

Abstract
Nanoparticles with tunable pharmacokinetics are desirable for various biomedical applications. Poly(ethylene glycol) (PEG) is well known to create “stealth” effects to stabilize and extend the blood circulation of nanoparticles. In this work, poly(carboxybetaine) (PCB), a new non-fouling polymer material, was incorporated as surface-grafted coatings, conjugated onto degradable shell crosslinked knedel-like nanoparticles (dSCKs) composed of poly(acrylic acid)- based shells and poly(lactic acid) (PLA) cores, to compare the in vivo pharmacokinetics to their PEG-functionalized analogs. A series of five dSCKs was prepared from amphiphilic block copolymers, having different numbers and lengths of either PEG or PCB grafts, by supramolecular assembly in water followed by shell crosslinking, and then studied by a lactate assay to confirm their core hydrolytic degradabilities. Each dSCK was also conjugated with 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) macrocyclic chelators and tyramine moieties to provide for 64Cu and/or radiohalogen labeling. The high specific activity of 64Cu radiolabeling ensured nanogram administration of dSCKs for in vivo evaluation of their pharmacokinetics. Biodistribution studies demonstrated comparable in vivo pharmacokinetic profiles of PCB-grafted dSCKs to their PEG-conjugated counterparts. These results indicated that PCB-functionalized dSCKs have great potential as a theranostic platform for translational research.
Keywords: Pharmacokinetic, biodistribution, radiolabeling, poly(carboxybetaine), poly(ethylene glycol), nanomedicine, shell crosslinked knedel-like nanoparticles
Over the past decade, nanoparticles have been increasingly employed as delivery vehicles for diagnostic and therapeutic applications, owing to the multifunctionality and multivalency.1–8 For different biomedical applications, it is of paramount importance to control the pharmacokinetics and biodistribution of nanoparticles, which can be achieved through tuning their physicochemical properties, such as shapes, sizes, internal compositions, external surface charges and functionalities.9–13 It's well known that nanoparticles are prone to be sequestrated by the mononuclear phagocyte system (MPS), which is triggered by the adsorption of plasma proteins onto nanoparticles.14 Thus, “stealth” nanoparticles are needed to minimize the protein adsorption and avoid the MPS clearance. Currently, the most widely used strategy to empower nanoparticles with “stealthy” functions is grafting poly(ethylene glycol) (PEG), which is non-toxic, water-soluble and of low immunogenic response, onto the nanoparticle surface to prevent protein binding.15–17 Although the mechanism is still controversial, studies suggested that it is the water barrier, created by strong hydrogen bonding with water molecules from its ether groups that resists proteins from approaching the surface.
However, the PEG coating has been reported with disadvantages for in vivo applications, such as the lack of functional groups for post-modification, interference with cell uptake and endosome escape of nanoparticles,18 accelerated blood clearance after the second dose caused by the development of anti-PEG antibodies by the immune system,19,20 potential in vivo degradation through oxidation by superoxide anions and H2O2 generated by polymorphonuclear leukocytes (PMN) and macrophages,21–23 and the loss of biological activity of PEGylated therapeutic proteins.24–26 Therefore, there have been continuous efforts to develop new materials, which are non-toxic, less immune-toxic, functionalizable and simple to synthesize, to outperform PEG. Among the many investigated materials, poly(zwitterions)s, especially poly(carboxybetaine)s (PCB) have attracted tremendous attention, due to its superior resistance to non-specific protein adsorption.27–32 It was reported that PCB-grafted gold nanoparticles showed better in vitro stability than the PEGylated counterparts.33 More importantly, the PCB coating could retain the bioactivity of the biomolecules, more effectively than could PEG, indicating the great potential of using PCB-modified nanoparticles for pre-clinical and translational research.34–36
Among various types of nanoparticles, shell crosslinked knedel-like (SCK) nanoparticles (SCK) have drawn particular attention, owing to the high loading capacity in the hydrophobic core and controlled payloads release from the hydrophilic shell, together with the unique attributes of stability and controlled shell transport that arise from the crosslinking.37–42 Recently, we have developed a facile and efficient “pre-grafting” methodology to incorporate mPEG and (DOTA) chelator with quantitatively-controlled densities onto SCKs for tunable in vivo pharmacokinetics and radiolabeling specific activities,43,44 which therefore was employed in this study for PCB and PEG conjugation onto SCKs.
In the development of nanomaterials for personalized medicine, concerns have increased consistently on the in vivo fate, clearance, and toxicity of nanoparticles in nanomedicine health risk evaluation, which makes biodegradable polymeric nanoparticles the most attractive candidates for potential clinical applications. Poly(lactic acid) (PLA) is a commonly-used material for nanoparticle construction, largely because of its degradability via hydrolysis and biosafety of the degradation product, lactic acid.45–47
Driven by our interest in developing multivalent nanoparticles with controlled in vivo pharmacokinetics, while retaining high bioactivity for theranostic applications, a diblock amphiphilic copolymer poly(acrylic acid)-b-poly(lactide) (PAA-b-PLA) was grafted with PCB or PEG chains and sites for radiolabeling, assembled into micelles and shell crosslinked to prepare PLA-core degradable SCKs (dSCK). This robust “pre-grafting” strategy involved combinations of reversible addition-fragmentation chain transfer (RAFT) polymerization, ring-opening polymerization (ROP), supramolecular assembly and chemical conjugation. The degradability of dSCKs was analyzed with lactate assay. The in vivo biodistribution profiles of PCB- or PEG-conjugated dSCKs were tracked by 64Cu radiolabeling through DOTA chelator conjugated within the PAA shell. Specifically, this work provides a new platform with tunable properties for nanoparticle translational research.
RESULTS AND DISCUSSION
The amphiphilic diblock copolymer PAA75-b-PLA33, which served as the precursor to all of the dSCK materials, was obtained by sequential RAFT polymerization of tert-butyl acrylate (tBA) and ROP of lactide (LA), followed by acidolysis upon the treatment of trifluoroacetic acid (TFA). The order of two polymerizations was chosen to enable fine tuning of the hydrophobic PLA chain length, while maintaining the length of PAA segment constant. This combined RAFT polymerization and ROP required a bifunctional RAFT chain transfer agent (CTA) 1, having a trithiocarbonate for RAFT polymerization and a terminal hydroxyl group that can facilitate ROP. The CTA was synthesized via esterification of 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid and 1,5-pentanediol, as shown in Figure 1. Proton NMR spectroscopy of 1 showed a series of characteristic chemical shifts, including those of methylene protons adjacent to the ester group at 4.10 ppm, methylene protons next to the hydroxyl group at 3.63 ppm, and the methyl group from C12 chain of the CTA at 0.88 ppm, and the integration intensity ratios agreed with the theoretical values (see experimental section for details). The poly(tert-butyl acrylate) with a terminal hydroxyl group (PtBA75-OH, 2) was then afforded by RAFT polymerization of tBA by using 1 as the chain transfer agent, 2,2'-azobis(isobutyronitrile) (AIBN, 10 mol %) as the thermal radical initiator and 2-butanone (50 vol %) as the solvent at 56 °C, with the feed ratio of [tBA]:[1] = 100:1. The polymerization was quenched after 4.5 h when the monomer conversion was measured to be 75% by 1H NMR spectroscopy. The well-defined structure of 2 was verified by gel permeation chromatography (GPC) analysis, with a monomodal molecular weight distribution (MWD), a polydispersity index (PDI) of 1.12, and a number-average molecular weight (MnGPC) of 11000 Da, which is close to the theoretical molecular weight (Mntheo. = 10050 Da) calculated by 1H NMR spectroscopy. The diblock copolymer, PtBA75-b-PLA33 (3) was afforded by chain extension of LA from macroinitiator 2 via 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU)-catalyzed ROP in dichloromethane ([2]:[LA]:[DBU] = 1:38:1.2). The organo-catalyzed ROP was chosen to construct the PLA second block is due to the high initiation and polymerization efficiency at room temperature, as well as the absence of any metal contamination that might cause problems for 64Cu radiolabeling (vide infra).48,49 The polymerization was quenched after 1 h by adding several drops of acetic acid when monomer conversion was determined to be 85 % by 1H NMR spectroscopy. The successful growth of the second block was verified by the observation of a complete shift of the GPC traces from the retention time of the first block 2, to the diblock copolymer 3 at shorter retention time. The well-defined structure of the diblock copolymer was also confirmed by the good agreement between molecular weight determined by GPC to the theoretical value (Mntheo. = 14400 Da, MnGPC = 16200 Da), and a monomodal MWD and narrow PDI of 1.08. 1H NMR spectroscopy further demonstrated the construction of the PLA block by comparing the integration ratio between methylene protons (2.36-2.13 ppm) from PtBA and methine protons (5.23-5.10 ppm) from PLA. Complete removal of the tert-butyl groups of 3 was achieved by TFA treatment for 2 h at room temperature, affording the amphiphilic diblock copolymer PAA75-b-PLA33, 4.
Figure 1.

Synthetic scheme for the preparation of amphiphilic diblock copolymer PAA75-b-PLA33, 4, and GPC traces of intermediates PtBA75-OH, 2, and PtBA75-b-PLA33, 3.
By simple amidation, polymer grafts as well as small molecule moieties with primary amine groups can be incorporated onto PAA. In order to prepare dSCKs with comparable molecular weight PEGs and PCBs as polymer grafts, monoamine-functionalized PCB samples having molecular weights (MW) of 2 kDa and 5 kDa, equivalent MW to commercially-available monoamine-functionalized, methoxy-terminated poly(ethylene glycol) (2 kDa and 5 kDa), were synthesized by RAFT polymerizations. A similar PCB polymer with a terminal amine group was reported by Jiang et al. by applying atom transfer radical polymerization (ATRP) of 2-tert-butoxy-N-(2-(methacryloxy)ethyl)-N,N'-dimethyl-2-oxoethanaminium bromide (CB-tBu monomer) with 2-aminoethyl 2-bromoisobutyrate trifluoroacetate as the initiator, and 1,1,4,7,10,10-hexamethyltriethylenetetramine (HMTETA)/CuBr as the catalyst.31 However, in our study, any trace amount of copper salt impurities might cause inefficient radiolabeling of 64Cu, thus, in order to overcome the potential issue of metal contamination, we chose RAFT polymerization to construct PCB grafts with terminal amine groups to be used as polymer grafts. Our initial model study showed that RAFT polymerization of N-(2-(acryloyloxy)ethyl)-2-(tert-butoxy)-N,N-dimethyl-2-oxoethanaminium bromide with 1 as the chain transfer agent, AIBN as the initiator, N,N-dimethylformamide (DMF) as the solvent at 70 °C could afford well-defined PCB polymers with controllable MWs, however conjugation of the hydroxyl group-terminated PCB grafts onto PAA-b-PLA copolymer was not successful, due to the relatively low reaction efficiency of esterification compared to amidation. To introduce terminal primary amine groups into PCBs, we designed and synthesized a functionalized RAFT chain transfer agent, 2-aminoethyl-2-(dodecylthiocarbonothioylthio)-2-methylpropanoate trifluoroacetate (5), by esterification between 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid and N-(tert-butoxycarbonyl) ethanolamine, followed by TFA treatment (Figure 2). The structure was confirmed by 1H NMR, 13C NMR, and IR spectroscopies, and high-resolution mass spectrometry. The RAFT polymerizations were conducted under optimized conditions, [5]:[monomer] = 1:20, in DMF at 70 °C. The polymerizations were quenched after 0.7 h and 3 h, after monomer conversions had reached 40 % and 90 %, respectively. The well-defined structures of PCBs (6: 2 kDa PCB; 7: 5 kDa PCB) were confirmed by GPC analyses showing monomodal MWDs and narrow PDIs of 1.16 for 6 and 1.08 for 7, respectively (Figure 2). The well-controlled characteristic was also confirmed by the agreement of the degree of polymerization (DP) values that were calculated from monomer conversion and chain end analysis integration ratio of the methyl protons resonating at 0.88 ppm from the C12 chain of the CTA unit vs. the acrylate methylene protons α- to the ammonium group (-CH2N(CH3)2CH2CH2OOC-) observed at 4.20-3.87 ppm by 1H NMR spectroscopy.
Figure 2.

Synthetic scheme and GPC traces of poly(carboxybetaine) (PCB) grafts, 6 and 7.
To minimize the water exposure time, which may cause premature hydrolysis of the PLA segment, conjugations of 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester), tyramine, mPEG-NH2 (2 kDa and 5 kDa) or PCB-NH2 (6 and 7) were conducted in a one-step manner by standard amidation chemistry in DMF at room temperature, as shown in Figure 3. DOTA was selected as the chelator for 64Cu because of its FDA approval and broad applications in clinical trials,50 and tyramine is well-known to be labeled effectively by radioactive halogens.51,52 The amounts of functional moieties were controlled by adjusting the stoichiometry of each molecule relative to 4. The actual incorporation of each unit was determined by 1H NMR spectroscopy, by calculating from the integrals corresponding to characteristic protons from each unit. It should be noted, however, that the conjugates of 4 with PCB grafts exhibited poor solubility in DMF, DMSO and water, probably due to strong electrostatic interactions between PAA carboxylic acid groups and the PCB quaternary ammonium groups, thus no characterization was performed before the following deprotection step. The tert-butyl groups of the DOTA units and PCB grafts were subsequently removed by acidolysis, upon treatment with TFA. The polymers after deprotection were purified by dialysis against nanopure water followed by lyophilization, affording PEG-grafted block terpolymers 8, 9, and 10, with five PEG of 2 kDa MW (5 PEG2k), five PEG grafts of PEG 5 kDa (5 PEG5k), and ten PEG grafts of 5 kDa (10 PEG5k), respectively, and PCB-grafted block terpolymers 11 and 12, with five PCB grafts of 2 kDa and 5 kDa (5 PCB2k and 5 PCB5k), respectively.
Figure 3.

Synthetic scheme of grafted polymers 8–12.
The overall strategy for the preparation of the series of dSCKs, having variable lengths and densities of PEG or PCB surface grafts is illustrated in Figure 4. A rapid, direct dissolution method was applied to prepare micelles from the grafted block terpolymer precursors, 8–12, to avoid hydrolysis of PLA, as may occur during the conventional solvent displacement method that involves relatively long water exposure times. To facilitate radiolabeling, 1.0 mg/mL micelle solutions were prepared by directly dissolving each polymer precursor (8–12) into nanopure water, followed by extensive stirring for 2 h. Then, the dSCKs were obtained by crosslinking of the hydrophilic shell domain through amidation of approximately 20% of the carboxylic acid groups by reaction with crosslinker 2,2'-(ethylenedioxy) bis(ethylamine) (EDDA), followed by purification by dialysis overnight. The dSCKs were characterized by transmission electron microscopy (TEM), dynamic light scattering (DLS) and zeta potential (ζ-potential) measurements. As shown in Table 1, each dSCK1-5 displayed < 100 nm size, as measured in the dry state by TEM and in aqueous solution by DLS (number average) (Figure 5). Moreover, with increased PEG chain length from 2 kDa (dSCK1) to 5 kDa (dSCK2) the ζ-potential increased from −41 ± 4 mV to −28 ± 3 mV, and with further increased density of 5 kDa PEG to 10 per polymer chain (dSCK3), the ζ-potential remained similar (−25 ± 4 mV). This trend indicated that increasing the length and density of the PEG grafts, more effectively shielded the negative charge character of the PAA shell, driving the ζ-potential of the dSCK nanoparticles closer to neutral. In contrast to the shielding effects from PEG, zwitterionic PCB grafts provided the dSCKs (dSCK4 and dSCK5) with strong negatively-charged characteristic, which is in agreement with reported results.31
Figure 4.

A schematic illustration for the overall strategy for the preparation of degradable SCK (dSCK) by self assembly of multifunctional block graft polymers 8–12 into micelles, followed by crosslinking to afford dSCK1-5, having different lengths and numbers of PEG vs. PCB grafts per block copolymer chain to afford different PEG vs. PCB surface coverage of the dSCKs.
Table 1.
Physicochemical properties, 64Cu radiolabeling specific activity of dSCK1-5.
| dSCK1 | dSCK2 | dSCK3 | dSCK4 | dSCK5 | |
|---|---|---|---|---|---|
| Polymer precursor | 8 | 9 | 10 | 11 | 12 |
| Polymer grafts | 5 × PEG2k | 5 × PEG5k | 10 × PEG5k | 5 × PCB2k | 5 × PCB5k |
| Dh (nm)a | 40 ± 2 | 30 ± 9 | 32 ± 9 | 29 ± 8 | 65 ± 19 |
| Diameter (nm)b | 18 ± 3 | 15 ± 4 | 14 ± 4 | 16 ± 4 | 32 ± 6 |
| Zeta potential (mV) | −41 ± 4 | −28 ± 3 | −25 ± 4 | −50 ± 5 | −58 ± 4 |
| 64Cu Specific Activity (μCi/μg) | 105.5 | 154.9 | 79.2 | 81.8 | 139.0 |
measured by DLS, number average
measured by TEM.
Figure 5.
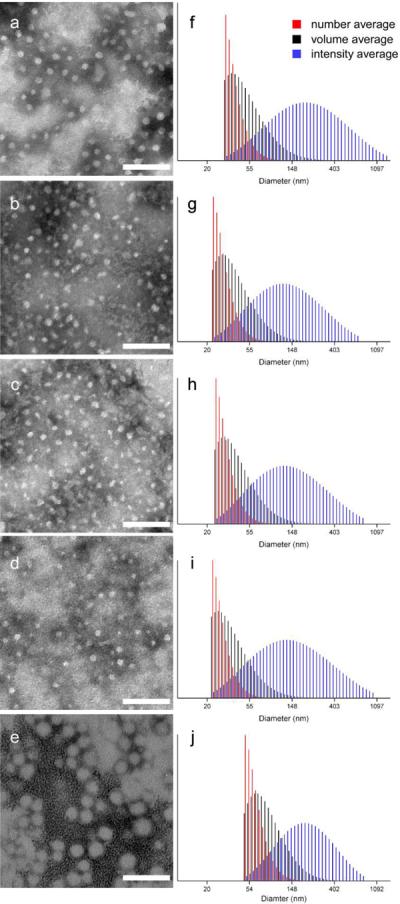
Left column: TEM images of (a) dSCK1; (b) dSCK2; (c) dSCK3; (d) dSCK4; (e) dSCK5. Right column: DLS histograms of (f) dSCK1; (g) dSCK2; (h) dSCK3, (i) dSCK4; (j) dSCK5. Scale bar = 100 nm.
Nanoparticle stability in serum is an important criterion for their in vivo biomedical applications. By monitoring the hydrodynamic diameter changes for the dSCKs over time in the presence of 10% bovine serum albumin (BSA), we investigated the stabilities of dSCK1-5 with PEG or PCB grafts in 10 % BSA solution at 37 °C. As shown in Figure 6, all of the dSCKs were stable over 12 h with only slight changes of their sizes, except for dSCK5, which exhibited ca. 15 % size increase. The subtle instability behavior of the nanoparticle presenting the longer PCB chains is under further investigation to determine the potential causes and biological effects in vitro and in vivo. Overall, the results are consistent with a previous study that found that both the presence of PEG and PCB could inhibit protein adsorption onto nanoparticles, which can potentially extend the blood circulation time.
Figure 6.
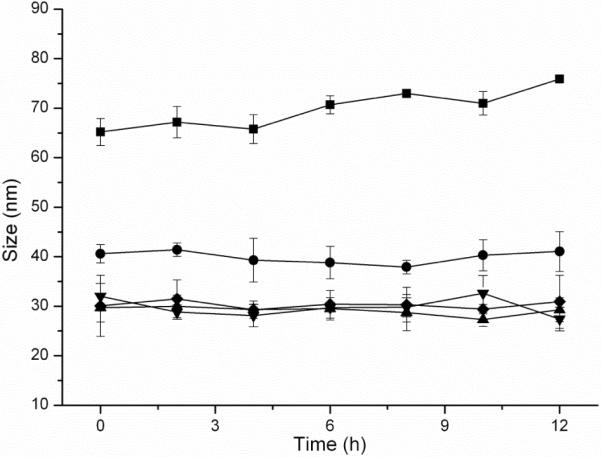
dSCK1-5 in vitro size stability measured by DLS in triplicate in 10 % bovine serum albumin (BSA) solution at 37 °C. (●) dSCK1; (◆) dSCK2; (▲) dSCK3; (▼) dSCK4; (■) dSCK5.
To evaluate the degradability of dSCKs, we studied the degradation of the dSCKs at 37 °C in pH 7.4 and pH 5.0 PBS, which represent physiological pH and endosomal/lysosomal or tumor tissue pH, respectively. The degradation was measured by the production of lactic acid, which is the final degradation molecule resulting from the hydrolysis of PLA, using a commercially-available lactate assay tool kit. As shown in Figure 7, after 15 days both PEG-grafted and PCB-grafted dSCKs showed certain extents of LA production, with PCB-grafted dSCKs having faster degradation than the PEG-grafted dSCKs. More specifically, at pH 7.4, dSCK1 and dSCK4 having 5 PEG2k and 5 PCB2k per polymer chain, underwent 10% and 19% LA release; with 5 PEG5k grafted dSCK2 and 5 PCB5k grafted dSCK5, 14% and 22% LA release was observed, respectively. We propose that the differences in degradation rates originated from the nature of PEG and PCB. PEG obtains its water solubility from the ability of binding water molecules by hydrogen bonding of its ether groups, while zwitterionic PCB, bearing both positive and negative charges, gains the non-fouling property by strong ionic structuring of water molecules, which may transfer water-soluble molecules, such as lactic acid and oligo-PLA faster than PEG, resulting in faster degradation of the PLA core or the release of the degradation product out of the dSCKs. In addition, dSCKs with longer polymer grafts (dSCK2 vs. dSCK1; dSCK5 vs. dSCK4) or higher density of polymer grafts on the shell (dSCK3 vs. dSCK2) showed more LA release over 15 days. With similar trend of degradation profiles at pH 7.4, the LA release in PBS 5.0 buffer was slower, which was probably due to a combination of factors, such as slower release of oligo-PLA from the core through the crosslinked PAA-based shell barrier and slower hydrolysis of PLA inside/outside the core region. Slower rates of PLA hydrolysis within the pH range of ca. 5 have been reported for hydrophobic PLA bulk samples and surface grafts.53–56 The differences in degradation rates for these core-shell dSCK nanoparticles having PCB vs. PEG surface grafts and under physiological vs. acidic pH may be utilized to control the release of imaging or therapeutic payloads.
Figure 7.
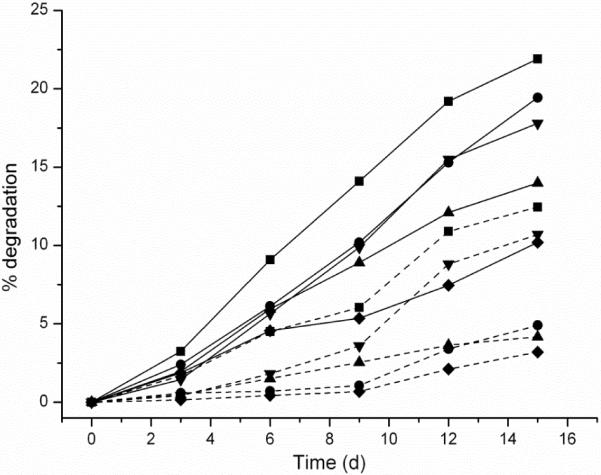
In vitro degradation of dSCKs in PBS pH = 7.4 buffer (solid lines) and PBS pH = 5.0 buffer (dashed lines) measured by lactate assay. (◆) dSCK1; (▲) dSCK2; (▼) dSCK3; (●) dSCK4; (■) dSCK5.
We next studied the 64Cu radiolabeling efficiency of dSCKs. All the dSCKs could be labeled with high specific activity through the DOTA chelator on the PAA shell (Table 1). Serum stability studies showed that after size exclusion column purification, dSCK1-5 had high (>95%) radiochemical purity. During the 24 h incubation with mouse serum, there was almost no decrease of radiochemical purity for each individual dSCK. All the radiochemical purities of the 64Cu-dSCKs were still more than 93%, as shown in Figure 8.
Figure 8.
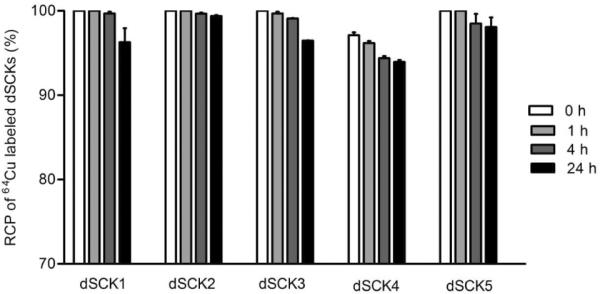
The radiochemical stability of 64Cu-dSCKs in mouse serum at 45 °C.
The effect of PEG chain length grafted to dSCKs on their in vivo pharmacokinetics is clearly depicted in Figure 9. With the increasing MW of PEG from 2 kDa to 5 kDa, the blood retention of dSCK2 was significantly (p<0.05, n=4) enhanced at each individual time point in contrast to dSCK1 (3 fold at 1 h, 6 fold at 4 h and 24 h). The hepatic and splenic accumulations were both greatly reduced, while the kidney clearance was increased at the same time. Interestingly, the bone and gastrointestinal (GI) tract (stomach and intestine) uptakes of dSCK2 were also elevated during the study, reasonably due to the increased blood retention. However, the further increase of number of 5 kDa PEG from 5 to 10 per dSCK polymer precursor chain did not enhance the blood retention proportionally. As shown in Figure 8, the biodistribution of dSCK3 was very similar to that of dSCK2, with a slightly better renal clearance.
Figure 9.

Biodistributions of (A) dSCK1, (B) dSCK2, and (C) dSCK3.
Although dSCK1 (2 kDa PEG) and dSCK4 (2 kDa PCB) had different surface coatings, they had similar physicochemical properties, including size and surface charge. The pharmacokinetic evaluation displayed similar initial blood retention with about 10%ID/g at 1 h post injection in C57BL/6 mice (Figure 10). However, the 2 kDa PEG grafted dSCK1 had a faster clearance, leading to a quick drop of blood retention to less than half of that obtained with 2 kDa PCB conjugated dSCK4 at 4 h p.i. Also, the MPS accumulations (liver and spleen) of dSCK1 were all significantly (p<0.05, n=4) higher than those acquired with dSCK4, although both particles maintained constant MPS uptakes during the 24 h study. Interestingly, in contrast to the main hepatic and splenic clearance of dSCK1, the major excretion pathway for dSCK4 was through kidney (>16% ID/g at 24 h). Further, both particles had similar distributions in other organs, including bone and gastrointestinal (GI) tract (Figure 10).
Figure 10.

Biodistributions of (A) dSCK4 and (B) dSCK5.
In contrast to dSCK4, the 5 kDa PCB grafted dSCK5 showed greatly improved blood retention at 1 h although its size was about twice big as dSCK4. However, it rapidly dropped to a level similar to dSCK4 at later time points (4 h and 24 h). The liver accumulations were slightly higher, while the spleen uptakes were very similar. Also, the kidney clearance was significantly (p<0.05, n=4) decreased during the 24 h period, reasonably owing to the size effect.
Compared to dSCK5, dSCK2 had extended blood pool retention including blood, lung and heart, comparable liver and spleen accumulations, but enhanced kidney clearance. Again, this could be due to the effect of dSCK5's size. Interestingly, consistent with the comparison between dSCK1 and dSCK4, the bone uptake for dSCK2 increased over time and was significantly (p<0.05, n=4) higher than that acquired with dSCK4 at 4 h and 24 h p.i.
CONCLUSIONS
In summary, a series of dSCK1-5 nanoparticles was prepared through a “pre-grafting” strategy with controlled sizes and surface charges, by designing, synthesizing and coupling an amphiphilic diblock copolymer of acrylic acid and lactide, and complementary monoamino-terminated zwitterionic PCB polymer grafts of varying lengths or comparable commercially-available monoamino-PEG grafts. All dSCKs were radiolabeled with 64Cu in high specific activities and showed extended serum stabilities. The in vivo pharmacokinetic evaluation indicated that the 2 kDa PCB-grafted dSCK4 had a slightly better biodistribution profile than did the 2 kDa PEG-grafted analog dSCK1, while longer chain (5 kDa) PEG grafts imparted dSCK2 with superior biodistribution to that of the corresponding 5 kDa PCB-grafted nanoparticles, dSCK5. All the dSCKs demonstrated pH-dependent degradation kinetics in vitro, with the PCB-grafted nanoparticles undergoing faster hydrolysis. These results suggest that PCB, as a new type of functionalizable anti-protein adsorption material, demonstrates comparable effects in tuning degradable polymer nanoparticle biodistributions. Further studies will be performed to compare their immunotoxicities and also the targeting efficiency of PCB-grafted dSCKs vs. PEG-functionalized dSCKs with positron emission tomography. The outcome from these studies may lay the foundation for using PCB as a versatile platform to design degradable nanoparticles for in vivo theranostics.
EXPERIMENTAL SECTION
Instrumentation
1H NMR and 13C NMR spectra were recorded on Varian Inova 300 MHz or Varian Mercury 300 MHz spectrometers interfaced to a UNIX computer using VnmrJ software. Chemical shifts were referred to the solvent resonance signals. IR spectra were recorded on an IR Prestige 21 system (Shimadzu Corp., Japan) and analyzed using IRsolution v. 1.40 software.
Tetrahydrofuran (THF) Gel permeation chromatography (GPC) was performed on a Waters Chromatography, Inc., 1515 isocratic HPLC pump equipped with an inline degasser, a model 2414 differential refractometer (Waters, Inc.), and four PLgel polystyrene-co-divinylbenzene gel columns (Polymer Laboratories, Inc.) connected in series: 5 μm Guard (50 × 7.5 mm), 5 μm Mixed C (300 × 7.5 mm), 5 μm 104 (300 × 7.5 mm), and 5 μm 500 Å (300 × 7.5 mm) using the Breeze (version 3.30, Waters, Inc.) software. The instrument was operated at 35 °C with THF as eluent (flow rate set to 1.0 mL/min). Polymer solutions were prepared at a concentration of ca. 3 mg/mL and an injection volume of 200 μL was used. The system was calibrated with polystyrene standards (Polymer Laboratories, Amherst, MA). The DMF gel permeation chromatography (GPC) was conducted on a Waters Chromatography, Inc. (Milford, MA) system equipped with an isocratic pump model 1515, a differential refractometer model 2414, and a four-column set of 5 μm Guard (50 × 7.5 mm), Styragel HR 4 5 μm DMF (300 × 7.5 mm), Styragel HR 4E 5 μm DMF (300 × 7.5 mm), and Styragel HR 2 5 μm DMF (300 × 7.5 mm). The system was equilibrated at 70 °C in prefiltered DMF containing 0.05 M LiBr, which served as polymer solvent and eluent (flow rate set to 1.00 mL/min). Polymer solutions were prepared at a concentration of ca. 3 mg/mL and an injection volume of 200 μL was used. Data collection and analysis were performed with Empower 2 v. 6.10.01.00 software (Waters, Inc.). The system was calibrated with poly(ethylene glycol) standards (Polymer Laboratories, Amherst, MA).
Glass transition temperatures (Tg) were measured by differential scanning calorimetry on a Mettler-Toledo DSC822e (Mettler-Toledo, Inc., Columbus, OH), with a heating rate of 10 °C/min. Measurements were analyzed using Mettler-Toledo Stare v. 10.00 software. The Tg was taken as the midpoint of the inflection tangent, upon the third heating scan. Thermogravimetric analysis was performed under N2 atmosphere using a Mettler-Toledo model TGA/DSC 1, with a heating rate of 5 °C/min. Measurements were analyzed using Mettler-Toledo Stare v. 10.00 software.
TEM samples were prepared by depositing ca. 5 μL of sample to carbon coated copper grids. Excess sample was wicked off using filter paper and the grids were allowed to dry in air for 10 min. The grids were then stained with 5 μL of 1% phosphotungstic acid (PTA) and excess stain was wicked off using filter paper. Specimens were observed on a JEOL 1200EX transmission electron microscope operating at 100 kV and micrographs were recorded at calibrated magnifications using an SIA-15C CCD camera. The number-average particle diameters (Dav) and standard deviations were generated from the analysis of particles from at least two different micrographs.
Dynamic light scattering (DLS) measurements were conducted using Delsa Nano C from Beckman Coulter, Inc. (Fullerton, CA) equipped with a laser diode operating at 658 nm. Size measurements were made in water (n = 1.3329, η = 0.890 cP at 25 ± 1 °C; n = 1.3293, η = 0.547 cP at 50 ± 1 °C; n = 1.3255, η = 0.404 cP at 70 ± 1 °C). Scattered light was detected at 165° angle and analyzed using a log correlator over 70 accumulations. The photomultiplier aperture and the attenuator were automatically adjusted to obtain a photon counting rate of ca. 10 kcps. The calculations of the particle size distribution and distribution averages were performed using CONTIN particle size distribution analysis routines. The samples in the glass sizing cell were equilibrated at the desired temperature for 5 min before measurements were made. The peak average of histograms from intensity, volume, or number distributions out of 70 accumulations was reported as the average diameter of the particles. The particle zeta potentials were determined by a Delsa Nano C particle analyzer (Beckman Coulter. Fullerton, CA) equipped with a 30 mW dual laser diode (658 nm). The zeta potential of the particles in suspension was obtained by measuring the electrophoretic movement of charged particles under an applied electric field. Scattered light was detected at a 30° angle at 25 °C. The zeta potential was measured at five regions in the flow cell and a weighted mean was calculated. These five measurements were used to correct for electroosmotic flow that was induced in the cell due to the surface charge of the cell wall. All determinations were repeated 5 times.
Materials
All Chemicals and reagents were purchased from Aldrich Chemical Co. and used as received, unless otherwise noted. Tert-butyl acrylate was passed through a neutral alumina column to remove the inhibitor before use. 2,2'-Azobis(isobutyronitrile) (AIBN) was recrystallized twice from methanol before use. Lactide was recrystallized twice from hexanes/ethyl acetate before use. Dichloromethane (CH2Cl2) was distilled over CaH2 and stored under N2 before use. The lactate colorimetric assay kit (ab65331) was purchased from Abcam®. 2-Aminoethyl-mono-amide-DOTA-tris(t-Bu ester) was purchased from Macrocyclics, Inc. Monoamine-functionalized, methoxy-terminated poly(ethylene glycol)s were purchased from Rapp Polymere The. Spectra/Por dialysis membrane tubes were purchased from Spectrum Medical Industries Inc. Nanopure water (18 MΩ·cm) was acquired by means of a Milli-Q water filtration system, Millipore Corp. (Bedford, MA).
5-hydroxypentyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate (1)
To a 250 mL RB flask equipped with a stir bar was placed 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (3.08 g, 8.45 mmol), 1,5-pentanediol (4.28 g, 41.1 mmol) and 4-(dimethylamino)pyridine (0.205 g, 1.67 mmol). Dry THF (50 mL) was then added into the flask to dissolve all reagents followed by dropwise addition of a solution of N,N**′-dicyclohexylcarbodiimide (2.05 g, 9.85 mmol) in THF. The reaction mixture was allowed to stir at room temperature for 12 h. The solid was removed by filtration, and the filtrate was concentrated under vacuum and purified by silica gel flash chromatography eluting with ethyl acetate/hexanes (1:2). The product was afforded as a yellow oil. Yield: 62%. IR (cm−1): 3547–3180, 2921, 2848, 1728, 1458, 1381, 1258, 1157, 1126, 1064, 810. 1H NMR (300 MHz, CDCl3, δ, ppm) 4.10 (t, 2H, J = 6.3 Hz, CH2OC(O)), 3.63 (t, 2H, J = 6.5 Hz, CH2OH), 3.26 (t, 2H, J = 7.3 Hz, CH2SC(S)), 1.72–1.58 (m, 12H, HOCH2CH2CH2CH2CH2OC(O), (CH3)2C, CH2CH2S), 1.47–1.29 (m, 20H, HO(CH2)2CH2(CH2)2OC(O), S(CH2)2(CH2)9CH3), 0.88 (t, 3H, J = 6.6 Hz, CH3CH2). 13C NMR (75 MHz, CDCl3, δ, ppm) 221.6, 173.1, 66.1, 62.8, 56.1, 37.0, 32.4, 32.0, 29.8, 29.7, 29.6, 29.5, 29.2, 29.1, 28.3, 28.0, 25.5, 22.8, 22.4, 14.3. HRMS (m/z): calcd for C22H42O3S3, 450.2296; found, 451.2374 [M + H]+.
Synthesis of PtBA75-OH (2)
To a 25 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with tBA (8.52 g, 66.6 mmol), 5-hydroxypentyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate (300 mg, 0.666 mmol), AIBN (10.9 mg, 0.0664 mmol, 10 mol %), and 10 mL of 2-butanone as the solvent. After three cycles of freeze-pump-thaw, the flask was placed in an oil bath at 56 °C. The polymerization was quenched after 4.5 h when the monomer conversion was measured to be 75% by 1H NMR. The polymer solution was precipitated three times in methanol/H2O (2:1). The product was collected and dried under vacuum for 24 h at room temperature to afford 2 as a yellow solid. Yield: 4.85 g (72%, based on 75% conversion of tBA). Mntheo. = 10050 Da, MnGPC = 11000 Da, Mw/Mn = 1.12. IR (cm−1): 3070–2792, 1728, 1450, 1365, 1249, 1141, 840, 748. 1H NMR (300 MHz, CDCl3, ppm) δ 4.04 (t, -CH2OC(O)), 3.65 (t, -CH2OH), 3.32 (t, -CH2SC(S)-), 2.36–2.13 (br, >CHC(O)- of polymer backbone), 1.92–1.09 (br, alkyl chain of CTA, >C(CH3)2 of CTA, HOCH2CH2CH2CH2CH2O-, -CH2- of polymer back bone, tert-butyl group of tBA units), 0.87 (t, -CH3 of CTA chain end). 13C NMR (75 MHz, CDCl3, ppm) δ 174.4–173.9, 80.6–80.2, 42.6–41.8, 37.5–34.9, 28.3. Tg = 41 °C. TGA in N2: 195–215 °C, 40% mass loss; 215–440 °C, 40% mass loss.
Synthesis of PtBA75-b-PLA33(3)
To a 250 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with PtBA75-OH (2) (2.04 g, 0.203 mmol), D,L-lactide (1.15 g, 7.79 mmol), and 80 mL of dry dichloromethane as the solvent. The polymerization was initiated by adding DBU (36.4 mg, 0.239 mmol) stock solution via N2-washed syringe. The polymerization was quenched after 1 h by adding several drops of acetic acid when the monomer conversion was measured to be 85% by 1H NMR. The polymer solution was precipitated three times in methanol/H2O (2:1). The product was collected and dried under vacuum for 24 h at room temperature to afford 3 as a yellow solid. Yield: 2.40 g (80%, based on 85% conversion of D,L-LA). Mntheo. = 14400 Da, MnGPC = 16200 Da, Mw/Mn = 1.08. IR (cm−1): 3055–2824, 1743, 1728, 1450, 1366, 1250, 1142, 1088, 849, 748. 1H NMR (300 MHz, CDCl3, ppm) δ 5.23–5.10 (m, >CHCH3 of PLA), 4.20–4.01 (m, -CH2OC(O)), 3.32 (t, -CH2SC(S)-), 2.36–2.13 (br, >CHC(O)- of PtBA polymer backbone), 1.92–1.09 (br, alkyl chain of CTA, >C(CH3)2 of CTA, HOCH2CH2CH2CH2CH2O-, -CH2- of polymer back bone, tert-butyl group of tBA units, -CH3 of PLA), 0.87 (t, -CH3 of CTA chain end). 13C NMR (75 MHz, CDCl3, ppm) δ 174.4–173.9, 169.8–169.4, 80.6–80.4, 69.3–67.0, 42.6–41.8, 37.6–36.0, 28.2, 16.9–16.8. Tg(PtBA) = 40 °C, Tg(PLA) = 51 °C. TGA in N2: 200–240 °C, 25% mass loss; 240–360 °C, 40% mass loss.
Synthesis of PAA75-b-PLA33 (4)
To a 250 mL flamed-dried RB flask with a stir bar was charged with PtBA75-b-PLA30 (3) (2.00 g, 0.0694 mmol). Trifluoroacetic acid (TFA, 150 mL) was added to dissolve the polymer and the reaction was allowed to stir 2 h at room temperature, after which the TFA was removed under vacuum. The crude product was dissolved in 100 mL DMF and transferred to a pre-soaked dialysis tubing (MWCO 6–8 kDa), and dialysis against nanopure water for two days. The aqueous was lyophilized to yield yellow solid of 4. Yield: 1.10 g, 78%. Mntheo. = 10200 Da. IR (cm−1): 3579–2731, 1720, 1643, 1442, 1381, 1180, 1249, 1087, 918, 864, 802. 1H NMR (300 MHz, DMSO-d6, ppm) δ 12.42–12.10 (br, -COOH), 5.23–5.10 (m, >CHCH3 of PLA), 4.20–4.07 (m, -CH2OC(O)), 3.32 (t, -CH2SC(S)-), 2.28–2.03 (br, >CHC(O)- of PtBA polymer backbone), 1.84–1.09 (br, alkyl chain of CTA, >C(CH3)2 of CTA, HOCH2CH2CH2CH2CH2O-, -CH2- of polymer back bone, -CH3 of PLA), 0.81 (t, -CH3 of CTA chain end). 13C NMR (75 MHz, CDCl3, ppm) δ 175.9–175.6, 169.3–169.0, 69.1–68.1, 16.5. Tg(PLA) = 50 °C, Tg(PAA) = 120 °C. TGA in N2: 195–215 °C, 40% mass loss; 215–370 °C, 30% mass loss.
2-(tert-butoxycarbonylamino)ethyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate
To a 100 mL RB flask equipped with a stir bar was placed 2-(dodecylthiocarbonothioylthio)-2- methylpropionic acid (2.14 g, 5.87 mmol), N-(tert-butoxycarbonyl)ethanolamine (0.946 g, 5.87 mmol) and 4-(dimethylamino)pyridine (0.143 g, 1.17 mmol). Dry CH2Cl2 (30 mL) was then added into the flask to dissolve all reagents followed by dropwise addition of a solution of N,N'-dicyclohexylcarbodiimide (1.45 g, 7.03 mmol) in CH2Cl2. The reaction mixture was allowed to stir at room temperature for 12 h. The solid was removed by filtration, and the filtrate was concentrated under vacuum and purified by silica gel flash chromatography eluting with ethyl acetate/hexanes (1:8). The product was afforded as a yellow oil. Yield: 78%. IR (cm−1): 3471–3279, 2924, 2854, 1712, 1504, 1458, 1365, 1250, 1157, 1064, 818. 1H NMR (300 MHz, CDCl3, δ, ppm) 4.73 (br, 1H, NHC(O)), 4.16 (t, 2H, J = 4.8 Hz, CH2OC(O)), 3.36 (t, 2H, J = 4.8 Hz, CH2NH), 3.26 (t, 2H, J = 7.5 Hz, CH2SC(S)), 1.69 (s, 9H, (CH3)3C), 1.45–1.40 (m, 12H, HOCH2CH2CH2CH2CH2OC(O), (CH3)2C, CH2CH2S), 1.32–1.17 (m, 18H, S(CH2)2(CH2)9CH3), 0.88 (t, 3H, J = 6.6 Hz, CH3CH2). 13C NMR (75 MHz, CDCl3, δ, ppm) 222.3, 173.1, 155.9, 79.6, 65.5, 56.1, 39.7, 37.2, 32.1, 29.8, 29.6, 29.5, 29.3, 29.1, 28.6, 28.0, 25.6, 22.9, 14.3. HRMS (m/z): calcd for C24H45NO4S3, 507.2511; found, 530.2448 [M + Na]+.
2-aminoethyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate trifluoroacetate (5)
At 0 °C, trifluoroacetic acid (8.10 mL, 105 mmol) was added to a solution of 2-(tert-butoxycarbonylamino)ethyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate (525 mg, 1.034 mmol) in dry CH2Cl2 (5 mL) and the reaction mixture was stirred at this temperature for 30 min and at room temperature for another 30 min. The solvent and TFA were removed under vacuum and purified by silica gel flash chromatography eluting with MeOH/CH2Cl2 (1:5). The product was afforded as a yellow oil. Yield: 69%. IR (cm−1): 2924, 2854, 2345, 1728, 1674, 1458, 1257, 1188, 1141, 1064, 817. 1H NMR (300 MHz, CDCl3, δ, ppm) 8.08 (br, 3H, TFA− ·NH3+CH2), 4.37 (t, 2H, J = 5.1 Hz, CH2OC(O)), 3.35–3.17 (m, 4H, CH2SC(S), TFA−·NH3+CH2CH2), 1.72–1.61 (m, 8H, (CH3)2C, CH2CH2S), 1.38–1.21 (m, 18H, S(CH2)2(CH2)9CH3), 0.88 (t, 3H, J = 6.6 Hz, CH3CH2). 13C NMR (75 MHz, CDCl3, δ, ppm) 222.7, 173.7, 165.2, 105.2, 62.4, 56.1, 39.4, 37.5, 32.1, 29.8, 29.7, 29.6, 29.5, 29.3, 29.2, 27.9, 25.2, 22.9, 14.3. HRMS (m/z): calcd for C21H38F3NO4S3, 521.1915; found, 520.9540 [M - H]−.
N-(2-(acryloyloxy)ethyl)-2-(tert-butoxy)-N,N-dimethyl-2-oxoethanaminium bromide
2-(dimethylamino)ethyl acrylate (10.0 g, 69.8 mmol), tert-butyl bromoacetate (19.7 g, 101 mmol) were reacted in 40 mL acetonitrile for 24 h at 50 °C under N2. The product was yielded as white solid by slowly adding 500 mL ethyl ether to the reaction mixture. The solvent was removed under vacuum for 24 h at room temperature. Yield: 83%. IR (cm−1): 3610–3255, 3008, 2924, 1720, 1635, 1458, 1396, 1242, 1188, 1141, 979, 918, 810. 1H NMR (300 MHz, D2O, δ, ppm) 6.48 (dd, 1H, J = 17 and 1.0 Hz, cis CHH=CHCO-), 6.22 (dd, 1H, J = 17 and 9.0 Hz, CH2=CHCO-), 6.06 (dd, 1H, J = 9.0 and 1.0 Hz, CHH=CHCO-), 4.67 (t, 2H, J = 4.2 Hz, -CH2N(CH3)2CH2CH2OOC-), 4.33 (s, 2H, -CH2N(CH3)2CH2CH2OOC-), 4.04 (t, 2H, J = 4.2 Hz, -CH2N(CH3)2CH2CH2OOC-), 3.37 (s, 6H, -CH2N(CH3)2CH2CH2OOC-), 1.52 (s, 9H, (CH3)3C-). 13C NMR (75 MHz, DMSO-d6, δ, ppm) 164.7, 163.9, 132.4, 127.7, 84.0, 62.1, 61.4, 57.9, 51.7, 27.6. HRMS (m/z): calcd for C13H24BrNO4, 337.0889; found, 336.0790 [M – H]−.
Synthesis of 2 kDa PCB (6)
To a 25 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with 2-aminoethyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate trifluoroacetate (CTA) (110 mg, 0.192 mmol), N-(2-(acryloyloxy)ethyl)-2-(tert-butoxy)-N,N-dimethyl-2-oxoethanaminium bromide (1.43 g, 4.23 mmol), AIBN (6.91 mg, 0.0421 mmol, 20 mol %), and 10 mL of DMF as the solvent. After three cycles of freeze-pump-thaw, the flask was placed in an oil bath at 70 °C. The polymerization was quenched after 40 min when the monomer conversion was measured to be 40 % by 1H NMR. The polymer solution was precipitated three times in ethyl acetate. The product was collected and dried under vacuum for 24 h at room temperature to afford PCB-2K as a yellow solid. Yield: 0.35 g (61%, based on 40% conversion of monomer). Mntheo. = 3200 Da, Mw/Mn = 1.16. IR (cm−1): 3672–3217, 2924, 1728, 1627, 1458, 1373, 1250, 1149, 987, 841. 1H NMR (300 MHz, DMSO-d6, ppm) δ 8.10 (br, TFA−·NH3+CH2), 4.83–4.37(br, TFA−·NH3+CH2CH2OC(O), -CH2N(CH3)2CH2CH2OOC-), 4.20–3.87 (br, -CH2N(CH3)2CH2CH2OOC-), 3.56–3.24 (br, -CH2N(CH3)2CH2CH2OOC-, CH2SC(S), TFA−·NH3+CH2CH2), 2.47–2.30 (br, >CHC(O)- of polymer backbone), 1.93–1.16 (m, alkyl chain of CTA, >C(CH3)2 of CTA, -CH2- of polymer back bone, tert-butyl groups), 0.85 (t, -CH3 of CTA chain end). 13C NMR (75 MHz, MeOD, δ, ppm) 166.5, 165.4–164.9, 86.5, 64.8–64.0, 53.3, 37.1, 31.8–31.0, 28.5. Tg = 116 °C
Synthesis of 5 kDa PCB (7)
To a 25 mL Schlenk flask with a stir bar and sealed by a rubber septum was charged with 2-aminoethyl 2-(dodecylthiocarbonothioylthio)-2-methylpropanoate trifluoroacetate (CTA) (100 mg, 0.192 mmol), N-(2-(acryloyloxy)ethyl)-2-(tert-butoxy)-N,N-dimethyl-2-oxoethanaminium bromide (1.29 g, 3.81 mmol), AIBN (6.28 mg, 0.0383 mmol, 20 mol %), and 9 mL of DMF as the solvent. After three cycles of freeze-pump-thaw, the flask was placed in an oil bath at 70 °C. The polymerization was quenched after 3.0 h when the monomer conversion was measured to be 90 % by 1H NMR. The polymer solution was precipitated three times in ethyl acetate. The product was collected and dried under vacuum for 24 h at room temperature to afford PCB-5K as a yellow solid. Yield: 0.47 g (40%, based on 90% conversion of monomer). Mntheo. = 6600 Da, Mw/Mn = 1.08. Tg = 130 °C.
Synthesis of PEG/PCB, DOTA, tyramine grafted PAA-b-PLA (8–12)
Grafting PEG/PCB, 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester), tyramine onto PAA75-b-PLA33 (4) involved the following: to a anhydrous N,N-dimethylformamide (DMF) solution of 4, 1-[3'-(dimethylamino)propyl]-3-ethylcarbodiimide methiodide (EDCI), and 1-hydroxylbenzotriazole (HOBt) were added. The mixtures were allowed to stir for 1 h at room temperature, followed by the addition of mono amine PEGs or PCBs. After 30 min, DMF solution of the mixture of 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester), tyramine and N,N-diisopropylethylamine (DIPEA) was added, followed by further stirring for 30 h. The relative feed ratios of DOTA and tyramine to 4 were kept constant at 5:5:1, whereas the PEG/PCB to 4 feed ratios were 7:1 when targeting five grafts per polymer chain, and 20:1 when targeting ten grafts per polymer chain, respectively. The grafted polymers were purified by dialyzing against nanopure H2O (18.2 MΩ-cm) for 3 d to remove organic solvent and byproducts. The aqueous solutions were then lyophilized to afford the products as white solid with yield of 60–80%. PCB grafted polymer were not characterized due to the poor solubility. 1H NMR (300 MHz, DMSO-d6, ppm) δ 7.05–6.87 (br, aromatic protons from tyramine), 6.73–6.59 (br, aromatic protons from tyramine), 5.24–5.11 (m, >CHCH3 of PLA), 3.56–3.19 (br, -OCH2CH2O- from PEG backbone, -CH2CO- from DOTA, -CH2CH2NH- from tyramine), 2.40–2.05 (br, >CHC(O)- of polymer backbone, >NCH2CH2N< from DOTA), 1.67–0.98 (br, alkyl chain of CTA, >C(CH3)2 of CTA, -CH2- of polymer back bone, tert-butyl groups from DOTA, -CH3 of PLA).
Graft polymer precursor for 8 with five 2 kDa PEG
PAA75-b-PLA33 (30 mg, 2.9 μmol), EDCI (17 mg, 57 μmol), HOBt (7.9 mg, 58 μmol), tyramine (2.0 mg, 14 μmol), 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester) (10 mg, 14 μmol), mPEG2000-NH2 (41 mg, 20 μmol), DIPEA (19 mg, 0.15 mmol). Yield: 70%.
Graft polymer precursor for 9 with five 5 kDa PEG
PAA75-b-PLA33 (30 mg, 2.9 μmol), EDCI (17 mg, 57 μmol), HOBt (7.9 mg, 58 μmol), tyramine (2.0 mg, 14 μmol), 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester) (10 mg, 14 μmol), mPEG5000-NH2 (103 mg, 20.6 μmol), DIPEA (19 mg, 0.15 mmol). Yield: 80%.
Graft polymer precursor for 10 with ten 5 kDa PEG
PAA75-b-PLA33 (30 mg, 2.9 μmol), EDCI (17 mg, 57 μmol), HOBt (7.9 mg, 58 μmol), tyramine (2.0 mg, 14 μmol), 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester) (10 mg, 14 μmol), mPEG5000-NH2 (294 mg, 58.8 μmol), DIPEA (19 mg, 0.15 mmol). Yield: 73%.
Graft polymer precursor for 11 with five 2 kDa PCB
PAA75-b-PLA33 (30 mg, 2.9 μmol), EDCI (17 mg, 57 μmol), HOBt (7.9 mg, 58 μmol), tyramine (2.0 mg, 14 μmol), 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester) (10 mg, 14 μmol), 6 (64 mg, 20.6 μmol), DIPEA (45 mg, 0.35 mmol).
Graft polymer precursor for 12 with five 5 kDa PCB
PAA75-b-PLA33 (30 mg, 2.9 μmol), EDCI (17 mg, 57 μmol), HOBt (7.9 mg, 58 μmol), tyramine (2.0 mg, 14 μmol), 2-aminoethyl-mono-amide-DOTA-tris(t-Bu ester) (10 mg, 14 μmol), 7 (136 mg, 20.6 μmol), DIPEA (45 mg, 0.35 mmol).
Deprotection to afford polymer 8–12
Polymers and TFA (200 equiv. to tert-butyl groups) were allowed to stir for 2 h, followed by removal of TFA under vacuum, dialysis against nanopure H2O and lyopholization to afford final polymer precursors as white solid with yield of 70–80%. 1H NMR of PEG-grafted polymers (300 MHz, DMSO-d6, ppm) δ 7.05–6.87 (br, aromatic protons from tyramine), 6.73–6.59 (br, aromatic protons from tyramine), 5.41–5.26 (m, >CHCH3 of PLA), 4.81–3.92 (br, -OCH2CH2O- from PEG backbone, -CH2CO- from DOTA, -CH2CH2NH- from tyramine), 2.40–2.05 (br, >CHC(O)- of polymer backbone, >NCH2CH2N< from DOTA), 1.67–0.98 (br, alkyl chain of CTA, >C(CH3)2 of CTA, -CH2- of polymer back bone). 1H NMR of PCB-grafted polymers (300 MHz, TFA-d, ppm) δ 7.13–6.07 (br, aromatic protons from tyramine), 6.90–6.80 (br, aromatic protons from tyramine), 5.24–5.11 (m, >CHCH3 of PLA), 3.56–3.19 (br, -CH2N(CH3)2CH2CH2OOC-, -CH2N(CH3)2CH2CH2OOC, -CH2CO- from DOTA, -CH2CH2NH- from tyramine), 2.73–2.36 (br, >CHC(O)- of polymer backbone, >NCH2CH2N< from DOTA), 1.95–1.25 (m, alkyl chain of CTA, >C(CH3)2 of CTA, -CH2- of polymer back bone, -CH3 of PLA), 0.86–0.79 (br, -CH3 of CTA chain end).
Preparation of dSCK1-5
The micelles were prepared by direct dissolving polymer precursors into nanopure H2O with polymer concentration ca. 1 mg/mL. The SCKs were prepared by adding 2,2'-(ethylenedioxy)-bis(ethylamine) (EDDA) in nanopure H2O dropwise. The reaction was allowed to stir for 2 h at room temperature. EDCI in nanopure H2O was then added to the solution dropwise. The stoichiometry applied to achieve 20% nominal crosslinking was 10:1:2.2 for carboxylic acid unites/EDCI/EDDA. The crosslinking reactions were further stir at room temperature for 12. The final SCKs were obtained after dialyzing against nanopure water overnight.
dSCK2 with five 5 kDa PEG
(Dh)n(DLS) = 30 ± 9 nm, (Dh)v(DLS) = 45 ± 27 nm, (Dh)i(DLS) = 162 ± 125 nm; ζ-potential: −28 ± 1.5 mV.
dSCK1 with five 2 kDa PEG
(Dh)n(DLS) = 40 ± 12 nm, (Dh)v(DLS) = 61 ± 39 nm, (Dh)i(DLS) = 279 ± 238 nm; ζ-potential: −41 ± 1.3 mV.
dSCK3 with ten 5 kDa PEG
(Dh)n(DLS) = 32 ± 9 nm, (Dh)v(DLS) = 47 ± 28 nm, (Dh)i(DLS) = 175 ± 141 nm; ζ-potential: −25 ± 2.2 mV.
dSCK4 with five 2 kDa PCB
(Dh)n(DLS) = 65 ± 19 nm, (Dh)v(DLS) = 94 ± 49 nm, (Dh)i(DLS) = 223 ± 143 nm; ζ-potential: −58 ± 4.8 mV.
dSCK5 with five 5 kDa PEG
(Dh)n(DLS) = 29 ± 8 nm, (Dh)v(DLS) = 43 ± 27 nm, (Dh)i(DLS) = 195 ± 173 nm; ζ-potential: −50 ± 2.6 mV.
dSCK in vitro size stability study
dSCKs were incubated with 10 wt% BSA solution at 37 °C under gentle stirring. At each time point, an aliquot of SCK solution was collected to measure the size using dynamic light scattering (DLS).
dSCK in vitro degradation study
dSCK solutions in PBS 7.4 buffer and PBS 5.0 buffer were prepared by dissolving lyophilized dSCK powder by the buffer solution with concentrations of ca. 1 mg/mL. The solutions were incubated at 37 °C. At each time point, the lactic acid levels were measured by lactate assay kit (abcam®, ab65331), the degradation percentages were calculated using calibration curve with DL-lactic acid as the standard. The experiments were performed in duplicates.
64Cu radiolabeling dSCKs
64Cu (half-life = 12.7 h, β+ = 17%, β− = 40%) was produced on the Washington University Medical School CS-15 cyclotron by the 64Ni (p,n) 64Cu nuclear reaction at a specific activity of 50–200 mCi/mg at the end of bombardment.59 Degradable dSCK1-5 (~4.5 μg) were incubated with 18.5 MBq of 64Cu in 100 μL of 0.1 M pH 5.5 ammonium acetate buffer at 45°C for 1 h (n = 3). After ethylene diamine tetraacetic acid (EDTA) challenge (10 mM in 50 mM, pH 7.4, phosphate buffer), the radiochemical purities of the radiolabeled nanoparticles were measured by radioactive thin-layer chromatography (Bioscan, Washington DC), followed by the purification with 7k Zeba spin desalting column (Piercenet, Rockford, IL).
Serum stability study
The radiochemical purities of 64Cu radiolabeled dSCKs were measured before the addition of mouse serum with 1:1 volume ratio. The 64Cu-dSCKs and serum mixture was incubated in microcentrifuge tubes at 45°C. At each time point (1 h, 4 h, and 24 h post injection, p.i.), 10 μL of the sample was removed and incubated at 45°C with 5 μL of EDTA (10 mM in 50 mM pH 7.4 phosphate buffer) for 5 min. The radiochemical purities of the samples (n=3 for each dSCK) were tested using radioactive thin-layer chromatography (Bioscan, Washington DC).
In vivo biodistribution study
All animal studies were performed in compliance with guidelines set forth by the NIH Office of Laboratory Animal Welfare and approved by the Washington University Animal Studies Committee. In vivo biodistribution studies were performed using 185 kBq of 64Cu-dSCKs (32–63 ng/mouse ) in 100 μL saline (APP pharmaceuticals, Schaumburg, IL) injected via the tail vein of C57BL/6 mice weighing 20–25 g (n=4/group) under inhaled isoflurane. The mice were euthanized by cervical dislocation at each time point (1 h, 4 h, and 24 h p.i.). Organs of interest were collected, weighed, and counted in a well gamma counter (Beckman 8000). Standards were prepared and measured along with the organs to calculate the average and standard deviation of the percentage of the injected dose per gram of tissue (%ID/g).58
ACKNOWLEDGMENT
This material is based upon work supported by the National Heart Lung and Blood Institute of the National Institutes of Health as a Program of Excellence in Nanotechnology (HHSN268201000046C). The Welch Foundation is gratefully acknowledged for support through the W. T. Doherty-Welch Chair in Chemistry, Grant No. A-0001. The authors thank Dr. M. Elsabahy for insightful discussions.
REFERENCES
- 1.Nyström AM, Wooley KL. The Importance of Chemistry in Creating Well-Defined Nanoscopic Embedded Therapeutics: Devices Capable of the Dual Functions of Imaging and Therapy. Acc. Chem. Res. 2011;44:969–978. doi: 10.1021/ar200097k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Brigger I, Dubernet C, Couvreur P. Nanoparticles in Cancer Therapy and Diagnosis. Adv. Drug Deliv. Rev. 2002;54:631–651. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 3.Torchilin VP. Multifunctional Nanocarriers. Adv. Drug Deliv. Rev. 2006;58:1532–1555. doi: 10.1016/j.addr.2006.09.009. [DOI] [PubMed] [Google Scholar]
- 4.Wang AZ, Langer R, Farokhzad OC. Nanoparticle Delivery of Cancer Drugs. Annu. Rev. Med. 2012;63:185–198. doi: 10.1146/annurev-med-040210-162544. [DOI] [PubMed] [Google Scholar]
- 5.Liu Y, Welch MJ. Nanoparticles Labeled with Positron Emitting Nuclides: Advantages, Methods, and Applications. Bioconjug. Chem. 2012;23:671–682. doi: 10.1021/bc200264c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elsabahy M, Wooley KL. Design of Polymeric Nanoparticles for Biomedical Delivery Applications. Chem. Soc. Rev. 2012;41:2545–2561. doi: 10.1039/c2cs15327k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schober O, Rahbar K, Riemann B. Multimodality Molecular Imaging - from Target Description to Clinical Studies. Eur. J. Nucl. Med. Mol. Imaging. 2009;36:302–314. doi: 10.1007/s00259-008-1042-4. [DOI] [PubMed] [Google Scholar]
- 8.Gref R, Minamitake Y, Peracchia MT, Trubetskoy V, Torchilin V, Langer R. Biodegradable Long-Circulating Polymeric Nanospheres. Science. 1994;263:1600–1603. doi: 10.1126/science.8128245. [DOI] [PubMed] [Google Scholar]
- 9.Khlebtsov N, Dykman L. Biodistribution and Toxicity of Engineered Gold Nanoparticles: A Review of In Vitro and In Vivo Studies. Chem. Soc. Rev. 2011;40:1647–1671. doi: 10.1039/c0cs00018c. [DOI] [PubMed] [Google Scholar]
- 10.Owens DE, Peppas NA. Opsonization, Biodistribution, and Pharmacokinetics of Polymeric Nanoparticles. Int. J. Pharm. 2006;307:93–102. doi: 10.1016/j.ijpharm.2005.10.010. [DOI] [PubMed] [Google Scholar]
- 11.Li SD, Huang L. Pharmacokinetics and Biodistribution of Nanoparticles. Mol. Pharmaceutics. 2008;5:496–504. doi: 10.1021/mp800049w. [DOI] [PubMed] [Google Scholar]
- 12.Venkataraman S, Hedrick JL, Ong ZY, Yang C, Ee PLR, Hammond PT, Yang YY. The Effects of Polymeric Nanostructure Shape on Drug Delivery. Adv. Drug Deliv. Rev. 2011;63:1228–1246. doi: 10.1016/j.addr.2011.06.016. [DOI] [PubMed] [Google Scholar]
- 13.Elsabahy M, Wooley KL. Strategies Toward Well-Defined Polymer Nanoparticles Inspired by Nature: Chemistry versus Versatility. J. Polym. Sci., Part A: Polym. Chem. 2012;50:1869–1880. doi: 10.1002/pola.25955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hume DA. The Mononuclear Phagocyte System. Curr. Opin. Immunol. 2006;18:49–53. doi: 10.1016/j.coi.2005.11.008. [DOI] [PubMed] [Google Scholar]
- 15.Vonarbourg A, Passirani C, Saulnier P, Benoit JP. Parameters Iinfluencing the Stealthiness of Colloidal Drug Delivery Systems. Biomaterials. 2006;27:4356–4373. doi: 10.1016/j.biomaterials.2006.03.039. [DOI] [PubMed] [Google Scholar]
- 16.Moghimi SM, Hunter AC, Murray JC. Long-Circulating and Target-Specific Nanoparticles: Theory to Practice. Pharmacol. Rev. 2001;53:283–318. [PubMed] [Google Scholar]
- 17.Peracchia MT, Vauthier C, Desmaële D, Gulik A, Dedieu JC, Demoy M, d'Angelo J, Couvreur P. Pegylated Nanoparticles from a Novel Methoxypolyethylene Glycol Cyanoacrylate-Hexadecyl Cyanoacrylate Amphiphilic Copolymer. Pharm. Res. 1998;15:550–556. doi: 10.1023/a:1011973625803. [DOI] [PubMed] [Google Scholar]
- 18.Mintezer MA, Simanek EE. Nonviral Vectors for Gene Delivery. Chem. Rev. 2009;109:259–302. doi: 10.1021/cr800409e. [DOI] [PubMed] [Google Scholar]
- 19.Ganson NJ, Kelly SJ, Scarlett E, Sundy JS, Hershfield MS. Control of Hyperuricemia in Subjects with Refractory Gout, and Induction of Antibody Against Poly(ethylene glycol) (PEG), in a Phase I Trial of Subcutaneous PEGylated Urate Oxidase. Arthritis Res. Ther. 2006;8:R12. doi: 10.1186/ar1861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Armstrong JK, Hempel G, Koling S, Chan LS, Fisher T, Meiselman HJ, Garratty G. Antibody Against Poly(ethylene glycol) Adversely Affects PEG-Asparaginase Therapy in Acute Lymphoblastic Leukemia Patients. Cancer. 2007;110:103–111. doi: 10.1002/cncr.22739. [DOI] [PubMed] [Google Scholar]
- 21.Ostuni E, Chapman RG, Holmlin RE, Takayama S, Whitesides GM. A Survey of Structure-Property Relationships of Surfaces that Resist the Adsorption of Protein. Langmuir. 2001;17:5605–5620. doi: 10.1021/la0015258. [DOI] [PubMed] [Google Scholar]
- 22.Shen M, Martinson L, Wagner MS, Castner DG, Ratner BD, Horbett TA. PEO-Like Plasma Polymerized Tetraglyme Surface Interactions with Leukocytes and Proteins: In Vitro and In Vivo Studies. J. Biomater. Sci., Polym. Ed. 2002;13:367–390. doi: 10.1163/156856202320253910. [DOI] [PubMed] [Google Scholar]
- 23.Li L, Shen S, Jiang S. Protein Interactions with Oligo(ethylene glycol) (OEG) Self-Assembled Monolayers: OEG Stability, Surface Packing Density and Protein Adsorption. J. Biomater. Sci., Polym. Ed. 2007;18:1415–1427. doi: 10.1163/156856207782246795. [DOI] [PubMed] [Google Scholar]
- 24.Gaberc-Porekar V, Zore I, Podobnik B, Menart V. Obstacles and Pitfalls in the PEGylation of Therapeutic Proteins. Curr. Opin. Drug Discov. Devel. 2008;11:242–250. [PubMed] [Google Scholar]
- 25.Veronese FM, Mero A. The Impact of PEGylation on Biological Therapies. Biodrugs. 2008;22:315–329. doi: 10.2165/00063030-200822050-00004. [DOI] [PubMed] [Google Scholar]
- 26.Chapman AP. PEGylated Antibodies and Antibody Fragments for Improved Therapy: A Review. Adv. Drug Deliv. Rev. 2002;54:531–545. doi: 10.1016/s0169-409x(02)00026-1. [DOI] [PubMed] [Google Scholar]
- 27.Vaisocherova H, Yang W, Zhang Z, Cao ZQ, Cheng G, Piliarik M, Homola J, Jiang SY. Ultralow Fouling and Functionalizable Surface Chemistry Based on a Zitterionic Polymer Enabling Sensitive and Specific Protein Detection in Undiluted Blood Plasma. Anal. Chem. 2008;80:7894–7901. doi: 10.1021/ac8015888. [DOI] [PubMed] [Google Scholar]
- 28.Zhang Z, Chen SF, Jiang SY. Dual-Functional Biomimetic Materials: Nonfouling Poly(carboxybetaine) with Active Functional Groups for Protein Immobilization. Biomacromolecules. 2006;7:3311–3315. doi: 10.1021/bm060750m. [DOI] [PubMed] [Google Scholar]
- 29.Jiang S, Cao Z. Ultralow-Fouling, Functionalizable, and Hydrolyzable Zwitterionic Materials and Their Derivatives for Biological Applications. Adv. Mater. 2010;22:920–932. doi: 10.1002/adma.200901407. [DOI] [PubMed] [Google Scholar]
- 30.Carr LR, Zhou Y, Krause JE, Xue H, Jiang S. Uniform Zwitterionic Polymer Hydrogels with a Nonfouling and Functionalizable Crosslinker Using Photopolymerization. Biomaterials. 2011;32:6893–6899. doi: 10.1016/j.biomaterials.2011.06.006. [DOI] [PubMed] [Google Scholar]
- 31.Cao Z, Yu Q, Xue H, Cheng G, Jiang S. Nanoparticles for Drug Delivery Prepared from Amphiphilic PLGA Zwitterionic Block Copolymers with Sharp Contrast in Polarity Between Two Blocks. Angew. Chem. Int. Ed. 2010;49:3771–3776. doi: 10.1002/anie.200907079. [DOI] [PubMed] [Google Scholar]
- 32.Abraham S, So A, Unsworth LD. Poly(carboxybetaine methacrylamide)-Modified Nanoparticles: a Model System for Studying the Effect of Chain Chemistry on Film Properties, Adsorbed Protein Conformation, and Clot Formation Kinetics. Biomacromolecules. 2011;12:3567–3580. doi: 10.1021/bm200778u. [DOI] [PubMed] [Google Scholar]
- 33.Yang W, Zhang L, Wang S, White AD, Jiang S. Functionalizable and Ultra Stable Nanoparticles Coated with Zwitterionic Poly(carboxybetaine) in Undiluted Blood Serum. Biomaterials. 2009;30:5617–5621. doi: 10.1016/j.biomaterials.2009.06.036. [DOI] [PubMed] [Google Scholar]
- 34.Keefe AJ, Jiang SY. Poly(zwitterionic) Protein Conjugates Offer Increased Stability without Sacrificing Binding Affinity or Bioactivity. Nature Chem. 2012;4:60–64. doi: 10.1038/nchem.1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Veronese FM, Pasut G. PEGylation, Successful Approach to Drug Delivery. Drug Discov. Today. 2005;10:1451–1458. doi: 10.1016/S1359-6446(05)03575-0. [DOI] [PubMed] [Google Scholar]
- 36.Bailon P, Palleroni A, Schaffer CA, Spence CL, Fung WJ, Porter JE, Ehrlich GK, Pan W, Xu ZX, Modi MW, et al. Rational Design of a Potent, Long-Lasting Form of Interferon: A 40 kDa Branched Polyethylene Glycol-Conjugated Interferon α-2a for the Treatment of Hepatitis C. Bioconjugate Chem. 2001;12:195–202. doi: 10.1021/bc000082g. [DOI] [PubMed] [Google Scholar]
- 37.Zhang L, Gu FX, Chan JM, Wang AZ, Langer RS, Farokhzad OC. Nanoparticles in Medicine: Therapeutic Applications and Developments. Clin. Pharmacol. Ther. 2008;83:761–769. doi: 10.1038/sj.clpt.6100400. [DOI] [PubMed] [Google Scholar]
- 38.Torchilin VP. Nanoparticles in Cancer Therapy and Diagnosis. Adv. Drug Deliv. Rev. 2002;54:235–252. doi: 10.1016/s0169-409x(02)00044-3. [DOI] [PubMed] [Google Scholar]
- 39.Thurmond KB, Kowalewski T, Wooley KL. Shell Cross-linked Knedels: A Synthetic Study of the Factors Affecting the Dimensions and Properties of Amphiphilic Core-Shell Nanospheres. J. Am. Chem. Soc. 1997;119:6656–6665. [Google Scholar]
- 40.Samarajeewa S, Shrestha R, Li YL, Wooley KL. Degradability of Poly(Lactic Acid)-Containing Nanoparticles: Enzymatic Access through a Cross-Linked Shell Barrier. J. Am. Chem. Soc. 2012;134:1235–1242. doi: 10.1021/ja2095602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang SY, Li Z, Samarajeewa S, Sun GR, Yang C, Wooley KL. Orthogonally Dual-Clickable Janus Nanoparticles via a Cyclic Templating Strategy. J. Am. Chem. Soc. 2011;133:11046–11049. doi: 10.1021/ja203133h. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lin LY, Lee NS, Zhu JH, Nyström AM, Pochan DJ, Dorshow RB, Wooley KL. Tuning Core vs. Shell Dimensions to Adjust the Performance of Nanoscopic Containers for the Loading and Release of Doxorubicin. J. Controlled Release. 2011;152:37–48. doi: 10.1016/j.jconrel.2011.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sun G, Hagooly A, Xu J, Nyström AM, Li Z, Rossin R, Moore DA, Wooley KL, Welch MJ. Facile, Efficient Approach to Accomplish Tunable Chemistries and Variable Biodistributions for Shell Cross-Linked Nanoparticles. Biomacromolecules. 2008;9:1997–2006. doi: 10.1021/bm800246x. [DOI] [PubMed] [Google Scholar]
- 44.Sun G, Xu J, Hagooly A, Rossin R, Li Z, Moore DA, Hawker CJ, Welch MJ, Wooley KL. Strategies for Optimized Radiolabeling of Nanoparticles for In Vivo PET Imaging. Adv. Mater. 2007;19:3157–3162. [Google Scholar]
- 45.Tian HY, Tang ZH, Zhuang XL, Chen XS, Jing XB. Biodegradable Ssynthetic Polymers: Preparation, Functionalization and Biomedical Application. Prog. Polym. Sci. 2012;37:237–280. [Google Scholar]
- 46.Anderson JM, Shive MS. Biodegradation and Biocompatibility of PLA and PLGA Microspheres. Adv. Drug Deliv. Rev. 1997;28:5–24. doi: 10.1016/s0169-409x(97)00048-3. [DOI] [PubMed] [Google Scholar]
- 47.Middleton JC, Tipton AJ. Synthetic Biodegradable Polymers as Orthopedic Devices. Biomaterials. 2000;21:2335–2346. doi: 10.1016/s0142-9612(00)00101-0. [DOI] [PubMed] [Google Scholar]
- 48.Lohmeijer BGG, Pratt RC, Leibfarth F, Logan JW, Long DA, Dove AP, Nederberg F, Choi J, Wade C, Waymouth RM, et al. Guanidine and Amidine Organocatalysts for Ring-Opening Polymerization of Cyclic Esters. Macromolecules. 2006;39:8583. [Google Scholar]
- 49.Kiesewetter MK, E. J., Shin EJ, Hedrick JM, Waymouth RM. Organocatalysis: Opportunities and Challenges for Polymer Synthesis. Macromolecules. 2010;43:2093–2107. [Google Scholar]
- 50. http://clinicaltrials.gov/ct2/results?term=dota.
- 51.Almutairi A, Rossin R, Shokeen M, Hagooly A, Ananth A, Capoccia B, Guillaudeu S, Abendschein D, Anderson CJ, Welch MJ, et al. Biodegradable Dendritic Positron-Emitting Nanoprobes for the Noninvasive Imaging of Angiogenesis. Proc. Natl. Acad. Sci. U.S.A. 2009;106:685–690. doi: 10.1073/pnas.0811757106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Adam MJ, Wilbur DS. Radiohalogens for Imaging and Therapy. Chem. Soc. Rev. 2005;34:153–163. doi: 10.1039/b313872k. [DOI] [PubMed] [Google Scholar]
- 53.Makino K, Arakawa M, Kondo T. Preparation and In Vitro Degradation Properties of Polylactide Microcapsules. Chem. Pharm. Bull. 1985;33:1195–1201. doi: 10.1248/cpb.33.1195. [DOI] [PubMed] [Google Scholar]
- 54.Belbella A, Vauthier C, Fessi H, Devissaguet JP, Puisieux F. In vitro Degradation of Nanospheres from Poly(D,L-lactides) of Different Molecular Weights and Polydispersities. Int. J. Pharm. 1996;129:95–102. [Google Scholar]
- 55.Jung JH, Ree M, Kim H. Acid- and Base-Catalyzed Hydrolyses of Aliphatic Polycarbonates and Polyesters. Catal. Today. 2006;115:283–287. [Google Scholar]
- 56.Xu LB, Crawford K, Gorman CB. Effects of Temperature and pH on the Degradation of Poly(lactic acid) Brushes. Macromolecules. 2011;44:4777–4782. [Google Scholar]
- 57.McCarthy DW, Shefer RE, Klinkowstein RE, Bass LA, Margeneau WH, Cutler CS, Anderson CJ, Welch MJ. Efficient Production of High Specific Activity 64Cu Using a Biomedical Cyclotron. Nucl. Med. Biol. 1997;24:35–43. doi: 10.1016/s0969-8051(96)00157-6. [DOI] [PubMed] [Google Scholar]
- 58.Liu YJ, Ibricevic A, Cohen JA, Cohen JL, Gunsten SP, Fréchet JMJ, Walter MJ, Welch MJ, Brody SL. Impact of Hydrogel Nanoparticle Size and Functionalization on In Vivo Behavior for Lung Imaging and Therapeutics. Mol. Pharmaceutics. 2009;6:1891–1902. doi: 10.1021/mp900215p. [DOI] [PMC free article] [PubMed] [Google Scholar]