

Abstract
This unit describes a method for quantifying various cellular features (e.g., volume, total and subcellular fluorescence localization) from sets of microscope images of individual cells. It includes procedures for tracking cells over time. One purposefully defocused transmission image (sometimes referred to as bright-field or BF) is acquired to segment the image and locate each cell. Fluorescent images (one for each of the color channels to be analyzed) are then acquired by conventional wide-field epifluorescence or confocal microscopy. This method uses the image processing capabilities of Cell-ID (Gordon et al., 2007, as updated here) and data analysis by the statistical programming framework R (R-Development-Team, 2008), which we have supplemented with a package of routines for analyzing Cell-ID output. Both Cell-ID and the analysis package are open-source.
Keywords: image processing, fluorescence microscopy, Cell-ID, R
INTRODUCTION
This unit describes a method for quantifying various cellular features from individual cells using sets of microscope images. It includes procedures for tracking cells over time. Using this method, the user can measure total and subcellular localization (nuclear, plasma membrane) of fluorescence for multiple fluorescence channels (Colman-Lerner et al., 2005; Yu et al., 2008). This method uses the image processing capabilities of Cell-ID (Gordon et al., 2007) and data analysis routines written in the by the statistical programming framework R. Both Cell-ID and R are open source. The first step for successful cytometry entails acquiring at least one set of images for each field of cells. Each set is composed of one purposefully defocused transmission image (referred to as bright-field, or BF) that will be used to locate each cell, and one fluorescent image for each of the color channels to be analyzed (xFP, where x designates the color of the fluorescent protein). Fluorescence images may be acquired by conventional wide-field epifluorescence or confocal microscopy. Cell-ID processes the images and outputs annotated TIFF images and a tab-delimited file with information extracted from each cell, for each time point and each fluorescence channel. Finally, the user analyzes the data using R (R-Development-Team, 2008), supplemented with a package design to analyze Cell-ID output.
Like the original Cell-ID 1.0 (Gordon et al., 2007), Cell-ID version 1.4 supports a full command-line interface, which allows sophisticated automatic analyses using scripts; it can be compiled to run on most computer systems. In addition, Cell-ID 1.4 incorporates a graphical user interface (VCell-0.1), which provides easy testing options that help the user choose the correct parameters to process the experiment in batch mode, as well as bug fixes and improvements. Although Cell-ID was originally tailored for yeast cells, it works well with non-adhering mammalian cells (Gordon et al., 2007).
R is an environment for statistical analysis, data manipulation, calculation, and graphical display (http://www.r-project.org). Rcell (author, give version number?) is a package that was created to aid in the analysis of Cell-ID output. Rcell vx contains functions to load the dataset into R, filter and visualize the data. The use of R allows all of R’s statistical capabilities to be applied on the data.
This unit focuses on procedures for measuring fluorescent protein–based reporters. However, the investigator can use nearly identical approaches when measuring signals from fluorescent antibodies or light-emitting enzymes. The Basic Protocol presents the methodology used to acquire, process, and analyze quantitative single-cell data extracted from time-course experiments. The Alternate Protocol describes a procedure for quantifying measurements obtained in intensity-based fluorescence resonance energy transfer (FRET) experiments. Support Protocol 1 provides information about obtaining and installing necessary software. Support Protocol 2 presents a short guide for preparing yeast and mammalian cells for imaging. Support Protocol 3 describes how to perform FRET calculations using split images and how to measure FRET in the nucleus and plasma membrane of yeast.
BASIC PROTOCOL
EXTRACTING QUANTITATIVE INFORMATION FROM SINGLE CELLS
In this protocol, Cell-ID and R are employed to process and analyze cell images in time-course experiments, where the same cells at one or more positions are followed over time. Here, a “position” corresponds to a defined xy coordinate for the microscope stage. All images of the same cells should have the same position number, and different positions should not be of the same cells. For each time point, the user will acquire one set of images. The simplest case is when there is only one time point; in this case, the user collects only one image set per position. If the cells at the different time points are not the same (e.g., when taking different samples from a culture at different times) each set should be labeled with a different position number (see below, under section how to label files), since in this type of experiment one will not be following the same cells over time, but rather different cells from the same population over time.
The combination of Cell-ID and R is most powerful when following the same cells in time series, since it allows the user to measure dynamic processes in living cells, e.g., reporter protein expression, relocalization of proteins in response to specific signals, correlations between fluorescent protein levels in single cells, changes in protein conformation and oligomerization state by FRET (fluorescence resonance energy transfer; see Alternate Protocol), and protein and mRNA degradation rates or growth rates (Colman-Lerner et al., 2005; Gordon et al., 2007; Yu et al., 2008). Time series are especially useful for measuring small signals close to the levels of noise, which are best detected as changes over time.
Materials
Cells of interest affixed to the bottom of multiwell glass-bottom plates or on slides (Support Protocol)
Optically appropriate support for cells (multiwell plates preferred over slides and cover slips for easy access)
For oil immersion with high-NA objectives: glass-bottom, coverslip-thin 96- or 384-well plates (available from Arctic White)
For air or water immersion objectives: plastic or glass-bottom multiwell plates
Hardware
- Fluorescence microscope with the following features:
- Inverted (to allow access to live cells during imaging over time), preferable
- Stage with motorized z control, preferable to manual focus for convenience and reproducibility
- Stage with motorized xy control (for live imaging of multiple fields of view, e.g., in neighboring wells with cells treated differently), optional
- Motorized shutter (for precisely controlling exposure time of fluorescence illumination), optional for bright-field illumination
- High-numerical-aperture (NA; e.g., >1.2 for 60× objective), chromatically corrected (at least PlanApo) objective to capture as much light as possible, especially important for work that requires subcellular colocalization
- Black-and-white (not color for quantitative imaging), cooled CCD camera (when using non-confocal microscope)
- Appropriate filter cubes or wheels (motorized filter cube turret for complete automation of imaging)
- PC to run microscope
- Acquisition software, e.g., Metamorph (Molecular Devices), ImagePro (Media Cybernetics), or µmanager (UNIT 14.20, open-source microscopy software initiative at UCSF; http://www.micro-manager.org)
PC workstation with UNIX, LINUX, or Windows XP or higher operating systems; or Apple workstation with Mac OS X operating systems, Xcode and X11 installed
Software
VCell-ID and Cell-ID (Gordon et al., 2007) (included in windows distribution of VCell-ID)
R (R-Development-Team, 2008) and the package Rcell (see Support Protocol)
Image files
Images in TIFF format (gray-level TIFF files of 8 and 16 bits); time-related information can be extracted from the Metamorph-generated image files (see below)
Determine experimental settings
-
1Determine the correct magnification for your application.One can normally use a 60× oil-immersion or 40× air objective for yeast and a 20× air objective for mammalian cells. The advantage of 20× magnification is that each image contains many more cells, and the air objective makes glass-bottom plates unnecessary; the disadvantage is that the objective captures less light and produces images with lower spatial resolution.
-
2
Prepare cells with the lowest expected fluorescence levels for each channel (e.g., cells not expressing any fluorophore) and cells with the maximum levels for a given experiment (e.g., fully-induced cells when using fluorescent protein–based transcriptional reporters).
-
3
Place these cells, either affixed to the bottom of multiwell glass-bottom plates or on slides, in the microscope and find the focus using one of the fluorescence channels.
-
4
At this z position, move the stage to a fresh unexposed field of cells and, starting with the lowest energy (longest wavelength) channel, acquire a series of images with increasing exposure times. Determine the shortest exposure time necessary to visualize the cells that do not express fluorescent protein.
-
5Repeat step 4 using cells expressing the reporter in the appropriate fluorescence channel, with different exposure times, until pixel intensities become saturated.In many applications (e.g., ImageJ and Metamorph), moving the cursor over an image will display the xy coordinates and the intensity of the pixel. Values for saturated pixels depend on the image depth: 255 for 8-bit and 65535 for 16-bit images.The times determined in steps 4 and 5 are the minimum and maximum exposure times. In some cases, the exposure time determined in step 4 is higher that of step 5. In this case, you need a CCD camera with larger dynamic range, or you will have to change exposure time during the experiment (this option will complicate analysis).Exposure time is a critical parameter. If exposure is high, sample photobleaching and phototoxicity increase. This might be an issue when performing a time-course experiment. On the other hand, if exposure is too low, the signal-to-noise ratio might be too low. Choose an exposure time appropriate for the particular experiment.
Acquire images
- Steps 6 and 7 represent images from one set of images, for one position and one time point.
-
6Acquire the bright-field image:
- Take an image with the cells at the focal plane using transmitted light.
- Move the z knob so that the objective moves slightly down (away from the stage) until a dark ring surrounds the cells (Fig. 14.18.1).
- Record how much the stage was moved.For a 60× objective, this is usually 1µm.
- Take another image.This is the bright-field image that Cell-ID uses to find the cells.
- Label the image BF_Position[X].tif, where [X] is a number. Make sure all of numbers [X] have the same number of digits, e.g., label images between 1 and 99 as 01, 02, 03, and so on; label images between 1 and 999 as 001,002,003 and so on. If following the same cells over time and taking images for multiple time points per position, name the file BF_Position[X]_time[Y].tif; in this case, also make sure that the numbers [Y] have the same number of digits.If your acquisition software saves the images with a defined pattern, you can rename them in bulk using applications like Bulk Rename Utility (http://www.bulkrenameutility.co.uk) or custom scripts.To be able to find the cells with Cell-ID, a bright-field image is required, which can be obtained with a transmission channel in the confocal setup. If these images are noisy because of the low integration time, you can improve them by averaging several images of the time course together.Cell-ID works better with homogenously illuminated bright-field images. To correct for uneven illumination you can apply a band pass filter on the BF images, a preprocessing step that greatly enhances the performance of Cell-ID. An ImageJ macro to do this correction is provided (http://sourceforge.net/projects/cell-id/files/)
-
7Acquire fluorescence images:
- Move the z position again to the focal plane (that is, move it up the same distance you moved it down).
- Acquire one fluorescence image for each channel required. Using the settings determined in steps 4 and 5, take images from lowest to highest energy (from red to violet) to minimize cross-photobleaching of the sample.
- Label images according to the color, in a stereotyped way, as in step 6. For example, RFP_Position01_time001.tif, YFP_Position01_time001.tif, and CFP_Position01_time001.tif.
-
8Repeat steps 6 and 7 for as many positions and time points as needed. Number each image set consecutively.Acquiring images with a confocal microscope has several advantages over epifluorescence. In first place, it eliminates out-of-focus light and has better spatial resolution. These assets could be useful for the quantification of fluorescence in subcellular compartments. In addition, the illumination intensity can be fine-tuned with great precision, allowing the optimization of the fluorescence signal while maintaining acceptable photobleaching levels.
-
9Optional: If you want Cell-ID to subtract a background image for every fluorescent color, then take an image with the shutter closed with the appropriate exposure time determined for each color in steps 4 to 6. Label these images appropriately, e.g., YFP-dark.tif, CFP-dark.tif, and RFP-dark.tif for YFP, CFP, and RFP image exposure times, respectively.
-
10Optional: If you want Cell-ID to correct for uneven illumination, take images with each filter cube and exposure settings in areas with medium but without cells. Label these images, e.g., YFP-flat.tif, CFP-flat.tif, and RFP-flat.tif.Usually rich culture medium (e.g., YPD) is autofluorescent enough to give a large enough signal for Cell-ID to calculate a correction. If the illumination is uneven, the fluorescence emitted will be too, and Cell-ID will attempt to apply a flattening correction based on these images. Additionally, if the medium is significantly autofluorescent, care should be taken to avoid xy positions that are near the edges of the wells.
-
11Place all images in the same folder.
-
6
Figure 14.18.1.

Examples of yeast and mammalian cells processed by Cell-ID. (A) From left to right: yeast in focus, slightly defocused (note the dark ring on the border of the cells), and the same cells after Cell-ID has identified each one and traced their borders correctly. Bar = 5 µm. Magnification: 60×. (B) HEK293 cells fixed, trypsinized, and imaged (see Support Protocol 2 and the Basic Protocol), before (left) and after (right) Cell-ID has located them in the image. Bar = 15 µm. Magnification: 20×.
Process images with Cell-ID
-
12
Start a VCell-ID session. PC users can click on the VCell-ID icon. For Linux and Macintosh users, start VCell-ID from an X11 terminal, typing at the prompt: vcellid &. The VCell-ID window will appear. The main window has several tab buttons across the menu.
-
13Select Setup→Load Images. The Load Images window will appear (Fig. 14.18.2).
- In the drop-down menu below “path”, select the folder containing the images from all the channels (including BF).
- In the drop-down menu next to “group images by,” select how Cell-ID should map bright-field images to fluorescence images. Select “Time (Metamorph)” only if Metamorph was used to acquire the images and NO preprocessing of the image was done (e.g. correction for uneven illumination of BFs). Otherwise, select the default “file name pattern.”With the Metamorph option, Cell-ID will use the bright-field image closest in time to a given fluorescence image based on its internal TIME TAG. This information is embedded in each Metamorph TIFF image(it is not the file creation time shown by the computer) and is usually erased by any kind of preprocessing of the image. Otherwise Cell-ID will use the last bright-field image acquired before the given fluorescence image, based on the files NAMES. For example BF_Position03_time005.tif will be used to process YFP_Position03_time005.tif.
- For images not named as suggested in steps 6 and 7, change the “position” and “time” token, and the “separator character” to match those of the images.
- For an experiment without time points, uncheck “time token.”
- Optional: For dark and flat images, include the tokens “camera background correction image” and “uneven illumination correction image” (see examples in steps 9 and 10).
- Click OK to load the images and to close this window.
-
14
Use the experiment tree shown on the left panel in the main window to explore the images in your experiment. Click on the triangles to expand a node. The experiment is now organized based on positions. Click on an image file to load it on the central window. You can zoom in or out using the buttons in the toolbar
-
15Optional: Select Setup→Image Setup. The Image Setup window will appear (Fig. 14.18.3).
- For bright-field images to be treated as fluorescence images, check “bf as fl.”This is sometimes useful to filter out out-of-focus cells, since these cells appear brighter in the bright-field image.
- To find fluorescently labeled nuclei with Cell-ID, check “nucleus from channel” and type the image token corresponding to it, e.g., YFP.
- To analyze a split image FRET experiment, check “split fret image” and proceed to the Alternate Protocol.
- To analyze the data with PAW (http://www.cern.ch/paw), select “PAW friendly output.”PAW (Physics Analysis Workstation) is an interactive system based on several components of the CERN Program Library. The rest of this chapter assumes you selected the default “R friendly output”.
- Type the string you want Cell-ID to add to the name of the output data file (default is “out”).
-
16Select one bright field image on the tree and right click (option-click in Mac’s X11). Select “Show Test Dialog” on the pop-up menu. The segmentation window will appear.The choice of values in this window will profoundly affect how Cell-ID segments the images.
Set the following parameters:
“max dist over waist” (D/W)
“max split over minor”These two parameters were designed to determine if a budding cell should be divided into two, a mother and a daughter cell. The defaults work well with yeast. If the bud´s perimeter (dist) divided by the width of the neck (waist) is more than “max dist over waist”, Cell ID will try splitting them. The split will be done only if the waist of the original cell (i.e., the minor distance between any two points of the cell boundary) divided by the smallest of the minor axis of both new cells is less than the value for” max split over minor”.“min pixels per cell”
“max pixels per cell”Lower and upper boundaries for the size of a cell, in number of pixels. This parameter is useful for limiting the number of spurious cells (false positives)identified by Cell-ID. The actual number depends on the magnification of the objective and the size of pixels in the chip of the CCD camera. For example, in images taken with an oil-immersion 60× objective on a CCD camera with 13-µm pixels, exponentially growing yeast have an average area of 400 pixels, and limits between 200 and 600 work well to include all cells.“background reject factor”Cell-ID´s segmentation algorithm makes an initial decision about the intensity of the boundary pixels. To do this, it takes the mean intensity of the bright-field images and subtracts a defined number of standard deviations. It then starts by considering all intensities below this value as being parts of the cell borders (black halos in the BFs images). The number of subtracted standard deviations is the parameter “background reject factor.” Usual values are between 0.4 and 1.2.Bright-field images taken slightly out of focus may do better with higher values (i.e., higher values will better avoid spurious cells), but if the cell boundaries in the image are too narrow, a smaller value may be necessary, which might have the consequence of increasing the level of incorrectly segmented cells.“Tracking comparison”Cell-ID attempts to track cells over time. The value of this parameter is the minimal fractional overlap between two cells in consecutive time points for them to be considered the same cell. The default value is 0.2.“Frame alignment”Cell-ID can perform image registrations, moving the the frame in XY to align it to a reference image. If “align FL to BF” is selected the bright field image is used as reference. If “align FL to first” is selected the first fluorescence image is used as reference. These options are especially useful when sampling different positions, as repositioning of the microscope stage might introduce some displacement between consecutive images.“Cell alignment”If “align individual cells” is selected Cell-ID will move the contour of each cell found on the bright-field image to match the cells in the fluorescence images. This feature is useful when cells are not affixed and move slightly between image acquisitions. This options significantly increases the execution time of Cell-ID and should not be used if the cells are affixed. -
17
After selecting values for these options, click “Test Cell-ID”. Cell-ID will do a test on the selected bright-field image, and it will display the output files on the main screen (Fig. 14.18.4).. Click on OK to return to the main menu.
-
18In the bright-field image, check if Cell-ID appropriately found the majority of the cells.Depending on how clean the background of the bright-field image is, Cell-ID might also have found structures in the image that were “mistaken” as cells. These incorrectly found “spurious cells” can be eliminated later during analysis by appropriately applying filters (see “cuts” below). If Cell-ID did not find the cells correctly change the parameters in the Segmentation Setup, specially the “background reject factor” (Fig. 14.18.5).
-
21
If the test was satisfactory, try it on another BF
-
22
Once you found adequate segmentation parameters for your images, right click on the tree view “root” node, and select “Show Run Dialog”. If not already visible, the Segmentation Run Dialog will appear (it might be behind the main window). Click on the Run button to process the whole experiment. Cell-ID will create new images named identically with the added “.out.tif” file extension (e.g., the processed version of BF_Position01_time01.tif will be called BF_Position01_time01.tif.out.tif). In addition, new folders will also be created with input and generated output files.
Figure 14.18.2.
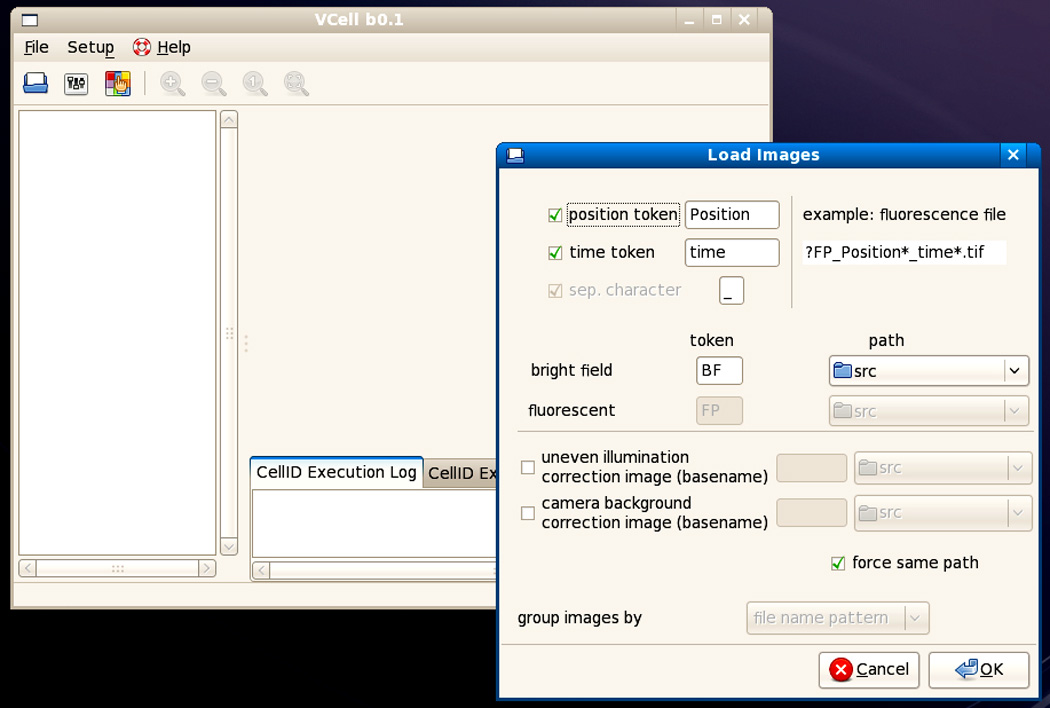
The main VCell-ID window with the Load Images window open, before selecting the folder containing the images.
Figure 14.18.3.
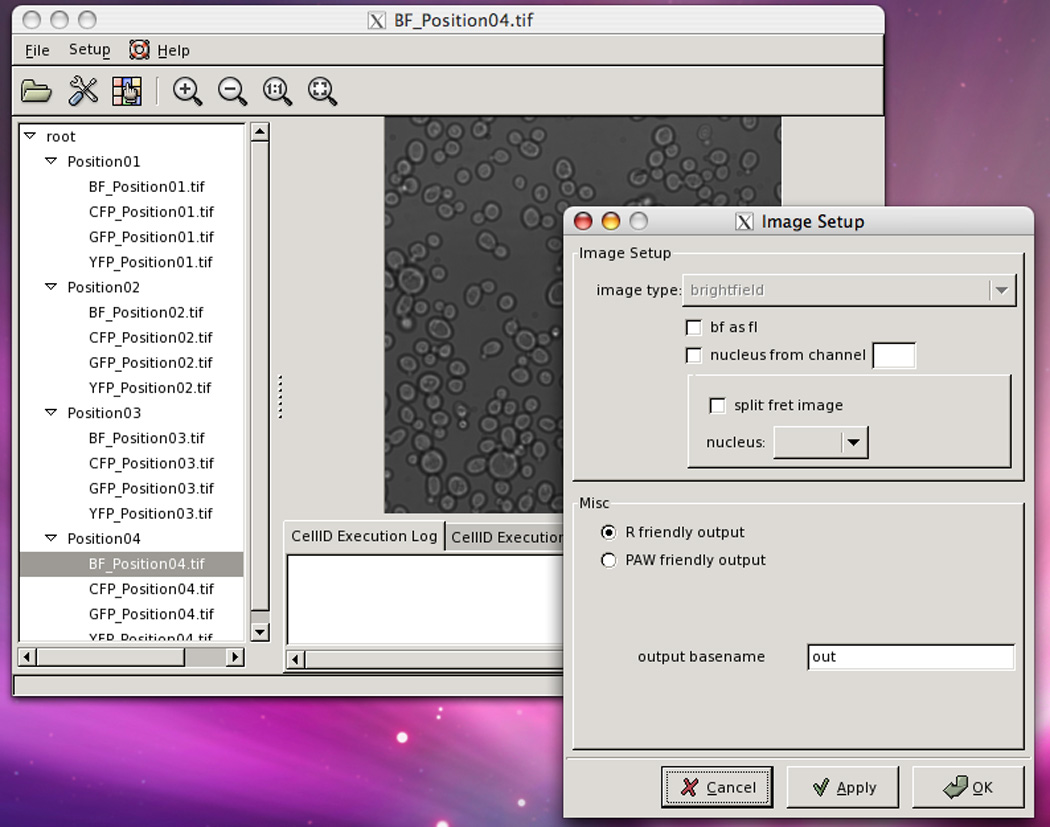
The main VCell-ID window with the Image Setup window open, after applying the settings shown in Figure 14.18.2. On the main window at the left, is the directory tree with BF_Position04.tif selected. That image is shown in the central window.
Figure 14.18.4.
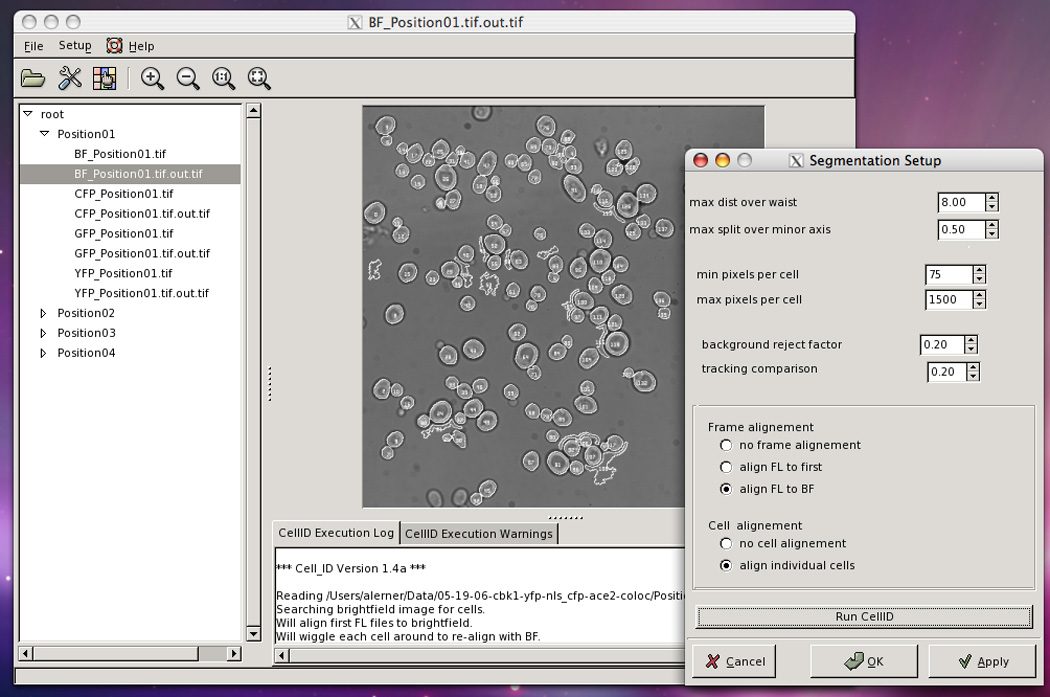
The main VCell-ID window with Segmentation Setup open, after running Cell-ID with the parameters shown. Note that on the tree directory there are new images, the out.tif files created by Cell-ID. On the central window the cells have been found by Cell-ID. Note the presence of several structures in the background wrongly identified by Cell-ID as cells.
Figure 14.18.5.

The effect of changing the value of “background reject factor.” The same image was processed by Cell-ID using two different values for this variable, 0.2 and 0.8 (left and right, respectively). Note that with 0.8 fewer spurious cells were found.
Analyze data with R
- Cell-ID will have created one folder named Position[X] for each position in the experiment and within each of these folders it will have created a file with the data called out_all. In this TAB delimited file, the columns contain features calculated by Cell-ID (Table 14.18.1). You can use the program of your choice to analyze it. This section presents an explanation of how to use the R package “Rcell” to analyze this data.
-
23Open R and load the R package Rcell by typing library(Rcell) in the R console, or selecting from the “packages > load package” menu if using an R-GUI.You can read a tutorial on the package by typing vignette("Rcell") in the console.
-
24Type setwd("c:/MyData/ExpFolder") in the console, using the path to the directory that contains the images and the Cell-ID output folders. This will set the working directory. Alternatively you can do it from the “file > change dir” menu in Windows.In R the backslash “\” is a reserved character. Use forward slash to specify the path for the working directory.
-
25Open the Cell-ID output files with the command X<-load.cellID.data(). This will load the out_all files and restructure the data for efficient filtering and plotting. The function returns an R object of class “cell.data” with all the required information associated with the experiment.All the functions included in the package operate over this object and its components should not be modified directly, but through the provided functions. You can name it as you prefer; throughout this example it is called X.The restructuring of the data modifies Cell-ID output variables and creates a few extra ones (Table 14.18.1). It appends a channel dependent postfix (the shortest unambiguous string from the channel token, in lower case). For example, if one has YFP and CFP channels, the total fluorescence for each channel becomes f.tot.y and f.tot.c, respectively. Analogously, f.bg becomes f.bg.y and f.bg.c, f.nucl becomes f.nucl.y and f.nucl.c, etc.Nonfluorescence-related variables are not renamed. For example, a.tot, pos and t.frame remain as such (Table 14.18.1).The R commands entered in the console window for data loading might look like this:
#Load the R package Rcell. library(Rcell) #Set the working directory. setwd("C:/Path/…/MyData") #Load the data for all the positions. X<-load.cellID.data()
-
23
Table 14.18.1.
Cell-ID Output Variables
| Variable Name | Description | |
|---|---|---|
| General measurements | ||
| 1 | pos | Position number (i.e., id of image field); same for a single cell through every set of images of a time course. This variable is created by Rcell when the dataset is loaded. |
| 2 | cellID | Cell identification number. In FRET experiments the cells in the upper and lower part of the split image are differentiated by an offset of 1000 added to this value. |
| 3 | ucid | Unique cell id. A variable that identifies cells across different positions; defined as pos*offset+cellID, where offset is 100000. This variable is created by Rcell when the dataset is loaded. |
| 4 | t.frame | Time frame of the cell (0 through n − 1 where n = number of points in the time course). Not every cell is necessarily found in every time point. |
| 5 | time | Time of that time frame in seconds. The time unit is an absolute number of seconds from some time in the distance past, but the time elapsed between time frames is more meaningful (only meaningful if using Metamorph images). |
| 6 | xpos | x coordinate of the centroid of the cell |
| 7 | ypos | y coordinate of the centroid of the cell |
| 8 | f.tot | Sum of the fluorescence image for all the pixels found in that cell |
| 9 | a.tot | Area of the cell in pixels |
| 10 | fft.stat | Statistic derived from the one-dimensional fast-Fourier-transform (FFT) of the function: radius vs. angle, where the radius is the distance from the cell centroid to the boundary at a given angle. Its value is the root of the squared sum of the ratio FFT(ω)/FFT(0) over all ω>0; for a perfect circle FFT_stat is 0, and we interpret this statistic as a measure of non-circularity. |
| 11 | perim | Circumference of the cell in pixel units |
| 12 | maj.axis | Length of the major axis in pixel units |
| 13 | min.axis | Length of the minor in pixel units |
| 14 | flag | Indicates the image type. For example, for YFP and CFP images, all YFP images would be flag 0 and all CFP would be flag 1, assuming that the YFP image was taken earliest in time |
| 15 | rot.vol | Volume of rotation of the cell around its major axis |
| 16 | con.vol | Volume of the cell as determined by the conical volume method (Gordon et al., 2007) |
| 17 | a.vacuole | Vacuole area: calculated from the region inside the cell that is less brightly fluorescent. In exponentially growing cells expressing fluorescence localized to the cytoplasm, this “dark” region corresponds to the vacuole. |
| 18 | f.vacuole | Vacuole fluorescence: calculated from the region inside the cell that is less brightly fluorescent. In exponentially growing cells expressing fluorescence localized to the cytoplasm, this “dark” region corresponds to the vacuole. |
| 19 | f.bg | Fluorescence background level; the mode of the distribution of all fluorescence pixels not associated with any cell |
| To calculate membrane proximal fluorescence (for relocalization experiments) | ||
| 20 | f.tot.p1 | Fluorescence of all the pixels interior to the boundary that is one pixel wider than the cell boundary. Numbers thus include the original cell plus an annular region one pixel around the outside of the cell. |
| 21 | a.tot.p1 | Area of all the pixels interior to the boundary that is one pixel wider than the cell boundary. Numbers thus include the original cell plus an annular region one pixel around the outside of the cell. |
| 22 | f.tot.m1 | Fluorescence of all pixels interior to the boundary that is one pixel smaller than the cell boundary |
| 23 | a.tot.m1 | Area of all pixels interior to the boundary that is one pixel smaller than the cell boundary |
| 24 | f.tot.m2 | Fluorescence of all pixels interior to the boundary that is two pixels smaller than the cell boundary |
| 25 | a.tot.m2 | Area of all pixels interior to the boundary that is two pixels smaller than the cell boundary |
| 26 | f.tot.m3 | Fluorescence of all pixels interior to the boundary that is three pixels smaller than the cell boundary |
| 27 | a.tot.m3 | Area of all pixels interior to the boundary that is three pixels smaller than the cell boundary |
| Information obtained from the “nuclear image” type (Variables contain the area and fluorescence of concentric disks of user-defined radius.) | ||
| 28 | f.nucl | Total fluorescence in the found nucleus. To find the nucleus, Cell-ID moves a disc with a radius of two pixels around the interior of the cell and finds the location where the disc has the maximum total fluorescence. From that location it calculates the fluorescence within a circle of four pixels of radius. This process is done for every fluorescence image. If some pixels of the disc fall outside the cell boundary, they are not used in the quantification. |
| 29 | a.nucl | Area of the found nucleus |
| 30 | f.nucl1 to f.nucl6 | Same as f.nucl, but using a disc of increasing radius to calculate the fluorescence for each image. f.nucl1 uses a disc of 2 pixels of radius, f.nucl2 uses a disc of 3 pixels, and so forth up to f.nucl6 which uses a disc of 7 pixels of radius. |
| 31 | a.nucl1 to a.nucl6 | The area corresponding to f.nucl1 to f.nucl6 |
| 32 | f.nucl.tag1 to f.nucl.tag6 | Same as f.nucl1 to f.nucl6, but the fluorescence is calculated from the nuclear tagged fluorescent channel, specified in step 15b. If no nuclear channel is specified, these variables are equal to f.nucl1 to 6. |
| More background information | ||
| 33 | f.local.bg | Measure of the background level at pixels located 5 radial pixels further out than the cell boundary; thus, a measure of the local fluorescence background level, the average fluorescence per pixel. Only pixels along the annular boundary NOT associated with ANY cell are included; background level here is the mean of the pixels. |
| 34 | a.local.bg | The number of pixels used in the background calculation for local.bg |
| 35 | a.local | Total number of pixels along the annular region, including all pixels (i.e., pixels associated with cells and pixels not associated with any cell) |
| 36 |
f.local2.bg a.local2.bg a.local2 |
Same information as the previous three variables, but using the background level at pixels located x radial pixels outward of the cell boundary, where x is one half of the minor axis of the cell. |
| More volume measurements | ||
| 37 | a.surf | Surface area as calculated by the union of spheres method (Gordon et al., 2007) |
| 38 | sphere.vol | Volume a measured by the union of spheres method (Gordon et al., 2007) |
Perform quality control
- The algorithm used by Cell-ID to find the cells can occasionally make mistakes in the assignation of the cell boundaries and produce badly found and spurious cells (i.e. image structures erroneously scored as cells). One can minimize these errors by adjusting Cell-ID´s parameters (step 16), but usually some badly found cells remain. Furthermore, the program cannot discriminate out-of-focus and dead cells.
-
26Filter out spurious cells from the dataset.This first filtering step is intended to eliminate from the dataset, spurious, out-of-focus and dead cells. If analyzing a time course, it filters the same cell in different time points independently, reflecting the fact that a cell can move out of focus, get its boundary found differently, or even die between time points. You can, however, require that a given cell appears in all the time points, or in more than a given fraction of time points.
-
bSpurious cells are usually odd shaped. The variable fft.stat (a meassure of non-cicularity) for those cells will be higher than for real, more spherical cells. Plot an histogram of this variable with cplot(X,~fft.stat) to see the distribution. Using the QC.filter function apply a cut over this variable: fft.stat < [your chosen value].For yeast and mammalian cells (e.g., HEK293 cells) a cut with fft.stat<0.3 is usually sufficient to eliminate most spurious cells.
-
cMany times, dead cells exhibit unusually bright fluorescence in one or more channels. Look for these cells and create a cut to limit the maximum fluorescence density (for YFP: f.tot.y/a.tot-f.bg.y<[your chosen value]).
-
dPerform additional cuts. You can plot the distribution of any variable with the cplot function. For example, for the background corrected fluorescence density in YFP, use cplot(X,~ f.tot.y/a.tot-f.bg.y). Select the cut limits and apply them using the QC.filter function.
-
eIf the “bf as fl” option was selected when running Cell-ID, use the intensity of the bright-field image in the analysis. Out-of-focus cells usually show a high intensity in the bright-field image, and thus can easily be removed by including in the cut f.tot.b/a.tot<[your chosen value] for this channel.
-
fIf you are analyzing a time course it is a good idea to filter by n.tot, the total amount of time frame in which a given cell appears. Spurious cell usually appear in a single image. First, update the value of n.tot as it depends on the previously applied filters. Do this by typing X<-update.n.tot(X) in the console. Apply a filter over n.tot as you would for any other variable, for example X<-QC.filter(X,n.tot==10) if your time course has 10 frames.Note the use of the comparison operator “==” when defining a cut. The single equal “=” is the assignation operator and will result in an error if used in the argument of QC.filer.A quality control cut might look like this:
#plot a histogram for fft.stat. cplot(X,~fft.stat) #filter the cells by circularity. X<-QC.filter(X,fft.stat<0.3) #cut on fluorescence density of the YFP channel. X<-QC.filter(X, f.tot.y/a.tot-f.bg.y<10000) #filter on n.tot. X<-update.n.tot(X) X<-QC.filter(X, n.tot>10)
The QC.filter function is cumulative: each time one applies a QC.filter function, it adds to the previous QC.filter; it does not replaced previous QC.filter. To undo one filtering step you can use the undo.QC function (typing X<-undo.QC(X)). To list the filters you have applied so far use the summary funtion (typing summary(X)). If you want to reset all the filters use the reset.QC function (typing X<-reset.QC(X)).
-
b
-
27Verify that quality control and cell selection were adequate using the show.img function.show.img displays the bright-field image generated by Cell-ID for a given position and time frame with the cells that were filtered out from the dataset marked with a cross (Fig. 14.18.6, left).
#show a image of Position 1 with the filtered #cells crossed. show.img(X,pos=1)
-
26
Figure 14.18.6.

Plotting with R. The upper left window contains an output of the show.img function. The upper right window shows a plot created with cplot. The bottom window shows the R console, with executed commands. For the color version of this figure go to http://www.currentprotocols.com.
Plot data
- Once the data is loaded in R and filtered, it can be readily plotted. R has extensive plotting capabilities, and there are several ways in which you can use them to obtain a particular graph. The easiest way is to use the Rcell package plotting functions, explained below.
-
28Plot the YFP total fluorescence versus the time frame for the first position, typing cplot(X,f.tot.y~t.frame,subset=pos==1) in the R console.Using cplot function you can do a lot of different plots. To read a tutorial on it type vignette("cplot") in the R console. Here we are plotting the data contained in the X object, with f.tot.y in the y axis and t.frame in the×axis, i.e. a scatter plot. The third argument, subset, is used to define the subset of the dataset that is going to be plotted. If the second argument has only a right term in it (ie, cplot(X,~f.tot.y,subset=pos==1)), a histogram will be plotted instead (see above).
#plots the total fluorescence over time for pos 1. cplot(X,f.tot.y~t.frame,subset=pos==1) #plots cells bigger than 400 pixels only. cplot(X,f.tot.y~t.frame,subset=pos==1&a.tot>400)
You can use logical operators (AND:& OR:| NOT:!) to create complex selections in the subset argument. Note that the subsetting will only be applied to the current plot, and will not be saved. -
29If desired, export filtered and aggregated variables of your choice to a tab-delimited file, for further analysis with other programs using write.table.The table to be exported can be created with the double square bracket operator (e.g. X[[pos==1,c("cellID","f.tot.y")]]) or with the aggregate function, that calculates a summary statistic of choice from the data, usually the mean.
#export a table with t.frame and f.tot.y for the #filtered cells of position 1 to a file. write.table(X[[pos==1,c("cellID","t.frame","f.tot.y")]],f ile="myOutput.dat", sep="\t") #export a table with the mean data used by cplotmeans. write.table(aggregate(X,f.tot.y~t.frame,subset=pos==1),fi le="myOutput.dat", sep="\t") -
30If desired, create new variables, using transform and transform.by.Filtering and plotting can be done over the new variables just as over the default ones. The new variable can be a frequently used combination of other variables, thus reducing the amount of typing required to produce the desired output. Using transform.by you can create normalized variables, for example the fold increase of the fluorescence with respect to the initial value. For a tutorial on these variables type vignette(“transform”) in the R console.
#create the nucl.fraction.y variable X<-transform(X,nucl.fraction.y=f.nucl.y/f.tot.y) #calculate the fold increase of f.tot.y with respect to #the initial value X<- transform.by(X,.(pos,cellID),norm.f.tot.y=f.tot.y/f.tot.y [t.frame==0])
-
28
Calculate nuclear fraction of fluorescence
-
31To calculate the nuclear fraction for a given channel, determine the ratio of nuclear to total fluorescence ((f.nucl-a.nucl*[auto])/(f.tot-[auto]*a.tot)). Cell-ID calculates both the total fluorescence associated with a cell (f.tot), and the nuclear fluorescence (f.nucl) for each channel (e.g, for YFP, f.tot.y and f.nucl.y).The parameter “auto” is not a variable directly available to the user. It is the average intensity (f.tot/a.tot-f.bg) observed in cells that do not express fluorescent proteins in that channel, and it should be determined experimentally in each case. To do this, include in your experiment a position with cells devoid of fluorescent proteins.Be aware that in a confocal setup f.tot.y is not a good measure of the total fluorescence because part of it is out of focus. You should take this into account when calculating the nuclear fraction.
ALTERNATE PROTOCOL
MEASURING FRET IN SINGLE CELLS USING A BEAM SPLITTER
This protocol describes how to employ Cell-ID to quantify intensity-based fluorescence resonance energy transfer (FRET), using Cell-ID’s capabilities to extract data from split images containing simultaneous emission from donor and acceptor fluorophores during donor fluorophore excitation. This method has advantages over sequential image collection of both donor and acceptor emission after donor excitation: reduced error in donor and acceptor emission by elimination of variation in donor excitation during separate exposures (both in time period and lamp strength during the exposure), reduced number of operations and increased speed, reduced donor fluorophore photobleaching, and reduced phototoxicity.
The protocol below refers to FRET measurements that are made between CFP donor and YFP acceptor fluorophores. The actual colors depend on optimizing spectral characteristics of the excitation and emission filters and the dichroic mirror.
We have successfully used this protocol to measure changes in FRET in the nucleus and in the plasma membrane of yeast (Yu et al., 2008). To locate the nucleus, one can label the nucleus with a separate color such as RFP. Alternatively, if one of the FRET partners is localized to the nucleus, it is possible to use, for example, the YFP excitation/YFP emission image to locate the nuclei and simultaneously quantify the amount of acceptor fluorophore. In the case of the cell membrane FRET, Cell-ID locates the membrane pixels using the bright-field image.
We have measured FRET in particular locations using a separate color (e.g., nuclearly localized RFP to identify the nucleus) with Cell-ID and an image splitter configured as described below. To measure FRET in a location that is marked by a color distinct from the donor or acceptor fluorophores, one should perform sequential image processing with four different images: donor excitation/donor emission; donor excitation/acceptor emission (FRET); acceptor excitation/acceptor emission; and location label color (different from donor or acceptor excitation and emission wavelengths).
Additional Materials (also see Basic Protocol)
Hardware
Image splitter with an appropriate dichroic mirror and top and bottom emission filters, e.g., for CFP/YFP FRET, a Dual View image splitter (Optical Insights/Photometrics, http://www.photometrics.de) fitted with the following filters and dichroic mirrors (Chroma): CFP emission, 480/30M; YFP emission, 535/20M; dichroic mirror, DX505.
Fluorescence microscope: same as in Basic Protocol, except remove the emission filter from the cube used to excite the donor fluorophore (e.g., for CFP/YFP FRET, remove the emission filter from the CFP cube); referred to as the FRET cube
Set up image splitter
-
1
Attach the image splitter to the microscope, and attach the camera to the image splitter.
-
2Cell-ID expects the image to be horizontally split so that the sub-images are on the top and bottom. Orient the camera relative to the image splitter so that the top sub-image is CFP emission and bottom sub-image is YFP emission image (recommended).This orientation will be assumed for the rest of this protocol.
-
3Test the image orientation by taking test bright-field images with the YFP cube selected. Ensure that the top half of the image is dark and the bottom half is not. Take another test bright-field image with the FRET (modified CFP) cube.Now both the top and bottom half should have an image.
-
4
On the image splitter, adjust the positions of the two halves of the image so that a thin horizontal black region separates the two sub-images halfway between the top and bottom.
Prepare samples
-
5
Follow steps 1a to 9a, in 9a Support Protocol for preparing cell samples.
Acquire images
-
6Remove the rapidly photobleachable autofluorescence by performing a number of prebleaching image sets. Establish the number by comparing the photobleaching curve of the autofluorescence (using the parental yeast strain not expressing any fluorescent protein) with that of the strain to be used in the FRET experiment.This procedure can be useful in any number of other applications.Yeast contain various fluorescent compounds, which together constitute cellular autofluorescence; this reduces the ability to distinguish the real signal. Typically, there are two groups of autofluorescent compounds, those that photobleach faster than YFP or CFP and those that are essentially photobleaching-resistant. When the signal to be measured is small, as in a FRET experiment, it is a good idea to reduce autofluorescence as much as possible.
-
7
Follow steps 1 to 5 in the Basic Protocol for determining exposure settings. Keep in mind that each time point for each image field requires two exposures, a FRET exposure and an acceptor fluorophore exposure.
-
8Acquire a bright-field image, following step 6 in the Basic Protocol.
- Use the FRET cube to ensure that the cells appear in both the top and bottom image panels.
-
Label the image BF_Position[X].tif, where [X] is a number. Make sure all of numbers [X] have the same number of digits, e.g., for between 1 and 99 images, label them 01, 02, 03, etc.; for between 1 and 999 images, label them 001, 002, 003, etc.If taking multiple time points per position, name the file BF_Position[X]_time[Y].tif; in this case, also make sure that the numbers [Y] have the same number of digits.
-
9
Acquire an acceptor excitation image using the YFP cube. Name it YFP_Position[X].tif or YFP_Position[X]_time[Y].tif, if taking multiple time points. The fluorescence will only show on the bottom half of the image.
-
10
Acquire a donor excitation image using the FRET cube. Name it CFP_Position[X].tif or CFP_Position[X]_time[Y].tif; again, ensure that all [X] and [Y] values have the same number of digits.
-
11
For each FRET measurement desired, repeat steps 7 to 9 for each time point and image field.
-
12
After collecting all images for all positions and all time points, place all images into the same folder.
Process images with Cell-ID
-
13Proceed to load images as described in steps 12 to 14 in the Basic Protocol. For example, for a typical nuclear CFP/YFP FRET experiment set the following parameter values given filenames as described above:
- Position token: Position
- Time token: time
- sep. character: _
- brightfield: BF
- Fluorescent: FP
- nuclear image: YFP.
-
14Set other options as described in steps 14 and 15 in the Basic Protocol.NOTE: The “uneven illumination image” is generally ignored. The typical FRET metrics are ratiometric in nature (i.e., they take some ratio of CFP emission and YFP emission from the top and bottom halves of the FRET image, respectively), which reduces or eliminates effects from uneven illumination as a function of position in the image typically.
-
15Set up segmentation according to step 16 in the Basic Protocol, except that the background reject factor is better if set lower for identifying cells in split brightfield images. Start at values from 0.5 to 0.8.Spuriously identified cells can be filtered during later analysis.
-
16
Test to see if Cell-ID is properly identifying cells in the split brightfield image, as outlined in steps 17 to 20 in the Basic Protocol. Cell-ID by default tries to match pairs of equivalent cells in the top and bottom images, where the ids of the cells in the upper image are offset by 1000. If only one partner is found in one of the image halves, then Cell-ID creates the partner in the other image half by copying the outline of the found partner to the top. Cell-ID calculates a predicted position and offset (i.e., translation and rotation from the found pair) based on how matched top and bottom pairs relate to each other in the rest of the image.
-
17
Start batch calculation as described in step 22 in the Basic Protocol.
Analyze data with R
- When performing split image FRET data extraction, Cell-ID outputs essentially the same variables as described in Table 14.18.1 in the Basic Protocol. When loading the data in R make sure to set the split.image argument to TRUE:
X<-load.cellID.data(split.image=TRUE).
- When the output files are loaded in R, the variables are renamed taking into account the channel and sub-image type (except for pos, t.frame, and cellID). A two letter postfix is appended to the fluorescent related variable name, the first one indicating the fluorescence channel as explained above, and the second one indicating if it is a upper sub-image (u) or a lower sub-image (l) cell. For nonfluorescent related variables, only the sub-image identifier is appended. For example, for an experiment with brightfield, CFP, and YFP images, the f.tot is converted into four variables: f.tot.cu, f.tot.cl, f.tot.yu, and f.tot.yl. Some variables, such as a.tot are typically the same for a given cell in both sub-images. In this case, for analysis, simply use one (i.e., a.tot.u).
-
18Proceed with data analysis as outlined in steps 23 to 31 in the Basic Protocol to examine basic cell properties (total fluorescence, area, etc.).
-
18
SUPPORT PROTOCOL 1
OBTAINING AND INSTALLING CELL-ID AND R
Cell-ID, VCell-ID and R are free, open-source software that can be readily downloaded and installed in several platforms (Windows, Mac-OS, and Linux). Here we outline how to install these programs and required packages.
Materials
Hardware
PC workstation with UNIX, LINUX, or Windows XP or higher operating systems; or Apple workstation with Mac OS X operating systems and X11 installed
Install R and Rcell
-
1
Download and install the latest binary distribution of R appropriate for the operating system being used, from the download section of the R-project Web page (http://www.r-project.org).
-
2
Download and install the latest binary release of ImageMagic for the operating system being used from http://www.imagemagick.org. Make sure to download the dynamic (DLL) version an NOT the static one. When asked select the “Add application to system path” and “Install development headers and libraries for C and C++” options.
-
3
Download and install GTK+ all-in-one bundle from http://www.gtk.org. Add the GTK/bin directory to your environment PATH variable.
-
4Open the R Console. To install Rcell from online repositories, type the following:
install.packages("Rcell")Alternatively, install it using the Packages->Install Packages menu of the R GUI. -
5To install EBImage (required for image manipulations), type at the console:
source("http://bioconductor.org/biocLite.R") biocLite("EBImage")If there is a problem during the installation of the EBImage package, download it from http://www.bioconductor.org/packages/release/Software.html, where there is an installation guide and a description of the package.
Install Cell-ID and VCell
-
6Download source code or binary files for Cell-ID and VCell from Source Forge http://sourceforge.net/projects/cell-id/, along with required dependences and installation guides.An installation wizard for windows is also available.
SUPPORT PROTOCOL 2
PREPARING YEAST AND MAMMALIAN CELLS FOR IMAGING
The exact culture conditions depend on the experiment. Below are standard procedures for obtaining an exponentially growing yeast cell population or mammalian cell population that will display low autofluorescence. This protocol works for many mammalian cell types, including HEK293, 3T3, and HeLa.
Materials
Yeast cultures
Appropriate growth medium for required yeast auxotrophies
Synthetic medium (also know as complete minimal medium; see UNIT 13.1) supplemented with all amino acids (except for those omissions required to select for plasmids, if any) and 20 µg/ml of adenine
1 mg/ml concanavalin A (Sigma) in phosphate-buffered saline (PBS; APPENDIX 2)
CaCl2
MgCl2
Yeast cell culture medium containing an appropriate stimulus
2% or 4% (w/v) paraformadehyde in PBS (APPENDIX 2) pH 7, 4°C
LB medium (UNIT 1.1) or YPD medium (UNIT 13.1)
PBS (APPENDIX 2)
1 mg/ml poly-L-lysine solution (Sigma)
Mammalian cell cultures (e.g., HEK293, 3T3, HeLa)
Mammalian cell culture medium containing an appropriate stimulus
50-ml glass tubes, sterile
30°C incubator
Rotating wheel or shaker for test tubes
Imaging plate appropriate for microscope
96- or 384-well glass-bottom plates
96-well plastic cell culture plates
Temperature and gas-controlled incubator mounted on the microscope (see Basic Protocol) for imaging live mammalian cells
Additional reagents and equipment for trypsinizing adherent cells (e.g., see APPENDIX 3F)
Prepare yeast cells
-
1a
Two days before the experiment, grow yeast overnight at 30°C in the appropriate synthetic medium for the required auxotrophies.
-
2aIn the morning, dilute the cells to 0.05 OD600 in fresh medium. Incubate at least 6 hr at 30°C.The goal is to shift the culture to exponential growth.
-
3aDuring the incubation time, prepare four 50-ml glass tubes with 5 ml synthetic medium supplemented with appropriate amino acids and 20 µg/ml of adenine.Adenine will repress the adenine biosynthesis pathway, which in many strains results in fluorescent by products that accumulate in the vacuole.Avoid YPD or other rich media, since they are usually highly fluorescent.
-
4aMeasure the OD600 in the afternoon and dilute the sample to OD600= 1 × 10−3 in the first tube. Perform 1:10 serial dilutions in the three remaining tubes. Incubate the tubes overnight at 30°C on a rotating wheel or shaker.The goal is to have one of the tubes at OD600 0.1 the next morning.Assuming a normal doubling time of 90 min, 15 hr of shaking will increase the OD 210 (~1000) times. In this case, the 2nd tube, with a starting OD600=1 × 10−4 will be used later.
Image live yeast cells
- Use the steps immediately below to image live cells. To image fixed cells go to step 12a.
-
5aThe morning of the experiment, or the day before, add 100 µl of 1 mg/ml concanavalin A solution to a 96-well glass-bottom plates, or 25uL to a 384-well plate.. Incubate at least 30 min or up to overnight at room temperature.
-
6aWash the plates three times with water.These concanavalin A treated plates are good for at least 1 day if kept wet.
-
7aAdd Ca2+ and Mn2+ to a final concentration of 200 µM Ca2+ and 100 µM Mn2+ to the cells in the tubes.This will improve the interaction of concanavalin A with the cell wall sugars.
-
8aAdd 100 µl of yeast culture of OD600 0.1 to the wells. Wait 10 min and wash away unbound cells three times with fresh medium.
-
9aSecure the plate tightly in the microscope stage.This is very important if you want to stimulate or otherwise add or remove medium from the wells DURING an experiment.The cells should be ready for live microscopy.
-
10aTo stimulate cells, pipet out the medium by carefully placing the tip on one of the corners of the well, then quickly replace with medium containing the appropriate stimulus.
-
11aProcess and analyze the cell images as in the Basic Protocol or the Alternate Protocol.
-
5a
Image fixed yeast cells
-
12a
Perform the experiment as usual, and take samples at the desired time points. For every 1 ml of sample, add 1 ml of ice cold 4% paraformadehyde and incubate for 1 hour on ice.
-
13aAdd 50 µl of sterile LB (or YPD) to each sample.Yeast do not centrifuge well if the sample is diluted and the experiment was done using synthetic medium. Adding LB largely corrects this problem.
-
14a
Centrifuge 20 sec at top speed in a microcentrifuge. Discard the supernatant and add 500 µl PBS.
-
15a
Repeat step 14a two more times.
-
16a
Load the cells (100 µl or 20 µl if using a 96- or a 384-well plate, respectively) onto the imaging plate and process and analyze the cell images as in the Basic Protocol or the Alternate Protocol.
Prepare mammalian cells
-
1b
Add 100 µl of 1 mg/ml poly-L-lysine solution to each of the wells of a 96-well plate and incubate for at least 30 min at room temperature.
-
2b
Wash the plates with PBS and store up to several days at 4°C.
-
3bSplit the cell culture one or two days before the experiment, so that the cells are actively growing.Use cells that are not confluent, so that there is enough space between cells for Cell-ID to locate individual cells and to calculate the background fluorescence. Live imaging is limited to cells that can be located by Cell-ID in a brightfield image. It works well with cells that do not spread on surfaces (e.g., lymphocytes) but not with cells with extensive spreading and processes (e.g., epithelial or neuronal cells). Images of cells that spread on a two-dimensional surface do not produce enough contrast for cell border identification by Cell-ID. Tissue culture medium is usually fluorescent, thus reducing the ability to image low-level signals. Try different media (e.g., medium without phenol red) or use medium diluted to half with PBS.
Image live mammalian cells
- Use the steps immediately below to image live cells. Proceed to step 7b to image fixed cells.
-
4bSet up the plate with the cells on the microscope stage, making sure that it is tightly secured.This is very important if you want to stimulate or otherwise add or remove medium from the wells DURING an experiment.Cells are ready for imaging.
-
5bTo stimulate cells, pipet out the medium by carefully placing the tip on one of the corners of the well, and then quickly replace it with medium containing the appropriate stimulus.
-
6bProcess and analyze the cell images as in the Basic Protocol or the Alternate Protocol.
-
4b
Image fixed mammalian cells
-
7b
Perform the experiment as usual, and take samples at the desired time points.
-
8b
Remove the medium. Trypsinize and collect the cells (e.g., see APPENDIX 3F).
-
9b
Resuspend the cells in 1 ml ice-cold 2% paraformadehyde per million cells. Incubate 1 hr on ice.
-
10b
Remove the paraformaldehyde by centrifuging 10 min at 1000 rpm in a benchtop centrifuge.
-
11b
Discard the supernatant and wash the cells two times with 2 ml PBS, centrifuging as in step 10b after each wash.
-
12bResuspend the cells in 100 µl of PBS. Use 1 µl to count the cells.NOTE: You can store these cells at 4°C.
-
13bDilute the cells appropriately in 100 µl of PBS and load in one poly-lysine-precoated (step 1b) well of a 96-well plate.NOTE: Usually, 20,000 to 50,000 cells per well is a good density.
-
14b
Process and analyze the cell images as in the Basic Protocol or the Alternate Protocol.
SUPPORT PROTOCOL 3
CALCULATING NUCLEAR AND PLASMA MEMBRANE CFP-YFP FRET USING SPLIT IMAGES
There are a number of published formulas for calculating FRET based on intensities measured from microscopic images (Gordon et al., 1998; Xia and Liu, 2001).
Perform FRET calculation
-
1For situations in which the CFP and YFP are attached to different proteins with different abundances, use the NFRET metric (Xia and Liu, 2001):
where the first letter in CY, CC, and YY refers to the excitation wavelength and the second to the emission wavelength used, respectively.CC, CY, and YY are measured in the region in which one wants to calculate FRET, such as the membrane, nucleus, or for the whole cell (see below for more details). -
2Correct the three measures (CC, CY, and YY) for background and cross-talk, using the following equations:
- CY = CYobs − BCY − ACC → CY(CCobs − BCC)
- CC = CCobs − BCC − ACY → CC(CYobs − BCY), ACY → CC ~0, thus CC = CCobs − BCC
- YY = YYobs − BYY
BYY, BCY, and BCC are the background cellular fluorescence in each emission channel;
ACC → CY and ACY → CC are the cross-talk coefficients of CFP into the YFP channel and of YFP into the CFP channel (see step 3 for cross-talk coefficient calculations).For nuclear FRET, a good estimate of BYY, BCY, and BCC is the average fluorescence of non-nuclear pixels, defined by all intracellular pixels outside of a circle with a radius of 6 centered on the calculated center of the nucleus (or by the intracellular pixels 3 pixels away from the border of the cell). In our experience, these are good per cell representations of background signal. Note that the radius of the circle needed for nuclear fluorescence quantification may change between different cell types and microscope setups.For membrane FRET, the signal in each channel is less affected by background cellular fluorescence; in this case, local backgrounds away from the cell can be used.The cross-talk coefficients allow calculation of how much signal one should expect to observe, for example, in the YFP channel due to CFP protein emission. The exact magnitude of the cross-talk coefficients ACC→CY and ACY→CC depends on the particular illumination and filter cubes used. -
3To calculate the cross-talk coefficient of CFP into the YFP channel ACC→CY, use a solution of purified CFP and take the ratio of YFP emission channel signal to CFP emission channel signal in a split image obtained with the FRET cube.For the image splitter configured as outlined above, this is the ratio of the bottom image signal to the top image signal. Using recombinant CFP we obtained 0.99, which shows that there is significant emission cross-talk of CFP into the YFP emission channel due to its broad emission spectrum (spectral bleed-through). For example, an image of a sample containing purified CFP protein, using CFP excitation wavelengths, will show up as a measurable signal in the YFP emission channel. Conversely, the emission cross-talk of YFP protein into the CFP emission channel with the optics outlined here is negligible.
Measure nuclear FRET in yeast
-
4To measure nuclear FRET, use the following quantities in the FRET equation.
For CC: CCobs = f.nucl3.cu/a.nucl3.cu BCC = (f.tot.cu-f.nucl6.cu)/(a.tot.u-a.nucl6.cu) For CY: CYobs = f.nucl3.cl/a.nucl3.cl BCY = (f.tot.cl-f.nucl6.cl)/(a.tot.l-a.nucl6.cl) For YY: YYobs = f.nucl3.yl/a.nucl3.yl BYY = (f.tot.yl-f.nucl6.yl)/(a.tot.l-a.nucl6.yl)
Measure plasma membrane FRET in yeast
-
5To measure membrane FRET, use the following quantities in the FRET equation.
For CC: CCobs = (f.tot.cu-f.tot.m1.cu)/(a.tot.u-a.tot.m1.u) BCC = f.local.bg.cu For CY: CYobs = (f.tot.cl-f.tot.m1.cl)/(a.tot.l-a.tot.m1.l) BCY = f.local.bg.cl ACC→CY=0.99 For YY: YYobs = (f.tot.yl-f.tot.m1.yl)/(a.tot.l-a.tot.m1.l) BYY = f.local.bg.yl
COMMENTARY
Background Information
Measurements in single cells can reveal information obscured in population averages. For example, studies of variation in gene expression in single Escherichia coli and Saccharomyces cerevisiae (Elowitz et al., 2002; Raser and O’Shea, 2004) have shown that only a fraction of cell-to-cell variation in the expression of reporter genes results from stochastic fluctuations in the workings of the gene expression machinery (Colman-Lerner et al., 2005; Pedraza and van Oudenaarden, 2005; Rosenfeld et al., 2005), and they have identified other processes and genes that account for and control the bulk of the variation (Colman-Lerner et al., 2005).
One means for collecting single cell data is flow cytometry. The instrument interrogates cells as they pass individually through an apparatus that excites fluorophores with a laser and collects the emitted light. These instruments are powerful (Shapiro, 2003; Bonetta, 2005) because they can automatically measure many hundreds or thousands of cells per second. However, flow cytometry has disadvantages. Flow cytometry: (1) cannot interrogate individual cells repeatedly to produce time series for each cell; (2) cannot collect a great deal of light, due both to the short time (typically µsec) that the cell passes the detector, and to the low numerical aperture of the objective, which often collects less than 10% of the emitted light; and (3) typically do not capture images of the cells, making it difficult or impossible to analyze cell shape, size, and intracellular localization of fluorescence.
Optical microscopy complements some of the limitations of flow cytometry. Optical microscopy provides the ability to revisit individual cells over time, excite cells, and collect emitted light for long times, and capture cell images with high resolution. Automation by computer-aided cell tracking and image analysis enables generation of such data with high throughput.
Recent research-directed, automated, microscope-based cytometry outside of clinical and pharmaceutical applications has relied on commercial software packages, e.g., Metamorph (Molecular Devices; see Inoue and Inoue, 1986; Schnapp, 1986; Gonzales and Woods, 2002) and ImagePro (Media Cybernetics), to operate the microscopes, collect the images, and analyze images. These packages, often used together with general purpose analysis programs, such as Matlab (Gonzales et al., 2003) and Labview (Bishop and Inc, 2005) probably constitute the state of the art in commercial software used for these purposes. The ability to perform microscope-based cytometry has recently been aided by open-source projects, in particular the Open Microscopy Environment (OME), which provides file formats and metadata standards for microscope images (Swedlow et al., 2003; Goldberg et al., 2005); Image J, a Java-based package of microscope image analysis tools (Abramoff et al., 2004); and CellProfiler (UNIT 14.17).
This unit describes a suite of user-modifiable technical and analytical methods to facilitate accurate, high-throughput measurements from single cells over time. Although Cell-ID was originally tuned to work with yeast cells, it has readily identified live mammalian lymphoid cells and trypsinized, live, HEK293 cells. When used with inexpensive optical microscopes and high-quality CCD equipment, these methods enable fluorescence measurements slightly more sensitive than those from contemporary flow cytometers (e.g., BD LSRII) and have allowed scoring, quantification, and extraction of meaningful statistics from one or two fluorescent proteins molecules per pixel in single cell images (Gordon et al., 2007).
Because it is open source, Cell-ID can be extended to work with any cell type that has an optically identifiable cell boundary. Cell-ID should also be applicable to different image types, e.g., phase-contrast, differential interference contrast, or even fluorescence images, although for the last case our work suggests that a preprocessing step might be necessary to determine the boundaries of the fluorescent regions.
Critical Parameters
Acquisition
z control and the brightfield image
It is critical to find consistently the dark ring in a defocused image. For example, image-to-image differences in z position in a time course will result in differences in the area assigned to the same cell over time, even if the cell really did not change its volume.
Camera specifications
Dynamic range is critical for imaging cells with large differences in FP levels. Sensitivity is important for reducing photobleaching when measuring time courses.
Objective
High numerical aperture is essential in order to image low levels of light.
Exposure time
This is critical to balance photobleaching and phototoxicity against higher signal-to-noise ratio.
Cell-ID
The value for the “background reject factor” parameter might make a difference between Cell-ID finding or missing cells in the bright-field image, as can be seen in Figure 14.18.5.
Troubleshooting
If cells are not identified, try defocusing the bright-field image further to increase the width of the dark ring around the cell border. Also try reducing the background reject factor.
Anticipated Results
Cell ID should have a very high rate of picking new cells and tracking them through multiple images from a time course.
Acknowledgments
Work was supported by grant 1R01GM097479-01 and subaward 0000713502 to Roger Brent and Alejandro Colman-Lerner from the National Institute of General Medical Sciences (USA), and by grants PICT2007-847 and PICT2010-2248 to Alejandro Colman-Lerner from the National Agency for the Promotion of Science and Technology (Argentina). The contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIGMS.
Literature Cited
- Abramoff MD, Magelhaes PJ, Ram SJ. Image processing with Image. J. Biophotonics International. 2004;11:36–42. [Google Scholar]
- Bishop R, Inc NI. Labview(TM) 7.0 Express Student Edition with 7.1 Update. Upper Saddle River, N. J: Prentice Hall; 2005. [Google Scholar]
- Bonetta L. Flow cytometry smaller and better. Nat. Methods. 2005;2:785–795. [Google Scholar]
- Colman-Lerner A, Gordon A, Serra E, Chin T, Resnekov O, Endy D, Pesce CG, Brent R. Regulated cell-to-cell variation in a cell-fate decision system. Nature. 2005;437:699–706. doi: 10.1038/nature03998. [DOI] [PubMed] [Google Scholar]
- Elowitz MB, Levine AJ, Siggia ED, Swain PS. Stochastic gene expression in a single cell. Science. 2002;297:1183–1186. doi: 10.1126/science.1070919. [DOI] [PubMed] [Google Scholar]
- Goldberg IG, Allan C, Burel JM, Creager D, Falconi A, Hochheiser H, Johnston J, Mellen J, Sorger PK, Swedlow JR. The Open Microscopy Environment (OME) data model and XML file: Open tools for informatics and quantitative analysis in biological imaging. Genome Biol. 2005;6:R47. doi: 10.1186/gb-2005-6-5-r47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzales RC, Woods RE. Digital Image Processing (2nd Edition) New York: Prentice Hall; 2002. [Google Scholar]
- Gonzales RC, Woods RE, Eddins SL. Digital Image Processing Using MATLAB. New York: Prentice Hall; 2003. [Google Scholar]
- Gordon A, Colman-Lerner A, Chin TE, Benjamin KR, Yu RC, Brent R. Single-cell quantification of molecules and rates using open-source microscope-based cytometry. Nat. Methods. 2007;4:175–181. doi: 10.1038/nmeth1008. [DOI] [PubMed] [Google Scholar]
- Gordon GW, Berry G, Liang XH, Levine B, Herman B. Quantitative fluorescence resonance energy transfer measurements using fluorescence microscopy. Biophys. J. 1998;74:2702–2713. doi: 10.1016/S0006-3495(98)77976-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue S, Inoue TD. Computer-aided stereoscopic video reconstruction and serial display from high-resolution light-microscope optical sections. Ann. N.Y. Acad. Sci. 1986;483:392–404. doi: 10.1111/j.1749-6632.1986.tb34548.x. [DOI] [PubMed] [Google Scholar]
- Pedraza JM, van Oudenaarden A. Noise propagation in gene networks. Science. 2005;307:1965–1969. doi: 10.1126/science.1109090. [DOI] [PubMed] [Google Scholar]
- R-Development-Team. R: A language and environment for statistical computing. Vienna: The R Foundation for Statistical Computing; 2008. [Google Scholar]
- Raser JM, O’Shea EK. Control of stochasticity in eukaryotic gene expression. Science. 2004;304:1811–1814. doi: 10.1126/science.1098641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. Gene regulation at the single-cell level. Science. 2005;307:1962–1965. doi: 10.1126/science.1106914. [DOI] [PubMed] [Google Scholar]
- Schnapp B. Viewing Single Microtubules by Video Light Microscopy. Vol. 134. San Diego: Academic Press; 1986. [DOI] [PubMed] [Google Scholar]
- Shapiro HM. Practical flow cytometry. 4th ed. Hoboken, N. J: John Wiley & Sons; 2003. [Google Scholar]
- Swedlow JR, Goldberg I, Brauner E, Sorger PK. Informatics and quantitative analysis in biological imaging. Science. 2003;300:100–102. doi: 10.1126/science.1082602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xia Z, Liu Y. Reliable and global measurement of fluorescence resonance energy transfer using fluorescence microscopes. Biophys. J. 2001;81:2395–2402. doi: 10.1016/S0006-3495(01)75886-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu RC, Pesce CG, Colman-Lerner A, Lok L, Pincus D, Serra E, Holl M, Benjamin K, Gordon A, Brent R. Negative feedback that improves information transmission in yeast signaling. Nature. 456:755–761. doi: 10.1038/nature07513. [DOI] [PMC free article] [PubMed] [Google Scholar]