

Abstract
Staphylococcus aureus is a Gram-positive pathogen that causes devastating disease and whose pathogenesis is dependent on interactions with host cell factors. Staphylococcal clumping factor A (ClfA) is a highly conserved fibrinogen (Fg)-binding protein and virulence factor that contributes to host tissue adhesion and initiation of infection. ClfA is being investigated as a possible component of a staphylococcal vaccine. We report the development of an Fg-binding assay that is specific for ClfA-mediated binding. Using the assay, we show that despite the presence of anti-ClfA antibodies, human sera from unvaccinated subjects are unable to prevent the binding of S. aureus to an Fg-coated surface. In contrast, antibodies elicited by a recombinant ClfA-containing vaccine were capable of blocking the ClfA-dependent binding of a diverse and clinically relevant collection of staphylococcal strains to Fg. These functional antibodies were also able to displace S. aureus already bound to Fg, suggesting that the ligand-binding activity of ClfA can be effectively neutralized through vaccination.
INTRODUCTION
Staphylococcus aureus is a versatile, Gram-positive pathogen of growing medical significance because of the rising incidence of infection, the emergence of highly virulent antibiotic-resistant strains such as community-associated methicillin-resistant S. aureus (MRSA), and the associated increase in medical costs. With limited treatment options available and few new antibiotics in development, there is considerable interest in exploring vaccine-based approaches for controlling staphylococcal infections. The various clinical manifestations of invasive S. aureus disease are mediated by a diverse collection of virulence factors that represent potential antigen targets for new vaccines in clinical development (reviewed in references 3 and 6).
S. aureus clumping factor A (ClfA), a well-established virulence factor and vaccine candidate, is a member of a family of Gram-positive bacterial surface proteins known as microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) (35). The ligand for staphylococcal ClfA is the plasma protein fibrinogen (Fg), the principal component of the coagulation system. ClfA binds specifically to the C-terminal region of the Fg γ chain (23, 27). Fg-bound ClfA promotes the binding of S. aureus to platelets, resulting in thrombus (blood clot) formation (2, 36). It has been proposed that ClfA binding to Fg may also decrease opsonophagocytic killing by enhancing the binding of complement factor I to ClfA on the bacterial surface, which in turn promotes the cleavage of opsonin C3b into inactive fragments (12, 13). A genetic analysis of S. aureus MSCRAMMs identified ClfA as the single most important contributor in a mouse model of sepsis, where it is believed to cause thromboembolic agglutination through its association with polymerized fibrin fibers (25). The importance of ClfA as a virulence factor independent of other S. aureus surface adhesins has been experimentally demonstrated by using Lactococcus lactis. Heterologous expression of ClfA by this otherwise harmless Gram-positive bacterium confers the ability to colonize heart valve tissue in rats with experimental endocarditis (33). In addition, domain-swapping experiments in which the ClfA Fg-binding domain was exchanged with the analogous domain of fibronectin-binding protein A have demonstrated that the Fg-binding capacity of ClfA is sufficient to permit L. lactis colonization of damaged heart valve tissue (32).
A ClfA antigen is a component of an investigational S. aureus vaccine. Initial studies were conducted with an early triantigen vaccine (SA3Ag) that also includes glycoconjugates of capsular antigen types 5 and 8 (ClinicalTrials.gov registry number NCT01018641; reference 34). Studies continued with a final formulation that included the addition of a fourth component, MntC (1). This tetra-antigen vaccine is currently in first-in-human (phase 1/2) studies in the United States (ClinicalTrials.gov registry number NCT01364571). The recombinant ClfA antigen is based on an Fg-binding domain fragment (rClfA) previously shown to protect against septic arthritis in an active-vaccination mouse model of infection (18). The ClfA antigen (rClfAm) in the SA3Ag vaccine differs in that it harbors a mutation (Y338A) that abolishes Fg-binding activity and reduces the virulence of S. aureus strains carrying this mutation (19). In the context of the vaccine antigen, the mutation prevents rClfA from inhibiting the normal clumping of activated platelets and blood clotting. Potential interference with this process is due to the ability of rClfA to bind to and compete for the same C-terminal region of the Fg γ chain as the platelet integrin αIIbβ3 receptor (7, 23).
A desirable property of any vaccine component is the ability to elicit antibodies capable of neutralizing the pathogen, either by opsonophagocytic killing or by blocking the function of virulence factors. For a ClfA-containing vaccine, elicitation of functional antibodies that prevent the binding of S. aureus cells to immobilized Fg constitutes a critical measure of the neutralization of this virulence factor. Enzyme-linked immunosorbent assay (ELISA)-based whole-cell S. aureus ClfA-Fg binding assays have previously been used to define Fg γ-chain peptide and ClfA sequences responsible for S. aureus Fg binding and to identify a monoclonal antibody (MAb) that can interfere with this interaction (14, 15, 27). A radiometric assay to measure ClfA immune serum titers in response to a DNA vaccine has also been described (4). We developed a simpler, high-throughput Fg binding inhibition assay based on a luminescence readout to detect bound live bacteria. Here we demonstrate that the assay is specific for ClfA and that it can be used to measure a biologically relevant immune response to this antigen in sera from animals and human subjects. Our results uncover important differences in the level of functional ClfA antibodies resulting from natural exposure to S. aureus compared with titers following vaccination with a recombinant ClfA-containing vaccine.
MATERIALS AND METHODS
Strain stocks.
S. aureus strains were grown in tryptic soy broth (TSB; BD Biosciences) for 16 h at 37°C with shaking at 250 rpm. Cells were pelleted at 3,700 × g for 15 min, resuspended in 20% glycerol Dulbecco's phosphate-buffered saline (DPBS; Invitrogen), aliquoted into 2-ml cryotubes (Corning), and stored at −70°C. Invasive clinical strains from the previously described Diversity collection and the Centers for Disease Control and Prevention MRSA collection were used, which were selected to maximize the diversity of S. aureus sequence types and to represent MRSA disease isolates circulating in the United States during 2005 (29). A previously described clfA∷Tn917 mutant of strain PFESA0237 (Newman) was used as a negative control in the Fg binding inhibition assay (26). Additional knockout strains were constructed by transducing a clfA∷dhfr mutation constructed in strain RN4220 into strains PFESA0179 and PFESA0119 with bacteriophage 52A or 80α, respectively. Transductions were performed essentially as described previously (22), except that 17 mM sodium citrate was added to the medium used to select transductants. Transductants were shown to have the desired mutation by using diagnostic PCRs and to be of the expected strain lineage with a Qualicon RiboPrinter (DuPont, Wilmington, DE).
Recombinant proteins.
Recombinant polypeptides representing various segments of the A domain of ClfA from S. aureus strain PFESA0237 were expressed and purified as previously described (7, 14). The N1-N2-N3 fragment corresponds to amino acids 40 to 559, the N2-N3 fragment corresponds to amino acids 221 to 559, the N2 fragment corresponds to amino acids 221 to 369, and the N3 fragment corresponds to amino acids 370 to 559 (7). The DNA encoding these fragments was subcloned into Escherichia coli expression vector pQE-30 (Qiagen, Valencia, CA) for the expression of recombinant fusion proteins containing an N-terminal six-histidine tag. Recombinant proteins were purified from E. coli lysates by metal affinity chromatography on a chelating Sepharose Fast Flow resin (Amersham Biosciences, Piscataway, NJ) and further purified using anion-exchange chromatography on a Q Sepharose column (Amersham Biosciences, Piscataway, NJ). Additional recombinant ClfA proteins were expressed without a histidine tag using a T7 RNA polymerase E. coli expression vector and purified with a combination of hydrophobic and anion-exchange chromatography steps. These include functionally active wild-type T7ClfA-N1N2N3 (rClfA) and the Fg-binding mutant T7ClfA-N1N2N3 Y338A (rClfAm) (7). The sequence of both rClfA and rClfAm corresponds to amino acid residues 40 to 559. The concentration of purified proteins was determined with the bicinchoninic acid assay (Pierce Biotechnology, Inc., Rockford, IL). Protein purity was assessed by sodium dodecyl sulfate-polyacrylamide gel electrophoresis, and endotoxin levels were analyzed by using the Limulus amebocyte lysate assay (Charles River, Wilmington, MA).
Fg binding inhibition assay.
Microplate wells were coated with 1.7 μg/ml human Fg (Calbiochem) in binding buffer (200 mM sodium carbonate, pH 10.0), incubated with blocking solution (MBA; GE Healthcare) to reduce nonspecific binding, and rinsed in wash buffer (0.1% bovine serum albumin in DPBS). Bacteria (106 to 107 CFU) were preincubated separately with antisera (serially diluted in wash buffer), and samples were subsequently transferred to the Fg-coated plate. After 30 min at 37°C, adherent cells were washed and quantified using the luciferase-based BacTiter-Glo lysis reagent (Promega), which measures the ATP released from live bacteria and the associated luminescence. After incubation with reagent at 37°C for 15 min, the plate was sealed with tape and read on an Envision 2100 Multilabel Reader (Perkin-Elmer L100) or Spectramax luminometer model L (Molecular Devices). In later iterations of the assay, an automated plate washer (Aquamax; Molecular Devices) was found to significantly increase the assay signal relative to the nonspecific background. S. aureus strain PFESA0237 and an isogenic ClfA knockout strain were used initially for development purposes to identify conditions maximizing ClfA-specific binding of cells while minimizing nonselective binding.
Fg binding displacement assay.
The Fg binding displacement assay procedure was performed in the same way as the Fg binding inhibition assay procedure, except that bacteria were added directly to the Fg plate without preincubation with sera or purified antibodies. After washing of bound cells, antibody or Fg was added to the plate and incubated for 30 min at room temperature (RT). Released cells were recovered by transferring the well contents and subsequent DPBS rinses to a separate deep-well microplate. Serial dilutions of the eluted samples were plated on tryptic soy agar plates, and CFU were counted after overnight incubation at 37°C. Residual bacteria remaining bound to the Fg plate were quantified by luminescence (as described above) after additional DPBS washing.
Control and immune sera.
Animal sera were generated according to Pfizer's laboratory animal care and use policy, which reflects an absolute commitment that animals used are treated humanely and that such research is conducted only after appropriate ethical consideration and review. Human clinical sera were obtained from study NCT01018641 (http://clinicaltrials.gov/), which was designed and conducted in accordance with the highest scientific and ethical standards, as well as regulatory requirements; the studies conducted with the human serum were exploratory using nonqualified assays.
Rabbit or rhesus macaque immune serum of known titer was included on every assay test plate to monitor assay performance across independent experiments. Immune sera were generated by injecting rabbits with 50 μg of rClfAm Y338A antigen and 250 μg AlPO4 at weeks 0, 4, and 6 (2 × 0.25 ml given intramuscularly [i.m.]). Macaques were injected with 200 μg of rClfAm Y338A antigen or 2 × 108 heat-killed S. aureus (0.5 ml i.m.). In this case, both vaccines were formulated with 10 μg (10 ISCO units) of Iscomatrix adjuvant (CSL Biotherapies Inc., USA). Affinity-purified ClfA immune antibodies from human donors (Veronate; Inhibitex) were used to construct a proficiency control plate representing a low (1:160), medium (1:40), or high (1:10) starting dilution titer. ClfA MAb 12-9 was included as an additional reference control at a final starting concentration of 1.2 μg/ml. This control panel was used to validate critical assay reagents such as additional S. aureus strains, new cell batches, or Fg lots. For animal studies and for the testing of clinical sera, samples were rerun whenever control serum titers varied by more than 2-fold (or 1 dilution point) from benchmark historic values. As described above, serially diluted sera were incubated with cells prior to addition to the Fg plate. Testing of pre- and postvaccination human subject sera was done with the operator blinded to sample identity.
Serum titers were determined from dilution plots and represent the reciprocal of the lowest serum dilution at which >90% inhibition of Fg binding (EC90) was observed relative to the control with no added serum (maximum value). Test sera were routinely evaluated by starting at a 1:100 dilution (final concentration in the assay). A titer of 100 (1:100 dilution) was chosen as the lower limit of detection (LLOD), with an arbitrary titer of 50 (0.5 times the LLOD) defined as the baseline (negative) level.
Antibody depletion by rClfA.
The specificity of Fg binding inhibition by immune serum was examined by evaluating the effect of preincubating sera with recombinant ClfA antigens (rClfA, rClfAm). Undiluted ClfA rabbit antiserum (5 μl) was preincubated overnight with different antigen concentrations (1 μg, 0.3 μg, or 0.1 μg in 50 μl of DPBS). The treated serum was then serially diluted in a microtiter plate, and 3-μl volumes were combined with 297 μl of 1 × 106 PFESA0237 cells according to the assay procedure.
Antibody purification.
Total IgG was purified from crude serum using HiTrap protein G columns (GE Healthcare). Columns were equilibrated with binding buffer (0.1 M sodium phosphate [pH 7.4], 0.15 M sodium chloride) prior to applying 1 to 10 ml of serum (diluted 1:1 with binding buffer). The column was then washed with binding buffer, and bound IgG was eluted with 10 ml pH 2.8 IgG elution buffer (Thermo Scientific). Eluted IgG was collected in 1-ml fractions with 45 μl/ml of 1 M Tris, pH 9.0 (Calbiochem). Protein concentrations of IgG fractions were determined by measuring optical density at 280 nm (OD280) with an EC280 (molar extinction coefficient) of 210,000 M−1 cm−1. The second fraction from each set of purified IgG contained >96% of the total protein eluted from the column and was used in experiments. ClfA-specific IgG was subsequently purified by affinity chromatography on a ClfA-Sepharose column. The ClfA affinity resin was prepared by coupling of recombinant ClfA to cyanogen bromide-activated Sepharose 4B (GE Healthcare) according to the manufacturer's instructions. Elution was monitored by dual-wavelength UV at 210 and 280 nm. The total IgG fraction eluted from the HiTrap protein G column was extensively dialyzed at 4°C against PBS (pH 7.4) buffer and loaded onto a ClfA-Sepharose 4B affinity column (C 10/10; GE Healthcare) equilibrated with the same buffer. Bound ClfA-specific IgG was eluted using 100 mM Gly-HCl, pH 2.7, buffer. Eluted protein was collected as 1-ml fractions in tubes containing 0.15 ml 1 M Tris-HCl, pH 9.0. Collected ClfA-specific IgG fractions were pooled, dialyzed against PBS (pH 7.4) buffer, and stored at 4°C. From 10 ml of pooled human serum, 95 mg of total IgG was eluted from the HiTrap protein G column. From this material, 1.2 mg or 1.3% of ClfA-specific IgG was recovered by ClfA-Sepharose column chromatography.
MAb production.
Four-week-old BALB/c mice received 0.1-ml subcutaneous injections at 0, 3, and 6 weeks with 10 μg of wild-type recombinant ClfA (6×His-N1N2N3, rClfA) and 200 μg AlPO4 adjuvant. At week 10, mice were boosted intraperitoneally with the same antigen dose, and spleens were harvested 3 days later. Splenocytes were fused to a nonsecreting myeloma cell line (X63Ag.8.653; ATCC). Polyethylene glycol-induced cell fusion and cell cultivation methods were performed according to the MAb production protocol in reference 39. Resulting hybridomas were subsequently screened following fusion by ELISA for antibody recognition of recombinant rClfA as described previously (14) and by flow cytometry to confirm detection of ClfA on the bacterial surface. MAb-producing hybridomas for MAbs 12-9 and 15EC6 were obtained from Inhibitex (Alpharetta, GA).
SPR measurements.
Surface plasmon resonance (SPR) was used to test the recognition of different recombinant ClfA protein domains by ClfA MAbs as described previously (14). Briefly, binding analysis was performed on a BIAcore 3000 (Biacore, Piscataway, NJ) by the ligand capture method. Supernatants containing anti-ClfA MAbs were passed over a Biacore chip coupled to rabbit anti-mouse Fc capture antibody. Individual purified recombinant protein fragments prepared as 60-μg/ml solutions were passed over the chip, and the response was measured. Each MAb was evaluated for the ability to recognize four different protein fragments corresponding to the following ClfA domains, i.e., N1-N2-N3 (rClfA), N2-N3, N2, and N3. The data were processed using the Biacore evaluation software (version 3.1; Biacore, Piscataway, NJ).
Flow cytometry.
Antigen-specific MAbs recognizing capsular polysaccharide 5 (CP5) and CP8 and the ClfA and ClfB surface proteins were used as primary antibodies for flow cytometry. Overnight bacterial cultures were washed in PBS and killed by heating (1 h, 60°C). Cells were then fixed in 1% paraformaldehyde–PBS (10 min) and blocked with 10% porcine serum–PBS. MAbs were added (20 μg/ml) and incubated for 30 min on ice. Mouse IgG was included as a negative control. Cells were similarly incubated with biotinylated anti-mouse IgG, followed by streptavidin-phycoerythrin (PE; BD Biosciences) with intermittent washing in 10% porcine serum–PBS. After two final washes, cell pellets were resuspended in 1% paraformaldehyde. For each well, 20,000 events were acquired on a BD LSR II flow cytometer and analyzed using FlowJo v7 software (Treestar, Ashland, OR). The mean fluorescence intensity of the PE channel was determined for each sample after gating on bacterial cells in the logarithmic forward scatter versus side scatter dot plot.
ELISAs.
S. aureus cells were grown in 50-ml TSB cultures to an OD600 of ∼1.0/ml. The culture was centrifuged, and the cell pellet was resuspended in an equal volume of PBS. High-binding 96-well microtiter plates (Immulon-2; Thermo Scientific) were coated overnight at 4°C with 0.1 ml cells/well. Plates were washed three times with ELISA buffer (0.01 M Tris-buffered saline [pH 7.2], 0.1% Brij 35; Cellgro) using a BioTek plate washer (EL405) and blocked for 2 h at RT with 10% porcine serum in 1× PBS. After washing as before, duplicate 11-point 2-fold serial dilutions of test sera prepared in 10% porcine serum–PBS buffer were added to each plate row along with a no-serum blank (column 12). After 2 h of incubation at RT, the plates were washed again and incubated for 2 h at RT with 0.1 ml/per well of secondary alkaline phosphatase-conjugated goat anti-human IgG antibody (Southern Biotech) diluted 1:2,000 in 10% porcine serum–PBS buffer. After a final washing step, the plates were developed according to the manufacturer's directions using an alkaline phosphatase detection kit (KPL). EC50 titers were interpolated from dose-response curves fitted with GraphPad Prism5 (variable slope). ClfA ELISAs were done similarly, except that plates were coated with 2.5 μg/ml rClfAm and PBS–0.05% Tween 20 was used instead of porcine serum as a blocking agent and diluent.
ClfA MAb interference mapping and binding kinetics study.
Epitope binding of ClfA MAbs was measured with ForteBio's Octet Red96 instrument. Purified mouse MAbs raised against S. aureus rClfA were used in matrix interference mapping studies. Microplate wells contained a minimum volume of 200 μl for proper immersion of biosensors. Experiments were conducted at 30°C with shaking at 1,000 rpm. Software version 6.4.1 was used for data acquisition and analysis. ClfA MAbs at 25 μg/ml were captured for 5 min on biosensors coated with anti-murine Fv (IgG) antibodies (loading step 1), followed by buffer washing for 1 to 2 min. rClfA (50 μg/ml) was loaded onto captured antibodies for 5 min (loading step 2), followed by a second buffer wash for 1 to 2 min. Biosensors were then immersed in 25 μg/ml of MAb (secondary) for 5 min to study interference. To determine binding affinity, anti-ClfA MAbs (25 μg/ml) were allowed to saturate a row of eight commercially available biosensors (ForteBio) coated with murine anti-Fv IgG. Twofold serial dilutions of rClfA antigen from a starting concentration of 50 nM were allowed to associate for 10 min and dissociate for 15 min. Duplicate measurements were captured within each experiment by reusing the reagent samples with fresh biosensor tips. Kinetic measurements were repeated three times in independent experiments to determine average values. Each experiment included a noninterfering capture MAb as a negative control. Buffer-subtracted kinetic data were globally fitted with a 1:1 Langmuir model to obtain a dissociation rate constant (KD = koff/kon).
RESULTS
Antiserum to the rClfAm vaccine antigen inhibits binding of S. aureus to immobilized Fg.
A high-titer anti-ClfA immune serum selected from a group of rabbits immunized with rClfAm was found to inhibit the binding of S. aureus to Fg at serum dilutions of up to 1:20,000 (Fig. 1A). In contrast, preimmune serum from the same animal had no effect on Fg binding. An isogenic ClfA knockout (clfA∷Tn917) strain failed to bind Fg, indicating that under these assay conditions, Fg binding was dependent on ClfA. To assess whether the inhibition of S. aureus Fg binding was mediated by antibodies directed against ClfA, the serum was preincubated with various amounts of rClfAm or rClfA. As shown in Fig. 1B and C, preincubation of immune serum with these ClfA polypeptides restored Fg binding to levels that were approximately 70% of those observed with preimmune control sera at the highest concentration tested (1:2,000 dilution). The effect was concentration dependent, with the immunizing rClfAm antigen showing activity comparable to that of the wild-type rClfA protein in preventing inhibition. These results demonstrate that antibodies elicited by vaccination with rClfAm that inhibit S. aureus Fg binding are ClfA specific. We can also conclude that the Fg binding mutation present in rClfAm does not adversely affect ClfA antigenicity, as depletion of immune serum with rClfAm or rClfA blocks Fg binding to a similar extent.
Fig 1.

ClfA vaccination generates antibodies that inhibit the binding of S. aureus to immobilized Fg. Bacteria preincubated with serially diluted sera were added to Fg-coated microplate wells, and after washing, luminescence of live adherent cells was detected with luciferase reagent. (A) Pretreatment of strain PFESA0237 with rabbit rClfAm immune serum prevents Fg binding across 103 and 104 dilutions (week [wk] 10, open circles). In contrast, analogous dilutions of preimmune sera show minimal suppression (week 0, closed circles). An isogenic clfA knockout strain (clfA∷Tn917) fails to bind regardless of the serum dilution (dotted lines). (B, C) Preincubation of the week 10 immune serum with rClfAm (B) or rClfA (C) antigens results in a concentration-dependent reversal of the serum-mediated inhibition of S. aureus Fg binding. Error bars represent standard deviations of duplicate samples.
Characterization of functional antibodies.
To investigate the locations of the epitopes responsible for inhibition of ClfA-Fg interaction, a panel of anti-ClfA MAbs that recognize the Fg-binding domain was characterized. Results of Fg binding inhibition assays, plasmon resonance binding with ClfA subdomain fragments, and MAb interference mapping experiments are summarized in Fig. 2 and Table 1. We confirmed that reference MAb 12-9, which was previously shown to inhibit the binding of recombinant ClfA to Fg in an ELISA (14), also blocked binding in the Fg binding inhibition assay with an EC50 of 4 ng/ml. A second MAb, 15EC6, that was reported to be unable to prevent ClfA Fg binding in an ELISA (14) was found to be inactive in the Fg binding inhibition assay. ClfA MAb 12-9 recognizes an N3 subdomain fragment epitope, while MAb 15EC6 binds to an N2 domain epitope. Binding sites for five additional MAbs were inferred in pairwise binding competition experiments using the Octet BioLayer interferometer, which identified instances of interference or binding site overlap for each antibody relative to all of the others (see Materials and Methods; see also Table S1 in the supplemental material). Three of these were found to be inhibitory in the Fg binding inhibition assay but were less potent than MAb 12-9, exhibiting 3- to 12-fold higher EC50s. MAb 23-3 recognizes an N3 domain epitope and interferes with the binding of MAb 12-9, suggesting an adjacent or overlapping binding site. The second functional MAb, 33-32, binds strongly to an N2 domain site that does not overlap the binding sites of any of the other MAbs. The third functional MAb (32-105) binds to a site that overlaps that recognized by nonfunctional MAb 15EC6 but requires the presence of both the N2 and N3 domains for binding. Noninhibitory MAbs 11-9 and 43-6 also recognize the intact N2-N3 domain in a region that does not intersect with sites for any of the other MAbs. Collectively, these results demonstrate that only a subset of ClfA-specific antibodies recognizing spatially distinct epitopes within the N2-N3 domain are capable of preventing the binding of S. aureus to Fg.
Fig 2.
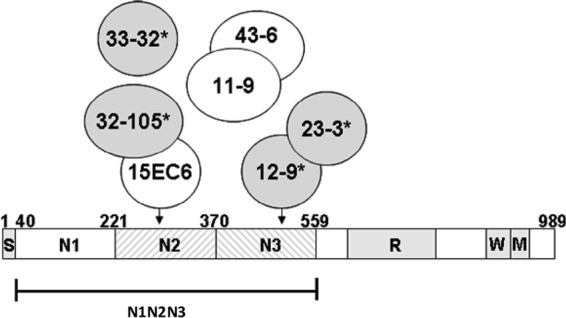
A subset of ClfA MAbs inhibit Fg binding. Schematic representation of S. aureus ClfA and the recombinant N1 to N3 vaccine antigen (rClfAm). S, signal peptide; N1 to N3 Fg-binding subdomains; R, serine-aspartate dipeptide repeat region; W, cell wall-spanning region; M, membrane-spanning region. Circles represent ClfA-specific MAbs that bind within the N2 and N3 domains. Their subdomain specificities, ClfA binding affinities, and abilities to interfere with S. aureus Fg binding are summarized in Table 1. Arrows reflect the specificities of MAbs 15EC6 and 12-9 for subdomain N2 and N3 epitopes. Intersecting circles indicate that epitopes recognized by MAbs overlap, as indicated by binding interference experiments (see Table S1 in the supplemental material). Four MAbs that prevent S. aureus Fg binding are shaded (with asterisks); three others are nonfunctional.
Table 1.
Summary of ClfA MAb properties
| MAb | Fg binding inhibition EC50 (ng/ml)a | Domain specificityb | ClfA affinity KD (nM)c |
|---|---|---|---|
| 12-9 | 3.8 ± 0.3 | N3 | 1.1 |
| 15EC6 | Inactive | N2 | 3.2 |
| 11-9 | Inactive | N2, N3 | 2.4 |
| 43-6 | Inactive | N2, N3 | 2.2 |
| 23-3 | 9.6 ± 0.6 | N3 | 24.7 |
| 33-32 | 47.2 ± 4.1 | N2 | 1.6 |
| 32-105 | 31.2 ± 18.7 | N2, N3 | 12.6 |
EC50s for Fg binding inhibition by MAbs are the averages of two experiments with standard deviations.
Specificity is based on SPR data obtained with recombinant subdomain fragments (see Materials and Methods).
MAb kinetic values were determined by BioLayer interferometry (ForteBio Octet instrument) with rClfA antigen in the absence of Fg.
Functional antibodies are elicited by rClfAm but not by heat-killed S. aureus.
To determine whether rClfAm could induce functional antibodies in nonhuman primates (NHPs), a group of rhesus macaques was vaccinated i.m. at week 0 and week 4 with 200 μg of rClfAm antigen and Iscomatrix adjuvant and immune sera were evaluated in the Fg binding inhibition assay (Fig. 3). Vaccination with 2 × 108 heat-killed S. aureus strain PFESA0164 bacteria also formulated with Iscomatrix adjuvant was included as a comparator to mimic infection with S. aureus. The inoculum was prepared from stationary-phase cells grown in TSB medium, and ClfA surface expression was confirmed by flow cytometry prior to injection (Table 2). None of the animals showed detectable preexisting Fg binding inhibition assay antibody titers prior to vaccination (Fig. 3A). Three of five rClfAm-vaccinated animals showed significant Fg binding inhibition antibody responses after 2 weeks, and all of the animals responded following the second dose (week 6). Fg binding inhibition assay serum antibody titers at weeks 6 and 10 were 40× and 60× higher than the baseline, respectively. In contrast, heat-killed bacteria failed to induce detectable functional antibodies in the ClfA Fg binding inhibition assay in any animal at any time point. However, weak ClfA-binding IgG responses were detected by ClfA ELISA at weeks 6 and 10 (Fig. 3B). Corresponding whole-cell ELISA titers confirmed the induction of an IgG response to bacterial vaccination: a 5-fold titer increase was observed by week 2, which declined following the second dose of antigen at week 4 to 3.5 times the baseline level by week 10 (Fig. 3C).
Fig 3.

Vaccination of NHPs with recombinant ClfA antigen but not heat-inactivated bacteria induces functional antibodies that inhibit binding of S. aureus to Fg. (A) Groups of five and four rhesus macaques were vaccinated with rClfAm antigen (200 μg) or heat-killed S. aureus strain PFESA0164 (2 × 108), respectively, and serum samples were collected at week (wk) 2, week 6, and week 10. Animals were dosed at week 0 and week 4. Bars reflect Fg binding inhibition titers (geometric mean titers) determined with strain PFESA0237, and error bars show 95% confidence intervals. Vaccination titers elicited by rClfAm were significantly different from those elicited by heat-killed cells (weeks 6 and 10) (P < 0.05, two-sided t test). The dotted line marks 0.5 times the LLOD. (B, C) ClfA and bacterial ELISA activities of sera from monkeys vaccinated with heat-killed bacteria show the kinetics of IgG responses. ClfA specificity of the response was demonstrated at all of the time points by complete inhibition in the presence of soluble rClfAm (not shown). Corresponding bacterial ELISA titers represent the reciprocal of EC50s calculated from serum dilution plots fitted with the four-parameter logistic equation (geometric mean titers, 95% confidence intervals).
Table 2.
Genotypes, in vitro surface antigen expression levels, and serum ClfA titers of S. aureus isolates evaluated in the Fg binding inhibition assay
| S. aureus strain | ST/CCa | ClfA variantb | % ClfA sequence identityc |
In vitro test result |
||||
|---|---|---|---|---|---|---|---|---|
| Surface antigen detectiond |
Fg binding inhibition titer (EC90)e |
|||||||
| ClfA | CP5 | CP8 | Rabbit serum | NHP serum | ||||
| PFESA0237 | 254/8 | ClfA_011 | 99.8 | +++ | + | − | 6,400 | 3,200 |
| PFESA0237 clfA∷Tn917f | − | + | − | No binding | No binding | |||
| PFESA0179 | 5/5 | ClfA_002 | 91.8 | +++ | +++ | − | 800 | 800 |
| PFESA0179 clfA∷dhfrf | − | +++ | − | No binding | No binding | |||
| PFESA0119 (MRSA) | 8/8 | ClfA_001 | 100 | + | − | − | 3,200 | 3,200 |
| PFESA0119 clfA∷dhfrf | − | − | − | No binding | No binding | |||
| PFESA0176 | 45/45 | ClfA_009 | 95 | ++ | − | +++ | 3,200 | 3,200 |
| PFESA0164 | 32/30 | ClfA_004 | 94.5 | ++ | − | +++ | 25,600 | 3,200 |
| PFESA0186 | 57/30 | ClfA_004 | 94.5 | ++ | − | +++ | 3,200 | 25,600 |
| PFESA0013 (MRSA) | 5/5 | ClfA_002 | 91.8 | +++ | − | − | 1,600 | 1,600 |
| PFESA0148 | 18/15 | ClfA_020 | 91.2 | + | − | − | 1,600 | 800 |
| PFESA0195 (MRSA) | 93/NC | ClfA_035 | 90.9 | + | − | + | 800 | 400 |
| PFESA0035 | 8/8 | ClfA_001 | 100 | ++ | − | − | 3,200 | 3,200 |
| PFESA0172 | 39/30 | ClfA_001 | 100 | ++ | − | +++ | 12,800 | 6,400 |
| PFESA0067 (MRSA) | 8/8 | ClfA_004 | 94.5 | ++ | − | − | 3,200 | 1,600 |
| PFESA0229 (MRSA) | 8/8 | ClfA_001 | 100 | +++ | − | − | 6,400 | 6,400 |
| PFESA0274 (MRSA) | 8/8 | ClfA_001 | 100 | + | − | − | 3,200 | 1,600 |
ST/CC denotes multilocus sequence type/clonal complex. NC, no clonal complex yet assigned.
ClfA genotypic variants are described in reference 29.
Percent identity of the strain's ClfA sequence with the immunizing antigen is indicated.
Antigen-specific MAb staining intensity is represented as the mean fluorescence intensity relative to that of a mouse IgG control and is scored by range as follows: <3×, −; 3× to 10×, +; 10× to 20×, ++; >20×, +++.
Fg binding inhibition EC90 values were calculated from dilution plots (binding activity versus fold serum dilution) as the reciprocal of the lowest 2-fold serum dilution at which >90% binding inhibition (EC90) is observed relative to the control maximum (no serum).
Isogenic ClfA knockout strain.
The binding of diverse S. aureus strains to Fg is inhibited by ClfA antibodies.
Additional S. aureus invasive clinical strains were evaluated in the Fg binding inhibition assay in order to examine the influence of surface antigen expression and ClfA sequence variation on ClfA-mediated Fg binding (Table 2). The strains evaluated were methicillin-susceptible S. aureus and MRSA strains from clonal complexes 5 (USA100) and 8 (USA300), the predominant genotypic groups circulating in the United States. Also tested were strains from clonal complex 30, the most prevalent S. aureus group in Europe, South America, and Asia, and clonal complex 45, another distinct genotypic cluster. In addition, strains expressing ClfA sequence variants with maximum amino acid sequence divergence (91%) relative to the immunizing antigen were included. Stationary-phase bacterial cultures were tested in the assay and analyzed in parallel by flow cytometry to measure levels of ClfA and capsular antigen expression.
As shown in Table 2, all of the cultured S. aureus clinical strains evaluated expressed ClfA on the bacterial surface. Serially diluted antisera to rClfAm from vaccinated rabbits and rhesus macaques were tested in the assay, and EC90s were determined. Although the level of ClfA was variable and did not strictly correlate with Fg-binding activity (based on the activity of no-serum controls [not shown]), the binding of every strain to Fg could be completely inhibited by ClfA immune sera, with EC90 titers varying from 1:400 to 1:25,600. In contrast, three different isogenic ClfA knockout control strains failed to show detectable ClfA expression or Fg binding. Coexpression of CP5 or CP8 did not prevent binding of S. aureus to Fg or appear to influence inhibition by antisera. For example, strains expressing the highest levels of surface CP5 or CP8 in vitro were as sensitive to Fg binding inhibition by ClfA antisera as strains expressing little or no capsule.
Inhibition of Fg binding by ClfA antisera was also observed with strains grown to mid-log phase (data not shown). However, the use of exponential-phase cells invariably resulted in higher levels of nonspecific binding (apparent at high concentrations of inhibitory ClfA antibody or with ClfA knockout strains) that we attribute to the expression of other Fg-binding surface adhesins such as ClfB (31). As a result, strains were routinely harvested from stationary-phase cultures for use in the assay.
Inhibition of S. aureus Fg binding by human sera from an experimental triantigen vaccine clinical study.
A phase 1 double-blind randomized placebo-controlled study of healthy volunteers was designed to assess the immunogenicity and safety of a three-antigen S. aureus investigational vaccine composed of CP5 and CP8 conjugated to CRM197 and rClfAm (SA3Ag; ClinicalTrials.gov number NCT01018641; reference 34). In this Australian first-in-human study, sera were sampled at various times after i.m. vaccination with a single dose of vaccine (one of three ascending dose levels). Prevaccination and week 2 postvaccination sera from a subset of donors (n = 20) injected with the midlevel dose (60 μg rClfAm and 30 μg of CP5- and CP8-CRM197 conjugates) were tested in the Fg binding assay using five S. aureus strains. In addition to the assay development strain (PFESA0237), four clinically relevant invasive strains, including three USA300 MRSA isolates, were evaluated. As shown in Fig. 4A, 65 to 85% of the vaccinated subjects showed Fg binding inhibition titers of 2- to 128-fold over the LLOD baseline of 100. In contrast, only a single subject showed a detectable prevaccination serum titer at the outset of the study (week 0). In this individual, prevaccination Fg binding inhibition titers were detected at 4 to 8 times the baseline levels and were subsequently boosted 2- to 8-fold following vaccination. Sera from an additional 27 placebo subjects evaluated with strain PFESA0237 also failed to show significant Fg binding inhibition assay titers (data not shown). Despite the lack of functional activity detected in sera from almost all prevaccinated volunteers, low-titer ClfA-binding antibodies were detected by ELISA (Fig. 4B). The specificity of this IgG for ClfA was demonstrated by blocking of ELISA activity with rClfAm (Fig. 4C).
Fig 4.

The SA3Ag vaccine generates antibodies that inhibit the Fg binding of invasive S. aureus clinical isolates. (A) Sera from 20 subjects vaccinated with a single dose of the triantigen vaccine (and sampled at the 2-week [wk] time point) were tested in the Fg binding inhibition assay with five S. aureus strains. Except for a single low-level preimmune Fg binding inhibition assay titer in one subject detected with four of five strains (marked with daggers), prevaccination titers were generally absent. (B) Corresponding ClfA IgG levels were determined by ELISA using affinity-purified ClfA IgG from pooled vaccinated subjects as the standard. (C) The presence of preexisting ClfA-specific IgG in sera of prevaccinated subjects (at a 1:20 dilution) was confirmed by demonstrating loss of ELISA activity upon depletion with free ClfA antigen. Lines and error bars reflect geometric mean titers and 95% confidence intervals, respectively.
ClfA-mediated S. aureus binding to Fg can be selectively released by functional antibodies.
ClfA binds to Fg through contacts between C-terminal amino acids of the Fg γ chain and ClfA amino acids within the intersecting cleft formed between the N2 and N3 domains (7, 10, 11). The mechanism by which antibodies interfere with this process is unknown. We explored whether the ClfA-mediated binding of S. aureus to an Fg-coated surface can be reversed by Fg ligand or by anti-ClfA antibodies. As shown in Fig. 5A, incubation with an excess of soluble Fg released the majority of the bound bacteria, with less than 20% of the bacteria remaining. By measuring the bacteria displaced in this way, the number of cells bound to the microwell surface was estimated to be 1.4 × 105 ± 0.6 × 105 [standard deviation] CFU (n = 7). Treatment of Fg-bound cells with MAb 12-9 released equivalent numbers of bacteria, while mock treatment released fewer than 25% of the Fg-bound cells. Antibody titrations demonstrated that antibody-induced release of bound bacteria was dependent on the IgG concentration and restricted to functional MAbs that selectively block Fg binding (Table 1; Fig. 5B). Nonfunctional MAbs 15EC6, 43-6, and 11-9 all failed to displace S. aureus bound to Fg. Corresponding luminescence data confirmed that displacement of bacteria from the Fg-coated microwell by functional antibodies is accompanied by a loss of bound cells from the same surface, which is not observed with nonfunctional antibodies (Fig. 5C).
Fig 5.

S. aureus is released from immobilized Fg by free ligand or by functional antibodies. (A) Washed PFESA0237 bacteria bound to Fg-coated microplate wells were incubated with Fg (1 mg/ml), PBS, or MAb 12-9 (2.4 μg/ml) for 30 min at RT. After mixing, cells released by treatment were counted by CFU plating (shaded bars, numbers of CFU released) and the remaining adherent cells were quantified by luminescence detection with values normalized relative to those of untreated wells (open bars, percentage of relative light units [RLU] retained). (B) Bacterial release following treatment with 10-fold dilutions of ClfA MAbs. Antibodies that are active in the Fg binding inhibition assay (closed symbols, Table 1) also reverse binding in a concentration-dependent manner. Inactive MAbs (open symbols, dotted lines) fail to release bound cells. (C) Companion luminescence data measuring bound cells remaining following MAb treatment and washing. (D) The activity of total IgG and ClfA-IgG purified from vaccinated (week [wk] 4) donors in releasing bacteria (in this case, PFESA0172) is compared with that of MAb 12-9. ClfA-IgG was purified from the total IgG pool by antigen affinity chromatography with recovery of 1.3%. Concentration-dependent release of bacteria was not observed with IgG purified from prevaccinated subjects (week 0) or with nonfunctional MAb 15EC6. Similar results were obtained in an independent experiment. Error bars represent standard deviations of duplicate samples.
A comparison of results from Fg-binding displacement experiments and Fg-binding inhibition experiments reveals that the concentrations of functional antibodies required to disrupt the S. aureus-Fg complex was greater than the levels sufficient to prevent binding to immobilized Fg. As an example, the concentration of MAb 12-9 that was able to displace 70 to 80% of the bound cells was approximately 13 times the concentration needed to prevent >90% binding. Disruption of the bacterium-Fg complex with the three other functional MAbs was achieved only at concentrations 32 to 64 times the amount required to prevent the interaction.
We next investigated whether polyclonal anti-ClfA immune sera generated against rClfAm were also able to reverse S. aureus binding to Fg and if the potency of mediating bacterial release was comparable between MAb and polyclonal immune sera. In this experiment, we utilized immune sera from the triantigen (SA3Ag) phase I clinical trial described earlier. Total IgG was purified from sera from two subjects sampled 4 weeks after vaccination with equivalent Fg binding inhibition assay titers (of 6,400) and from corresponding prevaccination sera. Next we purified ClfA-specific IgG by ClfA affinity chromatography. Dilutions of equivalent concentrations of MAb 12-9 IgG and the ClfA-specific preparation of human IgG were subsequently tested in the release assay. As shown in Fig. 5D, ClfA-specific polyclonal IgG was found to be more active than MAb 12-9 in releasing S. aureus from immobilized Fg. The total IgG from which the ClfA-IgG was recovered (at a yield of 1.3%) was also highly active. In contrast, the IgG purified from matched preimmune sera or nonfunctional MAb 15EC6 failed to release S. aureus from the Fg-coated microplate wells regardless of the antibody concentration.
DISCUSSION
In this report, we describe the antibody response induced by the ClfA antigen component of an exploratory vaccine to prevent S. aureus infections. An initial triantigen formulation was tested in phase 1 clinical trials in Australia (34). Later studies also included a fourth component, MntC (1), that is currently in phase 1/2 clinical trials in the United States (ClinicalTrials.gov registry number NCT01364571). ClfA is an important virulence factor with pathogenic properties that are dependent on its ability to bind Fg. As the efficacy of the ClfA vaccine antigen is expected to be largely dependent on the capacity to neutralize ClfA function, we developed a functional assay to measure the potency of the elicited response in preventing the binding of S. aureus to Fg immobilized on microtiter plate wells. Assay specificity was demonstrated by the ability of free antigen to block the inhibition of Fg binding by immune serum. The identification of a subset of ClfA MAbs that are capable of inhibiting S. aureus binding to Fg, as well as displacing Fg-bound bacteria, further illustrated the assay's selectivity. As with these functional MAbs, we demonstrate that immune sera from vaccinated volunteers were able to both prevent S. aureus Fg binding and displace bacteria already bound to immobilized Fg. Most importantly, antibodies elicited by the rClfAm antigen were more potent than a MAb which has shown efficacy in animal models of S. aureus infection. This mouse antibody is able to passively protect in a murine sepsis model, and the humanized equivalent (Aurexis) is protective against an intravenous challenge in a prophylactic rabbit model of infective endocarditis (8, 14). The clinical efficacy of Aurexis remains to be determined. Results of a phase IIa dose escalation study of Aurexis in S. aureus bacteremia patients undergoing vancomycin therapy were inconclusive with respect to the clinical outcome because of the small size of the patient population (n = 60) (38).
A variety of clinically relevant S. aureus strains, including invasive USA300 MRSA strains, can be used in the assay to detect ClfA neutralizing antibody responses (Table 2; Fig. 4). All strains expressed ClfA and were completely inhibited from binding to Fg by rabbit or NHP anti-ClfA immune serum. Although the lack of binding of isogenic knockout strains confirmed that ClfA expression is a prerequisite for Fg binding, no clear correlation between the level of ClfA surface antigen expression or the degree of ClfA sequence identity (relative to vaccine antigen) among strains and the assay inhibitory titer was observed. Coexpression of capsular antigens did not interfere with the ability of ClfA antibodies to inhibit Fg binding. This observation is consistent with previously published flow cytometry and opsonophagocytic assay data that show that antibodies can access ClfA and mediate opsonic killing in the presence of CP5 or CP8 (30). In the same study, accessibility of the ClfA antigen to antibodies was also observed in animal models of infection by immunofluorescence microscopy. ClfA expression was detected on the surface of six CP5-expressing and 12 CP8-expressing strains in both mouse bacteremia and wound models (30). Using the same technique, three additional strains listed in Table 2 were confirmed to express ClfA in the blood of infected animals (PFESA0067, PFESA0172, and PFESA0237; data not shown).
S. aureus ClfB is an MSCRAMM adhesin structurally related to ClfA that binds to the α chain of Fg (9) and has the potential to contribute to Fg binding in the assay. Significant levels of ClfB were not detected on the surfaces of the stationary-phase cells used in these experiments (not shown), an observation which is consistent with its expression primarily during the exponential phase of bacterial growth (31). We speculate that the apparent lack of interference in this in vitro assay by other known S. aureus Fg-binding adhesins such as FnbA (20, 24) and Bbp (37) may be similarly due to the use of stationary-phase cells. The relevance of Fg binding to the pathology of S. aureus infection has been demonstrated in a mouse abscess model in which ClfA or ClfB null mutants show a significant bacterial load reduction (5). These results suggest that Fg or fibrin binding by these proteins facilitates early dissemination of the pathogen in the blood. Compared with ClfA, the relatively high frequency of ClfB-encoding pseudogenes identified among unrelated S. aureus lineages limits this protein's utility as a vaccine antigen (29).
From the vaccine development standpoint, it is significant that nonvaccinated humans or animals rarely have preexisting functional antibodies to ClfA even though results reported here and by others (reviewed in reference 17) show that ClfA-binding antibodies are present in most humans. Here we demonstrate for the first time that such naturally acquired ClfA antibodies are unable to prevent ClfA from binding to immobilized Fg. In animals and humans, prevaccination Fg binding inhibition assay titers of antibody to ClfA antigen were mostly absent or close to the LLOD of the assay. In contrast, a single dose of rClfAm vaccine antigen is capable of generating antibodies that block Fg binding in both NHPs and human subjects. Also consistent with the lack of detectable preexisting functional ClfA antibodies in people is our demonstration that NHPs vaccinated with killed S. aureus cells expressing native ClfA antigen on the surface failed to generate functional ClfA antibodies. Whole-cell-based S. aureus vaccines have been used to treat bovine mastitis, but evidence of their efficacy is generally lacking (reviewed in reference 28). Similarly, antibody responses in patients with chronic staphylococcal infections who were vaccinated with autologous formalin-killed S. aureus had serum antibody levels only marginally higher than the preexisting levels (16). The inability of killed bacteria to elicit a robust functional response to a major virulence determinant such as ClfA raises doubts as to whether such S. aureus vaccines are capable of conferring protective efficacy. Recent experiments with mice confirm that S. aureus is able to actively suppress host immune responses through the expression of its surface-anchored virulence factors (21). In these studies, most live attenuated S. aureus strains failed to induce a robust humoral response and protection against subsequent infections. A prominent exception is an avirulent strain defective in sortase A, which failed to anchor surface MSCRAMM proteins to the cell wall. This strain elicits increased antibody responses directed toward several proteins, including ClfA, that are released from the bacterial surface because of the defect and can confer protective immunity to a subsequent challenge with virulent strains in a mouse abscess model (21). These results lend considerable support to the development of vaccines that target such protective antigens. Our ability to demonstrate a strong functional response to ClfA in NHPs and clinical subjects highlights the potential for a multicomponent subunit vaccine containing ClfA to elicit a protective anti-S. aureus immune response.
Supplementary Material
ACKNOWLEDGMENTS
We thank the following clinical investigators for the NCT01018641 clinical trial: Peter Richmond, University of Western Australia School of Pediatrics and Child Health & Telethon Institute for Child Health Research, Perth, Western Australia, Australia; Michael Nissen, QPID Clinical Trials Centre, Royal Children's Hospital and Children's Health Service District, Brisbane, Queensland, Australia; Helen Marshall, Vaccinology and Immunology Research Trials Unit, Women's and Children's Hospital and University of Adelaide, Adelaide, South Australia, Australia; Sepehr Shakib, CMAX, Adelaide, South Australia, Australia; Peter Hodsman, Nucleus Network, Melbourne, Victoria, Australia. We also thank Inhibitex team scientists Elena Gorovits, Perry S. Brignola, Bradley D. Prater, Jin Wang, Jeff Hutchins, and Joseph M. Patti for their help with SPR experiments. For their technical assistance, we thank Pfizer colleagues Luke Handke for constructing ClfA knockout strains, Stanley Mullen for help with FBI assay development, and Yekaterina Timofeyeva for immunofluorescence microscopy. We are grateful to Emilio Emini and other Pfizer colleagues for their review of the manuscript and to members of the clinical research team responsible for the SA3Ag study. Finally, we thank Magnus Hook and Emanuel Smeds (Texas A&M University) for sharing data and insightful discussions.
This study was sponsored by Pfizer Inc. John Vernachio is an employee of Inhibitex, which received financial support from Pfizer in connection with research conducted in this study and reported here. The remaining contributors are Pfizer employees.
Footnotes
Published ahead of print 15 August 2012
Supplemental material for this article may be found at http://cvi.asm.org/.
REFERENCES
- 1. Anderson AS, et al. 2012. Staphylococcus aureus manganese transport protein C is a highly conserved cell surface protein that elicits protective immunity against S. aureus and Staphylococcus epidermidis. J. Infect. Dis. 205:1688–1696 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bayer AS, et al. 1995. Staphylococcus aureus induces platelet aggregation via a fibrinogen-dependent mechanism which is independent of principal platelet glycoprotein IIb/IIIa fibrinogen-binding domains. Infect. Immun. 63:3634–3641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Broughan J, Anderson R, Anderson AS. 2011. Strategies for and advances in the development of Staphylococcus aureus prophylactic vaccines. Expert Rev. Vaccines 10:695–708 [DOI] [PubMed] [Google Scholar]
- 4. Brouillette E, et al. 2002. DNA immunization against the clumping factor A (ClfA) of Staphylococcus aureus. Vaccine 20:2348–2357 [DOI] [PubMed] [Google Scholar]
- 5. Cheng AG, et al. 2009. Genetic requirements for Staphylococcus aureus abscess formation and persistence in host tissues. FASEB J. 23:3393–3404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Daum RS, Spellberg B. 2012. Progress toward a Staphylococcus aureus vaccine. Clin. Infect. Dis. 54:560–567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deivanayagam CCS, et al. 2002. A novel variant of the immunoglobulin fold in surface adhesins of Staphylococcus aureus: crystal structure of the fibrinogen-binding MSCRAMM, clumping factor A. EMBO J. 21:6660–6672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Domanski PJ, et al. 2005. Characterization of a humanized monoclonal antibody recognizing clumping factor A expressed by Staphylococcus aureus. Infect. Immun. 73:5229–5232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ganesh VK, et al. 2011. Structural and biochemical characterization of Staphylococcus aureus clumping factor B/ligand interactions. J. Biol. Chem. 286:25963–25972 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ganesh VK, et al. 2008. A structural model of the Staphylococcus aureus ClfA-fibrinogen interaction opens new avenues for the design of anti-staphylococcal therapeutics. PLoS Pathog. 4:e1000226 doi:10.1371/journal.ppat.1000226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Geoghegan JA, et al. 2010. Molecular characterization of the interaction of staphylococcal microbial surface components recognizing adhesive matrix molecules (MSCRAMM) ClfA and Fbl with fibrinogen. J. Biol. Chem. 285:6208–6216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hair PS, et al. 2010. Clumping factor A interaction with complement factor I increases C3b cleavage on the bacterial surface of Staphylococcus aureus and decreases complement-mediated phagocytosis. Infect. Immun. 78:1717–1727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hair PS, Ward MD, Semmes OJ, Foster TJ, Cunnion KM. 2008. Staphylococcus aureus clumping factor A binds to complement regulator factor I and increases factor I cleavage of C3b. J. Infect. Dis. 198:125–133 [DOI] [PubMed] [Google Scholar]
- 14. Hall AE, et al. 2003. Characterization of a protective monoclonal antibody recognizing Staphylococcus aureus MSCRAMM protein clumping factor A. Infect. Immun. 71:6864–6870 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartford OM, Wann ER, Hook M, Foster TJ. 2001. Identification of residues in the Staphylococcus aureus fibrinogen-binding MSCRAMM clumping factor A (ClfA) that are important for ligand binding. J. Biol. Chem. 276:2466–2473 [DOI] [PubMed] [Google Scholar]
- 16. Holtfreter S, et al. 2011. Antibody responses in furunculosis patients vaccinated with autologous formalin-killed Staphylococcus aureus. Eur. J. Clin. Microbiol. Infect. Dis. 30:707–717 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Holtfreter S, Kolata J, Broker BM. 2010. Towards the immune proteome of Staphylococcus aureus—the anti-S. aureus antibody response. Int. J. Med. Microbiol. 300:176–192 [DOI] [PubMed] [Google Scholar]
- 18. Josefsson E, Hartford O, O'Brien L, Patti JM, Foster T. 2001. Protection against experimental Staphylococcus aureus arthritis by vaccination with clumping factor A, a novel virulence determinant. J. Infect. Dis. 184:1572–1580 [DOI] [PubMed] [Google Scholar]
- 19. Josefsson E, Higgins J, Foster TJ, Tarkowski A. 2008. Fibrinogen binding sites P336 and Y338 of clumping factor A are crucial for Staphylococcus aureus virulence. PLoS One 3:e2206 doi:10.1371/journal.pone.0002206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Keane FM, et al. 2007. Fibrinogen and elastin bind to the same region within the A domain of fibronectin binding protein A, an MSCRAMM of Staphylococcus aureus. Mol. Microbiol. 63:711–723 [DOI] [PubMed] [Google Scholar]
- 21. Kim HK, Kim H-Y, Schneewind O, Missiakas D. 2011. Identifying protective antigens of Staphylococcus aureus, a pathogen that suppresses host immune responses. FASEB J. 25:3605–3612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Lindsay JA. 2008. Staphylococcus: molecular genetics. Caister Academic Press, Norfolk, United Kingdom [Google Scholar]
- 23. Liu C-Z, Shih M-H, Tsai P-J. 2005. ClfA(221-550), a fibrinogen-binding segment of Staphylococcus aureus clumping factor A, disrupts fibrinogen function. Thromb. Haemost. 94:286–294 [DOI] [PubMed] [Google Scholar]
- 24. Matsuka YV, Anderson ET, Milner-Fish T, Ooi P, Baker S. 2003. Staphylococcus aureus fibronectin-binding protein serves as a substrate for coagulation factor XIIIa: evidence for factor XIIIa-catalyzed covalent cross-linking to fibronectin and fibrin. Biochemistry 42:14643–14652 [DOI] [PubMed] [Google Scholar]
- 25. McAdow M, et al. 2011. Preventing Staphylococcus aureus sepsis through the inhibition of its agglutination in blood. PLoS Pathog. 7:e1002307 doi:10.1371/journal.ppat.1002307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. McDevitt D, Francois P, Vaudaux P, Foster TJ. 1994. Molecular characterization of the clumping factor (fibrinogen receptor) of Staphylococcus aureus. Mol. Microbiol. 11:237–248 [DOI] [PubMed] [Google Scholar]
- 27. McDevitt D, et al. 1997. Characterization of the interaction between the Staphylococcus aureus clumping factor (ClfA) and fibrinogen. Eur. J. Biochem. 247:416–424 [DOI] [PubMed] [Google Scholar]
- 28. Middleton JR. 2008. Staphylococcus aureus antigens and challenges in vaccine development. Expert Rev. Vaccines 7:805–815 [DOI] [PubMed] [Google Scholar]
- 29. Murphy E, et al. 2011. Challenges for the evaluation of Staphylococcus aureus protein based vaccines: monitoring antigenic diversity. Hum. Vaccin. 7(Suppl):51–59 [DOI] [PubMed] [Google Scholar]
- 30. Nanra JS, et al. 2009. Heterogeneous in vivo expression of clumping factor A and capsular polysaccharide by Staphylococcus aureus: implications for vaccine design. Vaccine 27:3276–3280 [DOI] [PubMed] [Google Scholar]
- 31. Ní Eidhin D, et al. 1998. Clumping factor B (ClfB), a new surface-located fibrinogen-binding adhesin of Staphylococcus aureus. Mol. Microbiol. 30:245–257 [DOI] [PubMed] [Google Scholar]
- 32. Que YA, et al. 2005. Fibrinogen and fibronectin binding cooperate for valve infection and invasion in Staphylococcus aureus experimental endocarditis. J. Exp. Med. 201:1627–1635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Que YA, et al. 2001. Reassessing the role of Staphylococcus aureus clumping factor and fibronectin-binding protein by expression in Lactococcus lactis. Infect. Immun. 69:6296–6302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Richmond P, et al. 2011. A randomised, placebo controlled phase 1 first-in-human study of a novel 3-antigen Staphylococcus aureus vaccine in healthy adults, poster LB2769. Proceedings of the 21st European Congress of Clinical Microbiology and Infectious diseases, Milan, Italy [Google Scholar]
- 35. Rivas JM, Speziale P, Patti JM, Hook M. 2004. MSCRAMM-targeted vaccines and immunotherapy for staphylococcal infection. Curr. Opin. Drug Discov. Devel. 7:223–227 [PubMed] [Google Scholar]
- 36. Siboo IR, Cheung AL, Bayer AS, Sullam PM. 2001. Clumping factor A mediates binding of Staphylococcus aureus to human platelets. Infect. Immun. 69:3120–3127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Vazquez V, et al. 2011. Fibrinogen is a ligand for the Staphylococcus aureus microbial surface components recognizing adhesive matrix molecules (MSCRAMM) bone sialoprotein-binding protein (Bbp). J. Biol. Chem. 286:29797–29805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Weems JJ, Jr, et al. 2006. Phase II, randomized, double-blind, multicenter study comparing the safety and pharmacokinetics of tefibazumab to placebo for treatment of Staphylococcus aureus bacteremia. Antimicrob. Agents Chemother. 50:2751–2755 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yokoyama WM, Christensen M, Santos GD, Miller D. 2006. Production of monoclonal antibodies. Curr. Protoc. Immunol. Chapter 2:Unit 2.5 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.