

Abstract
We have achieved, to our knowledge, the first high-level heterologous expression of the gene encoding d-ribulose-5-phosphate 3-epimerase from any source, thereby permitting isolation and characterization of the epimerase as found in photosynthetic organisms. The extremely labile recombinant spinach (Spinacia oleracea L.) enzyme was stabilized by dl-α-glycerophosphate or ethanol and destabilized by d-ribulose-5-phosphate or 2-mercaptoethanol. Despite this lability, the unprecedentedly high specific activity of the purified material indicates that the structural integrity of the enzyme is maintained throughout isolation. Ethylenediaminetetraacetate and divalent metal cations did not affect epimerase activity, thereby excluding a requirement for the latter in catalysis. As deduced from the sequence of the cloned spinach gene and the electrophoretic mobility under denaturing conditions of the purified recombinant enzyme, its 25-kD subunit size was about the same as that of the corresponding epimerases of yeast and mammals. However, in contrast to these other species, the recombinant spinach enzyme was octameric rather than dimeric, as assessed by gel filtration and polyacrylamide gel electrophoresis under nondenaturing conditions. Western-blot analyses with antibodies to the purified recombinant enzyme confirmed that the epimerase extracted from spinach leaves is also octameric.
As a participant in the oxidative pentose phosphate pathway, Ru5P epimerase (EC 5.1.3.1), which catalyzes the interconversion of Ru5P and Xu5P, is widely distributed throughout nature. Beyond its catabolic role, the epimerase is also vital anabolically to photosynthetic organisms in the regenerative phase of the reductive pentose phosphate pathway (the Calvin cycle). In this capacity, Ru5P epimerase directs Xu5P, formed in two distinct transketolase reactions of the cycle, to Ru5P. Phosphorylation of the latter regenerates d-ribulose-1,5-bisphosphate, the substrate for net CO2 fixation. Because both the oxidative and reductive pentose phosphate pathways coexist in chloroplasts (Schnarrenberger et al., 1995), Ru5P epimerase and R5P isomerase facilitate partitioning of pentose phosphates between the two pathways, as dictated by the metabolic needs and redox status of the cell.
Scant structural and mechanistic information about Ru5P epimerase is available despite its inherent importance and dual metabolic roles. This neglect may in part reflect the low natural abundance of the enzyme. For example, achievement of electrophoretic homogeneity required a 2000-fold purification from yeast (Bär et al., 1996) and spinach (Spinacia oleracea L.) chloroplasts (Teige et al., 1998) and 9000-fold purification from beef liver (Terada et al., 1985). Although low overall recoveries (<10%) further limited the availability of pure material, molecular sieving and denaturing electrophoresis established that the epimerases from mammals (Wood, 1979; Karmali et al., 1983; Terada et al., 1985) and yeast (Bär et al., 1996) are homodimers of approximately 23-kD subunits, whereas the enzyme from spinach chloroplasts may be an octamer of 23-kD subunits (Teige et al., 1998). DNA-deduced amino acid sequences of Ru5P epimerases from both photosynthetic and nonphotosynthetic sources, which confirm this estimated subunit size, show greater than 50% similarities among the most evolutionarily distant species examined (Kusian et al., 1992; Blattner et al., 1993; Falcone and Tabita, 1993; Lyngstadaas et al., 1995; Nowitzki et al., 1995; Teige et al., 1995).
Although Ru5P epimerase has very recently been purified from a photosynthetic organism (spinach) for the first time (Teige et al., 1998), the low recovery (100 μg from 3.8 g of soluble chloroplast protein, representing an overall yield of 5%) imposes severe constraints on the directions of future experiments. Furthermore, despite successful cloning of cDNA fragments encoding Ru5P epimerase of several photosynthetic organisms (Kusian et al., 1992; Nowitzki et al., 1995; Teige et al., 1995), to our knowledge high-level heterologous expression and purification of enzymically active recombinant enzyme have not been achieved. Because of our interest in the regulation of photosynthetic carbon assimilation and the requisite need for ample supplies of the participant enzymes for use in mechanistic studies, we have attempted to optimize the heterologous expression of the spinach gene for Ru5P epimerase. In this paper we report cDNA clones that encode the mature chloroplastic enzyme or its cytoplasmic precursor. We also describe an efficient isolation procedure for the mature spinach enzyme synthesized in Escherichia coli and some of the properties of the purified enzyme. Contrasting features of the plant Ru5P epimerase, relative to the animal and yeast counterparts, include an octameric rather than a dimeric structure (also see Teige et al., 1998) and striking instability under routine laboratory conditions.
MATERIALS AND METHODS
Materials
Materials and vendors were as follows: a library of spinach (Spinacia oleracea L. cv Melody) cDNAs in λ ZAPII and Pfu DNA polymerase to use for PCR, Stratagene; [35S]dATP, New England Nuclear; T4 DNA ligase, New England Biolabs; nitrocellulose filters (82 mm) for plaque hybridization, Schleicher & Schuell; transketolase, triose phosphate isomerase, glycerophosphate dehydrogenase, thiamine pyrophosphate, G3P, R5P, Ru5P, leupeptin, and PMSF, Sigma; 4-(2-aminoethyl)benzenesulfonyl fluoride, Calbiochem; and 3,3′-diaminobenzidine tetrahydrochloride dihydrate, Bio-Rad. Common laboratory reagents for enzyme purification and assays were procured at the highest level of purity readily available. Phosphoribulokinase was prepared as described previously (Porter et al., 1986, 1988).
Rabbit serum containing polyclonal antibodies raised against purified recombinant spinach epimerase (a mixture of peaks I and II, see below) was prepared by Berkeley Antibody Company (Richmond, CA).
Cloning and Heterologous Expression of rpe, the Spinach Gene for Ru5P Epimerase
Oligonucleotide primers for PCR, mutagenesis, and dye-terminator sequencing were prepared with a PCR-Mate (Applied Biosystems) based on phosphoramidite chemistry. Synthesized PCR primers duplicated sequences of the potato rpe cDNA (Teige et al., 1995)2 that encode two highly conserved regions of the epimerase: PSILSANF (residues 17–24)3 and MSVNPGFG (residues 149–156)3 (see Nowitzki et al., 1995; Teige et al., 1995). These primers (ccg tcc atc ctt tct gct aac tt for the coding strand and cca aat cca ggg ttt aca gac at for the complementary strand) were used to amplify the 419-bp fragment, which brackets the two conserved regions, from an aliquot of the spinach cDNA library that contained 106 plaque-forming units. The PCR product was cloned into an SmaI site of plasmid pBS+ (Stratagene), sequenced to confirm anticipated homology to the potato rpe, and reamplified from the plasmid as a template. Subsequently, [35S]dATP was introduced into the reamplified PCR product with a random priming kit (United States Biochemical).
The labeled PCR product served as a probe in plaque-hybridization screens of the spinach cDNA library according to the manufacturer's protocol. Six positive clones were obtained from the 3 × 104 plaque-forming units screened. Two of these encompassed the entire coding sequence, inclusive of the N-terminal transit appendage, for spinach Ru5P epimerase, as shown by complete sequencing of both strands.
As an initial step in the construction of expression vectors, the 3′ untranslated region of the cDNA was truncated at the BamHI site. For generation of the transit form of the epimerase, an NcoI site was introduced by site-directed mutagenesis (Kunkel et al., 1987) at the Met initiation codon, coincidentally replacing the adjacent codon for Ser with a codon for Gly. For generation of the mature form of the epimerase, an NcoI site was introduced at the codon for Lys-48 of the transit protein, resulting in conversion to a Met initiation codon. These engineered clones were individually ligated as NcoI-BamHI fragments adjacent to the tac promoter of the expression vector pFL260 (Larimer et al., 1990) (Fig. 1). The ligated fragments of the expression vectors were sequenced to confirm the desired structures.
Figure 1.

Sequence of expression cassettes for spinach recombinant Ru5P epimerase. A, Transit protein expression cassette (accession no. AF070942). The tac promoter, operator, and ribosome-binding site (rbs) are shown in lowercase; the coding sequence is in uppercase. B, Mature epimerase expression cassette (accession no. AF070943).
An overnight culture (25 mL) of the appropriate expression vector in host strain MV1190 or XL 1-Blue, grown in 2× YT medium (Sambrook et al., 1989) containing 1% (v/v) glycerol and 50 μg mL−1 ampicillin at 37°C, was diluted 1:100 into the same medium and incubated for 4 h with vigorous shaking (250 rpm). Isopropyl β-d-thiogalactopyranoside was added to 0.1 mm, and the incubation was continued for an additional 3 h at 37°C followed by harvesting of the cells by centrifugation.
Purification of Recombinant Ru5P Epimerase
All steps of the purification were carried out at 2°C to 4°C, and all buffers were at pH 8.0. When G3P was the only buffer component, solutions of the commercial disodium salt were adjusted to pH 8.0 with 0.1 n HCl. Selection of fractions to pool from chromatographic steps was based on both epimerase assays and SDS-PAGE. Cell paste (approximately 15 g) from 1 L of culture was suspended in a buffer containing 50 mm Bicine, 10 mm G3P, 1 mm EDTA, 1 mm DTT, 10 μm leupeptin, 200 μm 4-(2-aminoethyl)benzenesulfonyl fluoride, 1 mm PMSF, and 5% (v/v) glycerol. The slurry was passed twice through a French pressure cell at 12,000 to 16,000 p.s.i. and ultracentrifuged at 100,000g for 1 h. The supernatant was adjusted to pH 8.0 with 1 m Tris (free base form), diluted 1.5-fold with water, and applied to a DE52 anion-exchange column (2.5 × 10 cm, Whatman) that had been equilibrated with 10 mm G3P. After the column was washed with 200 mL (about 4 column volumes) of 10 mm G3P to remove all unbound materials, elution was continued with 10 mm G3P containing 50 mm NaCl. Epimerase emerged as a rather sharp peak just beyond the breakthrough volume. Pooled fractions were concentrated (Centriprep-30, Amicon, Beverly, MA) to about 8 mL, adjusted to 5 mm potassium phosphate by the addition of 1% (v/v) 10 mm G3P/500 mm potassium phosphate, and filtered (0.45 μm, Gelman Acrodisc, Ann Arbor, MI). The filtered sample was applied to a 1.6- × 10-cm column of hydroxyapatite (ceramic hydroxyapatite type I, particle size of 40 μm, Bio-Rad), adapted to a fast-protein liquid chromatography unit (Pharmacia LKB Biotech Inc.), which had been equilibrated with 10 mm G3P/5 mm potassium phosphate. Upon isocratic elution of the column with equilibration buffer, epimerase emerged essentially with the solvent front. Peak fractions were pooled, concentrated (Centriprep-30) to about 8 mL, and dialyzed against 10 mm G3P/1 mm EDTA. Subsequent to dialysis, the sample was filtered (0.45 μm) and applied to a prepacked Mono-Q column (HR10/10), adapted to a fast-protein liquid chromatography unit, which had been equilibrated with 10 mm G3P/1 mm EDTA. Elution of the column with an 80-mL linear gradient of 0 to 100 mm NaCl in equilibration buffer resolved two major peaks of protein (Fig. 2) that contained epimerase activity. The major peak (75% of the total activity recovered) emerged at 40 mm NaCl, whereas the minor peak appeared at 65 mm NaCl; both peaks of epimerase had the same specific activity. Fractions from the two peaks were pooled separately, concentrated to >2 mg mL−1, and stored frozen at −80°C. A summary of the purification protocol is provided in Table I.
Figure 2.

Resolution of two peaks of epimerase by anion-exchange chromatography on Mono-Q. See text for additional details.
Table I.
Purification of spinach recombinant Ru5P epimerase
| Purification Step | Protein | Protein Yield | Activity | Activity Recovered | Specific Activity | Purification |
|---|---|---|---|---|---|---|
| mg | % | units | % | units mg−1 | -fold | |
| Centrifuged crude extract | 1054 | 100 | 265,766 | 100 | 252 | 1 |
| DE52 anion exchange | 36 | 3 | 212,368 | 80 | 5,899 | 23 |
| Hydroxyapatite | 12 | 1 | 149,065 | 56 | 12,422 | 49 |
| Dialysate | 11 | 1 | 122,066 | 46 | 11,097 | 44 |
| Mono-Q anion exchangea | 8 | 0.8 | 92,905 | 35 | 11,613 | 46 |
Epimerase eluted as two peaks of equal specific activity; of the total protein recovered, peak I represented 6 mg (69,909 units) and peak II represented 2 mg (22,996 units).
Ru5P Epimerase from Spinach
About 1 g of fresh spinach leaves was ground with a mortar and pestle in 1 mL of extraction buffer as used for Escherichia coli, and the resulting slurry was centrifuged at 4°C for 20 min at 4600g. The Ru5P epimerase in the crude supernatant was assessed by western-blot analyses of polyacrylamide gel electropherograms.
Protein and Enzyme Assays
Protein concentration was determined with Bradford's reagent (Bradford, 1976) with BSA as the standard. Ru5P epimerase activity was assayed spectrophotometrically at 340 nm and 25°C (Kiely et al., 1973). Assay solutions (120 μL) at pH 8.0 routinely contained 50 mm Bicine, 1 mm EDTA, 0.1 mm thiamine pyrophosphate, 0.25 mm NADH, 5 mm R5P, 1 mm Ru5P, 0.12 unit of transketolase, 2.4 units of triose phosphate isomerase, 0.24 unit of glycerophosphate dehydrogenase, and 1.6 × 10−3 to 2.4 × 10−3 units of Ru5P epimerase. Before the addition of the epimerase, the assay solution was preincubated for 5 min to ensure complete consumption of the Xu5P that was present as a contaminant in the commercial Ru5P preparation. One unit of epimerase activity is defined as the oxidation of 1 μmol NADH min−1 in the 120-μL assay solution.
In kinetic studies for Vmax, Km, and Ki determinations, the concentration of R5P in assay solutions was decreased from 5 to 2 mm, a change that did not diminish the measured epimerase activity. Use of a lower concentration of R5P was prompted by the finding that the commercial preparation contained 2% contamination by Ru5P, as estimated by analysis with phosphoribulokinase (Porter et al., 1986). Hence, at 5 mm R5P, Ru5P is concurrently introduced at 0.1 mm, a concentration that exceeds the lowest needed for reliable determination of its Km.
Molecular Mass Estimations
The subunit molecular mass of Ru5P epimerase was determined by SDS-PAGE at 15°C on 12.5% (w/v) PhastGels in conjunction with a PhastSystem apparatus (Pharmacia Biotech). Standards were provided by the low-molecular-mass calibration kit from Pharmacia Biotech. Gels were stained with silver or Coomassie blue according to the supplier's protocol.
The molecular mass of native Ru5P epimerase was estimated by gel filtration and by PAGE under nondenaturing conditions on gradient gels. A prepacked Superose 12 HR column (1.0 × 30 cm; Pharmacia Biotech) was equilibrated with a pH 8.0 buffer (10 mm G3P, 150 mm NaCl, and 1 mm EDTA) and calibrated with a gel-filtration calibration kit (inclusive of proteins from 25 to 440 kD) from the same vendor. The elution position of the epimerase (0.2 mg in 0.1 mL of equilibration buffer applied to the column) was then determined and assessed relative to the calibration profile. Nondenaturing PAGE was carried out at 15°C on 8% to 25% (w/v) gradient PhastGels; correlation of electrophoretic mobility with protein size was provided by the high-molecular-mass calibration kit from Pharmacia Biotech. Gels were fixed with TCA and stained with Coomassie blue according to the manufacturer's instructions.
pI of Ru5P Epimerase
IEF on polyacrylamide gels was used to assess the purity of epimerase preparations and also to determine the pI of the epimerase subunit under denaturing conditions and the pI of the holoenzyme under nondenaturing conditions. For the latter, PhastGels IEF 4 to 6.5 and the broad pI calibration kit were used according to the manufacturer's protocol, except that the temperature during focusing was maintained at 5°C to mitigate denaturation of the epimerase. IEF under denaturing conditions at 25°C was achieved with PhastGels IEF 5 to 8 that were presoaked for 10 min in 2% (v/v) ampholyte (pH 5.0–8.0) containing 9.5 m urea, quickly rinsed by dipping into water, and gently blotted to remove excess liquid before sample application. The epimerase sample was diluted with 2 volumes of 9.5 m urea containing 5% (v/v) 2-mercaptoethanol. Focused gels were stained with Coomassie blue in accordance with the manufacturer's instructions.
Equilibrium Constant
The equilibrium ratio of Ru5P to Xu5P, as established by the epimerase at 25°C and pH 8.0, was determined by the incubation of 2 mm Ru5P with either 0.03 or 3 μg of the purified enzyme in 1.2 mL of 50 mm Bicine and 1 mm EDTA. Periodically, aliquots (400 μL) were removed, deproteinated by centrifugation in Centricon-10 tubes (Amicon), and assayed for Ru5P with phosphoribulokinase (Porter et al., 1986) and for Xu5P with transketolase (the same assay solution used for epimerase activity but lacking the epimerase). The epimerase reactions were monitored until the concentrations of Ru5P and Xu5P were constant.
Western Blots
Ru5P epimerase was detected on polyacrylamide gels by western immunoblotting (Towbin et al., 1979; Burnette, 1981; Jahn et al., 1984; Johnson et al., 1984). Electrophoresis of samples under both denaturing and nondenaturing conditions and IEF under nondenaturing conditions relied on an XCell Mini-Cell apparatus (Novex, San Diego, CA) with gels, buffers, and prestained molecular-mass markers supplied by the same vendor. Denaturing electrophoresis at room temperature was achieved on 4% to 12% (w/v) NuPAGE Tris gels with Mes-SDS running buffer, and nondenaturing electrophoresis at 2°C was achieved on 8% to 16% (w/v) Tris-Gly gels with Tris-Gly native running buffer. IEF at 2°C entailed the use of pH 3.0 to 7.0 IEF gels with associated cathode and anode buffers. Proteins from nonstained gels were transferred to nitrocellulose membranes with an XCell Mini-Cell and Blot Module (Novex). The membranes were soaked for 10 min in 25% (v/v) isopropanol/10% (v/v) acetic acid, rinsed with water, blocked for 30 min with 5% (w/v) nonfat dry milk dissolved in pH 7.4 buffer (50 mm Tris, 200 mm NaCl), and placed in a 1:3000-fold dilution (with Tris buffer inclusive of 0.1% [v/v] Triton X-100) of the serum containing epimerase antibodies. After a 1-h incubation, the membranes were rinsed several times with the Tris-Triton X-100 buffer and then immersed for 30 min in peroxidase-conjugated goat anti-rabbit IgG (Bio-Rad), which was diluted 3000-fold with the Tris-Triton X-100 buffer. Color was developed in the Tris buffer containing 1.3 mm 3,3′-diaminobenzidine tetrahydrochloride dihydrate and 9 mm H2O2.
Sequencing
DNA sequencing was carried out on a sequencer (model 373A, Applied Biosystems) by use of either dye-primer or dye-terminator chemistries. Edman degradation of SDS-denatured Ru5P epimerase (a mixture of peaks I and II; 1 nmol) was accomplished with a gas-phase sequencer (model 470, Applied Biosystems) equipped with a 120A analyzer and 900A control module.
RESULTS
Cloning and Expression of rpe Spinach cDNA
Our clones of the rpe cDNA (accession no. AF070941) differ from that reported by Nowitzki et al. (1995) and the corresponding accession (no. L42328) only in the 5′ and 3′ untranslated regions, which may reflect the construction of cDNA libraries from different cultivars of spinach (cv Melody versus cv Monnopa). Our clones are longer on both ends compared with L42328; because we observe the same 5′ end in two independent clones, it may represent the true 5′ end of the mRNA without the cap. Poly(A+) tracts were not observed on the 3′ ends of any of the cDNA clones. In addition to construction of an expression vector for the mature form of Ru5P epimerase (Fig. 1B), we also designed one for the transit form of the enzyme (Fig. 1A), in anticipation of chloroplast import studies. However, based on western-blot analyses of crude extracts, E. coli completely processed the transit protein to mature epimerase (data not shown). Not surprisingly, epimerase activity levels in crude extracts of E. coli, whether transformed with the plasmid encoding the transit or the mature form of the enzyme, were found to be about the same. Thus, we have not addressed the question of intrinsic activity of the transit protein.
Assessment of Purity, Size, and Charge of Ru5P Epimerase
The isolation scheme summarized in Table I provides highly purified recombinant enzyme in reasonable overall recovery (35%). Based on purification level, the epimerase represents about 2% of the total soluble proteins extracted from the host. Beginning with the centrifuged crude extract from transformed E. coli, progression of the purification was readily monitored by SDS-PAGE (Fig. 3A). The DE52 column was particularly effective in rendering enrichment of the epimerase, which was only weakly adsorbed at pH 8.0 relative to most other components in the crude extract. Upon passage of the preparation through hydroxyapatite, the epimerase emerged with only minor contaminants and will thus be suitable for a variety of studies. Although these contaminants were effectively removed by Mono-Q chromatography, which resolved two peaks (I and II) of epimerase, the specific activity was somewhat compromised because of a modest loss of units during the preceding dialysis. Neither their specific activities (Table I) nor their mobilities during SDS-PAGE (Fig. 3B) distinguished the two peaks.
Figure 3.

Progression of purification of spinach recombinant epimerase as assessed by SDS-PAGE. A, Gel stained with Coomassie blue includes: lane 1, molecular mass markers (bands from top to bottom: phosphorylase b, 94 kD; albumin, 67 kD; ovalbumin, 43 kD; carbonic anhydrase, 30 kD; trypsin inhibitor, 20.1 kD; lactalbumin, 14.4 kD); lane 2, centrifuged extract (3 μg); lane 3, pool from DE52 (2 μg); and lane 4, pool from hydroxyapatite (0.5 μg). B, Gel stained with silver for increased sensitivity includes: lane 1, peak I from Mono-Q (20 ng); and lane 2, peak II from Mono-Q (20 ng).
The subunit size of the epimerase as estimated by SDS-PAGE (Fig. 3B) was 25,000 D, in close agreement with the 25,085 D tabulated from the DNA-deduced amino acid composition. Both peaks of epimerase were also identical and homogeneous, as judged by nondenaturing PAGE, and essentially co-migrated with beef liver catalase of 232,000 D (Fig. 4). Further examination by gel filtration, in parallel with appropriate standards, was consistent with a molecular mass of 200,000 D (data not shown). Taken together, these data demonstrate that the native epimerase is octameric.
Figure 4.
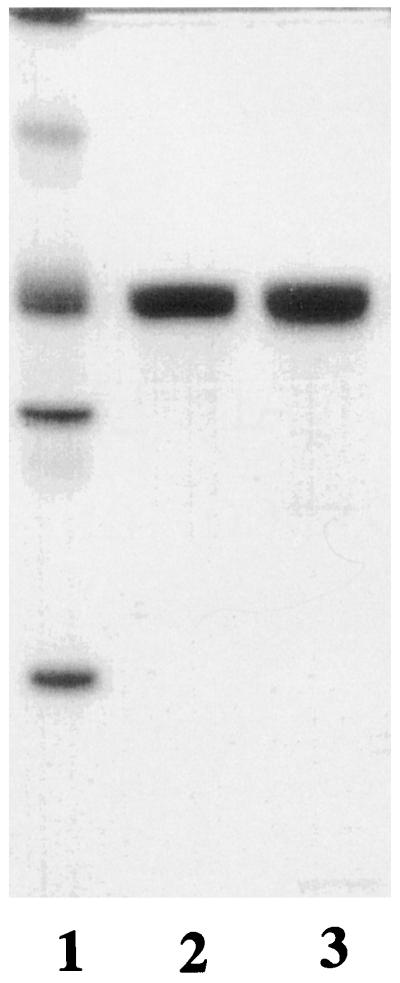
Purity and molecular mass of recombinant epimerase as assessed by PAGE under nondenaturing conditions. The Coomassie blue-stained gel includes: lane 1, molecular mass markers (bands from top to bottom: thyroglobulin, 669 kD; ferritin, 440 kD; catalase 232 kD; lactate dehydrogenase, 140 kD; albumin, 67 kD); lane 2, peak I from Mono-Q (1 μg); and lane 3, peak II from Mono-Q (1 μg).
Peaks I and II of the epimerase behaved as identical homogeneous species when subjected to IEF on gels under denaturing conditions (Fig. 5A). The experimentally determined pI was 6.3, based on the assumption that a linear pH gradient was established by the ampholytes, as compared with 6.0, as calculated from the amino acid composition. Peak I also focused primarily as a single compact species under nondenaturing conditions with a pI of 5.0, whereas peak II contained an additional compact species with a pI of 4.9 (Fig. 5B). These values were virtually the same (±0.1 pI unit) whether based on the assumption of a linear pH gradient or on the internal protein markers. Diffuse regions of staining bands were observed throughout the pH gradient at values exceeding pH 5.0, presumably indicative of dissociation and denaturation of the holoenzyme during IEF. These patterns visualized by nondenaturing IEF were not altered by rechromatography of the epimerase on Mono-Q, i.e. the single compact band of peak I and the compact doublet of peak II persisted.
Figure 5.

IEF of recombinant epimerase under denaturing (A) and nondenaturing (B) conditions. Lanes 1 and 2 in both Coomassie blue-stained gels represent peaks I and II, respectively, from Mono-Q chromatography (Fig. 2). Sample loads were 0.6 μg in A and 1.0 μg in B. pI markers in B include (from top to bottom) human carbonic anhydrase B (pI 6.55), bovine carbonic anhydrase B (pI 5.85), β-lactoglobulin A (pI 5.2), and soybean trypsin inhibitor (pI 4.55).
Stability of Ru5P Epimerase
Whether in crude extracts or highly purified, the recombinant epimerase is inherently unstable even at 2°C (Fig. 6). The instability did not correlate with epimerase concentration and was not affected by glycerol, exogenously added protein (BSA), cofactors (NADH, NAD, or ATP), divalent metal ions (Mg2+ or Zn2+), or EDTA (data not shown); instability was exacerbated by Ru5P. Ethanol and G3P stabilized the enzyme but were unable to reverse the spontaneous loss of activity.
Figure 6.

Inherent instability of recombinant epimerase and effects of additives. All incubations were at 2°C in a pH 8.0 buffer of 50 mm Bicine. With the exception the one data set depicting centrifuged extract (250 μg total protein mL−1) without any additives (○), all others depict purified enzyme (50 μg mL−1): no additives (▪), 10 mm G3P introduced at 0 time or after 2 h (□), 20% (v/v) ethanol (▵), 1 mm Ru5P (•), and 10 mm 2-mercaptoethanol (▴). See text for additional details.
In contrast to most intracellular enzymes, the epimerase was drastically destabilized by 2-mercaptoethanol (Fig. 6). DTT also destabilized the enzyme, but the rate of activity loss was less pronounced; GSH was without effect (data not shown). Neither G3P nor ethanol afforded protection against inactivation of the epimerase by 2-mercaptoethanol. Although sensitivity to thiols suggested reduction of a protein disulfide, neither GSSH nor dehydroascorbate reversed the inactivation by 2-mercaptoethanol (data not shown). Furthermore, spontaneous or 2-mercaptoethanol-induced inactivation of the epimerase did not alter its electrophoretic mobility or its IEF under nondenaturing conditions (data not shown).
Kinetic and Thermodynamic Parameters
Epimerase activity displayed a typical hyperbolic response to increasing concentrations of Ru5P, from which a Km of 0.22 mm and a Vmax of 17,000 units mg−1 were calculated. The corresponding kcat based on a subunit molecular mass of 25,000 D was 7083 s−1, thereby establishing kcat/Km as 3.2 × 107 m−1 s−1. G3P (examined at both 1 and 5 mm) behaved as a competitive inhibitor with a Ki of 0.9 mm.
At pH 8.0 and 25°C, the equilibrium ratio of Xu5P to Ru5P was 2.2, irrespective of epimerase concentration and preparation of epimerase used. Under very similar conditions, an equilibrium constant of 1.5 has been reported by use of impure epimerase from Lactobacillus pentosus (Hurwitz and Horecker, 1956).
Authenticity of Spinach Recombinant Ru5P Epimerase
Our construct of a cDNA clone encoding the mature form of Ru5P epimerase includes an initiation codon for Met in place of a codon for Lys at the cleavage site of the transit peptide. This forecasts an N-terminal sequence of MATSRVD for the recombinant epimerase, compared with TSRVD for the epimerase isolated from spinach chloroplasts (Teige et al., 1995, 1998). The N-terminal sequence of our purified recombinant enzyme (a mixture of peaks I and II from Mono-Q) as determined by Edman degradation was ATSRVD. Thus, the encoded N-terminal Met was removed in E. coli, so the recombinant enzyme differed from the authentic leaf protein merely by the presence of a single Ala residue as an appendage at the N terminus.
Western blots of denaturing and nondenaturing PAGE gels showed that the sizes of the recombinant epimerase subunit and the recombinant epimerase holoenzyme closely match those of the authentic enzyme present in extracts of fresh spinach leaves (Fig. 7, A and B). The antibody raised against recombinant spinach Ru5P epimerase (a mixture of peaks I and II) did not cross-react with the E. coli epimerase or with any other protein in extracts of nontransformed E. coli (Fig. 7B, lane 3), but did cross-react with the component unique to peak II from Mono-Q that was observed by IEF under nondenaturing conditions (Figs. 5B and 7C). Thus, this second component is a form of the recombinant epimerase and does not reflect contamination with constitutive E. coli epimerase. The diffuse staining regions that were detected with Coomassie blue (Fig. 5B) also cross-reacted strongly with the antibody and displayed a rather prominent band with a pI of 5.7 (Fig. 7C). Because this value approached that of the fully denatured subunit (pI 6.3), our previous supposition that the complex focusing pattern reflects partial dissociation and denaturation of the epimerase was reinforced.
Figure 7.

Western-blot analyses of purified recombinant epimerase, E. coli extracts, and spinach leaf extract subjected to SDS-PAGE (A), nondenaturing PAGE (B), or nondenaturing IEF (C). In A, samples are as follows: lane 1, peak I from Mono-Q (0.12 μg); lane 2, centrifuged extract from transformed E. coli (2.5 μg); lane 3, centrifuged extract from spinach (20 μg); and lane 4, prestained molecular mass markers (bands from top to bottom: myosin, 250 kD; phosphorylase b, 148 kD; glutamic dehydrogenase, 60 kD; carbonic anhydrase, 42 kD; myoglobin, 22 kD; lysozyme, 17 kD; aprotinin, 6 kD). In B, samples are as follows: lane 1, peak I from Mono-Q (15 ng); lane 2, peak II from Mono-Q (15 ng); lane 3, centrifuged extract from nontransformed E. coli (0.5 μg) (note the absence of cross-reactive material); lane 4, centrifuged extract from transformed E. coli (0.5 μg); and lane 5, centrifuged extract from spinach (15 μg). In C, samples are as follows: lane 1, peak I from Mono-Q (0.5 μg); and lane 2, peak II from Mono-Q (0.5 μg).
DISCUSSION
We elected to clone spinach rpe because previous clones (Kusian et al., 1992; Nowitzki et al., 1995; Teige et al., 1995) have not led to efficient overexpression in a foreign host. In the only case in which heterologous expression has been reported (Nowitzki et al., 1995), transformation of E. coli with a plasmid harboring spinach rpe resulted in only a 3-fold elevation of epimerase activity relative to activity indigenous to the host. This low level of activity may have reflected inherent instability of the recombinant enzyme rather than poor expression. By contrast, we observed a >350-fold elevation of activity with our expression system (0.7 unit mg−1 protein in extracts of nontransformed cells versus 250 units mg−1 protein in extracts of transformed cells after induction), in which recombinant Ru5P epimerase represents approximately 2% of the total soluble protein.
The extreme instability of the epimerase posed a hurdle to attaining a high level of purity while preserving the catalytic activity. Our discovery that G3P affords effective, long-term protection against spontaneous loss of activity enabled us to clear this hurdle. By a combination of maintaining the preparation at 2°C, including 10 mm G3P in all buffers used during the purification, and minimizing the time for the chromatographic steps, extensive loss of activity was averted. Some loss did occur during dialysis before Mono-Q chromatography (with the preparation summarized in Table I, this loss amounted to 11%, which is the worst case encountered among numerous preparations from independent cultures during the past 1.5 years). Final specific activities have ranged from 10,000 to 15,000 units mg−1. By comparison, the yeast and beef liver epimerases, which are not inherently unstable, have specific activities of approximately 7000 units mg−1. Apart from the dialysis step, the other variable with respect to final specific activity may be the intracellular status of the enzyme at the time of harvesting of the cultures. From sample application to pooling of fractions, the DE52, hydroxyapatite, and Mono-Q chromatographic steps were completed in 5, 1, and 2 h, respectively. The original high-speed supernatant and the concentrated pools from each of the three columns could be frozen in the absence of cryoprotectant and stored indefinitely.
The specific activity of Ru5P epimerase recently purified from spinach chloroplasts was observed to be only 180 units mg−1 (Teige et al., 1998). This extremely low value, <2% of that attainable under our protective conditions, probably reflected almost total inactivation of the enzyme wrought by 5 mm DTT, which was included in the initial extraction and column buffers.
Spontaneous and thiol-induced inactivation of Ru5P epimerase reflect distinct phenomena, because ethanol and G3P retard the former but not the latter. Despite a rather extensive effort, we have not identified any additives or conditions that even partially reverse spontaneous or thiol-induced inactivation of the epimerase. Enhanced stability of the enzyme in the presence of ethanol may be indicative of solvent-exposed hydrophobic patches, which in vivo serve as sites for association with other proteins or thylakoid membranes in situ. Although Ru5P epimerase has not been found in various multienzyme complexes of Calvin cycle enzymes (Sainis and Harris, 1986; Nicholson et al., 1987; Persson and Johansson, 1988; Rault et al., 1993; Süss et al., 1993; Anderson et al., 1995; Romanova and Pavolets, 1997), recent studies (Teige et al., 1998) using immunogold electron microscopy have localized the epimerase to thylakoid membranes. As judged by kinetic analyses, the binding of G3P and Ru5P by the epimerase is competitive. Therefore, the opposite effects of these ligands on enzyme stability is likely explained by their inducing different conformational changes. Although the thiol sensitivity of the enzyme immediately raises the prospect of a structurally or catalytically important disulfide, experimental data argue to the contrary. Thiol-induced inactivation is not reversed by customary oxidants, and the inactivation does not result from dissociation of subunits or any structural changes that can be detected by a variety of electrophoretic analyses. Furthermore, none of the three cysteinyl residues in each subunit of Ru5P epimerase are species invariant, as would be expected if any were critical to structure or catalysis.
Thiol sensitivity of the epimerase could be construed as reflective of in vivo redox regulation via the Fd-thioredoxin system, which has been well documented for several chloroplast enzymes (for reviews, see Buchanan, 1991; Jacquot et al., 1997). However, our inability to reverse the inactivation of Ru5P epimerase by 2-mercaptoethanol precludes further speculation on this issue.
Most species of Ru5P epimerase are homodimeric; the only previous exception is an octameric form of the enzyme isolated from spinach chloroplasts (Teige et al., 1998). However, the authors of that study cautioned that this may have been an artifact of purification because the epimerase activity released from freshly ruptured chloroplasts appeared to chromatograph as a dimer during gel filtration. Although we cannot reconcile these disparate observations, our finding that highly active recombinant spinach Ru5P epimerase is also octameric argues against an anomalous quaternary structure arising during purification. Furthermore, western-blot analysis of crude extracts of spinach leaves clearly shows that the authentic epimerase co-migrates with the recombinant form during nondenaturing PAGE (Fig. 7B), indicative of in vivo assembly of the 25,000-D subunit into an octameric holoenzyme. Whether octamers will prove typical of the epimerase, as found throughout the plant kingdom, must await future comparisons.
Despite the striking differences in subunit structure of spinach Ru5P epimerase and the corresponding yeast and mammalian enzymes, their kinetic parameters are rather similar. The Km (Ru5P) of 0.22 mm for the recombinant spinach enzyme compares with 0.2 to 1.5 mm for other species reported (Hurwitz and Horecker, 1956; Wood, 1979; Bär et al., 1996), and the Vmax of the former is about twice as great as the highest Vmax reported among the latter (Terada et al., 1985; Bär et al., 1996). The catalytically impaired preparation isolated from spinach chloroplasts exhibited a similar Km of 0.25 mm (Teige et al., 1998). Thus, the DTT-induced inactivation of the epimerase does not alter its apparent affinity for substrate.
We have not been able to determine the molecular basis of microheterogeneity displayed by the recombinant epimerase when chromatographed on Mono-Q (Fig. 2) or isoelectric focused under nondenaturing conditions (Figs. 5B and 7C). Obvious possibilities such as contamination by E. coli Ru5P epimerase or incomplete removal of the N-terminal Met residue encoded by the gene construct are excluded. With respect to the former, the antibody raised against the recombinant preparation does not cross-react with E. coli epimerase, and furthermore, >100 mm NaCl is required to elute the E. coli enzyme from the DEAE column compared with <50 mm NaCl for the recombinant enzyme. The only residue detected in the first cycle of Edman degradation is Ala, without even a trace of Met, thereby ruling out mixed N-terminal processing as a basis for microheterogeneity. We also dismiss partial dissociation of subunits as an explanation because the preparations always migrated as a single species during nondenaturing PAGE. If the microheterogeneity is caused by size or charge differences at the subunit level, they are below the threshold for detection by SDS-PAGE and IEF under denaturing conditions. The subtlety of the basis of microheterogeneity is emphasized by the very small difference of only 0.1 pI unit between the two bands (5.0 versus 4.9) observed by IEF of the native enzyme. In contrast, a difference of only one formal charge per subunit is calculated to alter its pI by 0.35 unit. We speculate that we may be encountering three discrete conformations of the enzyme, a pair that co-chromatograph on Mono-Q (the two components in peak II that focus with pI values of 5.0 and 4.9, respectively), and a different pair that co-focus on native IEF (the single component in peak I, which exhibits a pI of 5.0, and the component in peak II with a pI of 5.0). Future studies of reversible denaturation may provide an avenue for clarifying this situation.
In summary, to our knowledge we have designed the first high-level expression system for an rpe clone from any source. Subsequent development of a facile purification scheme for the recombinant spinach Ru5P epimerase enabled us to characterize structural, catalytic, and stability properties of the first highly active Ru5P epimerase of photosynthetic origin obtained.
Abbreviations:
- G3P
dl-α-glycerophosphate
- R5P
d-Rib-5-phosphate
- Ru5P
d-ribulose-5-phosphate
- Ru5P epimerase
d-ribulose-5-phosphate 3-epimerase
- Xu5P
d-xylulose-5-phosphate
Footnotes
This research was supported by the Office of Biological and Environmental Research, U.S. Department of Energy, under contract no. DE-AC0596OR22464 with the Lockheed Martin Energy Research Corporation.
cDNA for spinach rpe (Nowitzki et al., 1995) had not been reported at the time we initiated our studies.
Residue numbers refer to the mature recombinant enzyme, the N-terminal Ala of which corresponds to position 49 of the transit protein.
LITERATURE CITED
- Anderson LE, Goldhaber-Gordon IM, Li D, Tang X, Xiang M, Prakash N. Enzyme-enzyme interaction in the chloroplast: glyceraldehyde-3-phosphate dehydrogenase, triose phosphate isomerase and aldolase. Planta. 1995;196:245–255. doi: 10.1007/BF00201381. [DOI] [PubMed] [Google Scholar]
- Bär J, Naumann M, Reuter R, Kopperschläger G. Improved purification of ribulose 5-phosphate 3-epimerase from Saccharomyces cerevisiae and characterization of the enzyme. Bioseparation. 1996;6:233–241. [PubMed] [Google Scholar]
- Blattner FR, Burland V, Plunkett G, Sofia HJ, Daniels DL. Analysis of the Escherichia coli genome. IV. DNA sequence of the region from 89.2 to 92.8 minutes. Nucleic Acids Res. 1993;21:5408–5417. doi: 10.1093/nar/21.23.5408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- Buchanan BB. Regulation of the CO2 assimilation in oxygenic photosynthesis: the ferredoxin/thioredoxin system. Arch Biochem Biophys. 1991;288:1–9. doi: 10.1016/0003-9861(91)90157-e. [DOI] [PubMed] [Google Scholar]
- Burnette WN. “Western blotting”: electrophoretic transfer of proteins from sodium dodecyl sulfate-polyacrylamide gels to unmodified nitrocellulose and radiographic detection with antibody and radioiodinated protein A. Anal Biochem. 1981;112:195–203. doi: 10.1016/0003-2697(81)90281-5. [DOI] [PubMed] [Google Scholar]
- Falcone DL, Tabita FR. Complementation analysis and regulation of CO2 fixation gene expression in a ribulose 1,5-bisphosphate carboxylase-oxygenase deletion strain of Rhodospirillum rubrum. J Bacteriol. 1993;175:5066–5077. doi: 10.1128/jb.175.16.5066-5077.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurwitz J, Horecker BL. The purification of phosphoketopentoepimerase from Lactobacillus pentosus and the preparation of xylulose 5-phosphate. J Biol Chem. 1956;223:993–1008. [PubMed] [Google Scholar]
- Jacquot J-P, Lancelin J-M, Meyer Y. Thioredoxins: structure and function in plant cells. New Phytol. 1997;136:543–570. doi: 10.1046/j.1469-8137.1997.00784.x. [DOI] [PubMed] [Google Scholar]
- Jahn R, Schiebler W, Greengard P. A quantitative dot-immunobinding assay for proteins using nitrocellulose membrane filters. Proc Natl Acad Sci USA. 1984;81:1684–1687. doi: 10.1073/pnas.81.6.1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson DA, Gautsch JW, Sportsman JR, Elder JH. Improved technique utilizing nonfat dry milk for analysis of proteins and nucleic acids transferred to nitrocellulose. Gene Anal Tech. 1984;1:3–8. [Google Scholar]
- Karmali A, Drake AF, Spencer N. Purification, properties and assay of d-ribulose 5-phosphate 3-epimerase from human erythrocytes. Biochem J. 1983;211:617–623. doi: 10.1042/bj2110617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiely ME, Stuart AL, Wood T. Partial purification and kinetic properties of ribose-5-phosphate ketol-isomerase and ribulose-5-phosphate 3-epimerase from various sources. Biochim Biophys Acta. 1973;293:534–541. doi: 10.1016/0005-2744(73)90360-4. [DOI] [PubMed] [Google Scholar]
- Kunkel TA, Roberts JD, Zakour RA. Rapid and efficient site-specific mutagenesis without phenotypic selection. Methods Enzymol. 1987;154:367–382. doi: 10.1016/0076-6879(87)54085-x. [DOI] [PubMed] [Google Scholar]
- Kusian B, Yoo JG, Bednarski R, Bowien B. The Calvin cycle enzyme pentose-5-phosphate 3-epimerase is encoded within the cfx operons of the chemoautotroph Alcaligenes eutrophus. J Bacteriol. 1992;174:7337–7344. doi: 10.1128/jb.174.22.7337-7344.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larimer FW, Mural RJ, Soper TS. Versatile protein engineering vectors for mutagenesis, expression and hybrid enzyme formation. Protein Eng. 1990;3:227–231. doi: 10.1093/protein/3.3.227. [DOI] [PubMed] [Google Scholar]
- Lyngstadaas A, Løbner-Olesen A, Boye E. Characterization of three genes in the dam-containing operon of Escherichia coli. Mol Gen Genet. 1995;247:546–554. doi: 10.1007/BF00290345. [DOI] [PubMed] [Google Scholar]
- Nicholson S, Easterby JS, Powls R. Properties of a multimeric protein complex from chloroplasts possessing potential activities of NADPH-dependent glyceraldehyde-3-phosphate dehydrogenase and phosphoribulokinase. Eur J Biochem. 1987;162:423–431. doi: 10.1111/j.1432-1033.1987.tb10619.x. [DOI] [PubMed] [Google Scholar]
- Nowitzki U, Wyrich R, Westhoff P, Henze K, Schnarrenberger C, Martin W. Cloning of the amphibolic Calvin cycle/OPPP enzyme d-ribulose-5-phosphate 3-epimerase (EC 5.1.3.1) from spinach chloroplasts: functional and evolutionary aspects. Plant Mol Biol. 1995;29:1279–1291. doi: 10.1007/BF00020468. [DOI] [PubMed] [Google Scholar]
- Persson L-O, Johansson G. Studies of protein-protein interaction using countercurrent distribution in aqueous two-phase systems. Biochem J. 1988;259:863–870. doi: 10.1042/bj2590863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porter MA, Milanez S, Stringer CD, Hartman FC. Purification and characterization of ribulose-5-phosphate kinase from spinach. Arch Biochem Biophys. 1986;245:14–23. doi: 10.1016/0003-9861(86)90185-2. [DOI] [PubMed] [Google Scholar]
- Porter MA, Stringer CD, Hartman FC. Characterization of the regulatory thioredoxin site of phosphoribulokinase. J Biol Chem. 1988;263:123–129. [PubMed] [Google Scholar]
- Rault M, Giudici-Orticoni M-T, Gontero B, Ricard J. Structural and functional properties of a multi-enzyme complex from spinach chloroplasts. 1. Stoichiometry of the polypeptide chains. Eur J Biochem. 1993;217:1065–1073. doi: 10.1111/j.1432-1033.1993.tb18338.x. [DOI] [PubMed] [Google Scholar]
- Romanova AK, Pavolets VV. Supramolecular complexes of the enzymes participating in photosynthetic carbon dioxide assimilation. Russ J Plant Physiol. 1997;44:230–238. [Google Scholar]
- Sainis JK, Harris C. The association of ribulose-1,5-bisphosphate carboxylase with phosphoriboisomerase and phosphoribulokinase. Biochem Biophys Res Commun. 1986;139:947–954. doi: 10.1016/s0006-291x(86)80269-8. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, Maniatis T (1989) Molecular Cloning: A Laboratory Manual, Vol 3. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY, p A3
- Schnarrenberger C, Flechner A, Martin W. Enzymatic evidence for a complete oxidative pentose phosphate pathway in chloroplasts and an incomplete pathway in the cytosol of spinach leaves. Plant Physiol. 1995;108:609–614. doi: 10.1104/pp.108.2.609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Süss K-H, Arkona C, Manteuffel R, Adler K. Calvin cycle multienzyme complexes are bound to chloroplast thylakoid membranes of higher plants in situ. Proc Natl Acad Sci USA. 1993;90:5514–5518. doi: 10.1073/pnas.90.12.5514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teige M, Kopriva S, Bauwe H, Süss K-H. Chloroplast pentose-5-phosphate 3-epimerase from potato: cloning, cDNA sequence, and tissue-specific enzyme accumulation. FEBS Lett. 1995;377:349–352. doi: 10.1016/0014-5793(95)01373-3. [DOI] [PubMed] [Google Scholar]
- Teige M, Melzer M, Süss K-H. Purification, properties and in situ localization of the amphibolic enzymes d-ribulose 5-phosphate 3-epimerase and transketolase from spinach chloroplasts. Eur J Biochem. 1998;252:237–244. doi: 10.1046/j.1432-1327.1998.2520237.x. [DOI] [PubMed] [Google Scholar]
- Terada T, Mukae H, Ohashi K, Hosomi S, Mizoguchi T, Uehara K. Characterization of an enzyme which catalyzes isomerization and epimerization of d-erythrose 4-phosphate. Eur J Biochem. 1985;148:345–351. doi: 10.1111/j.1432-1033.1985.tb08845.x. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. Proc Natl Acad Sci USA. 1979;76:4350–4354. doi: 10.1073/pnas.76.9.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood T. Purification and properties of d-ribulose-5-phosphate 3-epimerase from calf liver. Biochim Biophys Acta. 1979;570:352–362. doi: 10.1016/0005-2744(79)90155-4. [DOI] [PubMed] [Google Scholar]