

Abstract
BACKGROUND AND PURPOSE
BAF312 is a next-generation sphingosine 1-phosphate (S1P) receptor modulator, selective for S1P1 and S1P5 receptors. S1P1 receptors are essential for lymphocyte egress from lymph nodes and a drug target in immune-mediated diseases. Here, we have characterized the immunomodulatory potential of BAF312 and the S1P receptor-mediated effects on heart rate using preclinical and human data.
EXPERIMENTAL APPROACH
BAF312 was tested in a rat experimental autoimmune encephalomyelitis (EAE) model. Electrophysiological recordings of G-protein-coupled inwardly rectifying potassium (GIRK) channels were carried out in human atrial myocytes. A Phase I multiple-dose trial studied the pharmacokinetics, pharmacodynamics and safety of BAF312 in 48 healthy subjects.
KEY RESULTS
BAF312 effectively suppressed EAE in rats by internalizing S1P1 receptors, rendering them insensitive to the egress signal from lymph nodes. In healthy volunteers, BAF312 caused preferential decreases in CD4+ T cells, Tnaïve, Tcentral memory and B cells within 4–6 h. Cell counts returned to normal ranges within a week after stopping treatment, in line with the elimination half-life of BAF312. Despite sparing S1P3 receptors (associated with bradycardia in mice), BAF312 induced rapid, transient (day 1 only) bradycardia in humans. BAF312-mediated activation of GIRK channels in human atrial myocytes can fully explain the bradycardia.
CONCLUSION AND IMPLICATIONS
This study illustrates species-specific differences in S1P receptor specificity for first-dose cardiac effects. Based on its profound but rapidly reversible inhibition of lymphocyte trafficking, BAF312 may have potential as a treatment for immune-mediated diseases.
Keywords: sphingosine 1-phosphate, BAF312, heart rate, lymphocyte trafficking, multiple sclerosis, translational pharmacology
Introduction
Sphingosine 1-phosphate (S1P) is a bioactive sphingolipid, found in body fluids and tissues and it regulates lymphocyte recirculation, cardiac function and a variety of other physiological processes (Spiegel and Milstien, 2003; Brinkmann, 2007; Mutoh et al., 2012). Most effects of S1P are mediated via five G-protein-coupled S1P receptor subtypes that are differentially expressed on various cell types, including lymphocytes and neural cells (Brinkmann, 2007). The therapeutic potential of S1P receptor modulation has been demonstrated using Gilenya™ (fingolimod), which has shown nanomolar affinity as an agonist at four of the five S1P receptors (S1P1, S1P3, S1P4 and S1P5; receptor nomenclature follows Alexander et al., 2011) and acts as a functional antagonist at S1P1 receptors, inducing receptor internalization and rendering T and B cells insensitive to a signal necessary for egress from secondary lymphoid tissues (Brinkmann et al., 2002; Baumruker et al., 2007; Cohen et al., 2010; Kappos et al., 2010). The anti-inflammatory effects of this class of agents involve modulation of S1P1 receptors on lymphocytes such as T and B cells due to persistent ligand-induced internalization of the receptor (Matloubian et al., 2004). The internalization of S1P1 receptors renders these cells unresponsive to S1P, depriving them of an obligatory signal to egress from lymphoid organs and recirculate to the periphery and the CNS (Matloubian et al., 2004; Cyster, 2005). Further immune cell functions have also been attributed to S1P1 receptor signalling, such as dendritic cell migration (Gollmann et al., 2008), human eosinophil chemotaxis and selective in vivo recruitment (Roviezzo et al., 2004), as well as mast cell degranulation and chemotaxis (Jolly et al., 2004). These functions are, however, less well characterized than the S1P1 receptor-mediated effects on T- and B-cell lymphocyte recirculation.
The existence of S1P receptors in the heart was first reported by Bünemann et al. (1995) and the subsequent demonstration that slowing of the sinoatrial node in rabbits was dependent on S1P (Guo et al., 1999) strongly implied that S1P was a naturally occurring regulator of heart rate (Peters and Alewijnse, 2007). In isolated cultured atrial myocytes, S1P activates muscarinic receptor-gated potassium channels. These studies led to the conclusion that activation of these ion channels by S1P is induced by binding to G-protein-coupled S1P receptors, triggering a Pertussis toxin-sensitive G-protein signalling cascade and resulting in the Gβγ-dependent activation of the muscarinic receptor-gated potassium channels (Bünemann et al., 1995). Gene deletion studies in mice further suggested that the transient decrease in heart rate was mediated by activation of the G-protein-coupled inwardly rectifying potassium (GIRK) channels (Koyrakh et al., 2005). In mice, S1P3 receptors were largely responsible for bradycardia (Forrest et al., 2004; Sanna et al., 2004) and this finding prompted the search for S1P receptor agonists devoid of S1P3 signalling (Gonzalez-Cabrera et al., 2008). However, the effects on heart rate in humans of S1P3-sparing S1P receptor modulators have not previously been reported.
In our search for S1P1 receptor modulators that are devoid of S1P3 receptor activity, we postulated that modifications of the hydrophobic alkyl chain in fingolimod would allow receptor sub-type selectivity. One aspect of this optimization was to introduce greater rigidity by replacement of the n-octyl moiety of fingolimod by a substituted benzyloxy oxime moiety. Further optimization effort was focused on using amino carboxylic acids to mimic the amino phosphate moiety in the active metabolite of fingolimod. Interestingly, analogues containing amino carboxylic acids generally showed shorter elimination half-lives in vivo. This effort led to the discovery of BAF312 (1-(4-{1-[(E)-4-cyclohexyl-3-trifluoromethylbenzyloxyimino]-ethyl}-2-ethylbenzyl)-azetidine-3-carboxylic acid) a selective modulator of S1P1 and S1P5 receptors (Figure 1), allowing S1P1 receptor-dependent modulation of lymphocyte traffic without producing S1P3 receptor-mediated effects.
Figure 1.
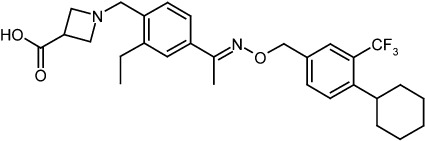
Chemical structure of BAF312.
BAF312 was furthermore designed to have a relatively short elimination half-life that provides a rapid recovery of blood lymphocyte counts on stopping treatment, but would allow once-daily oral dosing. BAF312 has been developed for the treatment of immune-mediated diseases, such as multiple sclerosis (MS), where redistribution of lymphocytes to secondary lymphoid organs and reduced infiltration of autoreactive cells into the target tissue should contribute to improvement of the disease.
Here, we present both preclinical and human phase I clinical data to (i) profile the effects of BAF312 on S1P1 receptors, resulting in reduction of peripheral lymphocyte counts and its preclinical efficacy in rat experimental autoimmune encephalomyelitis (EAE), a model for human MS; (ii) determine the effects of selective S1P1,5 receptor modulation by BAF312 on human peripheral blood lymphocyte counts and key leukocyte subsets; (iii) characterize the initial heart rate effect due to S1P1,5 receptor modulation without concomitant S1P3 receptor agonism and (iv) characterize the pharmacokinetic and initial safety profile of BAF312 in humans.
Methods
Animals
All animal care and experimental procedures for in vivo studies were in accordance with the Austrian Law on Animal Experimentation and the Novartis Animal Welfare Policy. All studies involving animals are reported in accordance with the ARRIVE guidelines for reporting experiments involving animals (Kilkenny et al., 2010; McGrath et al, 2010). For EAE, female Dark Agouti (DA) rats (200-250 g; total = 39) from Harlan Winkelmann (Borchen, Germany) were kept under standardized light- and climate-controlled conditions with free access to food and water.
Receptors and cell lines
Stable cell lines expressing human S1P receptors were prepared by transfecting cDNA encoding full-length S1P1, S1P2, S1P3, S1P4, and S1P5 receptors in CHO cells (ATCC, catalogue number: CCL-61). S1P1 and S1P3 receptors were constructed with an N-terminal Myc tag (EQKLISEEDL), while S1P4 receptor had a C-terminal Myc tag (EQKLISEEDL). cDNA for the 5 human receptors was amplified by polymerase chain reaction (PCR) from human tissues (spleen, brain, lung) and sequenced. After the insertion of the Myc tag, the final constructs were verified by DNA sequencing. Positive clones were selected based on functional receptors as described by Pan et al. (2006).
GTPγS-binding assay
The cells were homogenized and centrifuged at 26 900×g for 30 min at 4°C. Membranes were re-suspended in 20 mM HEPES (pH 7.4), 100 mM NaCl, 10 mM MgCl2, 1 mM EDTA and 0.1% fat-free BSA at 2–3 mg protein·mL−1. GTPγ[35S] binding assay was performed with the membranes (75 µg· protein mL−1 in 50 mM HEPES, 100 mM NaCl, 10 mM MgCl2, 20 µg·mL−1 saponin and 0.1% fat-free BSA (pH 7.4), 5 mg·mL−1 with wheat-germ agglutinin-coated scintillation proximity assay-bead (Amersham Biosciences, Little Chalfont, Buckinghamshire, UK), and 10 µM GDP for 10–15 min. The GTPγ[35S]-binding reaction was started by the addition of 200 pM GTPγ[35S] (Amersham; >1000 Ci·mmol−1). After 120 min at room temperature, the plates were centrifuged for 10 min at 300 x g and counted with a TopCount instrument (Packard Instruments, Meriden, CT, USA).
Agonist-mediated internalization of S1P1 receptors in CHO cells analysed by flow cytometry
Myc-tag hS1P1 cells were incubated for 1 h with agonist at 37°C in standard culture medium followed by a PBS wash. An aliquot was kept on ice for 3 h, while another aliquot was left for 3 (or 12) h in culture medium (no agonist) at 37°C. The cells were then incubated either with 4 µg·mL−1 monoclonal mouse anti C-myc IgG1 (Roche Applied Science, Mannheim, Germany) antibody or with isotype control mouse IgG1 (Pharmingen, BD Biosciences, Basel, Switzerland) for 60 min at 4°C, followed by an incubation with 1 µg·mL−1 of Alexa488-labelled goat anti-mouse secondary conjugates (Molecular Probes, Juro Supply, Luzern, Switzerland). The cells were subjected to flow cytometry measurements using 10 000 viable cells per sample.
Electrophysiological recordings of GIRK channels in human atrial myocytes
All samples of human tissue were obtained with full informed consent and after ethical review in accordance with Tulane University School of Medicine Institutional guidelines. Specimens of adult human right atrial appendage obtained from patients undergoing cardiopulmonary bypass surgery for the treatment of coronary artery disease. All samples were described as grossly normal in appearance and were obtained from patients without atrial dilation or P wave abnormalities. The procedures for the isolation of atrial myocytes have been described by (Crumb et al., 1995). Currents were measured using the whole-cell variant of the patch clamp method at a temperature of 37 ± 1°C. The external solution (bathing the cell) had an ionic composition of 120 mM NaCl, 20 mM KCl, 2 mM CaCl2, 1 mM MgCl2, 11 mM dextrose and 10 mM HEPES, adjusted to a pH of 7.4 with NaOH. The internal (pipette) solution had an ionic composition of: 100 mM potassium aspartate, 40 mM KCl, 5.0 mM magnesium ATP, 2.0 mM EGTA, 0.01 mM GTP-Tris, 10 mM HEPES, pH adjusted to 7.2 with KOH. In each cell, GIRK current elicited by rapid superfusion with 20 µM carbachol was measured by a current pulse to −100 mV from a holding potential of −90 mV. After a wash-out period, the test compound was superfused until an apparent steady-state effect was observed. Peak inward current induced by the test compound were normalized to the peak amplitude of current evoked by 20 µM carbachol.
EAE induction, drug treatment, clinical scoring and measurement of blood lymphocyte counts
Induction of EAE in the DA rat was as previously described (Lorentzen 1995, Adelmann 1995). Briefly, antigen was prepared by homogenization of lyophilized bovine spinal cord (19mg) in Arlacel A and DA rat brain (29mg) and spinal cord (19mg) homogenized in saline. These two mixtures were then added to an equal volume of complete Freund's adjuvant (CFA) containing 16.6mg/mL M. tuberculosis H37Ra antigen. The total volume was then homogenized, to provide a consistent and well mixed adjuvant with antigen. Rats were immunized at 9 weeks of age, s.c. in the tail base with 200µL of the antigen/adjuvant mix, while anaesthetized by isofluorane (0.5% Forane in air; Abbott Laboratories, Vienna, Austria). At the peak of the acute phase of disease, the animals were equally distributed to the experimental groups based on their initial onset and severity of disease in order to ensure consistency between each group: vehicle and BAF312 at 0.03, 0.3 and 3 mg kg-1. Dosing began at the peak of fully established disease on day 11 after inoculation and continued through to day 34. BAF312 was suspended in 1% aqueous carboxy-methylcellulose (C-4888, Sigma Chemical Co, St Louis, MO) and was administered orally once daily by gavage at a volume of 5 mL·kg−1 body weight. The animals were weighed and scored daily for neurological signs. The resultant chronic disease was evaluated using a numeric scale of progressive paralysis: 0, no paralysis; 1, loss of tail tonicity; 2, hind limb weakening or ataxia; 3, hind limb paralysis with or without urinary incontinence; 4, hind limb and fore limb paralysis/moribund; 5, death. DA rats with a score of 4 were killed if weight loss indicated little chance of recovery, in accordance with animal welfare standards. Mortality due to killing or spontaneous EAE-related death was recorded as a 4, which continued to be included in the clinical assessment; body weight measurements were not carried forward.
Human multiple-dose study
All subjects gave written informed consent before study participation. The study was conducted through Parkway Research Center Inc. (North Miami Beach, FL) after approval by the local ethics committee. The study was a phase I, randomized, parallel, double-blind, placebo-controlled, time-lagged, ascending, multiple-dose, pharmacokinetic, pharmacodynamic, safety and tolerability study of BAF312 in healthy subjects (n= 48), using a placebo control and five ascending dose levels of BAF312 (0.3, 1, 2.5, 10 and 20 mg BAF312). After a screening period of 21 days, treatment with BAF312 or matching placebo was over 28 days followed by a follow-up period of 14 (for cohorts 0.3, 1 and 2.5 mg) or 21 days (for cohorts 10 and 20 mg). The study population comprised healthy male and non-childbearing potential female subjects between the ages of 18 and 55 years of age inclusive. A total of six subjects were discontinued from the study, two of whom were discontinued due to severe adverse events (AEs) deemed not to be related to study drug by the principal investigator. These two subjects were replaced.
The primary objective of the study was to evaluate the safety and tolerability of once-daily, oral doses of BAF312 in healthy volunteers. Secondary objectives included the characterization of the once-daily, oral dose pharmacokinetics of BAF312 and the measurement of the effect of once-daily, oral dose BAF312 on lymphocyte counts, leukocyte subsets and the lymphocyte recovery period.
AEs were recorded throughout the study. Heart rate was assessed using 24 h Holter monitoring on days −1, 1, 2, 13 and 27. To ensure safety, telemetry was also utilized for real-time monitoring. For pharmacodynamic assessments on blood cells, 5 mL blood was collected into EDTA vacuum tubes at screening, on days −1, 1, 3, 5, 7, 14, 21, 28, 35, 42 and 49 (only for cohorts 4 and 5). Absolute lymphocyte count (ALC) and granulocytes were assessed as part of standard haematology tests processed centrally through Laboratory Corporation (Hollywood and Tampa, FL). Further leukocyte subpopulations were measured in whole blood by flow cytometry using the following fluorescent conjugated antibodies (all from Becton-Dickinson & Co, Cockeysville, MD): CD3-peridinin chlorophyll protein, CD4-phycoerythrin (PE), CD8-allophycocyanin (APC), CD45RA-fluorescein isothiocyanate (FITC), CCR7-PE-Cyanin7 (Cy7), CD16-FITC, CD14-PE, CD56-PECy7, CD19-FITC, CD138-APC, mouse-anti-human lineage cocktail-FITC monoclonal antibody mix (Becton-Dickinson # 340546), HLA-DR-APC and CD11c-PE. The antibody mixes were added to whole blood at the clinical site, incubated for 30 min at room temperature in the dark and fixed and frozen. After thawing, the samples were washed and analysed with a FACS-Canto flow cytometer (Becton-Dickinson).
Further assessments included routine clinical biochemistry and haematology laboratory tests, vital signs, standard ECG evaluations, 24 h ambulatory blood pressure monitoring (ABPM), pulmonary function tests (PFTs) and neurological examinations (Mini Mental State Examination and trail-making tests). Visual examinations included visual acuity (Snellen score) and ocular coherence tomography performed for both the right and left eyes.
Pharmacokinetic assay
For pharmacokinetic assessments in the phase I multiple dose study, 2 mL blood was collected into EDTA tubes on days 1, 2, 4, 6, 7, 8, 11, 14, 17, 20, 23, 26, 28, 29, 30, 32, 35, 38, 42 and 49. BAF312 was quantified in plasma using a validated specific LC/MS/MS bioanalytical method for pharmacokinetic assessments. The lower limit of quantitation was 0.02 ng·mL−1 for 0.1 mL plasma. Pharmacokinetic data analysis was conducted using a standard non-compartmental approach, and parameters were derived using WinNonlin Pro version 5.2 (Pharsight Corp, Mountain View, CA).
Statistical analysis
For the chronic EAE studies, area under the concentration–time curve (AUC) values of clinical grade scores were evaluated during days 12–34. anova was used to compare the AUC data sets using PRISM version 3.0 (GraphPad Software, Inc, San Diego, CA). Differences between groups were analysed using post hoc Dunn's multiple comparison test. For ALC and heart rate in the phase I multiple-dose study, the maximum attainable effect (Emax) was evaluated on pre-dose day −1 and pre-specified post-dose days. Treatment effects and treatment differences from the placebo group were analysed by analysis of covariance (ancova) and a linear mixed-effect model with day −1-value as covariate, treatment and day as fixed effects, and subject as random effect using Proc Mixed in SAS version 8.2 (SAS Institute, Cary, NC). To compare the effect of BAF312 on different leukocyte subsets in the 10 mg dose group, the fold decreases in cell counts compared with baseline (baseline value divided by post-baseline measurement) were assessed for each subset at the end of treatment (day 28) and the end of study (day 49). The distributions of fold changes for different leukocyte subsets were compared using the one-sided Wilcoxon rank sum test. P < 0.05 was considered significant.
Materials
BAF312 was synthesized by Novartis Pharma AG by the procedure of Pan et al., (2004) and was >99.8% pure. For the in vitro assays, BAF312 was dissolved in DMSO, to give stock solutions of 10 mM and stored at 4°C. Further dilutions for immediate use were made in the respective assay dilution buffers; the final concentration of DMSO in the assays did not exceed 0.3%. For early clinical use, BAF312 was formulated as drinking solution of a concentration of 0.5 mg·mL−1 BAF312 and oral capsules with dose strengths of 2.5 mg and 25 mg (Novartis Pharma AG, Switzerland).
Results
BAF312 is a selective agonist at S1P1 and S1P5 receptors and induces a profound and long-lasting internalization of S1P1 receptors
The selectivity of BAF312 for receptor subtypes S1P1 and S1P5, sparing activity on the S1P2, S1P3 and S1P4 receptors, was demonstrated by measuring agonist efficacy in the GTPγ[35S]-binding assay. The half-maximal effective concentration (EC50) values were in the sub-nanomolar range for S1P1 (0.39 ± 0.07 nM) and S1P5 (0.98 ± 0.43 nM) receptors, with Emax levels of approximately 100% (full agonist) (Table 1). Selectivity for S1P1 versus S1P2, S1P3, and S1P4 receptors was greater than 1000-fold.
Table 1.
Functional activity of BAF312 on human S1P receptors in the GTPγ[35S] binding assay
| Receptor subtype | EC50 (nM) ± SD | %Emax |
|---|---|---|
| S1P1 | 0.39 ± 0.07 | 91 ± 6 |
| S1P2 | >10 000 | – |
| S1P3 | >1 000 | – |
| S1P4 | 750 ± 487 | 90 ± 9 |
| S1P5 | 0.98 ± 0.43 | 112 ± 20 |
EC50 values (nM; mean ± SD) and percentage of maximum stimulation (%Emax) induced by BAF312 on human S1P1, S1P2, S1P3, S1P4 and S1P5 receptors.
To investigate the nature of S1P1 receptor modulation, we tested BAF312 on CHO cells over-expressing myc-tagged human S1P1 receptors (Cyster, 2005; Mullershausen et al., 2007; Marsolais and Rosen, 2009). Prominent internalization of S1P1 receptors was promoted by BAF312 (Figure 2). Internalization of S1P1 receptors by 91 ± 4% (mean ± SD, four independent experiments) was observed after incubation of cells for 1 h at 1 µM, compared with partial internalization (48 ± 12%; 12 experiments) of these receptors induced by the same concentration of S1P. When the incubation of cells with BAF312 was followed by a 3 h incubation in the absence of compound, still 87 ± 0.5% (three experiments) of the S1P1 receptors was localized intracellularly compared with almost complete return of the receptors to the cell surface after exposure to the endogenous agonist S1P (9 ± 9%; 6 experiments). The surface expression of S1P1 receptors in cells treated with BAF312 also remained low when they were incubated for up to 12 h without compound (data not shown). Lower concentrations of BAF312 of 100 and 10 nM still induced 99 ± 3% (2 experiments) and 86 ± 4% (2 experiments) internalization, respectively, after a 1 h incubation.
Figure 2.
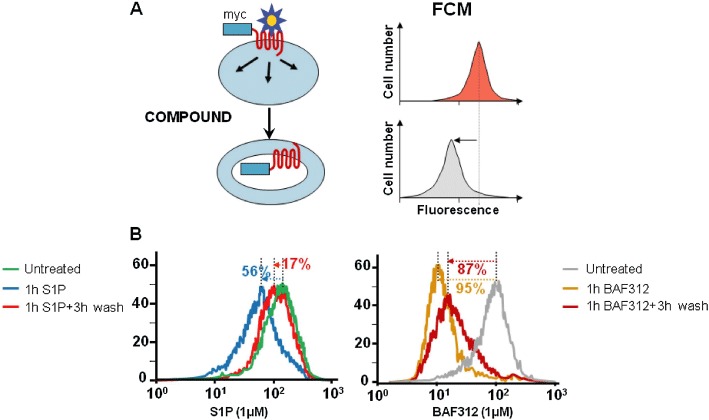
Internalization of human S1P1 receptors by S1P and BAF312. (A) Schematic representation of the internalization assay. The extent of surface expression of myc-tagged S1P1 receptors before and after incubation of the cells with compounds is determined by flow cytometry (FCM) using an anti-myc-epitope antibody. A reduction in fluorescence indicates disappearance of the receptor from the surface of the cell. (B) Representative experiment showing internalization of myc-tagged human S1P1 receptors in CHO cells mediated by 1 µM S1P or 1 µM BAF312 after a 1 h incubation, and after a 1 h incubation and a 3 h wash.
BAF312 suppresses ongoing disease symptoms in rat EAE
The model of chronic EAE in DA rats immunized with spinal cord homogenate was used to test the therapeutic efficacy of BAF312. Vehicle-treated animals developed an ascending and protracted disease starting on day 7 after immunization (Figure 3A), which was clinically characterized by progressive paraplegia and weight loss. Therapeutic intervention with BAF312, starting on day 11 at the peak of clinical symptoms, significantly suppressed established neurological deficits compared with sustained disease in the vehicle control group during the 24 day treatment period. Daily oral dosing with 0.3 or 3 mg·kg−1 of BAF312 resulted in a significant reduction of disease scores (AUC from days 12 to 34) compared with vehicle-treated control animals (Figure 3B), while the 0.03 mg·kg−1 dose had a borderline effect.
Figure 3.
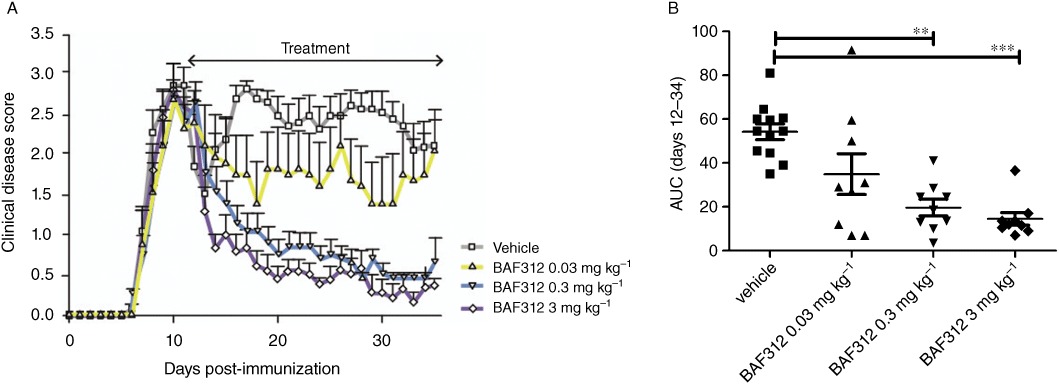
BAF312 suppresses ongoing disease symptoms in rat EAE. (A) EAE was induced and clinical disease was evaluated, the treatment period is indicated by the arrow (days 11–34 after immunization). Vehicle treatment group (n= 12, one moribund on day 20) and three treatment groups dosed with BAF312 (0.03, n= 7, one moribund on day 15, 0.3, n= 9, and 3 mg·kg−1, n= 9) are shown. BAF312 dose-dependently reduced clinical disease scores of established disease. (B) AUC of the disease scores during the treatment period (days 12–34); statistical significance in Dunn's multiple comparison test of BAF312 dosing groups versus vehicle-treated group. **P ≤ 0.01, ***P ≤ 0.001.
BAF312 activates the GIRK channel in atrial myocytes
GIRK activation has been identified as the mechanism by which fingolimod reduces heart rate in mice (Koyrakh et al., 2005). To investigate whether the S1P1,5-selective modulator BAF312 would still have a potential to induce a transient decrease in heart rate, GIRK activation in human atrial myocytes was measured. As indicated in Figure 4, BAF312 concentration-dependently increased GIRK current amplitude with an EC50 of 15.8 nM, reaching approximately 80% of the carbachol-induced maximal current activation. In the same assay, the endogenous agonist, S1P, was approximately 8 times more potent than BAF312 in activating GIRK current (EC50= 1.9 nM).
Figure 4.
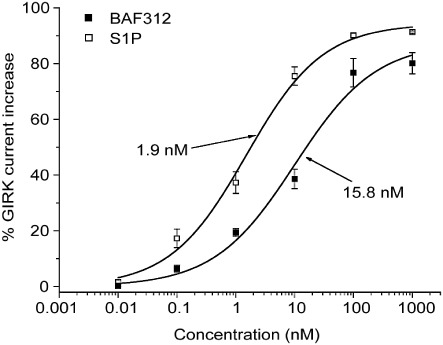
BAF312 activates human atrial myocytes via S1P1. Effects of BAF312 and for comparison S1P on GIRK current recorded from acutely isolated human atrial myocytes. Symbols are mean ± SEM (n= 4). Values were normalized to the current increase evoked by 20 µM carbachol.
Dose-dependent reduction of peripheral absolute lymphocyte counts by BAF312 in humans
BAF312 was tested in a phase I, randomized, double-blind, placebo-controlled trial in healthy subjects. BAF312 or placebo was administered once daily, for 28 days, to 48 subjects in five cohorts at the dose levels of 0.3, 1, 2.5, 10 and 20 mg. Six subjects per dose group received BAF312 and two subjects per dose group received placebo in the first three (0.3, 1 and 2.5 mg) cohorts, whereas nine subjects per dose group received BAF312 and three subjects per dose group received placebo in the last two (10 and 20 mg) cohorts. On day 1, the absolute lymphocyte count (ALC) declined with the maximal reduction observed at approximately 4–6 h post dose (Figure 5). Changes in ALC showed a dose-dependent decline for the BAF312 doses of 0.3 to 10 mg and maximal reduction was maintained throughout the 28 days of treatment. ancova, adjusted by baseline %Emax, showed that the differences in mean %Emax of BAF312 and placebo [with 90% confidence intervals (CIs)] were 32.8% (20.5–45.1), 61.6% (49.2–74.0), 56.7% (44.2–69.2), 74.6% (63.7–85.5) and 76.1% (64.9–87.3) for the 0.3, 1, 2.5, 10 and 20 mg dose groups respectively. Significant reductions in ALC reflected the pharmacodynamic effect of BAF312 and were not associated with any AEs.
Figure 5.
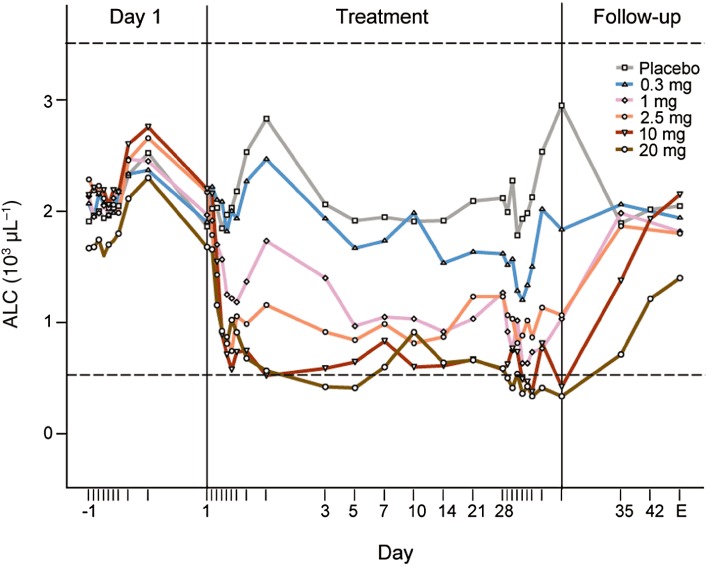
Mean changes in ALC after multiple daily doses of BAF312 in healthy subjects. Mean peripheral ALC trajectories are shown over the run-in (day −1), treatment phase (days 1–28) and during the follow-up period (until day 49) in subjects receiving placebo, 0.3, 1, 2.5, 10 and 20 mg·day−1 BAF312. End of study (EOS) is day 42 for cohorts 0.3, 1 and 2.5 mg, and day 49 for cohorts 10 and 20 mg. The normal range of ALC is indicated by dotted lines.
After treatment had been stopped on day 28, ALC returned to the normal range (1.0–3.5 × 103·µL−1) by day 35 for the dose groups 0.3–10 mg (Figure 5). A pharmacokinetic/pharmacodynamic analysis was performed to characterize the relationship between BAF312 plasma concentrations and changes in ALC over time in healthy subjects (data not shown). This model predicted that after stopping dosing at steady state, the average time to reach the lower limit of the normal ALC range (1.0 × 103·µL−1) is approximately 24 and 123 h (1 and 5.1 day) for the doses of 1 and 10 mg respectively. Recovery to baseline (day −1) levels of ALC was observed at the end of the study (day 49) for all except the 20 mg cohort, which showed a clear trend towards returning to baseline.
The effect of BAF312 on various leukocyte subsets, as estimated in the 10 mg dose group by the fold decrease in absolute count from baseline to the end-of-treatment values, was different for each subset (Figure 6). Little or no effect was seen for monocytes. In contrast, a strong reduction in cell counts was observed for both B and T cells, with more pronounced effect on CD4+ T cells [median fold decrease: 22.8 with interquartile range (IQR): 16.42–26.65] compared with CD8+ T cells [median fold decrease: 5.9; (IQR 4.3–6.1); P= 0.0003]. As shown in Figure 6, the effect of BAF312 on the CD4+ and CD8+ T-cell subtypes showed a consistent pattern of response strength in the following order (with median fold decreases): naïve T cells [for CD4+: 291.5 (IQR 143.9–320.3); for CD8+: 101.4 (IQR 68.9–2.7)] > central memory T cells [CD4+ TCM: 136.3 (IQR 119.3–150.7); CD8+ TCM: 44.9 (IQR 23.6–75.7)] > peripheral effector memory T cells [for CD4+ TPEM: 8.2 (IQR 6.2–13.1); for CD8+ TPEM: 3.3 IQR 2.7–4.8]. Three weeks after discontinuation of BAF312 therapy, all cell counts returned to baseline levels (data not shown).
Figure 6.
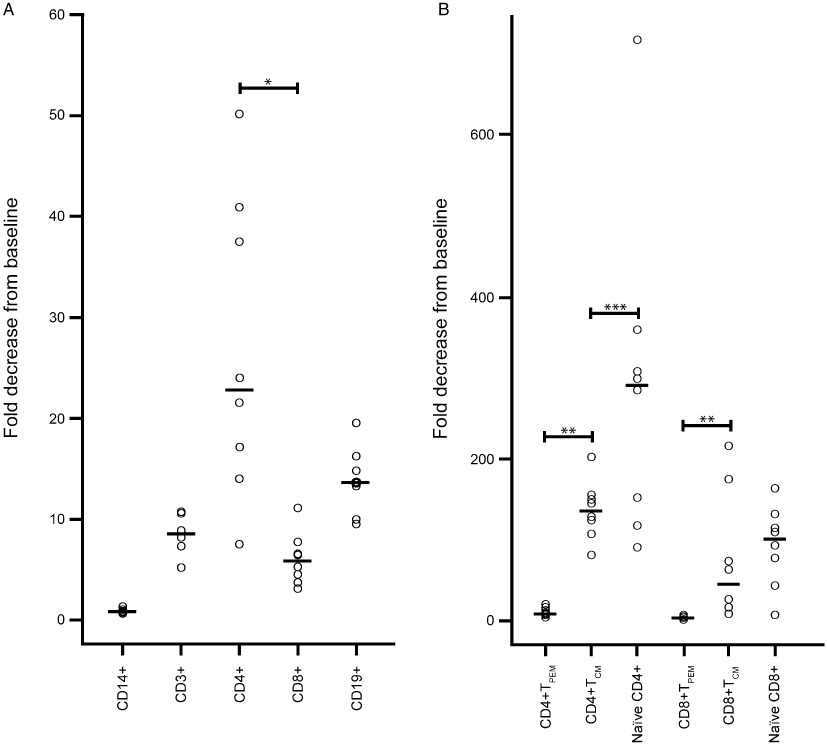
Effect of BAF312 on different leukocyte subsets after multiple daily doses of 10 mg BAF312 in healthy subjects. Fold decreases in the absolute peripheral counts are shown for different leukocyte subsets including (A) CD14+ monocytes, CD19+ B cells, CD3+, CD4+, CD8+ T cells as well as (B) naïve T cells, central memory T cells (TCM) and peripheral effector memory T cells (TPEM) of both CD4+ and CD8+ cells in individual subjects treated with 10 mg BAF312 for 28 days. Baseline values were divided by end of treatment (day 28) values. Grey bars represent median values. The effect of BAF312 estimated by the difference in fold decreases in ALC was more pronounced on CD4+ T cells than on CD8+ T cells (*P= 0.0003). The difference in fold decreases between TCM and TPEM was significant for both CD4+ and CD8+ T cells (**P < 0.0004). The difference between naïve T and TCM cells was marginally significant (***P= 0.04) for CD4+ T cells and not significant (P= 0.22) for CD8+ cells.
In humans, BAF312 induces maximal heart rate reductions at 2 h after the first dose, but these are not observed during continued once-daily oral dosing
GIRK current activation induced by BAF312 in atrial myocytes suggested that heart rate effects may also be demonstrated in humans by BAF312 despite its selectivity for S1P1 and S1P5 receptors. Indeed, a transient, dose-dependent decrease in mean ventricular heart rate was observed on day 1, with a maximum decrease at ∼2 h post dose for all (0.3–20 mg) doses in the phase I clinical trial (Figure 7). The maximum effect reached a plateau at the 10 mg dose of BAF312 on day 1 of treatment. For the two highest dose groups, BAF312 10 and 20 mg, the mean heart rate reduction from time-matched day −1 reached its maximum at 2 h post dose of day 1, approximately 24 ± 6 beats per minute (bpm) and 29 ± 10 bpm respectively. The decrease in mean Emax0-24 (bpm; [90% CI]) on day 1 in the BAF312-treated groups as compared with the placebo group on the same visit day, with the exception of the lowest 0.3 mg dose group (−0.92 [−4.70 to 2.86]) was significant in the 1 mg (−6.41 [−10.39 to −2.42]), 2.5 mg (−5.44 [−9.06 to −1.81]), 10 mg (−13.88 [−17.21 to −10.56]) and 20 mg (14.22 [−17.58 to −10.86]) dose groups. These transient, dose-dependent reductions in heart rate on day 1 were clearly due to BAF312 whereas other sporadic, non-dose-dependent decreases and increases in heart rate were also noted on days 2, 3 and 27 (data not shown), which could rather be attributed to physiological variability. All changes in heart rate described above were asymptomatic.
Figure 7.
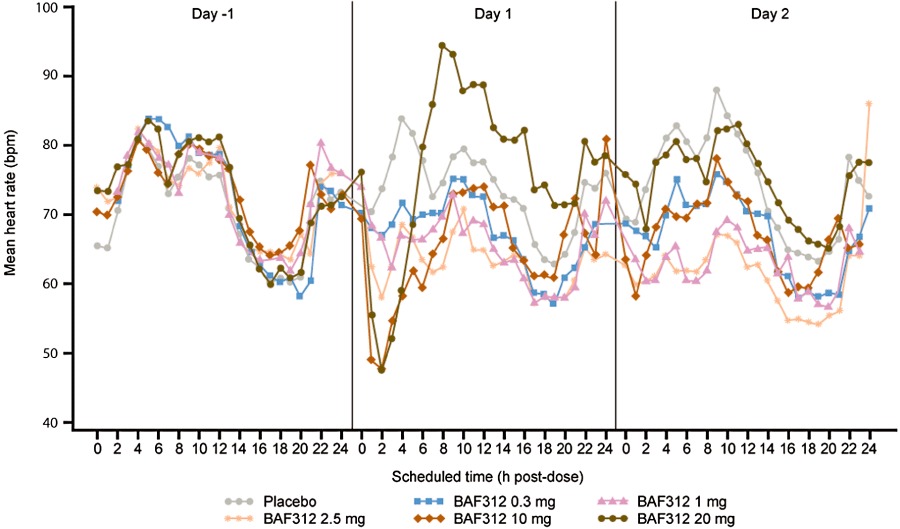
Mean changes in ventricular heart rate after multiple daily doses of BAF312 in healthy subjects. Mean supine heart rate during day −1, day 1 (first dosing day) and day 2 is shown in subjects receiving placebo, 0.3, 1, 2.5, 10 and 20 mg·day−1 BAF312.
In total, six subjects (three in the placebo and three in the active groups) were found on at least one occasion to have an asymptomatic second degree atrioventricular (AV) block that, in the placebo group, occurred sporadically on days −1, 2 and 27; whereas in the active group, each occurred on day 1 reflecting bradyarrhythmic effects of BAF312. All of the second degree AV blocks were considered by the investigator to be benign and not clinically significant, as the duration of these events was short and the conduction disturbances were not continuous. No Mobitz type 2, second degree or higher AV block occurred in the trial.
BAF312 has a favourable pharmacokinetic and initial safety profile that allows the wash-out of the compound within a week
In humans, BAF312 steady-state concentrations were achieved following approximately 6 days of multiple once-daily administration. Following once-daily administration over 28 days, the mean accumulation ratio was in the range of 1.9–2.7 (Table 2). Over a dose range of 0.3 to 20 mg, the pharmacokinetics of BAF312 appeared to increase proportionally with the dose. The effective elimination half-life based on drug accumulation at steady state (Boxenbaum and Battle, 1995) was 22–38 h (mean 30 h). A complete wash-out of the compound (defined as five effective half-lives) following treatment discontinuation is expected to take 6.3 days (150 h based on a 30 h half-life), which is consistent with the time needed for recovery of lymphocyte counts following last BAF312 dose administration.
Table 2.
Main pharmacokinetic parameters after multiple dose administration of BAF312 to healthy subjects under fasted conditions over 28 days
| Pharmacokinetic parameterA | Cohort 1 | Cohort 2 | Cohort 3 | Cohort 4 | Cohort 5 |
|---|---|---|---|---|---|
| 0.3 mg·day−1 | 1 mg·day−1 | 2.5 mg·day−1 | 10 mg·day−1 | 20 mg·day−1 | |
| Day 1 | |||||
| N | n= 6 | n= 6 | n= 7 | n= 9 | n= 9 |
| Tmax (h)B | 3.00 (3.00–6.00) | 4.50 (2.00–8.00) | 3.00 (3.00–6.00) | 3.00 (2.00–6.00) | 4.00 (3.00–8.00) |
| Cmax (ng·mL−1)C | 2.13 [13] | 8.01 [6] | 17.4 [88] | 84.1 [11] | 162 [20] |
| AUC0–24 h (h*ng·mL−1)D | 36.2 [10] | 136 [8] | 293 [85] | 1370 [11] | 2740 [14] |
| Day 28 | |||||
| n | n= 6 | n= 6 | n= 5 | n= 9 | n= 8 |
| Tmax,ss (h)B | 3.50 (3.00–12.00) | 3.00 (2.00–8.00) | 4.00 (3.00–8.00) | 3.00 (2.00–6.00) | 3.00 (3.00–4.00) |
| Cmax,ss (ng·mL−1)C | 5.31 [16] | 14.9 [10] | 38.3 [37] | 147 [24] | 359 [17] |
| AUCτ (h*ng·mL−1)C | 97.9 [19] | 282 [16] | 692 [45] | 2580 [24] | 6370 [23] |
| Cavg, ss (ng·mL−1)C | 4.08 [19] | 11.7 [16] | 28.8 [45] | 107 [24] | 265 [23] |
| Cmin,ss (ng·mL−1)C | 2.83 [27] | 7.74 [20] | 15.2 [120] | 70.6 [29] | 166 (88.0)D |
| Effective T1/2(h)C,E | 36.2 [19] | 25.2 [12] | 36.3 [47] | 21.9 [19] | 28.8 [23] |
| RaccC | 2.71 [19] | 2.07 [12] | 2.72 [47] | 1.88 [19] | 2.28 [23] |
Abbreviations and definitions for pharmacokinetic parameters: Tmax,ss: the time to reach maximum (peak) concentration following drug administration at steady state; Cmax,ss: the maximum (peak) observed steady-state drug concentration in the plasma during multiple dosing; AUCτ: area under curve during a dosing interval (τ) at steady state; Cavg,ss: the mean observed steady-state drug concentration in the plasma during multiple dosing; Cmin,ss: minimum (trough) observed steady-state drug concentration in the plasma during multiple dosing; T1/2: elimination half-life associated with the terminal slope (λz) of a semilogarithmic concentration-time curve; Racc: accumulation ratio.
Median (min-max).
Geometric mean [%CV geo mean].
Arithmetic mean (SD).
Effective elimination half-life based on drug accumulation at steady-state (Boxenbaum and Battle, 1995).
BAF312 was well tolerated, demonstrating a favourable safety profile for standard and special safety assessments including routine clinical biochemistry and haematology laboratory tests, vital signs, standard ECG evaluations, ABPM, telemetry, Holter monitoring and neurological exams.
As degradation of the S1P1 receptors was shown to lead to vascular leak in mice (Oo et al., 2011) and as macular oedema has been described as an AE for S1P receptor modulators in clinical studies (Cohen et al., 2010; Kappos et al., 2010), thorough visual examinations were performed. There were no notable changes found in the visual acuity scores over treatment days regardless of the dose of BAF312. Ocular coherence tomography showed no signs of macular oedema in the study.
In the pulmonary function tests, mild to moderate, statistically significant decreases were noted in the BAF312 treatment groups compared with placebo (data not shown); however, the magnitude of these changes seemed not to be related to dose and day in any treatment groups. Importantly, these changes in pulmonary function did not result in any AEs or clinical symptoms related to bronchoconstriction or dyspnoea.
There were no serious AEs in this trial. All AEs were mild or moderate in severity, except for those in two subjects who were excluded from the study due to severe, non-study drug-related AEs. One of these two subjects received a non-authorized massage therapy resulting in very high serum levels of creatine kinase and aminotransaminases, which interfered with the conduct of the study. The other excluded subject had had a preexisting neurological disorder that was hidden at screening but became apparent during the study. There were a total of 53 AEs reported in 26 (52%) subjects. AEs were experienced by three subjects in each of the 0.3 and 1 mg dose groups, four subjects in the 2.5 mg dose group, six subjects in each of the 10 and 20 mg dose groups, and four subjects in the placebo group. Of note, bradycardia as an AE was not reported for any of the subjects despite significant decreases in heart rate as these events were asymptomatic and judged as not clinically relevant by the investigator. The most frequently affected system organ class, which involved ≥5% of subjects, included nervous system disorders (16 subjects, 32%), investigations of clinically significant laboratory elevations (9 subjects, 18%), gastrointestinal disorders (5 subjects, 10%) and musculoskeletal and connective tissue disorders (3 subjects, 6%). The AEs reported by ≥5% of subjects were headache (14 subjects, 28%) and dizziness (3 subjects, 6%). Laboratory variables had sporadic out-of-range values but did not show an overall trend. Elevations in alanine aminotransferase level, however, occurred most frequently in subjects in the BAF312 20 mg dose group suggesting a dose–effect relationship.
Discussion and conclusions
The present findings in this paper characterize, to our knowledge for the first time, human data of lymphocyte and heart rate effects of a selective modulator of both S1P1 and S1P5 receptors. Interestingly, the lack of activity on S1P3 receptors and related cardiac effects seen in mice did not translate into humans, as treatment with BAF312 caused GIRK channel activation in human atrial myocytes and bradycardia in healthy subjects. The rapid attenuation of heart rate effects and the short elimination half-life, allowing both once-daily dosing and a potential for short compound wash-out, taken together with the initial human safety data, suggest a unique pharmacological profile, which may provide clinically relevant advantages over other compounds.
The decline in ALC in peripheral blood after application of BAF312 in human studies was observed at concentrations that caused long-lasting S1P1 receptor internalization. S1P1 receptors regulate lymphocyte egress from the thymus and lymph nodes by inducing a chemotactic response towards its natural ligand S1P (Fujino et al., 2003; Matloubian et al., 2004). This suggests that BAF312-treated T and B lymphocytes, lacking significant surface expression of S1P1 receptors and failing to respond to S1P, are retained within the thymus and lymph nodes. The preclinical efficacy of BAF312 in the EAE model can, to a large extent, be explained by reduced recirculation of autoreactive lymphocytes and prevention of their infiltration into the CNS (Fujino et al., 2003; Balatoni et al., 2007).
BAF312 effects in healthy volunteers were more pronounced for CD4+ than for CD8+ T cells, and preferential decreases in lymphocyte counts were noted for CD4+-naïve and CD4+ central memory (CCR7+) T cells (TCM), whereas CD4+ peripheral effector memory (CCR7-) T (TPEM) cells were affected to a much lesser extent. TCM are known to be involved in recall responses in secondary lymphoid organs, whereas TPEM are present primarily in peripheral tissues and are endowed with immediate effector functions (Sallusto and Lanzavecchia, 2009). However, the exact role of these T-cell subsets in the pathogenesis of human autoimmune diseases such as MS is unclear at present. Similar to our findings in healthy individuals, recent data in MS patients demonstrated that a therapeutic dose of fingolimod reduced peripheral naïve T cells and TCM, but not TPEM, in blood without affecting T-cell function (Mehling et al., 2008). As T cells and TCM express the homing receptor CCR7 and recirculate through secondary lymphoid tissues on a regular basis, these subtypes are particularly susceptible to being trapped in the lymph nodes by S1P1 receptor modulation. Similar results were recently reported for the S1P1 agonist, ponesimod, which shows 10- and 20-fold selectivity of S1P1 versus S1P5 (partial agonist) and S1P3 (full agonist) receptors, respectively. Ponesimod reduced αβ T cells, B cells, and γδ T cells, but not natural killer (NK) cells and monocytes in rats (Piali et al., 2011). The link between efficacy in MS and the effects of an S1P1 receptor modulator on different lymphocyte subsets still needs to be established.
Elimination of S1P receptor-induced heart rate reduction in S1P3 receptor knockout mice (Forrest et al., 2004; Sanna et al., 2004) led to the conclusion that eliminating activity of S1P3 receptors would help to avoid transient bradycardia effects. Evidence for S1P3 receptor-mediated activation of GIRK by S1P in human atrial cardiomyocytes (Himmel et al., 2000) seemed to support the relevant role of S1P3 receptors in mediating heart rate reductions. Our in vitro data demonstrated that the S1P1,5 receptor-selective agonist BAF312 also activated GIRK channels in human atrial myocytes, providing a possible cellular mechanism for, and indicating species-dependence of bradycardia.
The transient, dose-dependent bradycardia observed in healthy volunteers treated with BAF312 in our clinical trial demonstrated a dominant role of S1P1 receptors in mediating cardiac effects in humans. The rapid onset (reaching a maximum at 2 h post dose) on day 1, and the minimal or lack of heart rate reduction after dosing on day 2 demonstrate that desensitization to the heart rate-reducing effects of BAF312 develops quickly suggesting a unique pharmacological profile of BAF312. The profound and long-lasting internalization of S1P1 receptors induced by BAF312 and following receptor degradation, preventing further activation of GIRK channels, may be one of the mechanisms for this desensitization effect.
The data from the multiple ascending-dose study with BAF312 demonstrated good safety and tolerability in humans. Reversible, and in most cases clinically non-significant, liver function test elevations observed mainly at higher doses of BAF312 are in line with studies with another S1P receptor modulator, fingolimod, where similar elevations in liver enzyme levels were shown (Kappos et al., 2010). No increase in incidence of infection was seen in healthy volunteers, also in line with findings from the fingolimod Phase III FREEDOMS study (Kappos et al., 2010). The pharmacokinetic profile with a clinically relevant half-life of approximately 30 h supports once-daily administration and allows the complete wash-out of the compound within a week. In line with these findings, a relatively rapid recovery of the peripheral lymphocyte counts was seen, returning to the normal range within a week. In cases where fast lymphocyte recovery is desirable (e.g. during infections), this feature may be of considerable clinical benefit. Of note, the reversible lymphocyte redistribution, without destruction, to secondary lymphoid organs by BAF312 is distinct from the cytotoxic effects of several immunosuppressive agents (Gergely, 1999).
Overall, our findings suggest previously unknown species-specific responses to S1P1 receptor signalling on heart rate and a differentiated pharmacological profile of BAF312 regarding heart rate effects and lymphocyte recovery. Based on its efficacy in the EAE model and its capability of significantly modulating human T-cell and B-cell distribution, BAF312 was recently tested in a phase II clinical trial as an oral formulation for relapsing-remitting MS. Other chronic inflammatory diseases characterized by infiltration of autoreactive lymphocytes in specific target tissues may also serve as diseases that may benefit from the use of S1P receptor modulators such as BAF312.
Acknowledgments
The authors would like to thank the following at Novartis (Basel, Switzerland): Arndt Holger Brachat for his expertise in qualitative assessment of the human leukocyte subset analysis, Laurence Howard for his support with the EAE experiment, Markus Streiff for helping with the GTPγS-binding assay, Wenkui Li for analysis of human plasma concentrations, Marco Londei and Sohail Ahmed for their help with the phase I multiple-dose study design and Arthur Bertolino and Frank Dahlke for their critical review of the manuscript. The authors would also like to thank Helen Adams of iMed Comms Ltd for editing the manuscript. Research and clinical studies as well as editing work were funded by Novartis. Author contributions: PG (human study) and BNH (preclinical studies) designed and performed the studies, analysed the data, and wrote the manuscript. DG, VB, MT, CB, SP, NSG, KH, NGC, AG, AV, TS, OL, JY, AG, NW, WJC, MS and MR performed the studies. EW analysed the data and wrote the manuscript. PG and BNH contributed equally to this paper.
Glossary
- ABPM
ambulatory blood pressure monitoring
- AE
adverse event
- ALC
absolute lymphocyte count
- ancova
analysis of covariance
- AUC
area under the concentration–time curve
- AV
atrioventricular
- bpm
beats per minute
- BAF312
1-(4-{1-[(E)-4-cyclohexyl-3-trifluoromethylbenzyloxyimino]-ethyl}-2-ethylbenzyl)-azetidine-3-carboxylic acid CI, confidence interval
- DA
Dark Agouti
- EAE
experimental autoimmune encephalomyelitis
- GIRK
G-protein-coupled inwardly rectifying potassium
- IQR
interquartile range
- MS
multiple sclerosis
- S1P
sphingosine 1-phosphate
Statement of conflicts of interest
NSG's laboratory undertakes sponsored research from Novartis, and he is currently employed by Dana-Farber Cancer Institute, Harvard Medical School. KH is employed by Proteros Biostructures GmbH, Martinsried, Germany. SP is employed by the Genomics Institute of the Novartis Research Foundation and has no potential conflict of interest. MS and MR (Parkway Research Center, Inc) received research grant support (contract) from the sponsor (Novartis) for costs incurred for performing this clinical trial; otherwise, no conflict of interest exists. PG, BNH, DG, VB, MT, CB, NGC, AG, AV, TS, OL, JY, AG, NW and EW are employees of Novartis.
References
- Adelmann M, Wood J, Benzel I, Fiori P, Lassmann H, Matthieu JM, et al. The N-terminal domain of the myelin oligodendrocyte glycoprotein (MOG) induces acute demyelinating experimental autoimmune encephalomyelitis in the Lewis rat. J Neuroimmunol. 1995;63:17–27. doi: 10.1016/0165-5728(95)00124-7. [DOI] [PubMed] [Google Scholar]
- Alexander SPH, Mathie A, Peters JA. Guide to Receptors and Channels (GRAC), 5th Edition. Br J Pharmacol. 2011;164(Suppl. 1):S1–S324. doi: 10.1111/j.1476-5381.2011.01649_1.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balatoni B, Storch MK, Swoboda EM, Schönborn V, Koziel A, Lambrou GN, et al. FTY720 sustains and restores neuronal function in MOG-induced experimental autoimmune encephalomyelitis. Brain Res Bull. 2007;74:307–316. doi: 10.1016/j.brainresbull.2007.06.023. [DOI] [PubMed] [Google Scholar]
- Baumruker T, Billich A, Brinkmann V. FTY720, an immunomodulatory sphingolipid mimetic: translation of a novel mechanism into clinical benefit in multiple sclerosis. Expert Opin Investig Drugs. 2007;16:283–289. doi: 10.1517/13543784.16.3.283. [DOI] [PubMed] [Google Scholar]
- Boxenbaum H, Battle M. Effective half-life in clinical pharmacology. J Clin Pharmacol. 1995;35:763–766. doi: 10.1002/j.1552-4604.1995.tb04117.x. [DOI] [PubMed] [Google Scholar]
- Brinkmann V. Sphingosine 1-phosphate receptors in health and disease: mechanistic insights from gene deletion studies and reverse pharmacology. Pharmacol Ther. 2007;115:84–105. doi: 10.1016/j.pharmthera.2007.04.006. [DOI] [PubMed] [Google Scholar]
- Brinkmann V, Davis MD, Heise CE, Albert R, Cottens S, Hof R, et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J Biol Chem. 2002;277:21453–21457. doi: 10.1074/jbc.C200176200. [DOI] [PubMed] [Google Scholar]
- Bünemann M, Brandts B, zu Heringdorf DM, van Koppen CJ, Jakobs KH, Pott L. Activation of muscarinic K+ current in guinea-pig atrial myocytes by sphingosine-1-phosphate. J Physiol. 1995;489:701–707. doi: 10.1113/jphysiol.1995.sp021084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen JA, Barkhof F, Comi G, Hartung HP, Khatri BO, Montalban X, et al. Oral fingolimod or intramuscular interferon for relapsing multiple sclerosis. TRANSFORMS Study Group. N Engl J Med. 2010;362:402–415. doi: 10.1056/NEJMoa0907839. [DOI] [PubMed] [Google Scholar]
- Crumb WJ, Jr, Pigott JD, Clarkson CW. Description of a non-selective cation current in human atrium. Circ Res. 1995;77:950–956. doi: 10.1161/01.res.77.5.950. [DOI] [PubMed] [Google Scholar]
- Cyster JG. Chemokines, sphingosine-1-phosphate, and cell migration in secondary lymphoid organs. Annu Rev Immunol. 2005;23:127–159. doi: 10.1146/annurev.immunol.23.021704.115628. [DOI] [PubMed] [Google Scholar]
- Forrest M, Sun S-Y, Hajdu R, Bergstrom J, Card D, Doherty G, et al. Immune cell regulation and cardiovascular effects of sphingosine 1-phosphate receptor agonists in rodents are mediated via distinct receptor subtypes. J Pharmacol Exp Ther. 2004;309:758–768. doi: 10.1124/jpet.103.062828. [DOI] [PubMed] [Google Scholar]
- Fujino M, Funeshima N, Kitazawa Y, Kimura H, Amemiya H, Suzuki S, et al. Amelioration of experimental autoimmune encephalomyelitis in Lewis rats by FTY720 treatment. J Pharmacol Exp Ther. 2003;305:70–77. doi: 10.1124/jpet.102.045658. [DOI] [PubMed] [Google Scholar]
- Gergely P. Drug-induced lymphopenia: focus on CD4+ and CD8+ cells. Drug Saf. 1999;21:91–100. doi: 10.2165/00002018-199921020-00003. [DOI] [PubMed] [Google Scholar]
- Gollmann G, Neuwirt H, Tripp CH, Mueller H, Konwalinka G, Heufler C, et al. Sphingosine-1-phosphate receptor type-1 agonism impairs blood dendritic cell chemotaxis and skin dendritic cell migration to lymph nodes under inflammatory conditions. Int Immunol. 2008;20:911–923. doi: 10.1093/intimm/dxn050. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Cabrera PJ, Jo E, Sanna MG, Brown S, Leaf N, Marsolais D, et al. Full pharmacological efficacy of a novel S1P1 agonist that does not require S1P-like headgroup interactions. Mol Pharmacol. 2008;74:1308–1318. doi: 10.1124/mol.108.049783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo J, MacDonell KL, Giles WR. Effects of sphingosine 1-phosphate on pacemaker activity in rabbit sino-atrial node cells. Pflügers Arch. 1999;438:642–648. doi: 10.1007/s004249900067. [DOI] [PubMed] [Google Scholar]
- Himmel HM, Meyer Zu Heringdorf D, Graf E, Dobrev D, Kortner A, Schüler S, et al. Evidence for Edg-3 receptor-mediated activation of I(K.ACh) by sphingosine-1-phosphate in human atrial cardiomyocytes. Mol Pharmacol. 2000;58:449–454. doi: 10.1124/mol.58.2.449. [DOI] [PubMed] [Google Scholar]
- Jolly PS, Bektas M, Olivera A, Gonzalez-Espinosa C, Proia RL, Rivera J, et al. Transactivation of sphingosine-1-phosphate receptors by FcepsilonRI triggering is required for normal mast cell degranulation and chemotaxis. J Exp Med. 2004;199:959–970. doi: 10.1084/jem.20030680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappos L, Radue EW, O'Connor P, Polman C, Hohlfeld R, Calabresi P, et al. A placebo-controlled trial of oral fingolimod in relapsing multiple sclerosis. N Engl J Med. 2010;362:387–401. doi: 10.1056/NEJMoa0909494. [DOI] [PubMed] [Google Scholar]
- Kilkenny C, Browne W, Cuthill IC, Emerson M, Altman DG. NC3Rs Reporting Guidelines Working Group. Br J Pharmacol. 2010;160:1577–1579. doi: 10.1111/j.1476-5381.2010.00872.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyrakh L, Roman MI, Brinkmann V, Wickman K. The heart rate decrease caused by acute FTY720 administration is mediated by the G protein-gated potassium channel IKACh. Am J Transplant. 2005;5:529–536. doi: 10.1111/j.1600-6143.2005.00754.x. [DOI] [PubMed] [Google Scholar]
- Lorentzen JC, Issazadeh S, Storch M, Mustafa MI, Lassman H, Linington C, et al. Protracted, relapsing and demyelinating experimental autoimmune encephalomyelitis in DA rats immunised with syngeneic spinal cord and incomplete Freund's adjuvant. J Neuroimmunol. 1995;63:193–205. doi: 10.1016/0165-5728(95)00153-0. [DOI] [PubMed] [Google Scholar]
- Marsolais D, Rosen H. Chemical modulators of sphingosine-1-phosphate receptors as barrier-oriented therapeutic molecules. Nat Rev Drug Discov. 2009;8:297–307. doi: 10.1038/nrd2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matloubian M, Lo CG, Cinamon G, Lesneski MJ, Xu Y, Brinkmann V, et al. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor 1. Nature. 2004;427:355–360. doi: 10.1038/nature02284. [DOI] [PubMed] [Google Scholar]
- McGrath J, Drummond G, McLachlan E, Kilkenny C, Wainwright C. Guidelines for reporting experiments involving animals: the ARRIVE guidelines. Br J Pharmacol. 2010;160:1573–1576. doi: 10.1111/j.1476-5381.2010.00873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mehling M, Brinkmann V, Antel J, Bar-Or A, Goebels N, Vedrine C, et al. FTY720 therapy exerts differential effects on T cell subsets in multiple sclerosis. Neurology. 2008;71:1261–1267. doi: 10.1212/01.wnl.0000327609.57688.ea. [DOI] [PubMed] [Google Scholar]
- Mullershausen F, Craveiro LM, Shin Y, Cortes-Cros M, Bassilana F, Osinde M, et al. Phosphorylated FTY720 promotes astrocyte migration through sphingosine-1-phosphate receptors. J Neurochem. 2007;102:1151–1161. doi: 10.1111/j.1471-4159.2007.04629.x. [DOI] [PubMed] [Google Scholar]
- Mutoh T, Rivera R, Chun J. Insights into the pharmacological relevance of lysophospholipid receptors. Br J Pharmacol. 2012;165:829–844. doi: 10.1111/j.1476-5381.2011.01622.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oo ML, Chang SH, Thangada S, et al. Engagement of S1P-degradative mechanisms leads to vascular leak in mice. J Clin Invest. 2011;121:2290–2300. doi: 10.1172/JCI45403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan S, Gao W, Gray N, Mi Y, Fan Y. 2004. Preparation of benzylaminopropionic acid derivatives as immunosuppressants. Patent No. WO04103306.
- Pan S, Mi Y, Pally C, Beerli C, Chen A, Guerini D, et al. A monoselective sphingosine-1-phosphate receptor-1 agonist prevents allograft rejection in a stringent rat heart transplantation model. Chem Biol. 2006;13:1227–1234. doi: 10.1016/j.chembiol.2006.09.017. [DOI] [PubMed] [Google Scholar]
- Peters SL, Alewijnse AE. Sphingosine-1-phosphate signaling in the cardiovascular system. Curr Opin Pharmacol. 2007;7:186–192. doi: 10.1016/j.coph.2006.09.008. [DOI] [PubMed] [Google Scholar]
- Piali L, Froidevaux S, Hess P, Nayler O, Bolli MH, Schlosser E, et al. The selective sphingosine 1-phosphate receptor 1 agonist ponesimod protects against lymphocyte-mediated tissue inflammation. J Pharmacol Exp Ther. 2011;337:547–556. doi: 10.1124/jpet.110.176487. [DOI] [PubMed] [Google Scholar]
- Roviezzo F, Del Galdo F, Abbate G, Bucci M, D'Agostino B, Antunes E, et al. Human eosinophil chemotaxis and selective in vivo recruitment by sphingosine 1-phosphate. Proc Natl Acad Sci U S A. 2004;101:11170–11175. doi: 10.1073/pnas.0401439101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallusto F, Lanzavecchia A. Heterogeneity of CD4(+) memory T cells: functional modules for tailored immunity. Eur J Immunol. 2009;39:2076–2082. doi: 10.1002/eji.200939722. [DOI] [PubMed] [Google Scholar]
- Sanna MG, Liao J, Jo E, Alfonso C, Ahn M-Y, Peterson MS, et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J Biol Chem. 2004;279:13839–13848. doi: 10.1074/jbc.M311743200. [DOI] [PubMed] [Google Scholar]
- Spiegel S, Milstien S. Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol. 2003;4:397–407. doi: 10.1038/nrm1103. [DOI] [PubMed] [Google Scholar]