

Abstract
Whole-genome sequencing, transcriptomic analyses, and metabolic reconstruction were used to investigate Gordonia sp. strain KTR9's ability to catabolize a range of compounds, including explosives and steroids. Aspects of this mycolic acid-containing actinobacterium's catabolic potential were experimentally verified and compared with those of rhodococci and mycobacteria.
TEXT
Gordonia sp. strain KTR9 was isolated from soil at an explosives testing facility (19). Although the strain was reported to utilize hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) as a sole source of carbon and nitrogen under aerobic growth conditions, subsequent studies have replicated only its ability to utilize RDX as a sole source of nitrogen (9). KTR9 is similar to other mycolic acid-containing actinomycetes that degrade RDX (4, 14, 18) in that it utilizes XplA, a cytochrome P450, to catalyze the denitration of RDX (11, 17, 18). In KTR9, the xplAB genetic locus resides on a 182-kb plasmid, pGKT2, and is sufficient for the degradation of RDX in actinobacteria (9). More generally, Gordonia is a genus of catabolically versatile mycolic acid-containing actinobacteria known for their ability to degrade xenobiotics, recalcitrant polymers (2), and, as was found more recently, steroids (6). Here, we present the whole-genome sequencing, transcriptomic analyses, and metabolic reconstruction of KTR9, the first such studies for an explosives-degrading bacterium. Comparison with the genome sequences of other Gordonia (6, 7, 10) provides insight into the metabolism of this important genus of soil bacteria.
The KTR9 genome was sequenced using the Genome Sequencer FLX system (454 Life Science Corp.). In addition, a library of 1,536 fosmid clones (EpiFOS, Epicentre Biotechnologies) was constructed, fingerprint mapped, and end sequenced. The fosmid end reads and 454 Newbler assembly contigs were assembled using PHRAP (http://www.phrap.org/). Manual editing of the PHRAP assembly and integration with the physical map yielded 10 sequence scaffolds. To further improve the assembly, 38 fosmid clones were pooled and pyrosequenced using 454 technology (Genome Quebec, McGill University). The remaining four scaffolds were ordered using Gordonia bronchialis 3410T (10) as a reference genome and Mauve software (5). The four gaps between these scaffolds were closed by sequencing amplicons obtained using KTR9 chromosomal DNA and LongAmp Taq DNA polymerase (New England BioLabs).
Open reading frames (ORFs) were identified using FGENESB software (Softberry Inc., Mount Kisco, NY) and the RAST server (http://rast.nmpdr.org) (3). The genes that were identified were compared to databases of sequences using BLAST searches of the NCBI nonredundant (nr) database (1). The Clusters of Orthologous Groups (COG), Kyoto Encyclopedia of Genes and Genomes (KEGG), and RefSeq databases were searched for homologs using BLASTp and an e-value cutoff of 10−6. Automatic annotation results were manually verified.
The transcriptome of KTR9 was investigated using RNA-Seq. Briefly, KTR9 was grown on M9 minimal medium supplemented with trace elements and vitamin B1 (K. Swain, I. Casabon, L. D. Eltis, and W. W. Mohn, submitted for publication), 20 mM succinate, and either 0.9 mM NH4Cl or 0.3 mM RDX as the sole nitrogen source. Cells were harvested at mid-log phase (optical density at 600 nm [OD600] ∼ 0.9) and treated with RNAlater (Ambion). Total RNA was isolated using a RiboPure-Bacteria kit (Ambion, Austin, TX), treated with Turbo DNase (Ambion) for 1 h at 37°C, and purified using an RNeasy minikit (Qiagen, Valencia, CA). Purified RNA samples (∼10 μg) were sequenced by the Genome Science Center (Vancouver, BC). Paired-end reads (75 bp) from Illumina sequencing were mapped against the KTR9 genome, and RNA-Seq analysis was performed using the CLC Genomics workbench (CLC Bio). As listed in Table S3 in the supplemental material, a total of 433 genes were differentially regulated in RDX- versus NH4Cl-grown cells (>2-fold difference; P < 0.5), of which 249 were upregulated and 184 were downregulated.
The transcriptomic data were used to improve the annotation of the KTR9 genome. Briefly, the transcriptome reads were aligned to KTR9 genome sequence using CLC Genomics workbench. The output alignment, genome sequence, and genome annotation were loaded into Integrative Genomics Viewer software (16). The alignment between the transcriptome reads and the annotated genome was manually examined over the length of the genome. As summarized in Table 1, this alignment was used to (i) identify 919 transcripts, including start sites and untranslated regions; (ii) adjust predicted translation start sites; (iii) correct the list of predicted open reading frames (ORFs); and (iv) reveal the presence of potential sRNAs. As a result, 177 genes were identified that had not been detected using FGENESB software and the RAST server, and 77 genes were deleted from the annotation. Alterations of the ORFs previously predicted in pGKT2 (9) are listed in Table S1 in the supplemental material. Finally, the transcriptome sequence led to the correction of seven nucleotides, including two 1-bp insertions in pGKT3 and five 1-bp deletions in the chromosome, the latter of which led to four frameshifts.
Table 1.
KTR9 genome and its validation using transcriptomic analysis
| Genomic element | Size (bp) | G+C% | No. of ORFs | No. inserted/deleted | No. of UTRs identified |
No. of operons identified | |
|---|---|---|---|---|---|---|---|
| 5′a | 3′b | ||||||
| Chromosome | 5,441,391 | 67.8 | 4,741 | 107/64 | 1,141 | 136 | 874 |
| pGKT1 | 89,480 | 65.1 | 93 | 20/4 | 11 | 1 | 6 |
| pGKT2 | 182,454 | 62.7 | 178 | 27/6 | 28 | 0 | 21 |
| pGKT3 | 172,385 | 64.7 | 163 | 23/3 | 19 | 0 | 18 |
| Total | 5,885,710 | 177/77 | 1,199 | 137 | 919 | ||
Identifiable 5′-end untranslated region. (mRNA start site was annotated.)
Identifiable 3′-end untranslated region. (mRNA stop site was annotated.)
As summarized in Table 1, the 5.9-Mbp KTR9 genome consists of a 5.4-Mbp circular chromosome and three circular plasmids: pGKT1 (89 kbp), pGKT2 (182 kbp), and pGKT3 (172 kbp). The GC content of the plasmids was significantly lower than that of the chromosome. The annotated genome includes 45 tRNA genes representing all 20 amino acids, 9 rRNA genes (all of which occur in the chromosome), and 13 possible sRNAs (see Table S4 in the supplemental material). In addition, 9 pseudogenes were identified. Finally, while the gene densities of the chromosome and plasmids were similar, a significantly higher proportion of the coding sequences (CDSs) were annotated as hypothetical or conserved hypothetical proteins on the plasmids (56.4 to 78.7%) than on the chromosome (26.8%). These proportions are similar to those reported for RHA1 (13).
Metabolic reconstruction, performed using Pathway Tools (12), resulted in the prediction of 277 metabolic pathways in KTR9. These included 189 biosynthetic pathways, 117 degradation and assimilation pathways, 6 detoxification pathways, and 27 pathways involved in precursor metabolite production and energy generation. As part of this reconstruction, Pathway Hole Filler automatically reannotated 52 genes to complete pathways. The predicted pathways include those responsible for the de novo biosynthesis of all amino acids, nucleotides, coenzymes, and carbohydrates, consistent with the ability of KTR9 to grow on minimal medium supplemented with glucose and ammonium (9). These biosynthetic pathways were also examined manually, resulting in the reannotation of another 13 genes and the identification of others using tBLASTn to search the genome. Importantly, KTR9 harbors a large number of genes dedicated to fatty acid and lipid biosynthesis, as has been reported for rhodococci (8), and consistent with the ability of these bacteria to accumulate lipid bodies (22). Finally, although KTR9 is able to utilize RDX as the sole nitrogen source, the number of predicted amine and polyamine catabolic pathways is similar to that of other Gordonia and Rhodococcus species.
The nucleotide sequence of pGKT2 had previously revealed that xplA, encoding the RDX-transforming cytochrome P450, is tightly clustered with cyp151C and xplB, the latter of which is fused to a glnA ortholog. The transcriptomic data demonstrate that these three genes constitute an operon (Fig. 1). Moreover, the transcription start site mapped to 12 nucleotides upstream of the cyp151C start codon and is preceded by the previously predicted promoter and GntR-binding site (9). The xplR gene, encoding a GntR-type regulator and located just upstream of the xpl operon, constitutes a separate transcriptional unit. Interestingly, these genes were expressed in NH4Cl-grown cells and were only slightly upregulated in RDX-grown cells (Fig. 1). This is in contrast to our previous studies indicating that these genes are upregulated 3.7-fold in the presence of RDX (9). Time course studies under a variety of conditions are under way to better understand the dependence of xplA expression on the concentrations of RDX and other nitrogen sources.
Fig 1.
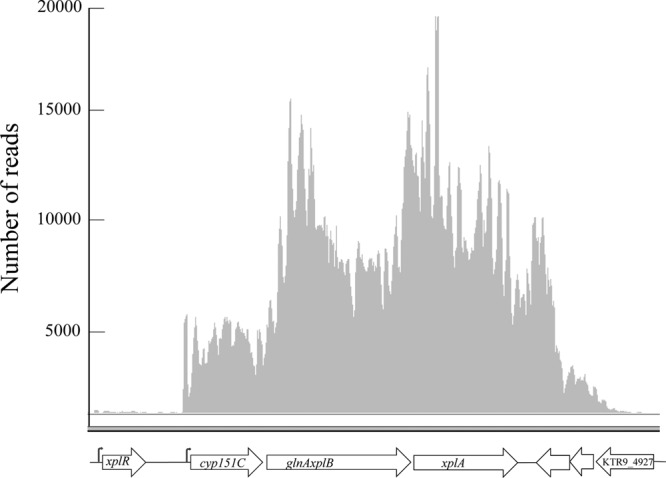
Structure of the xpl operon. The flags indicate the transcription start sites of xplR (nucleotide 156789) and cyp151C (nucleotide 158316), respectively, consistent with previously identified putative promoters (9). The graph indicates the depth of sequence coverage observed in RDX-grown cells. Upregulation of these genes in RDX-grown cells was −1.22-fold for xplR, 1.45-fold for cyp151C, 1.26-fold for glnA xplB, and 1.15-fold for xplA.
Overall, the transcriptomes of the RDX- and NH4Cl-grown cells were fairly similar, with few genes being differentially expressed more than 10-fold. By far the most highly upregulated genes in the RDX-grown cells, at ∼90-fold, were those of a three-gene operon predicted to encode a purine/allantoin permease (KTR9_0990) and a two-subunit N-methyl-hydantoinase/acetone carboxylase (KTR9_0991-0992). The upregulation of the permease gene is particularly intriguing considering that purines and allantoin are heterocyclic, like RDX. Nevertheless, it is unclear whether this gene is involved in RDX uptake or whether it is adventitiously induced by RDX.
A number of Gordonia strains have been studied for their ability to degrade cholesterol (6), but the catabolic pathway has not been well described in this genus. Given the importance of this pathway for the biotransformation of steroids (20) as well as the pathogenesis of Mycobacterium tuberculosis (15), we investigated its occurrence in KTR9. The KTR9 chromosome carries orthologs of all of the genes predicted to encode the pathway in Rhodococcus jostii RHA1 (21) and Mycobacterium tuberculosis (see Fig. S1 and Table S5 in the supplemental material). These genes include those encoding elements considered to be hallmarks of cholesterol catabolism in mycolic acid-containing actinobacteria, such as the Mce4 cholesterol transporter and a cytochrome P450 involved in initiating side chain degradation. Consistent with the predicted occurrence of a cholesterol catabolic pathway, KTR9 grew on M9 minimal medium supplemented with trace elements, vitamin B1, and 1 mM cholesterol as the sole organic substrate, essentially as described for RHA1 on cholate (Swain et al., submitted). In baffled flasks at 30°C, the doubling time of KTR9 on 1 mM cholesterol was ∼53 h compared to ∼9 h on 20 mM pyruvate. Nevertheless, the cholesterol catabolic genes were arranged quite differently in KTR9 than in RHA1 and M. tuberculosis. In the latter, the vast majority of the genes are clustered (21). In contrast, the genes are arranged into two distinct clusters in KTR9 (see Fig. S2 in the supplemental material). Moreover, three genes are found in separate locations of the genome. One of these is hsaD, which is found in an operon with hsaA, hsaB, and hsaC in rhodococci and mycobacteria. The arrangement of the cholesterol catabolic genes appears to be characteristic of Gordonia, as it is conserved in the strains sequenced to date (6, 7, 10).
Nucleotide sequence accession numbers and metabolic reconstruction.
The Gordonia sp. strain KTR9 genome is available in GenBank under accession numbers CP002907 (KTR9), CP002908 (pGKT1), CP002112 (pGKT2), and CP002909 (pGKT3). The metabolic reconstruction is available at BioCyc database (http://biocyc.org).
Supplementary Material
ACKNOWLEDGMENTS
This research was funded in part through grants from the U.S. Army Corps of Engineers Environmental Quality Research Program (project 08-34, K.J.I.), the Strategic Environmental Research and Development Program (project ER-1609, F.H.C.), and Genome BC (L.D.E., S.J.H., and W.W.M.). I.C. is the recipient of a postdoctoral fellowship from the Fonds de Recherche en Santé du Québec and the Michael Smith Foundation for Health Research.
Alice Lau and Amy E. Davis are thanked for manually checking each predicted open reading frame. Simon W. M. Eng and Charles G. Howes are thanked for assisting with running the annotation software and in writing the GenBank files.
Views, opinions, and/or findings contained herein are those of the authors and should not be construed as an official Department of the Army position or decision unless so designated by other official documentation.
Footnotes
Published ahead of print 24 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J. Mol. Biol. 215:403–410 [DOI] [PubMed] [Google Scholar]
- 2. Arenskotter M, Broker D, Steinbuchel A. 2004. Biology of the metabolically diverse genus Gordonia. Appl. Environ. Microbiol. 70:3195–3204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Aziz RK, et al. 2008. The RAST server: rapid annotations using subsystems technology. BMC Genomics 9:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Coleman NV, Nelson DR, Duxbury T. 1998. Aerobic biodegradation of hexahydro-1,3,5-trinitro-1,3,5-triazine (RDX) as a nitrogen source by a Rhodococcus sp., strain DN22. Soil Biol. Biochem. 30:1159–1167 [Google Scholar]
- 5. Darling AE, Mau B, Perna NT. 2010. progressiveMauve: multiple genome alignment with gene gain, loss, and rearrangement. PLoS One 5:e11147 doi:10.1371/journal.pone.0011147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ge F, et al. 2011. Draft genome sequence of Gordonia neofelifaecis NRRL B-59395, a cholesterol-degrading actinomycete. J. Bacteriol. 193:5045–5046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hiessl S, et al. 2012. Involvement of two latex-clearing proteins during rubber degradation and insights into the subsequent degradation pathway revealed by the genome sequence of Gordonia polyisoprenivorans strain VH2. Appl. Environ. Microbiol. 78:2874–2887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Holder JW, et al. 2011. Comparative and functional genomics of Rhodococcus opacus PD630 for biofuels development. PLoS Genet. 7:e1002219 doi:10.1371/journal.pgen.1002219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Indest KJ, et al. 2010. Functional characterization of pGKT2, a 182-kilobase plasmid containing the xplAB genes, which are involved in the degradation of hexahydro-1,3,5-trinitro-1,3,5-triazine by Gordonia sp. strain KTR9. Appl. Environ. Microbiol. 76:6329–6337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ivanova N, et al. 2010. Complete genome sequence of Gordonia bronchialis type strain (3410T). Stand. Genomic Sci. 2:19–28 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jackson RG, Rylott EL, Fournier D, Hawari J, Bruce NC. 2007. Exploring the biochemical properties and remediation applications of the unusual explosive-degrading P450 system XplA/B. Proc. Natl. Acad. Sci. U. S. A. 104:16822–16827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Karp PD, et al. 2010. Pathway Tools version 13.0: integrated software for pathway/genome informatics and systems biology. Brief Bioinform. 11:40–79 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. McLeod MP, et al. 2006. The complete genome of Rhodococcus sp. RHA1 provides insights into a catabolic powerhouse. Proc. Natl. Acad. Sci. U. S. A. 103:15582–15587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nejidat A, Kafka L, Tekoah Y, Ronen Z. 2008. Effect of organic and inorganic nitrogenous compounds on RDX degradation and cytochrome P-450 expression in Rhodococcus strain YH1. Biodegradation 19:313–320 [DOI] [PubMed] [Google Scholar]
- 15. Ouellet H, Johnston JB, Ortiz de Montellano PR. 2011. Cholesterol catabolism as a therapeutic target in Mycobacterium tuberculosis. Trends Microbiol. 19:530–539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Robinson JT, et al. 2011. Integrative Genomics Viewer. Nat. Biotechnol. 29:24–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sabbadin F, et al. 2009. The 1.5-Å structure of XplA-heme, an unusual cytochrome P450 heme domain that catalyzes reductive biotransformation of royal demolition explosive. J. Biol. Chem. 284:28467–28475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Seth-Smith HM, et al. 2002. Cloning, sequencing, and characterization of the hexahydro-1,3,5-trinitro-1,3,5-triazine degradation gene cluster from Rhodococcus rhodochrous. Appl. Environ. Microbiol. 68:4764–4771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Thompson KT, Crocker FH, Fredrickson HL. 2005. Mineralization of the cyclic nitramine explosive hexahydro-1,3,5-trinitro-1,3,5-triazine by Gordonia and Williamsia spp. Appl. Environ. Microbiol. 71:8265–8272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. van der Geize R, Dijkhuizen L. 2004. Harnessing the catabolic diversity of rhodococci for environmental and biotechnological applications. Curr. Opin. Microbiol. 7:255–261 [DOI] [PubMed] [Google Scholar]
- 21. van der Geize R, et al. 2007. A gene cluster encoding cholesterol catabolism in a soil actinomycete provides insight into Mycobacterium tuberculosis survival in macrophages. Proc. Natl. Acad. Sci. U. S. A. 104:1947–1952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Wältermann M, Steinbüchel A. 2005. Neutral lipid bodies in prokaryotes: recent insights into structure, formation, and relationship to eukaryotic lipid depots. J. Bacteriol. 187:3607–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.