

Abstract
Geldanamycin (GM) is a naturally occurring anticancer agent isolated from several strains of Streptomyces hygroscopicus. However, its potential clinical utility is compromised by its severe toxicity and poor water solubility. For this reason, considerable efforts are under way to make new derivatives that have both good clinical efficacy and high water solubility. On the other hand, glycosylation is often a step that improves the water solubility and/or biological activity in many natural products of biosynthesis. Here, we report the facile production of glucose-conjugated nonbenzoquinone GM analogs using the Bacillus UDP-glycosyltransferase BL-C. Five aglycon substrates containing nonbenzoquinone aromatic rings were chosen to validate the in vitro glycosylation reaction. Putative glucoside compounds were determined through the presence of a product peak(s) and were also verified using LC/MS analyses. Further, the chemical structures of new glucoside compounds 6 and 7 were elucidated using spectroscopy data. These glucoside compounds showed a dramatic improvement in water solubility compared with that of the original aglycon, nonbenzoquinone GM.
INTRODUCTION
Geldanamycin (GM) is a potent anticancer antibiotic; it is a benzoquinone ansamacrolide related to herbimycin, reblastatin, and macbecin (5). GM and its analogs bind to the N-terminal ATP binding pocket of heat shock protein 90 (Hsp90) (30). Hsp90 is a molecular chaperon that modulates oncogenic protein kinase activity in cells. While the anticancer potential of GM has long been recognized, clinical evaluation of GM has not been pursued due to GM's severe toxicity and poor water solubility (28). For this reason, efforts have been made to modify GM; these efforts have generated a number of analogs in attempts to increase GM's clinical efficacy and water solubility, including modifications of the C-17 benzoquinone ring position (27). Among the successful analogs is 17-allylaminogeldanamycin (17-AAG), which is currently under clinical development. The most advanced case currently is a phase III study of gastrointestinal stromal tumors (2, 22).
Although 17-AAG has demonstrated improved efficacy and relatively low toxicity, it appears that its hepatotoxicity and low water solubility may remain limiting factors for its clinical application (10, 13). 17-AAG and its benzoquinone GM analogs conjugate with sulfur-containing nucleophiles, such as glutathione, which leads to their cellular depletion (4). This conjugation with glutathione may contribute to the dose-limiting hepatotoxicity of these benzoquinone-containing compounds. For these reasons, new nonbenzoquinone GM analogs with improved pharmacological profiles are needed. Recently, we reported the development of nonbenzoquinone GM analogs using biosyntheic approaches (15, 16, 34). Moreover, a novel semisynthetic nonbenzoquinone GM showed improved efficacy (unpublished data). However, these nonbenzoquinone GM analogs still have formulation problems that predominantly result from their extremely low water solubility and, to a lesser extent, their cancer selectivity.
Commonly, glycosylation improves the water solubility and biological activity of many natural compounds (29). Several complementary strategies, including semisynthesis, pathway engineering, and in vitro enzymatic glycosylation techniques, have emerged from recent research as effective means of altering the natural product sugar structures (6, 8, 9, 11, 17, 32). A well-known example of the improvement in water solubility is the discovery of naturally occurring glycosides such as 7β-xylosyl-10 deacetyl paclitaxel, a taxane glycocojugate (20). This analog was also more than 2 orders of magnitude more water soluble than the original paclitaxel. Among the many taxane glycoconjugate prodrugs synthesized to date, dramatic improvements in solubility have been achieved without drastic reductions in potency. However, glycosyl-GM derivatives have never been isolated from microbial strains, though synthetic conjugates been reported recently (3). The galactose-amine GM derivative has been used as a prodrug that is specifically activated on targeted cancer cells through a tumor-associated glycoprotein (TAG-72) antibody-galactosidase conjugate (7). Thus, the glucose moiety of the glucose-amine GM conjugate was easily cut by the naturally existing β-glucosidase inside the cells (3). It is possible that the glucose-GM conjugate is activated for anticancer activity through the cleavage of the glucose moiety.
Our hypothesis is that the bulky glucose structure might be able to enhance drug solubility for formulation of clinical applications. Glycosyltransferases are an important class of enzyme and are essential for the biosynthesis of glycosylated natural products because they catalyze the attachment of a sugar to an aglycon (8, 19, 36). One glycosyltransferase, BcGT-3 from Bacillus cereus, is known to accept UDP-glucose; this has facilitated the in vitro glycodiversification of the aromatic ring moiety (1). In this report, we describe the function of the same glycosyltransferase (BL-C) from Bacillus licheniformis, which catalyzes the glycosylation of nonbenzoquinone GM analogs. Five putative glucoside GM analogs were found through the presence of the product peak(s) and were also verified via liquid chromatography-mass spectrometry (LC/MS) analyses. The glycosylated GM products that were generated by enzymatic reaction preferred to remain on the water layer.
MATERIALS AND METHODS
General.
The bacterial strain Escherichia coli BL21(DE3) was purchased from Stratagene. Plasmid pET302/NT-his was purchased from Invitrogen. All other chemicals and restriction enzymes were reagent grade and purchased from Fluka, New England BioLabs, or Sigma, unless otherwise stated. The primers were ordered from Genotech (Republic of Korea).
Cloning of the Bacillus glycosyltransferase gene.
The Bacillus BL-C gene (accession number AAU40842) was amplified from the genomic DNA of B. licheniformis (DSM13) using primer pair BLC-F (5′-GAA CTC GAG ATG GGA CAT AAA CAT ATC GCG-3′) (XhoI) and BLC-R (5′-CC GGA TCC TTA TTT TA CTC CTG CGG GTG-3′ (BamHI) (where the underlined sequences represent restriction enzyme recognition sites), 2 U Taq polymerase (Solgent, Republic of Korea), and 1× Taq DNA polymerase buffer. The PCR was performed in a total volume of 20 μl with 100 ng genomic DNA, 0.2 μM primer, and 200 nM deoxynucleoside triphosphate (dNTP) under the following conditions: denaturation at 94°C for 3 min, 25 cycles of denaturation at 94°C for 30 s, annealing at 52°C for 30 s, and extension at 72°C for 1 min. The PCR product of the BL-C gene (1,191 bp) was purified and cloned separately into pGEM-T Easy vectors for sequencing in order to confirm that no mutation had occurred during the PCR amplification. The XhoI-BamHI fragment of the BL-C gene was excised from the pGEM-T vector and cloned into the same sites of pET302/NT-his in order to generate the pET-GTC recombinant expression vector.
Expression and purification of Bacillus glycosyltransferases.
An overnight culture of E. coli bearing pET-GTC was diluted 100-fold in an LB medium supplemented with 100 μg/ml ampicillin and cultured at 37°C. The culture was cooled to 10°C, induced with isopropyl-β-d-thiogalactopyranoside (IPTG) (final concentration = 0.5 mM) when the optical density at 600 nm (OD600) reached 0.6, and incubated for another 12 h at 20°C. The cells were collected via centrifugation at 3,000 rpm for 10 min and stored at −80°C. The cell pellet was resuspended in buffer A (100 mM Tris and 10 mM imidazole, pH 8.0) at a concentration of 0.1 g/ml, and a protease inhibitor cocktail was added according to the manufacturer's instructions. The resuspended cells were sonicated, and the cell lysate was collected via centrifugation at 12,000 × g for 20 min. The pelleted cell lysate was resuspended with a nickel-nitrilotriacetic acid (Ni-NTA) resin equilibrated in buffer A for 30 min. The resin was then loaded into a column and washed with 10 column volumes of buffer A. Next, the Ni-NTA column was washed using a step gradient of 10, 50, 100, and 200 mM imidazole in buffer B (20 mM Tris and 300 mM NaCl, pH 8.0). The recombinant His6-BL-C was eluted from the Ni-NTA column using a linear gradient of 250 mM imidazole in buffer B. The fractions containing BL-C were pooled and concentrated using an Amicon Ultra-15 (Millipore) Centriprep ultrafilter. The concentrated fraction was dialyzed overnight at 4°C using a storage buffer containing 100 mM Tris-HCl (pH 8) and 20% (vol/vol) glycerol and stored at −80°C until use.
Isolation of nonbenzoquinone GM analogs.
18-Dehydroxy-17-demethoxyreblastatin (compound 1), 18-dehydroxy-17-O-demethyl reblastatin (compound 2), and 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin (compound 3) were isolated from the mutasynthetic approaches via a 3-amino benzoic acid feed on the culture plate of the 3-amino-5-hydroxybenzoic acid synthase gene disruption mutant (Fig. 1) (16). 17-Demethoxy-reblastatin (compound 4) was purified from the culture broths of S. hygroscopicus AC11, in which the benzoquinone ring oxidation gene (gel7) of the GM pathway was disrupted by the kanamycin resistance gene (15). 17-Demethoxy-15-hydroxylreblastatin (compound 5; DHQ3) was also purified from the culture broths of S. hygroscopicus AC15, a double mutation with the first dehydratase domain of the geldanamycin polyketide synthase (PKS) gene and the gel7 gene of S. hygroscopicus JCM4427, as previously reported (15, 34).
Fig 1.

Structures of nonbenzoquinone geldanamycin analogs (analogs 1 to 5), glucoside nonbenzoquinone geldanamycin analogs (analogs 6 to 10), and geldanamycin. Compound 1, 18-dehydroxy-17-demethoxyreblastatin; compound 2, 18-dehydroxy-17-O-demethylreblastatin; compound 3, 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin; compound 4, 17-demethoxy-reblastatin; compound 5, 17-demethoxy-15-hydroxylreblastatin.
In vitro glycosylation reaction.
Glycosylation reactions were performed in 1 ml reaction buffer (25 mM Tris-HCl, pH 8.0) containing 1 mg of the purified BL-C enzyme, 2 mM UDP-glucose, 1 mg nonbenzoquinone GM analogs, and 1 mM MgCl2. The mixture was incubated at 30°C for 12 to 15 h. The reaction was quenched and extracted with 1 ml ethyl acetate, and then the extracted reaction mixtures were analyzed using analytical reversed-phase high-performance LC (HPLC) with a YMC J'sphere ODS-H80 column (Tokyo, Japan) (150 by 4.6 mm) and a gradient of 20% to 100% CH3CN-H2O (0.05% trifluoroacetic acid [TFA]) for 25 min at 1 ml/min, with detection at 254 nm. The mass spectra were obtained using electrospray ionization and a Thermo LTQ Orbitrap XL mass spectrometer (Thermo Electron Co.) connected to a UV light-visible light (UV/vis) diode array detector. For the LC/MS analyses, the quenched reaction mixtures were analyzed using analytical reversed-phase ultraperformance LC (UPLC) with a Acquity BEH C18 column (1.7 μm pore size, 100 by 2.1 mm) and a gradient of 5% to 80% CH3CN-H2O (0.1% formic acid) for 10 min at 0.3 ml/min. In order to obtain nuclear magnetic resonance (NMR)-accessible amounts under the described reaction conditions, 50 mg of each of the nonbenzoquinone GM analogs (compounds 1 and 2) was used in a reaction mixture of increased volume. The reaction was quenched, and the reaction mixture was extracted with ethyl acetate and reduced in vacuo with a freeze dryer. The MeOH elute was further purified by reversed-phase high-performance liquid chromatography (HPLC) using a YMC-J'sphere ODS-H80 column (10 by 250 mm, 3 ml min−1) with 25% acetonitrile (CH3CN) to yield compounds 6 (8.5 mg) and 7 (5.0 mg). All NMR experiments were performed on a Varianunity Ionva-400 spectrometer or on a Varianunity 500 NMR spectrometer.
Cell viability assays and Western blot analysis.
The SK-Br3 human breast cancer cell line was obtained from a cell line bank in the Republic of Korea. The cells were seeded in 96-well plates at a concentration of 1 × 104 cells per well at 1 day before the start of treatment. We treated cells with compounds at various concentrations for 72 h. Cell viability was detected by the standard sulforhodamine B (SRB) procedures. Western blot analyses were performed as described previously (33).
Water solubility determination.
For the water solubility determination, the reaction mixture was quenched and extracted with 1 ml ethyl acetate and then centrifuged at 3,000 rpm for 5 min. Aliquots (100 μl) were removed from each water and solvent layer, and the aliquots were analyzed directly via HPLC as previously described. The HPLC peak areas were integrated, and the substrate and product concentrations were calculated as a percentage of the total peak area.
RESULTS
Heterologous expression and purification of Bacillus glycosyltransferase in E. coli.
The glycosyltransferase BL-C was amplified from a B. licheniformis strain and introduced in a pET302/NT-his expression vector in order to express the recombinant histidine-tagged protein in E. coli. Recombinant histidine-tagged Bacillus glycosyltransferase (BL-C) was purified by affinity chromatography. The molecular mass of the recombinant histidine-tagged BL-C was approximately 44.7 kDa (see Fig. S1 in the supplemental material). This family includes the BL-C, which is a group of homologous glycosyltransferases involved in the final stages of the biosynthesis of the antibiotics vancomycin and the related chloroeremomycin. In particular, BL-C is known as a UDP-glycosyltransferase that transfers UDP-activated sugar moieties to acceptor molecules containing an aromatic ring, such as flavonoids (1).
In vitro enzymatic glycosylation of nonbenzoquinone GM analogs.
The substrate flexibility of bacterial glycosyltransferases offers the possibility of producing antibiotics or other natural compounds with drastically changed glycosylation patterns (17, 32). Therefore, the exploitation of the substrate flexibility becomes an important focus in biosynthetic approaches to the chemical diversity of medically active compounds. On the basis of this precedent, we attempted the enzymatic glycosylation reaction using the purified Bacillus glycosyltransferase BL-C and various nonbenzoquinone GM aglycons and UDP-glucose in order to make novel glucoside products.
In order to complete an initial survey of the substrate flexibility of the BL-C enzyme, five nonbenzoquinone GM analogs (compounds 1 to 5 [18-dehydroxy-17-demethoxyreblastatin, 18-dehydroxy-17-O-demethylreblastatin, 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin, 17-demethoxy-reblastatin, and 17-demethoxy-15-hydroxylreblastatin]) and GM as a benzoquinone compound were used in the in vitro glycosylation reactions (Fig. 1). The incubation of the aglycons with UDP-glucose in the presence of BL-C led to the formation of a new product, which was detected by HPLC and LC-MS analyses (Fig. 2 and 3). The in vitro glycosylation reactions conducted in order to establish the optimal conditions of pH (range, approximately 6.5 to 9.5) and temperature (range, approximately 20 to 37°C) determined that the maximum activity occurred at pH 8.0 and 30°C.
Fig 2.

LC/MS analysis of the in vitro glycosylation reaction with 17-demethoxy-reblastatin. (A) Total ion chromatogram of in vitro glycosylation reaction mixture. The arrow indicates a new peak from the in vitro glycosylation reaction. The asterisk indicates 17-demethoxy-reblastatin. (B) MS spectra of selected ion peak at 4.92 min. The ion peak at m/z 679 [M-H]− (m/z 725 [M+HCOOH]−) corresponds to a glucoside form of 17-demethoxy-reblastatin as expected. (C) MS spectra of 17-demethoxy-reblastatin (m/z 517 [M-H]−) at 6.57 min.
Fig 3.

LC/MS analysis of the in vitro glycosylation reaction with 18-dehydroxy-17-O-demethylreblastatin. (A) Total ion chromatogram of the in vitro glycosylation reaction mixture. The arrows indicate new peaks from the in vitro glycosylation reaction; the asterisks indicate a nonbenzoquinone GM, 18-dehydroxy-17-O-demethylreblastatin. (B) MS spectra of 18-dehydroxy-17-O-demethylreblastatin (m/z 517 [M-H]−) at 6.50 min. (C) MS spectra of selected ion peak at 4.99 min. (D) MS spectra of selected ion peak at 4.58 min. (E) MS spectra of selected ion peak at 3.87 min.
The putative glucoside compound appeared as a new peak in the HPLC analyses, and the peak was accepted based on the mass spectrum data (an addition of 162 Da). The peak at m/z 679 [M-H]− (m/z 725 [M+HCOOH]−), which corresponded to compound 10 (a glucoside form of 17-demethoxy-reblastatin), was detected in significant amounts after the enzymatic reaction with 17-demethoxy-reblastatin (Fig. 2). A full scan and MSn mass spectral data for this putative glucoside product showed a loss of 43 Da (carbamoyl moiety [-CONH2]) from the parent ion, which is a distinguishing pattern of the GM compounds (16) (see Fig. S2 in the supplemental material for more details). In addition, we found three putative glucoside peaks in the HPLC analyses using 18-dehydroxy-17-O-demethylreblastatin (compound 2) as a substrate; each peak showed molecular ions at m/z 679 [M-H]−, m/z 725 [M+HCOOH]−, and m/z 887 [M+HCOOH]− (Fig. 3). The ion at m/z 887 [M+HCOOH]− indicated the addition of two glucoses from 18-dehydroxy-17-O-demethylreblastatin, which were presumably attached to the two hydroxyl groups at the C-11 and C-17 positions. Also, the two peaks at m/z 679 [M-H]− and m/z 725 [M+HCOOH]− made it difficult to determine the position of the hydroxyl group from the MSn fragmentation patterns. The full scan and MSn mass spectral data for these putative glucoside products demonstrated a loss of 43 Da from the molecular parent ion. This loss corresponds to the loss of the carbamoyl moiety (-CONH2) of the C-7 hydroxyl group on the GM backbone. Moreover, the fragmentation profile of these new products, with the loss of 162 Da, was comparable to that of the decarbamoyl GM, thus confirming that the products of the BL-C reactions were glucosylated compounds (see Fig. S3 in the supplemental material for more details). Also, 18-dehydroxy-17-demethoxyreblastatin (compound 1; molecular weight [MW] = 502) was converted to the expected glucoside peaks at m/z 663 [M-H]− and 709 [M+HCOOH]− (Table 1; see also Fig. S4 in the supplemental material for more details). Interestingly, 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin, 17-demethoxy-15-hydroxylreblastatin, and GM were not converted, as evidenced by the absence of a product peak and no significant decrease in the aglycon peak, as judged by the HPLC results (Table 1).
Table 1.
LC-MS/MS fragmentation analysis of the glucoside products via in vitro glycosylation reactiona
| Substrate (compound) | MW (predicted compound) | Product ion(s) (m/z) |
||
|---|---|---|---|---|
| MS [M-H]− | MS2 | MS3 | ||
| 18-Dehydroxy-17-demethoxyreblastatin (1) | 664 (6)b | 663, 709c | 620 | 588, 458 |
| 18-Dehydroxy-17-O-demethylreblastatin (2) | 680 (7)b | 679, 725c | 636, 474 | 442 |
| 680 (8) | 679 | 636 | 474 | |
| 842 (9) | 841, 887c | 636 | 474 | |
| 17-Demethoxy-reblastatin (4) | 680 (10) | 679, 725c | 636 | 604, 474 |
| 18-Dehydroxy-17-O-demethyl-4,5-dehydroreblastatin (3) | 678 | NDd | ||
| 17-Demethoxy-15-hydroxylreblastatin (5) | 696 | ND | ||
| Geldanamycin | 722 | ND | ||
MS/MS, tandem MS; MS2 and MS3 indicate two and three stages of separation, respectively.
NMR-assigned compounds.
[M+HCOOH]−.
ND, not detected (i.e., the predicted mass ion peaks were not detected).
In order to obtain NMR-accessible amounts from the enzymatic reactions, 50 mg of 18-dehydroxy-17-demethoxyreblastatin or 18-dehydroxy-17-O-demethylreblastatin was added to a 100-ml enzymatic reaction mixture. Overall, the 1H and 13C NMR spectra of compounds 6 and 7 were similar to those of the substrates that were added into the enzymatic reactions. However, the presence of signals that correspond to a glucose moiety and the presence of the anomeric configuration indicated that compounds 6 and 7 possess glucose. A complete analysis of the one-dimensional (1D) and 2D NMR spectroscopy data suggested that the structures of compounds 6 and 7 were 18-dehydroxy-17-demethoxyreblastatin 11-O-β-d-glucoside and 18-dehydroxy-17-demethylreblastatin 17-O-β-d-glucoside, respectively (see Table S1 and Fig. S5 in the supplemental material). The quantities of other purified glucoside compounds were not sufficient for structure elucidation.
Biological activities of glucoside compounds 6 and 7.
The antiproliferation activities of compounds 6 and 7 were tested via the SRB assay in Her2 highly expressed breast cancer cells (SK-Br3). As can be seen in Table 2, compound 7 exhibited weak antiproliferation activity against SK-Br3 cells, with a 50% inhibitory concentration (IC50) of 19.0 μM compared with that of the original substrates (9.4 μM). In contrast, GM showed submillimolar antiproliferation activity against several human cancer cells, including SK-Br3 cells (34). Compound 6 did not demonstrate antiproliferation activity within the experimental concentration ranges used in this study. This lack of activity might have been caused by the bulkiness of the glycosylation at the C-11 position, which is consistent with our previous results (18). Other structural modifications at the C-11 position, with an ester and N-methylcarbamate, failed to improve antiproliferative activities (18). In addition to the antiproliferation activity studies, we further examined whether Her2, Akt, and c-Raf, which are well-documented clients of Hsp90, were degraded by compound 7. As can be seen in Fig. 4, the Western blot analyses indicated that Her2, Akt, and c-Raf were degraded by compound 7 in a concentration-dependent manner. As expected from Hsp90 inhibitors, treatment by compounds 2 and 7 resulted in downregulation of key oncoproteins, including Her2, Akt, and c-Raf, in SK-Br3 cells.
Table 2.
Biological activity of the nonbenzoquinone GM and glucoside analogs 6 and 7
| Compound | Cell viability (SK-Br3 assay; IC50 [μM])a |
|---|---|
| 18-Dehydroxy-17-demethoxyreblastatin (1) | 13.26 ± 2.86 |
| 6 | >100 |
| 18-Dehydroxy-17-O-demethylreblastatin (2) | 9.43 ± 1.38 |
| 7 | 19.0 ± 3.12 |
Data represent means ± standard deviations of the results of three separate experiments. SK-Br3, human breast cancer cell line.
Fig 4.
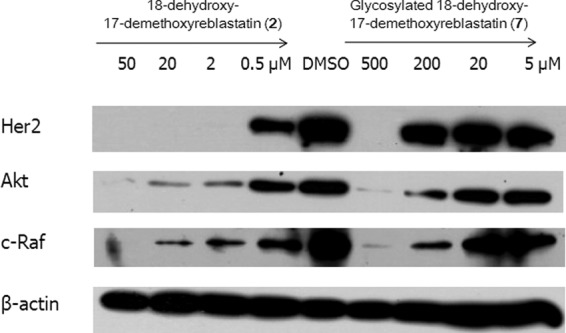
Western blot analysis of the indicated Hsp90 client protein expression. Each compound, at various concentrations, was evaluated for its ability to regulate several client proteins as described in Materials and Methods. (2), compound 2; (7), compound 7.
Water solubility determination.
For the water solubility determination, the glycosyltransferase reaction mixture was extracted with solvent ethyl acetate and then centrifuged to divide it into two layers. Each water and solvent layer was directly analyzed using HPLC, as described previously. The observed concentration of the conversion of the glucoside (compound 10) from its nonbenzoquinone GM (17-demethoxy-reblastatin) form as determined using the HPLC data for the reaction was approximately 40% compared with the original starting concentration of the nonbenzoquinone GM (Fig. 5). However, the glucoside compound was mostly located on the water layer and was not extracted to the ethyl acetate layer, while the substrate compound, nonbenzoquinone GM, preferred to remain in the solvent fraction. The other glucoside compounds were also similarly located on the water layer in glycoslyation reactions performed with other nonbenzoquinone GM analogs (data not showed). This result indicates that the glycosylated compound has dramatically improved water solubility compared to that of the original aglycon, nonbenzoquinone GM.
Fig 5.

Comparison of the HPLC data from analysis of the glycosylation reaction mixture. (A) Total enzymatic reaction mixture. The glycosylation conversion rate was approximately 40%. (B and C) The (B) solvent layer and (C) water layer after the ethylacetate extracted enzyme reaction mixture. The peaks at 7.1 min (S) represent nonbenzoquinone GM (17-demethoxy-reblastatin), and those at 5.5 min (P) represent glucoside product. The peaks at 1.77 min (A) and 1.80 min (C) represent water-soluble enzyme reaction material, i.e., UDP-glucose, enzyme, etc. mAU, milli-absorbance units.
DISCUSSION
Glycosylation is one of the most common and important reactions in biological systems. A number of bioactive compounds contain sugars attached to their aglycons. Glycosylated natural products include important antibiotics such as avermectin, erythromycin, vancomycin, and doxorubicin (12, 23). These sugars are often essential for the pharmacological properties, especially the water solubility and/or the biological activity, of the compounds. Several complementary strategies, including semisynthesis, pathway engineering, and in vitro enzymatic glycosylation techniques, have emerged from recent studies as effective means of altering the natural product sugar structures (9, 17, 31, 32).
The clinical evaluation of GM and some GM derivatives has not been pursued due to their severe toxicity and poor water solubility (22). For these reasons, we have developed new nonbenzoquinone GM analogs with reduced hepatotoxicity profiles (unpublished data). However, these nonbenzoquinone GM analogs still have formulation problems that predominantly result from their low solubility. Therefore, glycosylated GM could have improved water solubility and an extended spectrum for formulation. However, naturally occurring glycosylated GM analogs have not yet been reported; also, no sugar biosynthetic genes have been found in the GM biosynthetic gene cluster (14, 25, 26, 35). We chose in vitro enzymatic glycosylation techniques performed with Bacillus glycosyltransferase, which demonstrated broad substrate specificity with compounds containing aromatic rings (1).
Five aglycon substrates containing nonbenzoquinone aromatic rings were chosen in order to validate the proposed in vitro glycosylation reaction. Putative glucoside products were determined by the presence of a product peak(s) and were also verified through the LC/MS analyses (Table 1). These analyses revealed that three products (18-dehydroxy-17-demethoxyreblastatin, 18-dehydroxy-17-O-demethylreblastatin, and 17-demethoxy-reblastatin) were converted to the expected glucoside(s). Curiously, 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin and 17-demethoxy-15-hydroxylreblastatin were not converted (as evidenced by the absence of product peaks and no significant decrease in the aglycon peak, as determined using the HPLC), even though they did not have a significantly different structure from that of the glycosylated substrates. The only differences were that 18-dehydroxy-17-O-demethyl-4,5-dehydroreblastatin had a double bond in the C4-C5 position, compared to 18-dehydroxy-17-O-demethylreblastatin (16), and that 17-demethoxy-15-hydroxylreblastatin had a hydroxyl group in the C-15 position, compared to 17-demethoxy-reblastatin (15). Saturation of 4,5-olefin contributes to a higher population of the folded bound conformation in solution (21); thus, the high structural rigidity could be involved in the loss of substrate availability to the glycosyltransferase. Also, the analogs incorporated subtle changes within the GM macrocycle, which showed quite different effects on the binding to Hsp90 (21). The binding rate, which is sensitive to the presence of the Hsp90 protein, could analogize the uncertain substrate specificity of glycosyltrasferase. Interestingly, 18-dehydroxy-17-O-demethylreblastatin was converted to three different product peaks, as determined through the HPLC analysis. The full scan and MSn mass spectral data for these compounds showed a loss of 162 Da from the molecular parent ions of the glucose molecules. This loss corresponds to the loss of one or two glucoses; thus, it was expected that the product of the in vitro reaction would be C-11-glucoside, C-17-glucoside, or C-11, 17-bis-glucoside. Thus, 18-dehydroxy-17-demethoxyreblastatin, which does not have a hydroxyl group on the aromatic ring, could be converted to C-11-glucoside. The structures of purified glucoside compounds 6 and 7 were elucidated using spectroscopic methods. The anomeric configuration of compounds 6 and 7 was determined to be β on the basis of the large coupling constant values of the anomeric protons at δH 4.19 (J = 8.0 Hz) for compound 6 and δH 4.64 (J = 6.8 Hz) for compound 7 (see Table S1 in the supplemental material). The antiproliferative activities of glucoside compounds 6 and 7 (IC50s of more than 100 μM and 19 μM in SK-Br3 breast cancer cells, respectively) were found to be weaker than that of the original nonbenzoquinone GM (13.0 and 9.4 μM, respectively). Thus, the unfavorable antiproliferative activity of glucoside compound 6 might have been a consequence of the bulkiness of glycosylation at the C-11 position, which is consistent with our previous results (18). Also, glucoside compound 7, which has a glucose group at the C-17 hydroxyl group in the benzene ring, showed less activity than aglycone. The functionality of the benzene ring makes important hydrogen bonding networks at the active site of the Hsp90 protein (21, 24), suggesting that binding to Hsp90 is affected by the presence of the glucose moiety on the benzene ring; thus, inhibition of the molecular chaperone can be reduced. In order to further confirm that compound 7 interacts with Hsp90, we examined whether this compound could reduce the stability of the client oncogenic proteins. It is noteworthy that the compound demonstrated this activity even though its activity was weaker than that of the nonbenzoquinone GM control. Despite the fact that these glucoside compounds demonstrate weaker biological activity, the improvement in water solubility, due to the general properties of the carbohydrates, could be advantageous, with reduced hepatotoxicity from the nonbenzoquinone moiety (4). Therefore, a glucoside nonbenzoquinone-GM with improved efficacy can be exploited in prodrug approaches for formulation of clinical applications.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a Global R&D Center program and Global Frontier Project (ISBC 2011-0031947) funded by the NRF and by the Next-Generation BioGreen 21 Program (SSAC, PJ007999) funded by the RDA, Republic of Korea.
We thank Jong Suk Lee of the Gyeonggi Bio Center for providing the LC-MS measurements.
Footnotes
Published ahead of print 24 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Ahn BC, et al. 2009. Formation of flavone di-O-glucosides using a glycosyltransferase from Bacillus cereus. J. Microbiol. Biotech. 19:387–390 [DOI] [PubMed] [Google Scholar]
- 2. Biamonte MA, et al. 2010. Heat shock protein 90: inhibitors in clinical trials. J. Med. Chem. 53:3–17 [DOI] [PubMed] [Google Scholar]
- 3. Cheng H, et al. 2005. Synthesis and enzyme-specific activation of carbohydrate-geldanamycin conjugates with potent anticancer activity. J. Med. Chem. 48:645–652 [DOI] [PubMed] [Google Scholar]
- 4. Cysyk RL, et al. 2006. Reaction of geldanamycin and C17-substituted analogues with glutathione: product identifications and pharmacological implications. Chem. Res. Toxicol. 19:376–381 [DOI] [PubMed] [Google Scholar]
- 5. DeBoer C, Meulman PA, Wnuk RJ, Peterson DH. 1970. Geldanamycin, a new antibiotic. J. Antibiot. (Tokyo) 23:442–447 [DOI] [PubMed] [Google Scholar]
- 6. Erb A, Weiss H, Harle J, Bechthold A. 2009. A bacterial glycosyltransferase gene toolbox: generation and applications. Phytochemistry 70:1812–1821 [DOI] [PubMed] [Google Scholar]
- 7. Fang L, et al. 2006. Enzyme specific activation of benzoquinone ansamycin prodrugs using HuCC49ΔCH2-β-galactosidase conjugates. J. Med. Chem. 49:6290–6297 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fu X, et al. 2003. Antibiotic optimization via in vitro glycorandomization. Nat. Biotechnol. 21:1467–1469 [DOI] [PubMed] [Google Scholar]
- 9. Gantt RW, Peltier-Pain P, Thorson JS. 2011. Enzymatic methods for glyco(diversification/randomization) of drugs and small molecules. Nat. Prod. Rep. 28:1811–1853 [DOI] [PubMed] [Google Scholar]
- 10. Goetz MP, et al. 2005. Phase I trial of 17-allylamino-17-demethoxygeldanamycin in patients with advanced cancer. J. Clin. Oncol. 23:1078–1087 [DOI] [PubMed] [Google Scholar]
- 11. Griffith BR, Langenhan JM, Thorson JS. 2005. ‘Sweetening’ natural products via glycorandomization. Curr. Opin. Biotechnol. 16:622–630 [DOI] [PubMed] [Google Scholar]
- 12. Harle J, Bechthold A. 2009. Chapter 12. The power of glycosyltransferases to generate bioactive natural compounds. Methods Enzymol. 458:309–333 [DOI] [PubMed] [Google Scholar]
- 13. Heath EI, et al. 2008. A phase II trial of 17-allylamino-17-demethoxygeldanamycin in patients with hormone-refractory metastatic prostate cancer. Clin. Cancer Res. 14:7940–7946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Hong YS, et al. 2004. Inactivation of the carbamoyltransferase gene refines post-polyketide synthase modification steps in the biosynthesis of the antitumor agent geldanamycin. J. Am. Chem. Soc. 126:11142–11143 [DOI] [PubMed] [Google Scholar]
- 15. Kim W, et al. 2009. Rational biosynthetic engineering for optimization of geldanamycin analogues. ChemBioChem 10:1243–1251 [DOI] [PubMed] [Google Scholar]
- 16. Kim W, et al. 2007. Mutasynthesis of geldanamycin by the disruption of a gene producing starter unit: generation of structural diversity at the benzoquinone ring. ChemBioChem 8:1491–1494 [DOI] [PubMed] [Google Scholar]
- 17. Krauth C, Fedoryshyn M, Schleberger C, Luzhetskyy A, Bechthold A. 2009. Engineering a function into a glycosyltransferase. Chem. Biol. 16:28–35 [DOI] [PubMed] [Google Scholar]
- 18. Lee K, et al. 2008. Synthesis and anticancer activity of geldanamycin derivatives derived from biosynthetically generated metabolites. Org. Biomol. Chem. 6:340–348 [DOI] [PubMed] [Google Scholar]
- 19. Luzhetskyy A, Mendez C, Salas JA, Bechthold A. 2008. Glycosyltransferases, important tools for drug design. Curr. Top. Med. Chem. 8:680–709 [DOI] [PubMed] [Google Scholar]
- 20. Mandai T, et al. 2001. Synthesis and biological evaluation of water soluble taxoids bearing sugar moieties. Heterocycles 54:561–566 [Google Scholar]
- 21. Martin CJ, et al. 2008. Molecular characterization of macbecin as an Hsp90 inhibitor. J. Med. Chem. 51:2853–2857 [DOI] [PubMed] [Google Scholar]
- 22. Messaoudi S, Peyrat JF, Brion JD, Alami M. 2011. Heat-shock protein 90 inhibitors as antitumor agents: a survey of the literature from 2005 to 2010. Expert Opin. Ther. Pat. 21:1501–1542 [DOI] [PubMed] [Google Scholar]
- 23. Olano C, Mendez C, Salas JA. 2010. Post-PKS tailoring steps in natural product-producing actinomycetes from the perspective of combinatorial biosynthesis. Nat. Prod. Rep. 27:571–616 [DOI] [PubMed] [Google Scholar]
- 24. Prodromou C, Roe SM, Piper PW, Pearl LH. 1997. A molecular clamp in the crystal structure of the N-terminal domain of the yeast Hsp90 chaperone. Nat. Struct. Biol. 4:477–482 [DOI] [PubMed] [Google Scholar]
- 25. Rascher A, Hu Z, Buchanan GO, Reid R, Hutchinson CR. 2005. Insights into the biosynthesis of the benzoquinone ansamycins geldanamycin and herbimycin, obtained by gene sequencing and disruption. Appl. Environ. Microbiol. 71:4862–4871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Rascher A, et al. 2003. Cloning and characterization of a gene cluster for geldanamycin production in Streptomyces hygroscopicus NRRL 3602. FEMS Microbiol. Lett. 218:223–230 [DOI] [PubMed] [Google Scholar]
- 27. Schulte TW, Neckers LM. 1998. The benzoquinone ansamycin 17-allylamino-17-demethoxygeldanamycin binds to HSP90 and shares important biologic activities with geldanamycin. Cancer Chemother. Pharmacol. 42:273–279 [DOI] [PubMed] [Google Scholar]
- 28. Supko JG, Hickman RL, Grever MR, Malspeis L. 1995. Preclinical pharmacologic evaluation of geldanamycin as an antitumor agent. Cancer Chemother. Pharmacol. 36:305–315 [DOI] [PubMed] [Google Scholar]
- 29. Weymouth-Wilson AC. 1997. The role of carbohydrates in biologically active natural products. Nat. Prod. Rep. 14:99–110 [DOI] [PubMed] [Google Scholar]
- 30. Whitesell L, Mimnaugh EG, De Costa B, Myers CE, Neckers LM. 1994. Inhibition of heat shock protein HSP90-pp60v-src heteroprotein complex formation by benzoquinone ansamycins: essential role for stress proteins in oncogenic transformation. Proc. Natl. Acad. Sci. U. S. A. 91:8324–8328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Williams GJ, Yang J, Zhang C, Thorson JS. 2011. Recombinant E. coli prototype strains for in vivo glycorandomization. ACS Chem. Biol. 6:95–100 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Williams GJ, Zhang C, Thorson JS. 2007. Expanding the promiscuity of a natural-product glycosyltransferase by directed evolution. Nat. Chem. Biol. 3:657–662 [DOI] [PubMed] [Google Scholar]
- 33. Wu CZ, et al. 2010. 6-Alkylsalicylic acid analogues inhibit in vitro ATPase activity of heat shock protein 90. Arch. Pharm. Res. 33:1997–2001 [DOI] [PubMed] [Google Scholar]
- 34. Wu CZ, et al. 2011. New non-quinone geldanamycin analogs from genetically engineered Streptomyces hygroscopicus. J. Antibiot. (Tokyo) 64:461–463 [DOI] [PubMed] [Google Scholar]
- 35. Yin M, et al. 2011. The missing C-17 O-methyltransferase in geldanamycin biosynthesis. Org. Lett. 13:3726–3729 [DOI] [PubMed] [Google Scholar]
- 36. Zhang C, et al. 2006. Exploiting the reversibility of natural product glycosyltransferase-catalyzed reactions. Science 313:1291–1294 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.