Cardo C, Erdjument-Bromage H, Tempst P, Chi SG, Kim HJ, Misteli T, Jiang X, Pandolfi PP. 2007. Ubiquitination regulates PTEN nuclear import and tumor suppression. Cell 128:141–156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lindsay Y, McCoull D, Davidson L, Leslie NR, Fairservice A, Gray A, Lucocq J, Downes CP. 2006. Localization of agonist-sensitive PtdIns(3,4,5)P3 reveals a nuclear pool that is insensitive to PTEN expression. J Cell Sci 119:5160–5168 [DOI] [PubMed] [Google Scholar]
- 29. Liu JL, Sheng X, Hortobagyi ZK, Mao Z, Gallick GE, Yung WK. 2005. Nuclear PTEN-mediated growth suppression is independent of Akt down-regulation. Mol Cell Biol 25:6211–6224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ananthanarayanan B, Ni Q, Zhang J. 2005. Signal propagation from membrane messengers to nuclear effectors revealed by reporters of phosphoinositide dynamics and Akt activity. Proc Natl Acad Sci USA 102:15081–15086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kwon IS, Lee KH, Choi JW, Ahn JY. 2010. PI(3,4,5)P3 regulates the interaction between Akt and B23 in the nucleus. BMB Rep 43:127–132 [DOI] [PubMed] [Google Scholar]