

Abstract
Over eight different families of natural products, consisting of nearly seventy secondary metabolites, which contain the bicyclo[2.2.2]diazaoctane ring system, have been isolated from various Aspergillus, Penicillium, and Malbranchea species. Since 1968, these secondary metabolites have been the focus of numerous biogenetic, synthetic, taxonomic, and biological studies, and, as such, have made a lasting impact across multiple scientific disciplines. This review covers the isolation, biosynthesis, and biological activity of these unique secondary metabolites containing the bridging bicyclo[2.2.2]diazaoctane ring system. Furthermore, the diverse fungal origin of these natural products is closely examined and, in many cases, updated to reflect the currently accepted fungal taxonomy.
INTRODUCTION
Natural products derived from fungal sources have been an area of intense research for decades due to the vast structural diversity and biological activity of these secondary metabolites. Numerous fungal-derived natural products serve as clinical therapeutics for both human and animal diseases, and thus the search for new classes of bioactive secondary metabolites from terrestrial and marine fungal sources has garnered additional interest.1 Natural products containing a unique bicyclo[2.2.2]diazaoctane ring system are part of a diverse class of biologically active prenylated indole alkaloids isolated mainly from various Penicillium and Aspergillus species of fungi.2 The brevianamides,3 paraherquamides,4 stephacidins,5 notoamides,6 asperparalines,7 marcfortines,8 chrysogenamides,9 and malbrancheamides10 exhibit a range of interesting structural features, such as a bicyclo[2.2.2]diazaoctane nucleus and a complex, oxidized amino acid skeleton (Figure 1). Structurally, these natural products are comprised of tryptophan, a cyclic amino acid (proline, pipecolic acid or derivatives of proline or pipecolic acid), and one or two isoprene units. In addition to their structural diversity, a variety of bioactivities are also present for these compounds, including but not limited to, insecticidal, cytotoxic, anthelmintic, and antibacterial properties.
Figure 1.

Structures of several secondary metabolites that contain the core bicyclo[2.2.2]diazaoctane ring system.
Initial characterization of these types of natural products led to the conclusion that two isoprene units are present in these alkaloid families, in which one of the two isoprene units is involved in the formation of the bicyclo[2.2.2]diazaoctane ring system.11 In order to determine the biosynthetic pathway from which the isoprene units originate, [13C2]-acetate has been utilized in precursor incorporation studies with fungal cultures known to produce metabolites containing the bridging bicycle. Specific incorporation of intact C2 units, combined with the 13C coupling patterns observed by 13CNMR spectroscopy demonstrated that the isoprene units found in these metabolites arise from dimethylallyl pyrophosphate (DMAPP), a product of the mevalonic acid pathway (Scheme 1).11,12
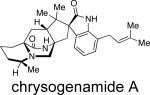
Of the natural products containing the unusual bridging bicycle, two distinct stereochemical configurations have been noted with respect to the relative configuration at the C-19 position (brevianamide numbering). The rare anti-configuration (1) and the more common syn-configuration (2) are determined by the position of the C-H proton at the bridgehead of the bicyclic core in relation to the bridging secondary lactam (Figure 2).13 The brevianamides are currently the only family of secondary metabolites to display the anti-configuration about the bridging bicycle; however, it was recently discovered that two novel metabolites, chrysogenamide A and versicolamide B, also display the rare anti-stereochemistry.6f,9 Contrary to this, all other known metabolites containing the core bridging bicycle display the syn-configuration.
Figure 2.

Anti- and syn- relationship of the bicyclo[2.2.2]diazaoctane core.
Since the first isolation of these bicyclo[2.2.2]diazaoctane-containing metabolites, substantial effort has focused on both the synthetic and the biosynthetic aspects of this unique ring system.14,15 However, although relatively little is known about the microbial assembly and modification of metabolites containing this core bridging bicycle, new genomic and biochemical approaches are beginning to clarify important aspects of these pathways. Originally proposed by Porter and Sammes in 1970, the biosynthetic formation of the bicyclo[2.2.2]diazaoctane ring system is believed to arise via an intramolecular hetero Diels-Alder reaction (IMDA) of a 5-hydroxypyrazine-2(1H)-one (Scheme 2).16 Extensive synthetic research carried out by Williams and co-workers has aimed at supporting a biosynthetic Diels-Alder reaction via the biomimetic syntheses of several advanced metabolites.15-17 Concurrently, substantial effort has been put forth to isolate and characterize the putative Diels-Alderase enzyme responsible for this biosynthetic transformation, but, to date, no such enzyme has been isolated.
Scheme 2.

Proposed biosynthetic Diels-Alder reaction.16
The Penicillium and Aspergillus fungal producers of secondary metabolites containing the bicyclo[2.2.2]diazaoctane ring have been defined taxonomically based on early phenotypic methods of Raper and Thom18 for Penicillium and Raper and Fennell19 for Aspergillus. However, several of these species have since been reclassified based on modern molecular methods. Extensive taxonomical research carried out by Frisvad and co-workers have provided new phylogenetic information for each fungal culture known to produce the bicyclo[2.2.2]diazaoctane ring system described in this review, and both the original and updated fungal strain nomenclature will be mentioned when applicable. Furthermore, numerous strains of these fungal cultures have been determined to produce the same sets of secondary metabolites (Table 1). Taxonomic revisions of species and re-identifications of the strains are based on a polyphasic approach, including morphological, physiological, chemical and DNA sequence data. Mostly sequences from β-tubulin and calmodulin genes have been valuable in assigning strains to new or known species.
Table 1.
Culture Collection Strains Producing Secondary Metabolites with a Bicyclo[2.2.2]diazaoctane Unit and Related Compounds (new data: Frisvad, unpublished).
| secondary metabolite | fungal species | producers: isolates available in international culture collections |
|---|---|---|
| malbrancheamides | Malbranchea aurantiacaa M. graminicola | (RRC1813)b (UCSC 086937A) |
| chrysogenamide | P. chrysogenum | (No. 005)b |
| brevianamides | P. viridicatum | IMI 039755ii,c IMI 351305, IMI 351306, NRRL 869, NRRL 870, NRRL 959, NRRL 962, NRRL 3586, NRRL 3600, NRRL A-15402, NRRL A-18563, NRRL A-26909 |
| P. brevicompactum | CBS 257.29,c CBS 256.31, CBS 316.59, CBS 317.59, CBS 480.84, CBS 100068, CBS 110069, NRRL 867, NRRL 32579 | |
| A. carlsbadensis | CBS 123894c | |
| deoxybrevianamides | A. pseudoustus | NRRL 5856,c MRC 096 |
| P. italicum | ATCC 48952, CBS 339.48, CBS278.58, CBS 349.75, CBS 489.84, | |
| P. ulaiense | CBS 136.41, CBS 210.92, CBS 722.92, CBS 262.94 | |
| paraherquamides | P. brasilianum | CBS 253.55,c CBS 338.59 |
| P. cluniae | CBS 276.89c | |
| P. glaucoroseum | NRRL 908c | |
| P. cf. canescens | ATCC 20841, IMI 332995 | |
| paraherquamide E | A. aculeatinus | CBS 204480 |
| A. fijiensis | CBS 313.89,c IMI 337664a | |
| asperparalines | A. aculeatinus | ATCC 204480 = JV-23, CBS 101.43, CBS 186.67, CBS 610.78, CBS 621.78, CBS 116.80, CBS 121060, CBS 121875 |
| A. fijiensis | CBS 313.89,c CBS 119.49, IMI 337664a | |
| notoamides (including (–)- versicolamide) | A. bridgeri | CBS 350.81c |
| A. melleus | CBS 546.65,c NRRL 394 | |
| A. ochraceus | CBS 108.08,c CBS 589.68, CBS 624.78, NRRL 6319, (CL41582) | |
| A. ostianus | CBS 103.07,c CBS 101.23, CBS 311.80, IMI 177964b, IMI 348937, NRRL 422, NRRL 5225, NRRL A-15156, (01F313) | |
| A. persii | CBS 112795c | |
| A. petrakii | CBS 105.57c | |
| A. sclerotiorum | CBS 549.65,c ATCC 18413, NRRL 5167, (CNC358) | |
| A. westerdijkiae | CBS 112803c = NRRL 3174, CBS 112791, CBS 112804, NRRL 5175, NRRL 5221, NRRL 5228, | |
| notoamides (including (+)- versicolamide) | A. protuberus | CBS 602.74,c CBS 601.74 |
| A. versicolor | CBS 118.50, CBS 106.57, CBS 599.65, IMI 096225, NRRL 35600 | |
| A. elongatus | CBS 387.75c | |
| austamide | A. pseudoustus | NRRL 5856,c MRC 096 |
| citrinadins | P. citrinum | CBS 139.45,c (N-059) |
| PF1270 citrinadins | P. westlingii | (PF1270) |
| talathermophilins | Talaromyces thermophilus | CBS 116.72, (YM 1-3) |
Possible misidentification.
Strain unavailable.
Type strain.
MALBRANCHEAMIDES
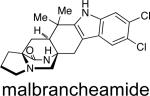
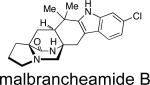
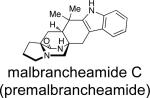
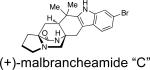
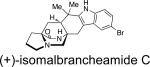
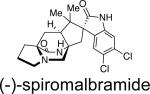
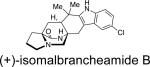
First identified in 2006 by Mata and co-workers, malbrancheamide and malbrancheamide B were isolated from Malbranchea aurantiaca RRC1813, a fungus obtained from bat guano found in a cave in Mexico (Table 2).10 Shortly following the isolation of malbrancheamide and malbrancheamide B, Mata and co-workers reported the presence of the nonhalogenated precursor malbrancheamide C, which was later named premalbrancheamide.10c This unique family of halogenated secondary metabolites has expanded recently with the isolation of two new chlorinated metabolites, (–)-spiromalbramide and (+)-isomalbrancheamide B, from Malbranchea graminicola, as reported by Crews and co-workers.10d Furthermore, two brominated analogues, (+)-malbrancheamide C (hereafter referred to as (+)-malbrancheamide “C”) and (+)-isomalbrancheamide C, were obtained from M. graminicola when the growth medium was enriched with bromine salts.
Table 2.
Fungal Origin of the Malbrancheamides.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
|
M. aurantiaca RRC1813 | calmodulin inhibitor | 10a, 20 |
|
M. aurantiaca RRC1813 | weak calmodulin inhibitor | 10b,c, 20 |
|
M. aurantiaca RRC1813 | 10c,f | |
|
M. graminicola UCSC 086937A | 10d | |
|
M. graminicola UCSC 086937A | 10d | |
|
M. graminicola UCSC 086937A | 10d | |
|
M. graminicola UCSC 086937A | 10d |
This family of natural products represent the only bicyclo[2.2.2]diazaoctane containing metabolites to be reported from a fungal source outside the genera Penicillium and Aspergillus. Taxonomically, the strain of Malbranchea aurantiaca (RRC1813) may either be that species or represent the species Malbranchea graminicola, since three strains of M. aurantiaca from the CBS collection (CBS 127.77, Utah, ex culture contaminant, type strain; CBS 759.74 and CBS 309.76, both ex soil, India) did not produce the malbrancheamides or penicillic acid.10e However, the isolation of (+)-malbrancheamide, (+)-malbrancheamide B, (+)-premalbrancheamide, and (–)-spiromalbramide from M. graminicola indicates that these compounds are common in the genus Malbranchea.
Structurally, the malbrancheamides are the first members of this class of metabolites to display halogenation of the indole moiety. While malbrancheamide was determined to contain chlorination at the C-5 and C-6 position of the indole, the exact structure of malbrancheamide B was unclear. By synthesizing both the C-5 chlorinated and C-6 chlorinated potential compounds, it was established that malbrancheamide B contains C-6 monochlorination.21 In conjunction with the unique indole chlorination, the malbrancheamides are also characterized by the syn-configuration about the bridging bicycle, as well as the presence of a monoketopiperazine moiety in the core bicycle. Biologically, the malbrancheamides were determined to display calmodulin inhibitory activity of CaM-dependent phosphodiesterase (PDE1).10c,f
Following the isolation of the malbrancheamides, interest was sparked in elucidating the biosynthetic timing of the indole chlorination. As shown in Scheme 3, the initially proposed biogenesis of the malbrancheamides begins with the condensation of tryptophan, proline, and dimethylallyl pyrophosphate (DMAPP), to afford the known natural product, deoxybrevianamide E. From here, two distinct biosynthetic pathways are possible to form the known metabolite premalbrancheamide. One potential pathway involves oxidation of deoxybrevianamide E to yield the Diels-Alder precursor, 5-hydroxypyrazin-2(1H)-one (4a), which would undergo [4+2] cycloaddition to afford keto-premalbrancheamide. Subsequent carbonyl reduction of the tertiary amide would provide premalbrancheamide. On the other hand, carbonyl reduction could occur prior to the IMDA reaction, in which 4b would serve as the key achiral intermediate and direct intermediate to premalbrancheamide. Two individual and concurrent halogenases would then lead from premalbrancheamide to malbrancheamide B and then to malbrancheamide.
Scheme 3.

Proposed biosynthesis of the malbrancheamides.
To test whether premalbrancheamide and/or keto-premalbrancheamide serve as biosynthetic precursors to the halogenated metabolites, doubly 13C-labeled pre-malbrancheamide and keto-premalbrancheamide were synthesized by Williams et al. and provided to M. aurantiaca in separate precursor incorporation studies (Scheme 4).10f Results from these feeding studies showed significant incorporation of [13C]2-pre-malbrancheamide into malbrancheamide B (5.5% incorporation), but surprisingly, [13C2]-keto-premalbrancheamide did not incorporate. The latter result suggests that the timing for the reduction of the tryptophan-derived amide carbonyl residue must occur prior to the [4+2] cycloaddition, thus the biosynthetic formation of pre-malbrancheamide most likely arises from the proposed intermediate 4b. Future genetic and biochemical analysis of the malbrancheamide biosynthetic pathway will undoubtedly resolve the mechanism of monoketopiperizine assembly by M. aurantiaca.
Scheme 4.

Precursor incorporation studies with M. aurantiaca.
CHRYSOGENAMIDE
In 2008, Zhu and co-workers were interested in identifying new neuroprotective compounds from endophytic fungi on the parasitic plant Cistanche deserticola collected in northwest mainland China (Table 3).9 From the roots of C. deserticola, three active strains were screened for neuroprotective effects on SH-SY5Y cells, and the fungal culture Penicillium chrysogenum No. 005 demonstrated significant activity. From P. chrysogenum, the novel metabolite, chrysogenamide A, was isolated. From a taxonomic perspective, even though the original culture was not available for examination, other secondary metabolites characteristic of P. chrysogenum were also isolated,9 supporting the identity.22-24
Table 3.
Fungal Origin and Bioactivity of Chrysogenamide A.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
|
P. chrysogenum No. 005 | neuroprotective effect on SH-SY5Y cells | 9 |
From a structural point of view, this novel metabolite is closely related to the marcfortines (discussed below), in that chrysogenamide A was found to contain a bicyclo[2.2.2]diazaoctane ring system, a spiro-oxindole moiety, and a pipecolic acid unit with C-17 methylation. Additionally, chrysogenamide A displays the rare anti- configuration about the bridging bicycle and an isoprene unit at the C-7 indole position, as opposed to the more commonly observed dioxepin and pyran rings. Chrysogenamide A is the first and only metabolite of these families of natural products to display neurocyte protection against oxidative stress-induced cell death.9
BREVIANAMIDES
Between 1969 and 1972, Birch and co-workers isolated six novel secondary metabolites, brevianamides A-F (Figure 3), from Penicillium brevicompactum.3 Of these six metabolites, four compounds were found to contain the unique bicyclo[2.2.2]diazaoctane core. Additional research revealed that brevianamides A and B are diastereoisomers, while brevianamides C and D are artifacts of isolation due to light irradiation.3c Structurally, the brevianamides were found to display the anti- configuration about the bicyclic core, as well as the spiro-indoxyl moiety, and an unsubstituted indole ring. Furthermore, the brevianamides are a class of natural products that display modest biological activities, in which brevianamides A and D have been shown to possess antifeedant and insecticidal effects.25
Figure 3.

Structures of the brevianamides and related metabolites.
As shown in Table 4, the brevianamides have been isolated predominately from two Penicillium species. The two main producers of brevianamide A are P. brevicompactum3,22-23 and P. viridicatum,3e,22-23 of which both are consistent producers of brevianamide A.26 The isolate of P. ochraceum reported to produce brevianamide A3d was determined to be a brown-spored mutant of P. viridicatum.26 Deoxybrevianamide E is produced by Penicillium italicum,27d-eP. ulaiense,22 and a fungus identified as Aspergillus ustus NRRL 5856.27a-c The latter isolate has been the basis for the description of a new species, Aspergillus pseudoustus;28 however, A. pseudoustus is the only species known to produce austamide (discussed below). The first biosynthetic precursor is the common dioxopiperazine brevianamide F (cyclo-l-Trp-l-Pro). This alkaloid has been shown to serve as the precursor for several related secondary metabolites, those lacking the bridging bicycle, such as the fumitremorgins and spirotryprostatins in Aspergillus fumigatus.29
Table 4.
Fungal Origin of the Brevianamides.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
| brevianamide A | P. brevicompactum, P. viridicatum, P. ochraceuma | antifeedant insecticidal |
3a-c, 3e, 3d, 25 |
| brevianamide B | P. brevicompactum | 3a-c | |
| brevianamide C | P. brevicompactum | 3a-c | |
| brevianamide D | P. brevicompactum | insecticidal | 3a-c, 25 |
| brevianamide Fb brevianamide E | A. versicolor, P. brevicompactum P. brevicompactum | antibacterial | 6f, 3c, 30 3a-c |
| deoxybrevianamide E | Aspergillus sp. MF297-2,cA. ustus,d P. ulaiense, P. italicum | 6a, 22, 27 |
Misidentified fungal culture; correct identification = P. viridicatum.
Widely distributed biosynthetic precursor, thus only relevant fungal origins are listed.
Undergoing re-identification (possibly A. sclerotiorum).
Reclassified as A. pseudoustus.
Following the isolation of these unique natural products in 1969, which were the first metabolites to be isolated to display the unusual bicyclo[2.2.2]diazaoctane ring system, Birch and co-workers carried out several radiolabeled precursor incorporation studies in order to elucidate early stages of the biosynthetic pathway of the brevianamides.3a-c,31 Birch demonstrated that the brevianamides are derived biogenetically from tryptophan, proline, and mevalonic acid, and later showed that radiolabled cyclol-Trp-l-Pro (brevianamide F) is also a precursor to brevianamide A in cultures of P. brevicompactum. Based on these results, Birch proposed that brevianamide F arises from the condensation of tryptophan and proline, which is followed by C-2 reverse prenylation to afford deoxybrevianamide E. Oxidation and cyclization of deoxybrevianamide E would give rise to brevianamides A and B (Scheme 5).
Scheme 5.

Birch's proposed biosynthesis of the brevianamides.
Williams later demonstrated through tracer incorporation studies with P. brevicompactum that radiolabeled deoxybrevianamide E incorporates into brevianamides A, B, and E in significant radiochemical yields.32 At the same time, Williams showed that neither tritium-labeled brevianamide E nor the proposed racemic, 13C-labeled cycloadduct 5 incorporates into brevianamides A or B when provided to P. brevicompactum in isotopically labled precursor incorporation studies (Scheme 6). These results led to the currently proposed biosynthetic pathway of brevianamides A and B, as shown in Scheme 6. Since cycloadduct 5 does not incorporate into the brevianamides, it was suggested that stereoselective indole oxidation must occur prior to the formation of the bicyclo[2.2.2]diazaoctane ring system. Thus, it was proposed that deoxybrevianamide E is first converted to 3R-hydroxyindolenine (6) via an R-selective indole oxidase, which could then proceed along one of three pathways. Following an irreversible nucleophilic addition, 6 could be readily converted to brevianamide E; or 6 could undergo a pinacol-type rearrangement to indoxyl 7. A second oxidation - enolization sequence would then yield the chiral and optically pure azadiene (3), followed by an IMDA reaction to directly give brevianamides A and B. It should be noted that, efforts to substantiate this alternative biosynthetic pathway remain elusive due to complications regarding the synthesis of 7. Despite extensive synthetic efforts, 7 appears to be unstable and spontaneously suffers a retro-Michael reaction to give the dehydroalanine piperazinedione and the reverse-prenylated indoxyl.
Scheme 6.

Alternative biosynthetic pathway for the brevianamides.
Alternatively, re-ordering these steps would involve the 3R-hydroxyindolenine (6) undergoing two-electron oxidation and enolization to the corresponding azadiene followed intramolecular Diels-Alder cycloadditon and final pincaol-type rearrangement of the Diels-Alder cycloadducts, each of which being constituted with a 3R-hydroxyindolenine.
MARCFORTINES
In 1980, Polonsky and co-workers reported the isolation of three new alkaloids from Penicillium roqueforti (later reclassified as P. paneum), an essential fungus used in the production of many varieties of blue cheese that contain mold.8a,b From the strain B26 of P. paneum, marcfortines A, B, and C were isolated (Figure 4). From a structural point of view, these metabolites contain the bicyclo[2.2.2] ring system with the syn-configuration and a monoketopiperazine ring. Structurally resembling chrysogenamide A, the marcfortines contain a spiro-oxindole moiety and an unsubstituted pipecolic acid unit. Furthermore, all three marcfortine natural products display substitution on the indole ring at the C-6 and C-7 positions; marcfortines A and B both contain a dioxepin ring, whereas macfortine C has a pyran moiety.
Figure 4.

The marcfortine family of natural products.
As shown in Table 5, the marcfortines were originally isolated from a strain identified as Penicillium roqueforti;8a,b however, this species was subsequently subdivided into three distinct species, P. roqueforti, P. carneum, and P. paneum,33 and it was determined that only the latter can produce marcfortines.8d,8e,22,23 Additionally, Aspergillus carneus has been reported to produce marcfortine A;4c however, the isolate producing marcfortine A and acyl aszonalenin differed from the culture ex type of A. carneus, which produced citrinin. Therefore, the identity of the marcfortine-producing Aspergillus is questionable.
Table 5.
Fungal Origin of the Marcfortines.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
| marcfortine A | A. carneus, P. paneum, P. roqueforti a | anthelmintic | 8c, 8d-e, 8a-b, 8f |
| marcfortine B | P. paneum, P. roquefortia | 8d-e, 8a-b | |
| marcfortine C | P. paneum, P. roquefortia | 8d-e, 8a-b |
Reclassified as Penicillium paneum.
The biosynthesis of marcfortine A has been investigated by Kuo et al., and, as shown in Scheme 7, this metabolite is derived from l-tryptophan (oxindole moiety), l-methionine (via SAM methylation of the monoketopiperazine N), l-lysine (pipecolate residue), and acetate (isoprene units).34 On the basis of these results, it is clear that the isoprene residues originate from mevalonic acid; however, there are two possible biosynthetic routes for the formation of pipecolic acid from l-lysine, as shown in Scheme 8.34b To determine which pathway metabolizes l-lysine to the pipecolic acid unit, Kuo and co-workers carried out separate feeding studies with [α-15N]- and [ε-15N]-l-lysine. NMR and MS analysis revealed that the [α-15N]-l-lysine was incorporated at the α, β, and γ nitrogen of marcfortine A, with a rate of 1.8%, 13%, and 9%, respectively. Contrary to this result, [ε-15N]-l-lysine showed an incorporation rate of 54%, 2.7%, and 3.9% respectively, for the three nitrogens, thus leading to the conclusion that the [ε-15N]-l-lysine gave a more specific and higher incorporation at the pipecolate nitrogen. Based on these data, pathway A is the preferred method of l-lysine metabolism to the pipecolic acid moiety.34b
Scheme 7.

Biosynthetic derivatives of marcfortine A.14
Scheme 8.

Two biogenetic routes from l-lysine to pipecolic acid.34b
PARAHERQUAMIDES
One of the largest group of metabolites containing the bicyclo[2.2.2]diazaoctane ring system is the paraherquamides, which were first isolated from Penicillium paraherquei,4a a synonym of P. brasilianum,35 and later from an isolate ATCC 20841, originally identified as P. charlesii.4h However, P. charlesii was stated incorrectly36 to be a synonym of P. fellutanum by Pitt,37 and ATCC 20841 has therefore been cited as belonging to one of these species in later publications. Consequently, this isolate and another similar isolate producing paraherquamides (IMI 332995)4e represent a new species closely related to P. canescens and will be called hereafter P. cf. canescens. Other producers of paraherquamides include Penicillium cluniae4d and Aspergillus aculeatinus.4j
Paraherquamide A (Figure 5) was the first member of this family to be isolated by Yamazaki et al. from Penicillium paraherquei (= Penicillium brasilianum).4a A decade later, paraherquamide A was isolated from a fungus identified as Penicillium charlesii (= P. fellutanum sensu Pitt37) along with six structurally related novel analogues, paraherquamides B-G.4b,c The original isolate (ATCC 20841) and a new isolate (IMI 332995) both represent a new species, provisionally called P. cf. canescens. In the following years, the paraherquamide family of prenylated indole alkaloids continued to expand with the isolation of paraherquamides H and I and several related compounds being reported, including VM55595, VM55596, VM55597, VM55599, SB200437, and SB203105.4 More recently, Sherman, Williams and co-workers reported the occurrence of pre-paraherquamide in cultures of the paraherquamide A-producing P. cf. canescens, as well as in the asperparaline A-producing fungus, Aspergillus japonicus (later identified as A. aculeatinus).4j Furthermore, it was determined that paraherquamides A and B are also produced by A. aculeatinus. P. brasilianum, P. cf. canescens, and P. cluniae, which are all soil fungi and the main producers of paraherquamides (Table 6).
Figure 5.

Structures of the paraherquamides and related compounds.
Table 6.
Fungal Origin of the Paraherquamides.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
| paraherquamide A | P. charlesii,a P. cluniae, P. brasilianum (= P. paraherquei), Penicillium sp. IMI 332995,a Penicillium species | anthelmintic antinematodal |
4b-c, 4d, 4a, 4e, 4g, 38-42 |
| paraherquamide B | P. charlesii,a P. cluniae | antinematodal | 4b-c, 4d |
| paraherquamide C | P. charlesiia | antinematodal | 4b,c,h |
| paraherquamide D | P. charlesiia | antinematodal | 4b,c,h |
| paraherquamide E | Aspergillus sp. IMI 337664, A. aculeatus, P. charlesii,a P. cluniae, Penicillium sp. IMI 332995,aPenicillium species | antinematodal | 4f, 4b,c,h,k 4d, 4e, 4g |
| paraherquamide F | P. charlesii,a Penicillium sp. IMI 332995, Penicillium species | antinematodal | 4b,c,h, 4e, 4g |
| paraherquamide G | P. charlesii,a Penicillium sp. IMI 332995,a Penicillium species | antinematodal | 4b,c,h, 4e, 4g |
| paraherquamide H | P. cluniae | 4d | |
| paraherquamide I | P. cluniae | 4d | |
| VM55595 | Penicillium sp. IMI 332995a | 4e | |
| VM55596 | P. cluniae | insecticidal | 4d |
| VM55597 | P. cluniae, Penicillium sp. IMI 332995a | insecticidal | 4d, 4e |
| VM55599 | P. paneum, Penicillium sp. IMI 332995,a A. japonicus,b P. charlesiia | 4i, k, 4e, 4j | |
| SB200437 | Aspergillus sp. IMI 337664c | anthelmintic | 4f |
| SB203105 | Aspergillus sp. IMI 337664c | anthelmintic | 4f |
| pre-paraherquamide | A. japonicus,b P. charlesiia | 4j |
Reclassified as a new species related to Penicillium canescens.
Reclassified as Aspergillus aculeatinus.
Reclassified as Aspergillus fijiensis.
Structurally, all of the paraherquamides contain the bicyclo[2.2.2]diazaoctane core with the syn-configuration; however, the paraherquamide family displays a plethora of skeletal diversity. For example, the paraherquamides exhibit varying substitution and oxygenation on not only the proline ring, but also the spiro-oxindole moiety, in that several of the paraherquamides contain a unusual dioxepin ring, while others contain a pyran moiety. Within this family of secondary metabolites, VM55599 and pre-paraherquamide are the only two compounds that do not contain either a spiro-oxindole functionality or an oxygenated indole ring system.
The paraherquamides have attracted considerable interest due to their potent anthelmintic activity. Parasitic nematodes cause severe health problems in humans and domestic animals, and the severity of this problem is increasing due to the development of drug-resistant strains of parasites.38 Initial anthelmintic activity was detected in Trichostrongylus colubriformis,39 and subsequentinvestigations showed that paraherquamide A is effective against strains of parasites that are resistant to broad-spectrum anthelmintics.40 However, the toxicity of paraherquamide A was determined to be too great for use as a broad-spectrum anthelmintic.41 To decrease the toxicity, but still maintain potency, several paraherquamide derivatives have been synthesized and tested for anthelmintic activity.38 Of these derivatives, 2-deoxyparaherquamide A (Figure 6), later named derquantel (Pfizer), displayed excellent activity in models of Haemonchus contortus, Trichostrongylus colubriformis, and Ostertagia ostertagi, while maintaining a safe level of toxicity. Manufacturing of 2-deoxyparaherquamide relies on a semi-synthetic procedure wherein paraherquamide is isolated from large-scale fermentation of P. simplicissimum, purified, and chemically converted to the 2-deoxyparaherquamide analogue.38 This new spiroindole was released in July 2010 as a combination treatment with abamectin, and is currently marketed under the brand name Startect® for use as an anthelmintic in sheep.42
Figure 6.
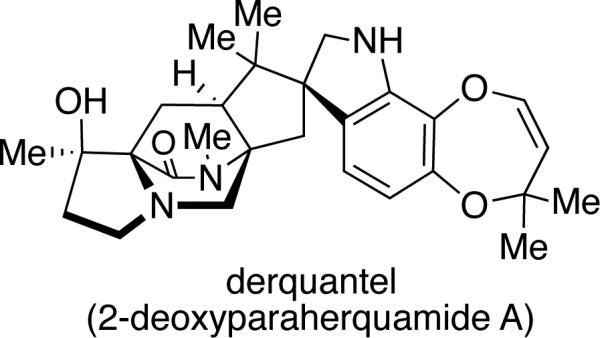
Structure of 2-deoxyparaherquamide A (derquantel).
Elucidating the biosynthesis of paraherquamide A has been an area of intense research for several years, largely by Williams et al. Based on the results obtained from the precursor incorporation studies of the brevianamides, this group postulated that the structurally similar paraherquamides follow a comparable biosynthetic pathway. To determine which amino acid residues are incorporated into paraherquamide A, feeding experiments were performed with P. cf. canescens using [1-13C]-l-tryptophan, [methyl-13C]-l-methionine, [1-13C]-l-proline, and [1-13C]-l-isoleucine (Scheme 9).43 Through 13CNMR spectroscopy and electrospray mass spectrometry, the position and percentage of 13C incorporation was analyzed, and as expected, [1-13C]-l-tryptophan incorporated at the C-12 (paraherquamide numbering) position of the oxindole ring with 2.5% incorporation. Moreover, similar to results obtained in the marcfortine biosynthesis, a small amount (0.6%) of [methyl-13C]-l-methionine was found to incorporate at the nitrogen methyl of the monoketopiperazine ring.
Scheme 9.

(A) 13C-labeled amino acid incorporation into paraherquamide A in Penicillium cf. canescens. (B) Proposed biosynthetic pathway for the conversion of l-isoleucine to 3(S)-methyl-l-proline.43
With regard to the biogenesis of the β–methyl-β-hydroxyproline ring, precedent established by Arigoni and co-workers in the biosynthesis of bottromycin revealed incorporation of [methyl-13C]-l-methionine into the natural product at the 3(R)-methyl group of the β–methyl proline ring of bottromycin.43b However, results from the precursor incorporation study with P. cf. canescens showed that neither [methyl-13C]-l-methionine nor [1-13C]-l-proline incorporated into the β–methyl-β-hydroxyproline ring of paraherquamide A. It was later found that [1-13C]-l-isoleucine exhibited incorporation to the extent of 3.3-3.7%, with the label at the C-18 position. To establish a potential mechanism of incorporation of isoleucine into paraherquamide A, [13C]-[2H]3-l-isoleucine was synthesized and fed to cultures of P. cf. canescens.43c,d Results showed that only one deuterium label was retained, thus confirming a four-electron oxidative cyclization pathway to form β–methyl-l-proline (Scheme 9B).
This possibility was supported by an additional 13C-labeling study, which revealed a higher percentage of 13C incorporation in the monoketopiperazine ring when l-isoleucine underwent oxidative cyclization to form the β–methyl-l-proline prior to the precursor incorporation study with P. cf. canescens. An incorporation of 14.6% was observed in this case, compared to the previously observed 3.7% incorporation of l-isoleucine.43c,d
Additional biosynthetic research, regarding the exact origin of the two isoprene units present in paraherquamide A, has been carried out and rigorously analyzed by Williams and colleagues. By providing [13C2]-acetate to P. cf. canescens it was demonstrated that the isoprene units in paraherquamide A were introduced in two distinct ways (Scheme 10).11,12 Through 13CNMR spectroscopy, the coupling of intact C2 units revealed that in the C5 fragment formed by C-19 to C-23, C-19 was coupled to C-20, while C-21 was coupled to C-22 and C-23, but not to both simultaneously. For the second C5 unit in paraherquamide A, C-24 to C-28, the coupling constants showed that C-24 was coupled to C-25, while C-26 was coupled to C-27, but not to C-28.11,12 On the basis of the observed coupling patterns, it was concluded that the introduction of the C-24 to C-28 isoprene unit must proceed through a direct prenyl transfer in which nucleophilic displacement occurs at the pyrophosphate-bearing methylene carbon while the hydrophobic tail of DMAPP is buried in the enzyme active site.
Scheme 10.

Relevant coupling patterns observed in the 13C NMR spectrum of paraherquamide A. Thick lines represent intact acetate units.12
The mechanistic formation of the C-19 to C-23 fragment was further complicated by the observed scrambling of the 13C labels of the geminal methyl groups at C-22 and C-23, thus indicating a loss of stereochemical integrity during the biosynthetic construction of the quaternary center.11,12 These unexpected results pointed to a non-face-selective reverse prenyl transferase. In this case, the olefinic π system of DMAPP is presented in such a manner that both faces are susceptible to attack by the 2-position of the indole moiety. As shown in Scheme 11, one possible explanation is that the pyrophosphate group is anchored in the enzyme active site with the hydrophobic isopropenyl moiety being presented in a conformationally flexible disposition (A ⇄ B).
Scheme 11.

Proposed biosynthetic mechanism of reverse prenylation in paraherquamide A to explain the scrambling of the geminal methyl 13C labels observed during the precursor incorporation study.12
As shown in Scheme 12, the advanced hexacyclic indole derivative, 13C-labeled preparaherquamide, was synthesized and provided to P. cf. canescens in a precursor incorporation study.44 Results showed that this material incorporated with the double label intact and 13C-incorporation of 0.72%, suggesting that pre-paraherquamide is in fact a pathway metabolite in paraherquamide biosynthesis. This information also indicates that formation of the dioxepin ring and the spiro-oxindole moiety occurs after the formation of this intermediate. Furthermore, the dioxopiperazine version of preparaherquamide (8) was also synthesized as an isotopomer and fed to P. cf. canescens; however, no incorporation was detected. Therefore, reduction of the tryptophan-derived carbonyl residue to the monoketopiperazine ring system must occur prior to formation of the core bridging bicycle.44 These results are also consistent with the similar observation discussed above, regarding the biosynthesis of malbrancheamide.
Scheme 12.

Incorporation of 13C-labeled pre-paraherquamide into paraherquamide A in P. cf. canescens.
Taken together, the data indicate that the sub-families of alkaloids bearing a monoketopiperazine ring system might derive from a non-ribosomal peptide synthase (NRPS) that reductively cleaves the dipeptide from the thiolation domain resulting in off-loading of the tryptophan-derived carboxyl group as an aldehyde. Ring closure of the cyclic amino acid secondary amine on this aldehyde results in imine formation followed by tautomerization to an azadiene (see Scheme 3). The azadiene intermediate then undergoes an IMDA to furnish the bicyclo[2.2.2]diazaoctane ring system. Conversely, the sub-families that are comprised of a dioxopiperazine-based bicyclo[2.2.2]diazaoctane ring core would arise via an oxidative process to generate an azadiene species (see Scheme 6).
ASPERPARALINES
Asperparaline A is an indole-derived alkaloid containing the core bridging bicycle that was first isolated in 1997 by Hayashi and co-workers from the fungus Aspergillus japonicus JV-23, in Sakai, Japan.7a However, the group of uniseriate black Aspergillus species has been extended from one species to seven species, and their chemical profiling has shown that the actual asperparaline producers are A. aculeatinus and A. fijiensis (Table 7).45,46 In 2000, the same group reported the isolation of two new related indole alkaloids, asperparalines B and C, from the same fungal strain.7b In separate work, Everett et al. reported the isolation of asperparaline A, named aspergillimide, from Aspergillus sp. IMI 337664.7c From the same fungal culture, most likely the new species Aspergillus fijiensis, the 16-keto derivative of asperparaline A was isolated and named SB202327. From a structural point of view, the asperparalines possess the core bicyclo[2.2.2]diazaoctane ring system. In addition, asperparalines A and B both contain a β-methyl proline ring, whereas asperparaline C is the C-3–desmethyl analog. The asperparalines also contain a unique 3-spiro-succinimide ring system, the orientation of which is consistent with the relative configuration of the spiro-oxindole ring system present in the paraherquamides.47
Table 7.
Fungal Origin and Bioactivity of the Asperparalines.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
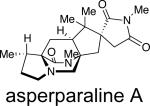
|
A. japonicus,a Aspergillus sp. IMI 337664b | paralytic to silkworms,Anthelmintic | 7a-b, 7c |
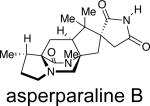
|
A. japonicusa | paralytic to silkworms | 7a-b |
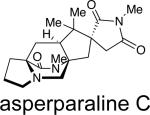
|
A. japonicusa | paralytic to silkworms | 7a-b |
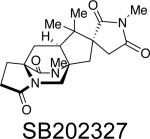
|
Aspergillus sp. IMI 337664b | anthelmintic | 7c |
Reclassified as A. aculeatinus.
This strain is probably A. fijiensis, which has pink sclerotia like IMI 337664.46f
All members of the asperparaline family have exhibited some form of biological activity. Asperparaline A exhibited paralysis against silkworms at a dose of 10 μg/g of the diet within 1 h and lasted for 7 to 10 h upon oral administration.7 Asperparalines B and C displayed almost the same effect on silkworms. Separately, asperparaline A and SB202327 were tested for anthelmintic activity against adult Trichostrongylus colubriformis infection in gerbils.7 Results from this experiment showed that asperparaline A displayed a 44% and 98% reduction in fecal egg count when dosed at 10 mg/kg and 20 mg/kg, respectively. SB202327 exhibited in vitro activity against Haemonchus contortus L3 larvae, but did not display in vivo activity against T. colubriformis in gerbils.
Since the asperparaline carbon skeleton closely resembles the paraherquamides, Williams and co-workers directed their attention toward elucidating the biosynthesis of the asperparalines. To determine the primary amino acid building blocks of asperparaline A [1,2-13C2]-acetate, [methyl-13C]-l-methionine, [1-13C]-l-isoleucine, [1-13C]-l-tryptophan, and [indole-2-13C]-l-tryptophan were employed in precursor incorporation studies (Scheme 13).47 The position of the 13C enrichment in asperparaline A was determined by 13CNMR and the percentage of labeled amino acid enrichment was determined by 13CNMR and electrospray mass spectrometry. Results showed that [1,2-13C2]-acetate incorporates into asperparaline A in an analogous manner to that of paraherquamide A, indicating that the isoprene units in asperparaline A also arise from the mevalonic acid pathway. A significant amount of incorporation from [methyl-13C]-l-methionine was observed at the C-29 (25.2%) and C-30 (26.9%) positions of asperparaline A, but no incorporation was observed in any part of the β-methyl proline ring. However, as expected, [1-13C]-l-isoleucine exhibited a 5.8% incorporation at the C-18 position of the β-methyl proline ring, which indicates that the β-methyl proline ring in both asperparaline A and paraherquamide A is biosynthesized along a similar, if not identical route. While the asperparalines appear at first glance to be the only prenylated indole alkaloids not derived from tryptophan, the precursor incorporation studies show otherwise. Both [1-13C]-l-tryptophan and [indole-2-13C]-l-tryptophan showed incorporation at the C-12 (6.4-7.2%) and C-2 (12.2%) positions of asperparaline A, respectively. This indicated that tryptophan is responsible for the construction of the spiro-succinimide ring found in the asperparalines.
Scheme 13.

Asperparaline A precursor incorporation study.
Results from the precursor incorporation studies, along with the recent detection of preparaherquamide in A. aculeatinus, provide indirect support that: (a) pre-paraherquamide is a precursor to asperparaline A; and (b) paraherquamide A and asperparaline A share a unified biosynthetic scheme.4j To address both of these hypotheses, a unified biosynthetic pathway has been proposed. As illustrated in Scheme 14, the proposed unified biogenesis of paraherquamide A and asperparaline A arises from preparaherquamide, which is derived from the common precursors l-tryptophan, l-isoleucine, and DMAPP.44,47 From preparaherquamide, the biosynthesis of paraherquamide A could arise via oxidation of the aromatic ring of preparaherquamide to the catechol derivative (9), followed by prenylation, dioxepin formation, and oxidation to the spiro-oxindole.12 Along this same pathway, formation of the spiro-succinimide ring of asperparaline A would occur via interruption of the paraherquamide A biosynthesis just before the prenylation step of the catechol derivative.47 This would provide a possible branching point for the oxidative cleavage of four carbons of the aromatic ring, thus resulting in the formation of the spiro-succinimide moiety.
Scheme 14.

Proposed unified biosynthetic pathway with branch-point to generate paraherquamides or asperparalines.47
STEPHACIDINS AND NOTOAMIDES
The stephacidins and notoamides comprise a family of prenylated indole alkaloids closely related to the paraherquamides. The first member to be isolated of what was to become the stephacidin and notoamide families, was sclerotiamide (Figure 7). This was identified in 1996 from Aspergillus sclerotiorum by Gloer and White, and was actually classified as a paraherquamide-type compound, despite containing a diketopiperazine as opposed to a monoketopiperazine as all paraherquamide family members are constituted.5a Structurally similar to the paraherquamides, sclerotiamide contains the core bicyclo[2.2.2]diazaoctane moiety, a spiro-oxindole ring system with a pyran group, and an unsubstituted proline ring. However, unlike the paraherquamides, sclerotiamide retains an unreduced dioxopiperazine unit and also contains a unique C-10 hydroxyl group (paraherquamide numbering). At the time, the unusual structural skeleton of sclerotiamide made it appear to be a member of the paraherquamides.
Figure 7.

Structures of the stephacidins and related compounds.
It was not until 1999 that other structurally related prenylated indole alkaloids were isolated, including (+)-stephacidin A, stephacidin B, and avrainvillamide.5b,c Stephacidins A and B were both isolated from the terrestrial-derived Aspergillus ochraceus WC76466 by a group at Bristol-Myers Squibb,5b,c while avrainvillamide was isolated independently from a marine-derived Aspergillus sp.CNC358 by Fenical and associates.5d Two years later, Sugie and co-workers reported the isolation of avrainvillamide, under the name of CJ-17,665, from A. ochraceus CL41582.5e
In 2007, Tsukamoto and co-workers isolated notoamides A-D (Figure 8) from a marine-derived Aspergillus sp. MF297-2, most likely Aspergillus sclerotiorum, found growing on the mussel, Mytilus edulis, collected off the Noto Peninsula in the Sea of Japan.6a Along with the notoamides, the known compounds sclerotiamide, (+)-stephacidin A, and deoxybrevianamide E were also obtained. Notoamides A-D contain the pyranoindole ring system common in the stephacidins and some of the paraherquamides. Notoamides A and B both contain the core bridging bicycle with C-12 oxidation (paraherquamide numbering), as well as the spiro-oxindole ring system. In 2005, a group of structurally similar indole alkaloids, named the norgeamides, was isolated at the Hans-Knöll Institute from a cold water Aspergillus sp.48 Minor metabolites of the notoamides continue to be isolated from marine-derived Aspergillus sp. MF297-2; notoamides E-R are known natural products while notoamides S and T are potential biosynthetic intermediates that have yet to be isolated.2,6b-e,49
Figure 8.

Structures of the notoamides and related compounds.
Stephacidin A and notoamide B were also isolated recently from the terrestrial fungus Aspergillus versicolor, along with a new metabolite, versicolamide B.6f This new secondary metabolite displays the rare anti-relative configuration at C-6 (stephacidin numbering) in the bicyclo[2.2.2]diazaoctane ring system, which is similar to that in brevianamides A and B. Following the isolation of versicolamide B from A. versicolor, it was found that this metabolite is also produced by the marine-derived Aspergillus sp. MF297-2.6d Further investigation of the metabolites isolated from A. versicolor and Aspergillus sp. MF297-2 led to the discovery of another unique trait within this family. (–)-Stephacidin A, (+)-notoamide B, and (+)-versicolamide B from A. versicolor are the corresponding enantiomers to (+)-stephacidin A, (–)-notoamide B, and (–)-versicolamide B produced by the marine-derived Aspergillus sp. MF297-2 (Figure 9).6f Additionally, (–)-stephacidin A is the enantiomer to (+)-stephacidin A isolated from A. ochraceus WC76466, a terrestrial fungus. The discovery of these enantiomeric metabolites produced by two different Aspergillus species represents the first known occurrence of reverse prenylated enantiomeric natural products.
Figure 9.

Structures of the enantiomeric metabolites produced by Aspergillus sp. MF297-2 and A. versicolor.
As shown in Table 8, numerous prenylated indole alkaloids belonging to the stephacidin and notoamide family have been isolated from various Aspergillus fungal cultures. Taxonomically, the notoamides (stephacidins, aspergamides, avrainvillamides, sclerotiamide, versicolamide, norgeamides) are produced by two groups of Aspergillus: species in sections Circumdati and Versicolores. In section Circumdati, Aspergillus sclerotiorum,5aA. ochraceus,5b-dA. ostianus,6g and A. westerdijkiae50 can produce notoamides. In addition, chemical screening has shown that A. melleus, A. persii, A. petrakii and A. bridgeri can also produce notoamides, while the remaining twelve species in section Circumdati do not appear to produce them. In Aspergillus section Versicolores, a strain of Aspergillus versicolor can produce the stereoisomers, as compared to those produced by species in section Circumdati of the notoamides.6f We have detected these compounds later in other strains of A. versicolor and in two strains of the closely related species Aspergillus protuberus.
Table 8.
Fungal Origin of the Stephacidins and Notoamides.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
| sclerotiamide | A. sclerotiorum, Aspergillus sp. MF297-2a | insecticidal antifeedant |
5a, 6a |
| (+)-stephacidin A | A. ochraceus WC76466, Aspergillus sp. MF297-2a | cytotoxic | 5b-c, 6a |
| (–)-stephacidin A | A. versicolor | 6f | |
| stephacidin B | A. ochraceus WC76466 | cytotoxic | 5b-c |
| avrainvillamide | A. ochraceus CL41582, Aspergillus sp. CNC358 | antibiotic cytotoxic |
5e, 5d |
| notoamide A | Aspergillus sp. MF297-2a | 6a | |
| (–)-notoamide B | Aspergillus sp. MF297-2a | 6a | |
| (+)-notoamide B | A. versicolor | 6f | |
| notoamide C | Aspergillus sp. MF297a | 6a | |
| notoamide D | Aspergillus sp. MF297a | 6a | |
| notoamide E | Aspergillus sp. MF297a | 6b | |
| notoamide F | Aspergillus sp. MF297,aA. ostianus | 6c, 6g | |
| notoamide G | Aspergillus sp. MF297a | 6c | |
| notoamide H | Aspergillus sp. MF297a | 6c | |
| notoamide I | Aspergillus sp. MF297a | 6c | |
| notoamide J | Aspergillus sp. MF297a | 6c | |
| notoamide K | Aspergillus sp. MF297a | 6c | |
| notoamide L | Aspergillus sp. MF297a | 6d | |
| notoamide M | Aspergillus sp. MF297a | 6d | |
| notoamide N | Aspergillus sp. MF297a | 6d | |
| notoamide O | Aspergillus sp. MF297a | 6e | |
| notoamide P | Aspergillus sp. MF297a | 6e | |
| notoamide Q | Aspergillus sp. MF297a | 6e | |
| notoamide R | Aspergillus sp. MF297,aA. ostianus | 6e, 6g | |
| (–)-versicolamide B | Aspergillus sp. MF297a | 6d | |
| (+)-versicolamide B | A. versicolor | 6f | |
| norgeamide A | Aspergillus species | cytotoxic | 48, 51 |
| norgeamide B | Aspergillus species | cytotoxic | 48, 51 |
| norgeamide C | Aspergillus species | cytotoxic | 48, 51 |
| norgeamide D | Aspergillus species, A. versicolor | cytotoxic | 48, 6f, 51 |
Undergoing re-identification (possibly A. sclerotiorum).
Biologically, the notoamides appear to have moderate and limited biological activity, while the structurally related norgeamides have all been shown to inhibit the growth of a variety of cancer cell lines with varying efficacies.51 Preliminary biological assays revealed that norgeamide A is the most cytotoxic against several carcinoma cell lines. However, sclerotiamide, avrainvillamide, and stephacidins A and B appear to be the most bioactive of the stephacidins and notoamides. These secondary metabolites display anti-insecticidal, antibacterial, and cytotoxic properties.5a,c,e
Sclerotiamide has been shown to exhibit potent activity against first instar larvae of the corn earworm Helicoverpa zea.5a When incorporated into standard test diets of 200 ppm, sclerotiamide resulted in a 46% mortality rate of H. zea. There was also a 98% reduction in growth rate, relative to controls, among the survivors. Several physiological effects, such as shriveling and blackening, were also observed in the larvae. In a feeding assay against adults and larvae of the fungivorous beetle Carpophilus hemipterus, at a 100-ppm dietary level, sclerotiamide reduced feeding rates by 44% and 40%, respectively.
Avrainvillamide is an antibiotic that inhibits the growth of multi-drug resistant Staphylococcus aureus, Streptococcus pyogenes, and Enterococcus faecalis with MICs of 12.5, 12.5, and 25 μg/mL, respectively.5e When tested against Escherichia coli, at 100 μg/mL it showed no antibacterial activity. Avrainvillamide also displays cytotoxic activity against a variety of tumor cell lines, including human colon HCT116 cell lines, melanoma MALME-3M cell lines, and two breast cancer cell lines, βT-549 and T-47D.5d
Stephacidins A and B both demonstrate cytotoxicity against various human tumor cell lines; however, stephacidin B exhibited more potent and selective antiproliferative activities, especially for testosterone-dependent LNCaP cells.5b,c The effects of stephacidins A and B indicate a novel mode of action since they are not mediated by p53, mdr, or bc12, and they are not tubulin- or topoisomerase II-mediated. Recent studies conducted by Myers et al. suggest that the growth-inhibitory activity of stephacidin B is a result of retrodimerization to form the monomer avrainvillamide, which may then act as a novel Michael acceptor via the α,β-unsaturated nitrone group.52
Following the isolation of the first set of enantiomeric reverse prenylated indole metabolites, attention has been directed towards elucidating the biosynthesis of the stephacidins and notoamides. In collaboration with the Sherman laboratory at the University of Michigan, additional insight into the biogenesis of the stephacidins and notoamides was obtained through genome sequencing of Aspergillus sp. MF297-2 and A. versicolor.53 Through data mining and deep annotation, the stephacidin and notoamide biosynthetic gene clusters have been identified and characterized. The target gene cluster in Aspergillus sp. MF297-2 was identified from an open reading frame (orf) named notE, using the fumitremorgin gene ftmA as an in silico probe.53a Bioinformatics analysis revealed a total of eighteen genes (notA-notR) encompassing ~44 kb of DNA (Figure 10), including a bimodular NRPS (NotE), and two aromatic prenyltransferases, notC and notF. These latter two genes were functionally expressed in E. coli, with NotC biochemically characterized as a normal prenyltransferase and NotF determined to be a reverse prenyltransferase.
Figure 10.

Notoamide (not) biosynthetic gene cluster from Aspergillus sp. MF297-2.
Based on the above genome sequencing, bioinformatic mining, and biochemical studies, a comprehensive stephacidin and notoamide biogenesis was proposed (Scheme 15). Following the condensation of tryptophan and proline via NotE to form the common precursor brevianamide F, NotF is then responsible for the C-2 reverse prenylation of brevianamide F to give deoxybrevianamide E.53 Subsequent oxidation at the C-6 indole position gives rise to 6-hydroxydeoxybrevianamide E, which has been confirmed recently to be the NotC substrate. The reaction of 6-hydroxydeoxybrevianamide E in the presence of NotC and DMAPP affords 6-hydroxy-7-prenyl-deoxybrevianamide E, which we have named notoamide S.
Scheme 15.

Currently postulated biosynthetic pathway of the stephacidins and notoamides in Aspergillus sp. MF297-2 and A. versicolor.
It is the oxidation and tautomerization of notoamide S that we have proposed presently to give rise to the putative achiral azadiene intermediate, which undergoes an enantiospecific IMDA reaction in the respective Aspergillus fungal cultures to provide the proposed metabolite, notoamide T. Oxidative cyclization of (+)-notoamide T in Aspergillus sp. MF297-2 would afford (+)-stephacidin A, which, following an oxidation and pinacol-type rearrangement to the spiro-oxindole, yields (–)-notoamide B. The enantiomerically opposite IMDA reaction in Aspergillus versicolor would yield (–)-notoamide T to then furnish (–)-stephacidin A and (+)-notoamide B.
Recent precursor incorporation studies carried out by Williams et al. have provided further validation for the proposed biogenesis of the stephacidins and notoamides. More specifically, (±)-[13C]2-stephacidin A was synthesized and provided to the marine-derived Aspergillus sp. MF297-2 and the terrestrial-derived A. versicolor (Scheme 16).54 Analysis of the metabolites revealed enantiospecific incorporation of (+)-stephacidin A into (–)-notoamide B and (–)-sclerotiamide in Aspergillus sp. MF297-2, and (–)-stephacidin A into (+)-notoamide B and (+)-sclerotiamide in A. versicolor. These results indicate the presence of stereospecific, yet complementary face-selective indole oxidases in the two fungal cultures.
Scheme 16.

(±)-[13C]2-stephacidin A precursor incorporation study with A. versicolor and Aspergillus sp. MF297-2.53
RELATED METABOLITES LACKING THE BICYCLO[2.2.2]DIAZAOCTANE RING SYSTEM
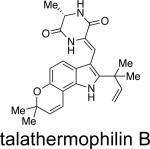
From a structural point of view, the austamides,27 citrinadins,55 and the PF1270 family of metabolites56 appear to be closely related to the previously discussed indole alkaloids, albeit lacking the core bicyclo[2.2.2]diazaoctane nucleus. As shown in Table 9, these related families of metabolites have been isolated from various Penicillium and Aspergillus species and also display a plethora of bioactivity. From a taxonomic perspective, the citrinidins are produced by Penicillium citrinum55 and the closely related PF1270 compounds are produced by Penicillium waksmanii;56 however it is more likely that Penicillium westlingii produces both families of compounds, as this species is closely related to P. citrinum.57P. westlingii was treated as a synonym of P. waksmanii by Pitt,37 but has later been shown to be a valid species.35a,57Talaromyces thermophilus strain YM 1-3 produces talathermophilins A and B,58 secondary metabolites related to notoamide E, except that glycine or alanine is replacing proline and the end-products have a double-bond conjugating one of the ketones in the dioxopiperazine to the indole part of the molecule. We have found that the glycine and alanine notoamide E analogues (without the extended conjugation) are also formed in Talaromyces thermophilus CBS 116.72, albeit accumulated in much smaller amounts compared to the end-products. Talaromyces thermophilus has recently been shown to be closely related to Thermomyces and will probably be transferred to that genus61c,d. This means that no strains of Talaromyces sensu stricto have been reported to produce compounds with the core bicyclo[2.2.2]diazaoctane nucleus yet.
Table 9.
Fungal Origin of Several Related Secondary Metabolites.
| compound | fungal species | bioactivity | reference |
|---|---|---|---|
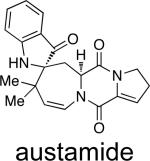
|
A. ustusa | toxic to ducklings | 27 |
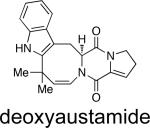
|
A. ustus,a P. italicum | toxic to ducklings | 27b,d |
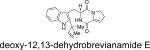
|
A. ustus,a P. italicum, P. ulaiense | toxic to ducklings | 27b,d,e, 22, 23 |
|
A. ustusa | toxic to ducklings | 27b |
|
A. ustusa | toxic to ducklings | 27c |
|
P. citrinum N059 | cytotoxic | 55 |
|
P. citrinum N059 | cytotoxic | 55b |
|
P. waksmanii PF1270b | histamine H3R inhibitor | 56 |
|
P. waksmanii PF1270b | histamine H3R inhibitor | 56 |
|
P. waksmanii PF1270b | histamine H3R inhibitor | 56 |
|
Talaromyces thermophilus | antinematodal | 58 |
|
Talaromyces thermophilus | antinematodal | 58 |
Reclassified as A. pseudoustus.
Probably P. westlingii, closely related to P. citrinum.
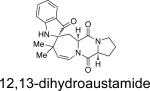
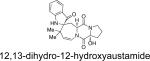
Structurally, austamide, 12,13-dihydroaustamide, and 12,13-dihydro-12-hydroxyaustamide all contain a spiro-indoxyl group similar to the brevianamides, however, these metabolites also contain a unique seven-membered spiro-indoxyl ring.27 Another metabolite isolated from A. pseudoustus is deoxyaustamide, which displays an eight-membered ring between the indole and dioxopiperazine ring, making it a potential metabolic precursor to the austamides. Furthermore, deoxybrevianamide E, the known biosynthetic precursor to the brevianamides, stephacidins, and notoamides, was also isolated from austamide-producing A. pseudoustus, thus indicating the possibility of a shared biogenesis between the brevianamides and austamides.27
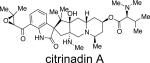
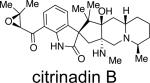
Citrinadins A and B were isolated from the fungus Penicillium citrinum, which was found growing on the red alga Actinotrichia fragilis.55 These metabolites appear to be structurally related to the marcfortine and chrysogenamide families of indole alkaloids, in that the citrinadins also contain a substituted pipecolate moiety, C-7 indole substitution, and a spiro-oxindole ring system. While the citrinadins do not contain the core bridging bicyclo[2.2.2]diazaoctane, Kobayashi and co-workers proposed that they could biosynthetically arise from a marcfortine-type skeleton.55b As shown in Figure 11, loss of the amide carbonyl carbon (C-18), followed by oxidation at C-15 and C-20 and methylation at C-17 could lead to an advanced citrinadin metabolite.
Figure 11.

Possible biosynthetic precursor of citrinadins.
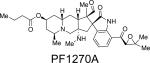
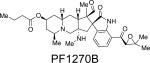
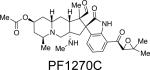
The PF1270 family of metabolites was isolated from a soil-borne fungal culture, Penicillium waksmanii = P. westlingii, in Japan.56 The carbon skeleton of these metabolites is closely related to the citrinadins, as well as the marcfortine and chrysogenamide families. Similar to the citrinadins, PF1270A, PF1270B, and PF1270C contain a substituted pipecolic acid residue, a C-7 substituted indole ring, and a spiro-oxindole moiety. Biosynthetically, this family could also arise from a marcfortine-type compound following loss of the amide carbonyl carbon.
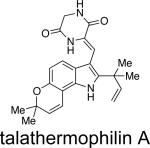
Recently, two novel metabolites, talathermophilins A and B, were isolated from the thermophilic fungus Talaromyces thermophilus.58a These new metabolites have been postulated to serve as potential precursors to the more advanced metabolites containing the bridging bicycle. Unlike notoamide B, metabolites tentatively identified by HPLC-MS as dihydrotalathermophilins A and B appear to be efficiently converted into the talathermophilins A and B, which, according to metabolite profiling, appear to be the end-products. Indeed these dihydrotalathermophilins A and B were recently structure elucidated58b and named talathermophilin C and D. There were no indications of other metabolites with a bicyclo[2.2.2]diazaoctane ring system and/or any of the spiro-compunds of the notoamide type in Talaromyces thermophilus, where glycine or alanine analogues of the proline-derived metabolites discussed here could theoretically have been secondary metabolites in this themophilic species.
CHEMOTAXONOMIC OVERVIEW
The bioactive prenylated brevianamide F-derived secondary metabolites, some of which have the unique bicyclo[2.2.2]diazaoctane ring system, have mostly been found in Aspergillus and Penicillium. The bicyclo[2.2.2]diazaoctane ring system has only been found in fungi that can produce prenylated tryptophan-proline (or pipecolic acid) dioxopiperazines, and not in any other tryptophan containing prenylated dioxopiperazines, which are also common in Aspergillus and Penicillium. However, Malbranchea is unrelated phylogenetically to Penicillium and Aspergillus, and thus the biosynthetic gene cluster for the bicyclo[2.2.2]diazaoctane ring system compounds could have been horizontally transferred, or established through convergent evolution. Dioxopiperazines may have been some of the first secondary metabolites to have been produced by microorganisms during evolution, as they are of widespread occurrence and very bioactive.59 More elaborate natural product structures, like those reviewed here, are of restricted taxonomic distribution. The genera Penicillium and Aspergillus are closely related, yet both contain species that can produce optical enantiomers of the same or nearly the same molecule. For example, the genus Aspergillus can be subdived into three major clades,60 all of which contain producers of brevianamide F. In the Neosartorya clade, the end-products based on brevianamide F (tryptophanyl-prolyl-dioxopiperazine) are the fumitremorgins and spirotryprostatins;29 in the Emericella clade, Aspergillus versicolor produce (+)-versicolamide B; and, in the Eurotium clade, Aspergillus sclerotiorum produce (–)-versicolamide B. Penicillium can roughly be subdivided into six clades,61a or 25 clades61b,c of which only two contain producers of the bicyclo[2.2.2]diazaoctane ring system secondary metabolites. Clade 5 contains Penicillium brasilianum, P. glaucoroseum and P. cluniae, all producing paraherquamides, while clade 6 contains all the remaining producers in Penicillium: P. paneum of marcfortines, P. chrysogenum of chrysogenamides, P. brevicompactum and P. viridicatum of brevianamides and the less related P. cf. canescens of paraherquamides. The food-borne species in Penicillium subgenus Penicillium appear to represent a hot-spot for producers of the bicyclo[2.2.2]diazaoctane ring system compounds, but even within this subgenus, the compounds are widely distributed among the subclades, even both as anti (P. chrysogenum) and syn (the other species) compounds. The production of paraherquamides by soil-borne types of Penicillium in both clade 5 (P. brasilianum) and 6 (P. cf. canescens) could indicate horizontal transfer of the whole biosynthetic gene cluster, or re-inventive evolution guided by selection. Thus, dioxopiperazines may have been vertically inherited from a common ancestor, and may be also simple dioxopiperazine prenylation, but the end-products of the biosynthetic routes appear to be unique and specific for the different species. Within closely related species the same secondary metabolites seem to be produced, and could thus be derived from a common ancestor. For example in Aspergillus section Circumdati, of the twenty different species known,50 eight produce notoamides, and twelve do not, but those that are produced are of the same chemical type. In section Circumdati, the notoamides are produced in two different clades, which are not sister clades.50 The “central” clade contains A. ochraceus, A. petrakii, A. ostianus, A. melleus and A. westerdijkiae,50 all producing notoamides, while in a second clade consisting of A. sclerotiorum, A. persii, A. bridgeri and A. sulphureus, the three first species produce notoamides.
In most other clades in Penicillium and Aspergillus one, or rarely two, out of all species in a series produce bicyclo[2.2.2]diazaoctane compounds (Table 10). Thus, metabolites containing the dioxopiperazine and prenylation are very widespread in Aspergillus and Penicillium, but the more complicated metabolites containing the bicyclo[2.2.2]diazaoctane ring system are only present in a few species. While it is only P. paneum that can produce marcfortines in Penicillium series Roqueforti, all species in the series can produce roquefortine C, another prenylated diketopiparazine (Trp + His). In the eight species in Penicillium series Viridicata it is only P. viridicatum that can produce brevianamide A, but all eight species produce other prenylated dioxopiperazines:22,23P. neoechinulatum, P. aurantiogriseum and P. freii produce aurantiamine (Val + His), P. viridicatum produces the closely related viridamine (Val + His), P. melanoconidium produces roquefortines (Trp + His), and P. cyclopium, P. polonicum, and P. tricolor produce verrucofortines (Trp + Leu) and puberulines (Trp + Phe). In the five species in Penicillium series Chrysogena it is only P. chrysogenum than can produce chrysogenamide, but all species produce prenylated dioxopiperazines or dioxopiperazines: P. chrysogenum, P. rubens and P. flavigenum produce roquefortine C (Trp + His), and P. dipodomyis and P. nalgiovense produce dipodazin (Trp + Gly, un-prenylated), and P. chrysogenum produce chrysogenazine (Trp + Ala, prenylated).62 The fact that prenylated dioxopiperazines are widespread in Penicillium and Aspergillus, and also occur outside these two genera, and the fact that the bicyclo[2.2.2]diazaoctane ring system-containing secondary metabolites are more restricted in their distribution, show that these latter natural products are advanced molecules that appeared later during evolution than other dioxopiperazines.
Table 10.
Production of Bicyclo[2.2.2]diazaoctane Compounds and Austamides in Different Ecological/ Phylogenetic Series of Aspergillus and Penicillium (Frisvad, unpublished and ref. 22, 23, 24, 26, 29, 45, 46, 50, based on the methods described in ref. 63)
| secondary metabolite group | section or series | species | production of bicyclo[2.2.2]diazaoctane compounds (and / or austamides) |
|---|---|---|---|
| brevianamides | P. series Olsonii | P. brevicompactum | + |
| P. astrolabium | - | ||
| P. bialowiezense | - | ||
| P. neocrassum | - | ||
| P. olsonii | - | ||
| brevianamides | P. series Viridicata | P. viridicatum | + |
| P. aurantiogriseum | - | ||
| P. cyclopium | - | ||
| P. freii | - | ||
| P. melanoconidium | - | ||
| P. neoechinulatum | - | ||
| P. polonicum | - | ||
| P. tricolor | - | ||
| marcfortines | P. series Roqueforti | P. paneum | + |
| P. carneum | - | ||
| P. roqueforti | - | ||
| P. psychrosexualis | - | ||
| marcfortines | Aspergillus section Flavipedes | A carneus | + |
| A niveus | - | ||
| A flavipes | - | ||
| chrysogenamides | P. series Chrysogena | P. chrysogenum | + |
| P. dipodomyis | - | ||
| P. flavigenum | - | ||
| P. nalgiovense | - | ||
| P. rubens | - | ||
| paraherquamides | P. series Canescentia | P. cf. canescens | + |
| P. canescens | - | ||
| P. flavidostipitatum | - | ||
| P. janczewskii | - | ||
| P. jensenii | - | ||
| P. murcianum | - | ||
| P. nigricans | - | ||
| P. radiatolobatum | - | ||
| paraherquamides | P. series Simplicissima | P. brasilianum | + |
| P.cluniae | + | ||
| P. glaucoroseum | + | ||
| P. janthinellum | - | ||
| P. piscarium | - | ||
| P. pulvillorum | - | ||
| P. simplicissimum | - | ||
| asperparalines | Aspergillus section Nigri series Aculeati | A. aculeatinus | + |
| A. fijiensis | + | ||
| A. aculeatus | - | ||
| A. indologenus | - | ||
| A. japonicus | - | ||
| A. uvarum | - | ||
| A. violaceofuscus | |||
| notoamides, stephacidins, aspergamides, avrainvillamides | Aspergillus section Circumdati | A. bridgeri | + |
| A. melleus | + | ||
| A. ochraceus | + | ||
| A. ostianus | + | ||
| A. persii | + | ||
| A. petrakii | + | ||
| A. sclerotiorum | + | ||
| A. westerdijkiae | + | ||
| A. auricomus | - | ||
| A. cretensis | - | ||
| A. elegans | - | ||
| A. flocculosus | - | ||
| A. insulicola | - | ||
| A. muricatus | - | ||
| A. neobridgeri | - | ||
| A. ochraceopetaliformis | - | ||
| A. pseudoelegans | - | ||
| A. roseoglobulosus | - | ||
| A. steynii | - | ||
| A. sulphureus | - | ||
| notoamides, opposite stereochemistry | Aspergillus series Versicolores | A. versicolor | + |
| A. protuberus | + | ||
| A. sydowii | - | ||
| austamides and deoxybrevianamides | Aspergillus section Usti | A. pseudoustus | + |
| A. amylovorus | - | ||
| A. calidoustus | - | ||
| A. californicus | - | ||
| A. carlsbadensis | (brevianamide A) | ||
| A. cavernicola | - | ||
| A. deflectus | - | ||
| A. egyptiacus | - | ||
| A. elongatus | (possibly notoamide E) | ||
| A. germanicus | - | ||
| A. granulosus | - | ||
| A. heterothallicus | - | ||
| A. insuetus | - | ||
| A. kassunensis | - | ||
| A. keveii | - | ||
| A. lucknowensis | - | ||
| A. monodii | - | ||
| A. pseudodeflectus | - | ||
| A. puniceus | - | ||
| A. subsessilis | - | ||
| A. turkensis | - | ||
| A. ustus | - | ||
| deoxybrevianamides | P. series Italica | P. italicum | + |
| P. ulaiense | + |
CONCLUSION
Natural products containing the unique bicyclo[2.2.2]diazaoctane ring system are intriguing from a synthetic, biosynthetic, evolutionary, and medicinal perspective. This unusual ring system is proposed to arise biosynthetically via an azadiene-mediated intramolecular hetero-Diels-Alder reaction that has been supported through biomimetic syntheses. An emerging body of evidence suggests that there are two distinct biogenetic familes: (1) those constituted of a dioxopiperazine as an integral part of the bicyclo[2.2.2]diazaoctane ring system, and (2) those comprised of a monooxopiperazine as an integral part of the bicyclo[2.2.2]diazaoctane ring system. Precursor incorporation experiments and recent genetic, bioinformatic and biochemical analyses further suggest that the dioxopiperazine-containing family members, such as stephacidins, arise by oxidative processing of C-2 reverse-prenylated indole dipeptides that culminate in azadiene formation and intramolecular Diels-Alder cyclization. Similarly, precursor incorporation experiments and recent analysis of the NRPS domains corresponding to the monooxopiperazine-containing family members indicate that natural products in the larger paraherquamide family arises by reductive processing of the initial indole dipeptides, to form a reduced azadiene species, which undergoes C-2 reverse prenylation and an intramolecular Diels-Alder cyclization. Whether or not the putative Diels-Alderases are none other than the oxidase and reductase domains of the NRPS modules that process the dipeptide intermediates at an early stage remains to be ascertained. At the heart of this central question is whether these redox functions catalyze the IMDA reaction through a pericyclic transition state, or whether the IMDA reaction is an “afterthought” or spontaneous construction immediately following the generation of the respective azadiene species that have been implicated. One of the most provocative findings in this context has been the discovery of the enantiomeric natural products stephacidin A, notoamide B, and versicolamide B as discussed above as well as the production of the diastereomeric and pseudo-enantiomeric substances VM55599 and preparaherquamide. These intriguing stereochemical puzzles remain to be solved and are the subject of central focus in our laboratories.
The fungal origin of secondary metabolites containing the bicyclo[2.2.2] nucleus is largely limited to species within the Aspergillus and Penicillium genera. Moreover, secondary metabolites with the bicyclo[2.2.2] nucleus appear to be different from the more widespread prenylated dioxopiperazines in that they have evolved later and followed recent speciation in Aspergillus and Penicillium. Comparison of the malbrancheamide biosynthetic genes in Malbranchea with the parallel ones in Penicillium and Aspergillus may help clarify whether secondary metabolite gene clusters are acquired vertically or horizontally or if they have developed independently during evolution. It is tempting to ponder if the putative Diels-Alderase genes have evolved at a different rate than the amino acid cyclases and prenyl transferases in the producing organisms, a hypothesis that can be tested using bioinformatics combined with taxonomy and organic chemistry. The key step that culminates in the formation of the bicyclo[2.2.2]diazaoctane ring system is also the stage at which the structural complexity of the simple prenylated dipeptide progenitors are vastly increased. That Nature has apparently evolved two distinct mechanistic manifolds (reductive and oxidative) to build this ring system was an unexpected yet wonderous discovery that was only made possible through the synthesis of isotopically labeled biosynthetic intermediates, whole genome sequencing and bioinformatic analyses of the respective gene clusters. The modern tools of whole genome sequencing, proteomics, bioinformatics, and functional expression of the biosynthetic genes are currently being deployed to gain a detailed understanding of the elusive mysteries of the biogenesis of these structurally complex secondary metabolites.
Scheme 1.

The classical mevalonic acid pathway showing the labeling pattern via 1,2-doubly labeled acetate (note the change in carbon numbering).
REFERENCES
- 1.Ding Y, de Wet JR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. J. Am. Chem. Soc. 2010;132:12733–12740. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sunderhaus JD, Sherman DH, Williams RM. Isr. J. Chem. 2011;51:442–452. doi: 10.1002/ijch.201100016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.a Birch AJ, Wright JJ. J. Chem. Soc. Chem. Commun. 1969:644–645. [Google Scholar]; b Birch AJ, Wright JJ. Tetrahedron. 1970;26:2329–2344. doi: 10.1016/s0040-4020(01)92812-1. [DOI] [PubMed] [Google Scholar]; c Birch AJ, Russel RA. Tetrahedron. 1972;28:2999–3008. [Google Scholar]; d Robbers JE, Straus JW, Tuite J. J. Nat. Prod. 1975;38:355–356. [PubMed] [Google Scholar]; e Wilson BJ, Yang DTC, Harris TM. Appl. Microbiol. 1973;26:633–635. doi: 10.1128/am.26.4.633-635.1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.a Yamazaki M, Okuyama E, Kobayashi M, Inoue H. Tetrahedron Lett. 1981;22:135–136. [Google Scholar]; b Ondeyka JG, Goegelman RT, Schaeffer JM, Kelemen L, Zitano L. J. Antibiot. 1990;43:1375–1379. doi: 10.7164/antibiotics.43.1375. [DOI] [PubMed] [Google Scholar]; c Goegelman R, Ondeyka J. 1987 U. S. Patent 4,873,247.; d Lopez-Gresa MP, Gonzalez MC, Ciavatta L, Ayala I, Moya P, Primo J. J. Agric. Food Chem. 2006;54:2921–2925. doi: 10.1021/jf0530998. [DOI] [PubMed] [Google Scholar]; e Blanchflower SE, Bank RM, Everett JR, Reading C. J. Antibiot. 1993;46:1355–1363. doi: 10.7164/antibiotics.46.1355. [DOI] [PubMed] [Google Scholar]; f Banks RM, Blanchflower SE, Everett JR, Manger BR, Reading C. J. Antibiot. 1997;50:840–846. doi: 10.7164/antibiotics.50.840. [DOI] [PubMed] [Google Scholar]; g Blanchflower SE, Banks RM, Everett JR, Manger BR, Reading C. J. Antibiot. 1991;44:492–497. doi: 10.7164/antibiotics.44.492. [DOI] [PubMed] [Google Scholar]; h Liesch JM, Wichmann CF. J. Antibiot. 1990;43:1380–1386. doi: 10.7164/antibiotics.43.1380. [DOI] [PubMed] [Google Scholar]; i Nielsen KF, Sumarah MW, Frisvad JC, Miller JD. J. Agric. Food Chem. 2006;54:3756–3763. doi: 10.1021/jf060114f. [DOI] [PubMed] [Google Scholar]; j Ding Y, Gruschow S, Greshock TJ, Finefield JM, Sherman DH, Williams RM. J. Nat. Prod. 2008;71:1574–1578. doi: 10.1021/np800292n. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Antia BS, Aree T, Kasettrathat C, Wiyakrutta S, Ekpa OP, Ekpe UJ, Mahindol C, Ruchirawat S, Kittakop P. Phytochemistry. 2011;72:816–820. doi: 10.1016/j.phytochem.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 5.a Whyte AC, Gloer JB, Wicklow DT, Dowd PF. J. Nat. Prod. 1996;59:1093–1095. doi: 10.1021/np960607m. [DOI] [PubMed] [Google Scholar]; b Qian-Cutrone J, Krampitz KD, Shu Y-Z, Chang L-P, Lowe SE. 2001 U. S. Patent 6,291,461.; c Qian-Cutrone J, Huang S, Shu Y-Z, Vydas D, Fairchild C, Menendez A, Krampitz K, Dalterio R, Klohr SE, Gao Q. J. Am. Chem. Soc. 2002;124:14556–14557. doi: 10.1021/ja028538n. [DOI] [PubMed] [Google Scholar]; d Fenical W, Jensen PR, Cheng XC. 2000 U. S. Patent 6,066,635.; e Sugie Y, Hirai H, Inagaki T, Ishiguro M, Kim Y-J, Kojima Y, Sakakibara T, Sakemi S, Sugiura A, Suzuki Y, Brennan L, Duignan J, Huang LH, Sutcliffe J, Kojima NJ. J. Antibiot. 2001;54:911–916. doi: 10.7164/antibiotics.54.911. [DOI] [PubMed] [Google Scholar]
- 6.a Kato H, Yoshida T, Tokue T, Nojiri Y, Hirota H, Ohta T, Williams RM, Tsukamoto S. Angew. Chem. Int. Ed. 2007;46:2254–2256. doi: 10.1002/anie.200604381. [DOI] [PubMed] [Google Scholar]; b Tsukamoto S, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. J. Am. Chem. Soc. 2009;131:3834–3835. doi: 10.1021/ja810029b. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Tsukamoto S, Kato H, Samizo M, Nojiri Y, Onuki H, Hirota H, Ohta T. J. Nat. Prod. 2008;71:2064–2067. doi: 10.1021/np800471y. [DOI] [PubMed] [Google Scholar]; d Tsukamoto S, Kawabata T, Kato H, Greshock TJ, Hirota H, Ohta T, Williams RM. Org. Lett. 2009;11:1297–1300. doi: 10.1021/ol900071c. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Tsukamoto S, Umaoka H, Yoshikawa K, Ikeda T, Hirota H. J. Nat. Prod. 2010;73:1438–1440. doi: 10.1021/np1002498. [DOI] [PubMed] [Google Scholar]; f Greshock TJ, Grubbs AW, Jiao P, Wicklow DT, Gloer JB, Williams RM. Angew. Chem. Int. Ed. 2008;47:3573–3577. doi: 10.1002/anie.200800106. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kito K, Ookura R, Kusumi T, Namikoshi M, Ooi T. Heterocycles. 2009;78:2101–2106. [Google Scholar]
- 7.a Hayashi H, Nishimoto Y, Nozaki H. Tetrahedron Lett. 1997;38:5655–5658. [Google Scholar]; b Hayashi H, Nishimoto Y, Akiyama K, Nozaki H. Biosci. Biotechnol. Biochem. 2000;64:111–115. doi: 10.1271/bbb.64.111. [DOI] [PubMed] [Google Scholar]; c Banks RM, Blanchflower SE, Everett JR, Manger BR, Reading C. J. Antibiot. 1997;50:840–846. doi: 10.7164/antibiotics.50.840. [DOI] [PubMed] [Google Scholar]
- 8.a Polonsky J, Merrien M-A, Prangé T, Pascard C, Moreau S. J. Chem. Soc. Chem. Commun. 1980:601–602. [Google Scholar]; b Prangé T, Billion M-A, Vuilhorgen M, Pascard C, Polonsky J. Tetrahedron Lett. 1981;22:1977–1980. [Google Scholar]; c Capon RJ, Skene C, Stewart M, Ford J, O'Hair RAJ, Williams L, Lacey E, Gill JH, Heiland K. Org. Biomol. Chem. 2003;1:1856–1862. doi: 10.1039/b302306k. [DOI] [PubMed] [Google Scholar]; d Nielsen KF, Sumarah MW, Frisvad JC, Miller JD. J. Agric. Food Chem. 2006;54:3756–3763. doi: 10.1021/jf060114f. [DOI] [PubMed] [Google Scholar]; e O'Brien M, Nielsen KF, O'Kiely P, Forristal PD, Fuller HT, Frisvad JC. J. Agric. Food Chem. 2006;54:9268–9276. doi: 10.1021/jf0621018. [DOI] [PubMed] [Google Scholar]; f Lee BH, Clothier MF, Kornis GI. In: Alkaloids: Chemical and Biological Perspectives. Chapter 6. Pelletier SW, editor. Vol. 12. Elsevier Science; Oxford: 1998. pp. 344–374. [Google Scholar]
- 9.Lin Z, Wen J, Zhu T, Fang Y, Gu Q, Zhu WJ. Antibiot. 2008;61:81–85. doi: 10.1038/ja.2008.114. [DOI] [PubMed] [Google Scholar]
- 10.a Marínez-Luis S, Rodrígues R, Acevedo L, Gonzáles MC, Lira-Rocha A, Mata R. Tetrahedron. 2006;62:1817–1822. [Google Scholar]; b Figueroa M, González MDC, Mata R. Nat. Prod. Res. 2008;22:709–714. doi: 10.1080/14786410802012361. [DOI] [PubMed] [Google Scholar]; c Figueroa M, Gonzales M, Mata R. Planta Med. 2008;74:PB92. [Google Scholar]; d Watts KR, Loveridge ST, Tenney K, Media J, Valeriote FA, Crews P. J. Org. Chem. 2011;76:6201–6208. doi: 10.1021/jo2009593. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Martínez-Luis S, González MC, Ulloa M, Mata R. Phytochemistry. 2005;66:1012–1016. doi: 10.1016/j.phytochem.2005.03.005. [DOI] [PubMed] [Google Scholar]; f Ding Y, Greshock TJ, Miller KA, Sherman DH, Williams RM. Org. Lett. 2008;10:4863–4866. doi: 10.1021/ol8019633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stocking EM, Sanz-Cervera JF, Williams RM. Angew. Chem. Int. Ed. 1999;38:786–789. doi: 10.1002/(SICI)1521-3773(19990315)38:6<786::AID-ANIE786>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- 12.Stocking EM, Williams RM, Sanz-Cervera JF. J. Am. Chem. Soc. 2000;122:9089–9098. [Google Scholar]
- 13.Williams RM, Cox RJ. Acc. Chem. Res. 2003;36:127–139. doi: 10.1021/ar020229e. [DOI] [PubMed] [Google Scholar]
- 14.Williams RM, Sanz-Cervera JF, Stocking EM. In: Topics in Current Chemistry, Volume on Biosynthesis-Terpenes and Alkaloids. Lepper F, Vederas JC, editors. Vol. 209. Springer; Berlin: 2000. pp. 97–173. [Google Scholar]
- 15.Miller KA, Williams RM. Chem. Soc. Rev. 2009;38:3160–3174. doi: 10.1039/b816705m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.a Porter AEA, Sammes PG. Chem. Commun. 1970:1103. [Google Scholar]; b Williams RM. Chem. Pharm. Bull. 2002;50:711–740. doi: 10.1248/cpb.50.711. [DOI] [PubMed] [Google Scholar]
- 17.a Williams RM, Sanz-Cervera JF, Sancenón F, Marco JA, Halligan K. J. Am. Chem. Soc. 1998;120:1090–1091. doi: 10.1016/s0968-0896(98)00102-3. [DOI] [PubMed] [Google Scholar]; b Williams RM, Sanz-Cervera JF, Sancenón F, Marco JA, Halligan K. Bioorg. Med. Chem. 1998;6:1233–1241. doi: 10.1016/s0968-0896(98)00102-3. [DOI] [PubMed] [Google Scholar]; c Greshock TJ, Grubbs AW, Tsukamoto S, Williams RM. Angew. Chem. Int. Ed. 2007;46:2262–2265. doi: 10.1002/anie.200604378. [DOI] [PubMed] [Google Scholar]; d Greshock TJ, Williams RM. Org. Lett. 2007;9:4255–4258. doi: 10.1021/ol701845t. [DOI] [PubMed] [Google Scholar]; e Greshock TJ, Grubbs AW, Williams RM. Tetrahedron. 2007;63:6124–6130. doi: 10.1016/j.tet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Miller KA, Tsukamoto S, Williams RM. Nature Chem. 2009;1:63–68. doi: 10.1038/nchem.110. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Domingo LR, Zaragoza RJ, Williams RM. J. Org. Chem. 2003;68:2895–2902. doi: 10.1021/jo020564g. [DOI] [PubMed] [Google Scholar]; h Stocking EM, Sanz-Cervera JF, Williams RM. J. Am. Chem. Soc. 2000;122:1675–1683. [Google Scholar]
- 18.Raper KB, Thom C. Manual of the Penicillia. Williams and Wilkins; Baltimore: 1949. [Google Scholar]
- 19.Raper KB, Fennell DI. The Genus Aspergillus. William and Wilkins; Baltimore: 1965. [Google Scholar]
- 20.Miller KA, Figueroa M, Valente MWN, Greshock TJ, Mata R, Williams RM. Bioorg. Med. Chem. Lett. 2008;18:6479–6481. doi: 10.1016/j.bmcl.2008.10.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Miller KA, Welch TR, Greshock TJ, Ding Y, Sherman DH, Williams RM. J. Org. Chem. 2008;73:3116–3119. doi: 10.1021/jo800116y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Frisvad JC, Smedsgaard J, Larsen TO, Samson RA. Stud. Mycol. 2004;49:201–241. [Google Scholar]
- 23.Frisvad JC, Samson RA. Stud. Mycol. 2004;49:1–173. [Google Scholar]
- 24.Houbraken J, Frisvad JC, Samson RA. IMA Fungus. 2011;2:87–95. doi: 10.5598/imafungus.2011.02.01.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.a Paterson RRM, Simmonds MSJ, Blaney WM. J. Invertebr. Pathol. 1987;50:124–133. [Google Scholar]; b Paterson RRM, Simmonds MSJ, Kemmelmeier C, Blaney WM. Mycol. Res. 1990;94:538–542. [Google Scholar]
- 26.Frisvad JC, Filtenborg O. Mycologia. 1989;81:836–851. [Google Scholar]
- 27.a Steyn PS. Tetrahedron. 1971;36:3331–3334. [Google Scholar]; b Steyn PS. Tetrahedron. 1973;29:107–120. [Google Scholar]; c Steyn PS, Vleggaar R. Phytochemistry. 1976;15:355–356. [Google Scholar]; d Scott PM, Kennedy BPC, Harwig J, Chen Y-K. Appl. Microbiol. 1974;28:892–894. doi: 10.1128/am.28.5.892-894.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Arai K, Miyajima H, Mushiroda T, Yamamoto Y. Chem. Pharm. Bull. 1989;37:3229–3235. [Google Scholar]
- 28.Samson RA, Varga J, Meijer M, Frisvad JC. Stud. Mycol. 2011;69:81–97. doi: 10.3114/sim.2011.69.06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Frisvad JC, Rank C, Nielsen KF, Larsen TO. Med. Mycol. 2009;47:S53–S71. doi: 10.1080/13693780802307720. [DOI] [PubMed] [Google Scholar]
- 30.Zhang D, Noviendri D, Nursid M, Yang X, Son BW. Nat. Prod. Sci. 2007;13:251–254. [Google Scholar]
- 31.Baldas J, Birch AJ, Russell RA. J. Chem. Soc., Perkin Trans. I. 1974:50–52. [Google Scholar]
- 32.Sanz-Cervera JF, Glinka T, Williams RM. Tetrahedron. 1993;49:8471–8482. [Google Scholar]
- 33.Boysen M, Skouboe P, Frisvad JC, Rossen L. Microbiology-UK. 1996;142:541–549. doi: 10.1099/13500872-142-3-541. [DOI] [PubMed] [Google Scholar]
- 34.a Kuo MS, Wiley VH, Cialdella JI, Yurek DA, Whaley HA, Marshall VP. J. Antibiot. 1996;49:1006–1013. doi: 10.7164/antibiotics.49.1006. [DOI] [PubMed] [Google Scholar]; b Kuo MS, Yurek DA, Mizsak SA, Cialdella JI, Baczynskyj L, Marshall VP. J. Am. Chem. Soc. 1999;121:1763–1767. [Google Scholar]
- 35.a Filtenborg O, Frisvad JC. In: Modern concepts in Penicillium and Aspergillus classification. Samson RA, Pitt JI, editors. Plenum Press; New York: 1990. pp. 159–170. [Google Scholar]; b Tuthill DE, Frisvad JC, Christensen M. Mycologia. 2001;93:298–308. [Google Scholar]
- 36.a Frisvad JC, Samson RA, Stolk AC. Persoonia. 1990;14:193–202. [Google Scholar]; b Frisvad JC, Samson RA, Stolk AC. Persoonia. 1990;14:209–232. [Google Scholar]
- 37.Pitt JI. The Genus Penicillium and its Teleomorphic States Eupenicillium and Talaromyces. Academic Press; New York: 1992. [Google Scholar]
- 38.a Byung HL, Clothier MF, Dutton FE, Nelson SJ, Johnson SS, Thompson DP, Geary TG, Whaley HD, Haber CL, Marshall VP, Kornis GI, McNally PL, Ciadella JI, Martin DG, Bowman JW, Baker CA, Coscarelli EM, Alexander-Bowman SJ, Davis JP, Zinser EW, Wiley V, Lipton MF, Mauragis MA. Curr. Top. Med. Chem. 2002;2:779–793. doi: 10.2174/1568026023393705. [DOI] [PubMed] [Google Scholar]; b Lee BH, Clothier MF, Johnson SS. Bioorg. Med. Chem. Lett. 2001;11:553–554. doi: 10.1016/s0960-894x(00)00698-3. [DOI] [PubMed] [Google Scholar]
- 39.Ostlind DA, Mickle WG, Ewanciw DV, Andriuli FJ, Campbell WC, Hernandez S, Mochales S, Munguira E. Res. Vet. Sci. 1990;48:260–261. [PubMed] [Google Scholar]
- 40.Zinser EW, Wolfe ML, Alexander-Bowman SJ, Thomas EM, Davis JP, Groppi VE, Lee BH, Thomphson DP, Geary TG. J. Vet. Pharmacol. Therap. 2002;25:241–250. doi: 10.1046/j.1365-2885.2002.00423.x. [DOI] [PubMed] [Google Scholar]
- 41.Shoop WL, Haines HW, Eary CH, Michael BF. Am. J. Vet. Res. 1992;53:2032–2034. [PubMed] [Google Scholar]
- 42.a Kaminsky R, Bapst B, Stein PA, Strehlau GA, Allan BA, Hosking BC, Rolfe PF, Sager H. Parasitol. Res. 2011;109:19–23. doi: 10.1007/s00436-010-2216-0. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Love S. AFBM J. 2010;7:45–52. [Google Scholar]; c Little PR, Hodges A, Watson TG, Seed JA, Maeder SJ. N. Z. Vet. J. 2010;58:121–129. doi: 10.1080/00480169.2010.67513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.a Stocking EM, Sanz-Cervera JF, Unkefer CJ, Williams RM. Tetrahedron. 2001;57:5303–5320. [Google Scholar]; b Kellenberger JL. Ph.D. Thesis. ETH Zurich; Zurich, Switzerland: 1997. The Stereochemical Course of the C-methylation Steps in the Biosynthesis of Bottromycin. [Google Scholar]; c Stocking EM, Sanz-Cervera JF, Williams RM, Unkefer CJ. J. Am. Chem. Soc. 1996;118:7008–7009. [Google Scholar]; d Stocking EM, Martinez RA, Silks LA, Sanz-Cervera JF, Williams RM. J. Am. Chem. Soc. 2001;123:3391–3392. doi: 10.1021/ja005655e. [DOI] [PubMed] [Google Scholar]
- 44.Stocking EM, Sanz-Cervera JF, Williams RM. Angew. Chem. Int. Ed. 2001;40:1296–1298. doi: 10.1002/1521-3773(20010401)40:7<1296::aid-anie1296>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 45.Noonim P, Mahakarnchanakul W, Varga J, Frisvad JC, Samson RA. Int. J. Syst. Evol. Microbiol. 2008;58:1727–1734. doi: 10.1099/ijs.0.65694-0. [DOI] [PubMed] [Google Scholar]
- 46.a Pareniková L, Skouboe P, Frisvad JC, Samson RA, Rossen L, Hoor Suykerbuyk M, Visser J. Appl. Environ. Microbiol. 2001;67:521–527. doi: 10.1128/AEM.67.2.521-527.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Samson RA, Houbraken JAMP, Kuijpers AFA, Frank JM, Frisvad JC. Stud. Mycol. 2004;50:45–61. [Google Scholar]; c Samson RA, Noonim P, Meijer M, Houbraken J, Frisvad JC, Varga J. Stud. Mycol. 2007;59:129–145. doi: 10.3114/sim.2007.59.13. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Perrone G, Varga J, Susca A, Frisvad JC, Stea G, Kocsubé S, Tóth B, Kozakiewicz Z. Int. J. Syst. Evol. Microbiol. 2008;58:1032–1039. doi: 10.1099/ijs.0.65463-0. [DOI] [PubMed] [Google Scholar]; e Sørensen A, Lübeck PS, Lübeck M, Nielsen KF, Ahring BK, Teller PJ, Frisvad JC. Int. J. Syst. Evol. Microbiol. 2011;61:3077–3083. doi: 10.1099/ijs.0.029884-0. [DOI] [PubMed] [Google Scholar]; f Varga J, Frisvad JC, Kocsubé S, Brancovics B, Tóth B, Szigeti G, Samson RA. Stud. Mycol. 2011;69:1–17. doi: 10.3114/sim.2011.69.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Gray CR, Sanz-Cervera JF, Silks LA, Williams RM. J. Am. Chem. Soc. 2003;125:14692–14693. doi: 10.1021/ja037687i. [DOI] [PubMed] [Google Scholar]
- 48.The structures of the norgeamides were published via the Internet detailing the research performed by the Hans-Knöll Institute. www.hki-jena.de
- 49.McAfoos TJ, Li S, Tsukamoto S, Sherman DH, Williams RM. Heterocycles. 2010;82:461–472. doi: 10.3987/COM-10-S(E)19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Frisvad JC, Frank JM, Houbraken JAMP, Kuijpers AFA, Samson RA. Stud. Mycol. 2004;50:23–43. [Google Scholar]
- 51.Grubbs AW, Artman GD, III, Williams RM. Tetrahedron Lett. 2005;46:9013–9016. [Google Scholar]
- 52.a Myers AG, Herzon SB. J. Am. Chem. Soc. 2003;125:12080–12081. doi: 10.1021/ja0372006. [DOI] [PubMed] [Google Scholar]; b Wulff JE, Herzon SB, Siegrist R, Myers AG. J. Am. Chem. Soc. 2007;129:4898–4899. doi: 10.1021/ja0690971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ding Y, de Wet JR, Cavalcoli J, Li S, Greshock TJ, Miller KA, Finefield JM, Sunderhaus JD, McAfoos TJ, Tsukamoto S, Williams RM, Sherman DH. J. Am. Chem. Soc. 2010;132:12733–12740. doi: 10.1021/ja1049302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Finefield JM, Kato H, Greshock TJ, Sherman DH, Tsukamoto S, Williams RM. Org. Lett. 2011:3802–3805. doi: 10.1021/ol201284y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.a Tsuda M, Kasai Y, Komatsu K, Sone T, Tanaka M, Mikami Y, Kobayashi J. Org. Lett. 2004;6:3087–3089. doi: 10.1021/ol048900y. [DOI] [PubMed] [Google Scholar]; b Mugishima T, Tsuda M, Kasai Y, Ishiyama H, Fukushi E, Kawabata J, Watanabe M, Akao K, Kobayashi J. J. Org. Chem. 2005;70:9430–9435. doi: 10.1021/jo051499o. [DOI] [PubMed] [Google Scholar]
- 56.a Kushida N, Watanabe N, Okuda T, Yokoyama F, Gyobu Y, Yaguchi T. J. Antibiot. 2007;60:667–673. doi: 10.1038/ja.2007.85. [DOI] [PubMed] [Google Scholar]; b Kushida N, Watanabe N, Yaguchi T, Yokoyama F, Tsujiuchi G, Okuda T. 2009 U. S. Patent 7,501,431.
- 57.a Houbraken J, Frisvad JC, Samson RA. Fungal Diversity. 2010;44:117–133. [Google Scholar]; b Houbraken J, Frisvad JC, Samson RA. Stud. Mycol. 2011;70:53–138. doi: 10.3114/sim.2011.70.02. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.a Chu Y-S, Niu X-M, Wang Y-L, Guo J-P, Pan W-Z, Huang X-W, Zhang K-Q. Org. Lett. 2010;12:4356–435. doi: 10.1021/ol101817g. [DOI] [PubMed] [Google Scholar]; b Guo J-P, Tan J-L, Wang Y-L-, Wu H-Y, Zhang C-P, Niu X-M, Pan W-Z, Huang X-W, Zhang K-Q. J. Nat. Prod. 2011;74:2278–2281. doi: 10.1021/np200365z. [DOI] [PubMed] [Google Scholar]
- 59.Davies J. Mol. Microbiol. 1990;4:1227–1232. doi: 10.1111/j.1365-2958.1990.tb00701.x. [DOI] [PubMed] [Google Scholar]
- 60.Peterson SW. In: Integration of Modern Taxonomic Methods for Penicillium and Aspergillus Classification. Samson RA, Pitt JI, editors. Harwood; Amsterdam: 2000. pp. 323–355. [Google Scholar]
- 61.a Peterson SW. In: Integration of Modern Taxonomic Methods for Penicillium and Aspergillus Classification. Samson RA, Pitt JI, editors. Harwood; Amsterdam: 2000. pp. 163–178. [Google Scholar]; b Houbraken J, Samson RA. Stud. Mycol. 2011;70:1–51. doi: 10.3114/sim.2011.70.01. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Samson RA, Yilmaz N, Houbraken J, Spierenburg H, Seifert KA, Peterson SW, Varga J, Frisvad JC. Stud. Mycol. 2011;70:159–183. doi: 10.3114/sim.2011.70.04. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Houbraken J, Spierenburg H, Frisvad JC. Antonie van Leeuwenhoek. 2012;101:403–421. doi: 10.1007/s10482-011-9647-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Naik CG, Devi P, Rodrigues EU. 2005 S. Patent 2005143392.
- 63.Nielsen KF, Månsson M, Rank C, Frisvad JC, Larsen TO. J. Nat. Prod. 2011;74:2338–2348. doi: 10.1021/np200254t. [DOI] [PubMed] [Google Scholar]