

Abstract
Pneumococcal surface protein A (PspA) is a surface molecule on pneumococci that is required for full virulence in mouse models of infection. PspA has been reported to inhibit complement deposition on the pneumococcal surface. It has been assumed that this decreased complement deposition results in the inefficient phagocytosis of wild-type pneumococci. However, an effect of PspA on phagocytosis had not been shown. Our present studies demonstrated that a loss of PspA by capsular type 3 strains WU2 and A66.1 led to enhanced complement-dependent phagocytosis of the pneumococci by the mouse macrophage cell line J774A.1. This observation was made using human complement as well as mouse complement. Since this enhanced phagocytosis could be blocked by antibody to complement receptor CR3 on J774A.1, it was concluded that PspA's effect on phagocytosis was due to its effect on the amount of deposited complement, which in turn helped opsonize the pneumococci for phagocytosis. Since these studies included new independent mutants lacking PspA, the results provide solid confirmation of the previously reported effects of PspA on pneumococcal virulence and complement deposition. Finally, we showed that antibody to PspA, which is also known to enhance complement deposition, also enhances the phagocytosis of pneumococci in a largely complement-dependent manner.
INTRODUCTION
The complement system and phagocytes are major components of host innate immunity to bacterial pathogens (35). Streptococcus pneumoniae is a major human pathogen. It causes the majority of cases of severe community-acquired bacterial pneumonia and is an important cause of otitis media, meningitis, and septicemia. Pneumococcal infections are more common in young children, the elderly, and immunocompromised patients than in healthy older children and young adults, who are fairly resistant to pneumococcal infection (17, 49). Invasive pneumococci are cleared from the host largely by the recognition of pathogen surface-bound complement by receptors on host phagocytic cells (13, 18). Pneumococci can be opsonized by complement proteins, especially complement protein 3 (C3), which is activated by either the classical or alternative pathway and then binds covalently to the pneumococcal surface (14, 33, 53). The surface-bound complement serves as a ligand to interact with complement receptors on host cells, facilitating the pneumococcal uptake by phagocytes (2, 16, 27, 33, 37). The importance of the complement system in protection against pneumococci is highlighted by the fact that humans with deficiencies of either complement protein 2 or complement protein 3 or factor B are more susceptible to pneumococcal infection than normal individuals (3, 38).
Pneumococcal surface protein A (PspA) is one of the pneumococcal surface molecules required for full virulence of pneumococci in mice (31, 43). Since PspA is expressed on all pneumococci from humans (15, 19), PspA is probably also important for virulence in humans. PspA attaches to pneumococci through interaction with surface choline residues, primarily those of the lipoteichoic acids (52). Previous studies showed that PspA− pneumococci are more susceptible than wild-type PspA+ pneumococci to being cleared from mouse blood (31, 41, 45), and mice infected with PspA− pneumococci have longer survival times than mice infected with a PspA+ strain (43). Moreover, PspA has been reported to interfere with complement deposition on the pneumococcal surface (42, 43) and does so primarily by interfering with antibody-independent activation of the classical pathway by pneumococci (25). In vitro, PspA− pneumococci bind more complement C3 than PspA+ strains (42, 43), and this result has been demonstrated using pre- and postimmune sera from humans immunized with PspA (39). In mice, more C3 is activated if the mice are infected with PspA− pneumococci than with wild-type pneumococci expressing PspA (41, 45). In mice deficient in C3, factor B, or factor D, PspA− pneumococci become as virulent as PspA+ wild-type pneumococci (41, 45). In addition, mice deficient in complement receptors are more susceptible to pneumococcal challenge than are wild-type mice (41). For humans but not mice, the interaction of pneumococci with phagocytes is enhanced by immune adherence mediated by complement deposition on pneumococci and the complement receptor 1 (CR1) present on red blood cells (26).
Although it was clear that antibody to PspA can protect mice from otherwise fatal infection, initial attempts failed to show any effect of antibody to PspA or mutations of PspA on phagocytosis and killing by phagocytes (9). There are reports that antibody to PspA could affect association of pneumococci with phagocytic cells, but the studies did not distinguish whether or not the bacteria were intracellular (4, 32). In the present study, we examined phagocytosis by adapting the technique of labeling viable pneumococci with fluorescein, opsonizing them with complement-containing human or mouse serum, and then allowing the bacteria to interact with J774A.1 mouse macrophages. The incubation of S. pneumoniae with phagocytes was carried out in the presence of trypan blue to quench fluorescence of fluorescein isothiocyanate (FITC)-labeled extracellular molecules (48), a property that was confirmed in our study. This approach permitted us to quantitate the pneumococci that became intracellular.
Most prior in vitro studies of PspA's effect on complement activation and deposition were conducted by us (25, 40–43, 45) and by others using capsular type 3 and type 2 strains, WU2 and D39 and their PspA− mutants. All of these prior complement activation studies either used PspA mutants that secreted the alpha-helical portion of PspA (36, 41–43, 45) or used a mutant that produced no PspA (41–43, 45) but which also had a 3-kb deletion at the 5′ end of the pspA gene (51). Therefore, it was important to know whether this 3-kb deletion is involved in the interaction between pneumococci and the complement system and whether it contributes to the lower virulence of strain JY1119 compared to that of WU2. To address this concern, a new WU2 PspA− mutant, BR260.1, without a deletion upstream of pspA, was constructed. This new mutant was compared to WU2 and JY1119 in assays of in vitro complement deposition, phagocytosis, and virulence in mice. To test whether the effect of PspA on bacterial virulence and innate immunity observed previously was dependent on the specific genetic background of the WU2 strain, we also constructed a similar PspA− mutant, BR240.1, of another capsular type 3 strain, A66.1, which exhibits up to 103-fold greater virulence than WU2 (11). To confirm that the phagocytosis observed was dependent on interactions between deposited complement and the phagocytic cells, we used a monoclonal antibody (MAb) to the CD11b chain of complement receptor 3 to attempt to block the phagocytosis.
MATERIALS AND METHODS
Bacterial strains, media, and growth conditions.
Pneumococci were grown at 37°C in Todd-Hewitt broth with 0.5% yeast extract (THY) or on blood agar plates. Escherichia coli strains were grown in Luria-Bertani (LB) broth or on LB plates with 1.5% agar. Antibiotics were used at the following concentrations: erythromycin, 0.3 μg/ml for S. pneumoniae and 200 μg/ml for E. coli; ampicillin, 100 μg/ml for E. coli. Wild-type pneumococci were transformed with either plasmid (extracted from E. coli) by insertion duplication mutagenesis or with chromosomal DNA of donor pneumococcal strains by procedures described previously (43). The transformants were selected on blood agar plates supplemented with antibiotics. All strains were grown in THY to exponential phase, collected, and used in the in vitro and in vivo assays.
Construction of new PspA-negative strains.
The strains, plasmids, and primers used in this study are listed in Table 1. Insertion duplication was used to inactivate pspA genes in the parental strains WU2 and A66.1. Primers LSM12 and BING3 (Table 1) were used to amplify a 382-bp fragment located in the N-terminal portion of the WU2 pspA gene adjacent to its start codon. The amplified PCR product was first cloned into pTOPO vector (Invitrogen, Carlsbad, CA), and then the insert was released from pTOPO by digestion with EcoRI and subcloned into the vector pJY4164 (43) carrying an erythromycin resistance gene. The constructed plasmid was designated pREN101, which was used to transform S. pneumoniae WU2 and A66.1. Erythromycin-resistant transformants were obtained after plasmid integration via homologous recombination into the chromosome at the site of the cloned fragment. Each new mutant was back transformed three times into the recipient strain to minimize the chance of retaining any unanticipated genetic changes. The integration of the plasmid in the correct locations in the pneumococcal chromosomes was confirmed with PCR and by the absence of PspA expression (of the expected 80-kDa fragment) as detected by Western blotting. The resultant pspA-inactivated strains made from WU2 and A66.1 were designated BR260.1 and BR240.1, respectively. The surface attachment of PspA to the wild-type and mutant strains was examined with polyclonal rabbit antibodies to PspA and FITC-conjugated goat anti-rabbit IgG. Surface binding of antibody to PspA was observed for WU2 and A66.1 strains but not for the three PspA− mutants, BR260.1, BR240.1, and JY1119 (data not shown).
Table 1.
Strains, plasmids, and primers used in this study
| Strain, plasmid, or primer | Relevant characteristics, sequence, or gene | Source or reference |
|---|---|---|
| S. pneumoniae | ||
| WU2 | Wild-type capsule serotype 3, PspA+ | 30 |
| JY1119 | WU2 transformed with chromosomal DNA of WG44.1 and backcrossed 3 times | 51 |
| BR260.1 | WU2 pspA::pREN101, PspA− | This study |
| A66.1 | Wild-type capsule serotype 3, PspA+ | 5, 10 |
| BR240.1 | A66.1 pspA::pREN101, PspA− | This study |
| E. coli | ||
| TOP10 | General cloning strain | Invitrogen |
| Plasmids | ||
| pJY4164 | 4.4 kb, Ermr, ColE1 ori | 51 |
| pREN101 | pJY4164 with an internal pspA fragment; Ermr | This study |
| Primers | ||
| LSM12 | CCGGATCCAGCGTCGCTATCTTAGGGGCTGGTT | 29 |
| BING3 | GCTCTCGCCTCATTTTCG |
The insertion of the vector pJY4164 into the chromosome of S. pneumoniae resulted in a stable cointegrants for which spontaneous deletions have not been observed, even when grown in the absence of antibiotic selection (6, 21, 43). When mice were infected intravenously with 1 × 106 CFU of these two mutant pneumococcal strains, no antibiotic-sensitive bacteria (indicative of the loss of the insert) were observed among ≥100 CFU examined per infected mouse daily for 2 days following infection. BR260.1 was used to infect CBA/N mice, and BR240.1 was used to infect BALB/c mice. No pneumococci of either mutant were observed in the circulation of the mice during the 2 days of observation, even though some BALB/c mice infected with BR240.1 became severely ill. We do not expect there to be polar effects of these mutations, since a previous study has shown that the transcript for PspA is monocistronic and that polar effects were not observed after insertion of pJY4164 immediately downstream of pspA (1).
Western blot analysis.
To verify the knockout of PspA protein in BR240.1 and BR260.1, the mutants and their parental wild-type strains were grown to exponential phase and harvested by centrifugation. The bacterial cell pellets were incubated with lysis buffer (0.01% sodium dodecyl sulfate, 0.1% sodium deoxycholate, 0.015 M sodium citrate) at 37°C for 10 min. Lysates from different strains were separated by electrophoresis on 10% polyacrylamide gels after being boiled in 0.01% sodium dodecyl sulfate. Both WU2 and A66.1 express family 1 PspA (20). After the proteins were transferred to a nitrocellulose membrane, the blot was probed with rabbit antisera specific to family 1 PspA (43). To develop the blots, goat anti-rabbit IgG (H+L)-biotin was used in conjunction with streptavidin alkaline phosphatase (Southern Biotechnology Associates, Inc., Birmingham, AL).
C3 deposition on pneumococcal surface.
To quantify the deposition of C3 on pneumococcal surfaces, pneumococci in mid-log phase were adjusted to an optical density at 600 nm (OD600) of 0.5, and 200 μl of this suspension was centrifuged and the pelleted bacteria washed with phosphate-buffered saline (PBS). The washed bacteria then were incubated with 100 μl of so-called human complement, which is different concentrations of fresh normal human sera (NHS) collected from a healthy volunteer (determined, as described previously [10], to have very low levels of antibody to PspA) and diluted in gelatin veronal buffer (GVB; Sigma, St. Louis, MO) for 30 min at 37°C. After washing with PBS, the bacteria were incubated with a 1/100 dilution in PBS of goat anti-human C3 IgG conjugated to FITC (ICN, Costa Mesa, CA) for 30 min on ice. C3 deposition on bacteria was examined using flow cytometry (FACScan; Becton, Dickinson), and the quantitation of C3 on the pneumococcal surface was presented as geometric mean fluorescence intensities (GMFI) for each sample. All flow cytometry evaluations were based on ≥20,000 gated events.
Phagocytosis assay.
Mid-log-phase pneumococci (<109 CFU) were incubated in a 1-ml solution of FITC isomer I (0.5 mg/ml; Sigma) in PBS (pH 8.4) at 37°C for 1 h with shaking. The FITC-labeled bacteria were washed and resuspended in sterile Hanks balanced salt solution (HBSS) with 0.25% bovine serum albumin (BSA). Glycerol was added to a final concentration of 10%, and the bacterial suspension was stored at −80°C until use. The phagocytosis assay was a modification of an earlier assay (4). Indicated numbers of pneumococci were incubated with 10% NHS, unless otherwise indicated, in round-bottom microtiter plates at 37°C for 30 min with shaking. The bacterial suspensions were then incubated for an additional 30 min at 37°C with the J774A.1 macrophage cell line (2 × 106 J774A.1 cells/well). Subsequently, the cells were washed with 0.25% BSA-HBSS, and surface adherence of FITC-labeled pneumococci was quenched by incubation with trypan blue solution (0.2 mg/ml trypan blue in PBS) (48). We verified that this concentration of trypan blue essentially eliminates fluorescence of the pneumococci in the absence of phagocytes. After being washed, samples were analyzed using flow cytometery. The geometric mean fluorescence intensity of macrophages containing fluorescent bacteria was used as a measurement of phagocytic activity.
To see if the observed phagocytosis was dependent on CR3, a blocking study was performed by addition of anti-CD11b MAb (blocking CR3) to the incubation of FITC-labeled bacteria and macrophages. Specifically, a series of dilutions of phycoerythrin (PE)-conjugated rat anti-mouse CD11b MAb (M1/70; 0.2 mg/ml; Pharmingen, BD Biosciences) (7) were incubated with macrophages in microtiter plates for 30 min. FITC-labeled pneumococci, which had been preincubated with NHS at 37°C for 30 min, were added to the plates (in the presence of the 10% NHS) with a bacterium/macrophage ratio of 60:1, and the mixture was incubated for an additional 30 min at 37°C. After incubation with trypan blue solution and washing with 0.25% BSA-HBSS, the samples were resuspended in PBS and analyzed using a flow cytometer. For each sample, ≥20,000 gated events were analyzed. The level of anti-CD11b antibody bound on the macrophage surface was represented as the fluorescence of PE-conjugated antibody, and the phagocytosis of pneumococci was represented as the fluorescence of the FITC-labeled pneumococci within the macrophages.
To study the effect of antibody to PspA on phagocytosis, immune serum to PspA was produced with BALB/c mice by immunization with purified recombinant PspA (rPspA) protein, UAB055 (12), which contains the full α-helical domain and the first 14 amino acids in the proline-rich region of PspA from strain Rx1 (family 1 PspA). Mice were immunized subcutaneously with 2 μg of purified protein emulsified with 50 μg of alum (Pierce, Rockford, IL) in 0.1 ml of Ringer's saline on days 1, 8, and 22. Immune serum was collected and pooled on day 29. The concentration of antibody to PspA was measured by enzyme-linked immunosorbent assay (ELISA). The phagocytosis assay was performed as described above, except that FITC-labeled pneumococci were opsonized in normal mouse serum or immune mouse serum. To dissect the role of complement versus antibody to PspA in mediating phagocytosis, normal mouse serum and immune mouse serum samples were heat inactivated at 56°C for 30 min prior to phagocytosis assay to abolish the effects of complement in mouse serum. The level of phagocytosis in different mouse serum preparations was evaluated by positive percentages of macrophages with ingested pneumococci. For each sample, ≥15,000 gated events were analyzed.
Infection of mice.
Female, 4- to 6-month-old, CBA/CaHN-Btkxid/J (CBA/N) or BALB/cByJ mice (Jackson Laboratories, Bar Harbor, ME) were used. Frozen infection stocks containing a known concentration of viable bacteria were diluted in lactated Ringer's injection solution (Abbott Laboratories, North Chicago, IL) to achieve the desired concentrations of bacteria. The numbers of bacteria injected were confirmed by plating on blood agar plates. For the survival studies, CBA/N mice were challenged intravenously (i.v.) with 200 CFU of pneumococci in 200 μl Ringer's solution per mouse. BALB/cByJ mice were similarly infected with 1 × 104 CFU/mouse. These mice were monitored every 6 h, and the time at which they became moribund was recorded (unresponsive to touch and/or body temperature below 26°C). Once they were moribund, mice were euthanized by CO2 narcosis and then cervically dislocated. Differences between groups in survival time to a moribund endpoint were analyzed using the Mann-Whitney two-sample rank test (two-tailed) and are written above plots of data with mutant strains. To assess the pneumococcal net growth/clearance in mouse blood in the early phase of infection, we challenged groups of 5 BALB/cByJ mice i.v. with 2 × 105 CFU per mouse. Blood samples were collected from the retro-orbital plexus at indicated time points and were serially diluted and plated on blood agar with or without erythromycin to determine the number of viable wild-type and mutant S. pneumoniae organisms, respectively. P values were determined at each time point by Student's t test (*, P < 0.05; **, P < 0.002). For survival and blood CFU data, no P value is given if the comparison was not statistically significant at P < 0.05.
Statistics.
Statistical differences in complement deposition or phagocytosis were calculated with the Student's t test. Statistical differences for time to being moribund were calculated with the Mann-Whitney test. In both cases only P < 0.05 was considered to be statistically significant. All flow cytometry evaluations were based on ≥20,000 gated events with the exception of the antibody to PspA-induced phagocytosis flow cytometry experiments, which were based on ≥15,000 gated events.
RESULTS
Increased C3 deposition on PspA− strains.
We previously showed that when incubated with normal human serum, PspA− JY1119 bound more C3 than did PspA+ WU2 (43). In our present studies we included the PspA− mutant strain JY1119 as well as two new independent mutants, BR260.1 and BR240.1, made in WU2 and A66.1, respectively. Both of these parent strains are capsular type 3, but A66.1 is about 1,000-fold more virulent than WU2 (11). In the present studies we observed that, like JY1119, both BR260.1 and BR240.1 showed more C3 deposition on their surfaces than did the parental PspA+ strains when incubated with 10% normal human serum (Fig. 1A). BR240.1 showed more C3 binding than did A66.1 at each concentration of normal human serum tested (Fig. 1B). Since the amounts of C3 on BR260.1 and JY1119 are similar and since BR240.1 also showed enhanced C3 deposition compared to A66.1, the data strongly indicate that the increased C3 deposition on these mutant strains was due to the pspA mutation and was not due to a genetic change unrelated to PspA expression.
Fig 1.
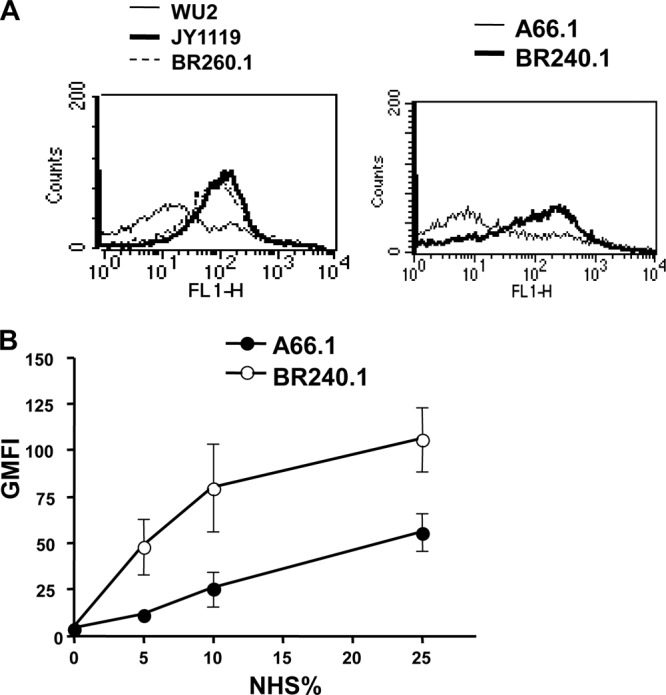
C3 deposition on PspA+ and PspA− pneumococci. (A) Pneumococcal strains were incubated with 10% normal human serum, and the relative amount of C3 deposited on their surfaces was measured using FITC-conjugated goat IgG anti-human C3. The geometric mean fluorescence intensities (GMFI) of samples of WU2, JY1119, and BR260.1 are 20.4, 97.3, and 89.5, respectively, and they are 15.5 and 56.1 for A66.1 and BR240.1, respectively. (B) A66.1 and BR240.1 were incubated with different concentrations of NHS, and the fluorescence intensities of each strain were plotted. The standard error bars from three individual experiments are indicated. All flow cytometry profiles and determinations were based on ≥20,000 events.
Increased uptake of PspA− pneumococci by macrophages.
Pneumococcal surface-bound C3 fragments can serve as ligands for phagocytic receptors on host phagocytes. To investigate whether the PspA-dependent reduction in complement deposition on the pneumococcal surface would decrease bacterial uptake by eukaryotic phagocytes, we examined the relative phagocytosis of PspA+ and PspA− pneumococcal strains opsonized in 10% NHS.
When WU2 and its PspA− mutants were examined, WU2 was phagocytosed less well than either of its two mutants, JY1119 and BR260.1, at all ratios (in CFU) of bacteria to macrophage tested (Fig. 2A). As we increased the ratio of bacteria to macrophage, we enhanced the average amount of WU2 and its mutants ingested into macrophage (Fig. 2A). A66.1 was also more resistant to phagocytosis by macrophage than its PspA− mutant, BR240.1 (Fig. 2B).
Fig 2.

Effect of PspA expression on the phagocytosis of pneumococci by macrophages. The phagocytosis of WU2 (A) and A66.1 (B) was examined at different ratios of bacterial CFU to macrophage in the presence of 10% normal human serum. FITC-labeled pneumococci were incubated with mouse macrophage cell line J774A.1 with a ratio of bacterial CFU to macrophage ranging from 240:1 to 4:1. Trypan blue solution was used to quench fluorescence from pneumococci adherent to the cell surface, thus only fluorescence from ingested pneumococci was measured. Each of these studies was repeated at least three times with similar results. Results of representative experiments are presented. To be able to evaluate these data statistically, the data points for each replicate were ranked. The ranked data points for each replicate for the portions of the curves that discriminated most strongly among the strains (bacterium/macrophage ratios of 30:240 for panel A and 60:24 for panel B) were pooled and analyzed by the Mann-Whitney two-sample rank test. In both experiments the differences between the wild-type and mutant strains were significant at P < 0.003. (C) Effect of the amount of available complement on pneumococcal phagocytosis of A66.1 and BR240.1. FITC-labeled bacteria were incubated with the indicated concentrations of normal human serum and then incubated with macrophage with ratios of bacterial CFU to macrophage of 60:1 and 120:1. Standard errors are indicated. The absence of visible standard error bars indicates that they were closer together than the width of the respective symbols. All flow cytometry data were based on ≥20,000 gated events.
Since we knew from the complement fixation assay that increasing the amount of available complement (Fig. 1B) enhanced C3 deposition, we determined the relationship between the phagocytosis of A66.1 and BR240.1 and the concentration of normal human serum used for incubation with pneumococci. With the ratio of bacteria CFU to macrophage at 60:1 and 120:1, we observed that the phagocytosis increased as the concentration of normal serum increased from 0 to about 20% for both BR240.1 and A66.1 (Fig. 2C). Although higher concentrations of normal serum actually resulted in somewhat less phagocytosis when the ratio of A66.1 bacterial CFU to phagocytes was 60:1, the level of C3 deposition on wild-type A66.1 pneumococci was still less on those lacking PspA (Fig. 2C). The decrease in bacteria associated with phagocytes at higher complement levels may be the result of more rapid destruction and shorter half-life of the bacteria within the phagocytes as the complement concentration is increased. No decrease in complement deposition was observed at high complement levels when the ratio of A66.1 to phagocytes was 120:1.
Anti-CR3 antibody inhibited pneumococcal phagocytosis.
It has been shown that the major cleavage product of C3 on capsular type 3 pneumococci is iC3b, which is one of the high-affinity ligands for complement receptor 3 on phagocytes (41). Moreover, mice deficient in CR3 are more susceptible to pneumococcal challenge than are wild-type mice (41). In this study, we evaluated the importance of CR3 on phagocytosis of pneumococci by macrophages using the in vitro phagocytosis assay. When the anti-CR3 monoclonal antibody M1/70 was added to the incubation mixture of pneumococci, macrophages, and normal human sera, it inhibited phagocytosis (Fig. 3). The level of phagocytosis (Fig. 3, solid circles and solid triangles) was inversely proportional to the amount of anti-CR3 added (Fig. 3, x axis). This inhibitory effect by anti-CR3 antibody was more accentuated for the PspA− strain BR240.1 than it was for the wild-type strain A66.1.
Fig 3.

Blocking of pneumococcal phagocytosis by anti-CR3 (anti-CD11b) antibody. Macrophages were preincubated with a series of dilutions of PE-conjugated anti-CR3 MAb, and then FITC-labeled pneumococci preopsonized with normal human serum were added, and the mixture was incubated for 30 min. Trypan blue solution was used to quench the fluorescence from pneumococci adherent to the cell surface. The level of the surface-bound anti-CR3 antibody is represented by the PE intensity (open symbols), and the amount of the ingested pneumococci is represented by FITC intensity (solid symbols). Samples of macrophage incubated with only anti-CR3 MAb or macrophage incubated only with pneumococci were included as controls. This study was repeated three times with similar results. The data were analyzed by ranking all of the data values in each experiment and comparing the ranked values for all three experiments at all dilutions of anti-CR3 that gave results that permitted comparisons either in the presence or absence of bacteria on anti-CD11b binding or the comparison of internalization of A66.1 bacteria with that of the BR2401 mutant. In the comparison of internalization of A66.1 versus BR240.1, the difference was significant at P < 0.0001. In the analysis of binding of anti-CD11b, it was observed that in the presence of bacteria (and 10% NHS) that significantly less (P < 0.0001) anti-CD11b bound compared to cultured macrophages than in the absence of anti-CD11b and NHS. All flow cytometry evaluations were based on ≥20,000 events.
Macrophages incubated with either BR240.1 or A66.1 showed similar levels of anti-CR3 MAb binding on their surfaces (Fig. 3, open circles and open triangles). However, almost twice as much anti-CR3 was bound to macrophages in the absence of bacteria and 10% NHS (Fig. 3, open squares) as in their presence (Fig. 3, open circles and open triangles). This effect could have been due to uncontrolled effects of NHS and bacterial products on the membrane turnover or CR3 turnover on the surface of the cultured bacteria.
Antibody to PspA-mediated uptake of pneumococci by phagocytes.
WU2 and PspA− mutant JY1119 pneumococci were incubated with either fresh normal mouse serum or fresh anti-PspA mouse serum prior to incubation of the opsonized pneumococci with phagocytes. With nonimmune (normal) mouse serum, we observed greater bacterial phagocytosis in the absence versus the presence of PspA. This was a repeat of what we had observed in Fig. 2 using normal human serum as the complement source and strengthens the overall observations. It was observed that the fraction of macrophages phagocytosing WU2 increased from 47 to 79% in the presence of antibody to PspA. In the presence of the immune serum to rPspA, similar numbers of macrophages contained PspA+ and PspA− pneumococci (Fig. 4).
Fig 4.
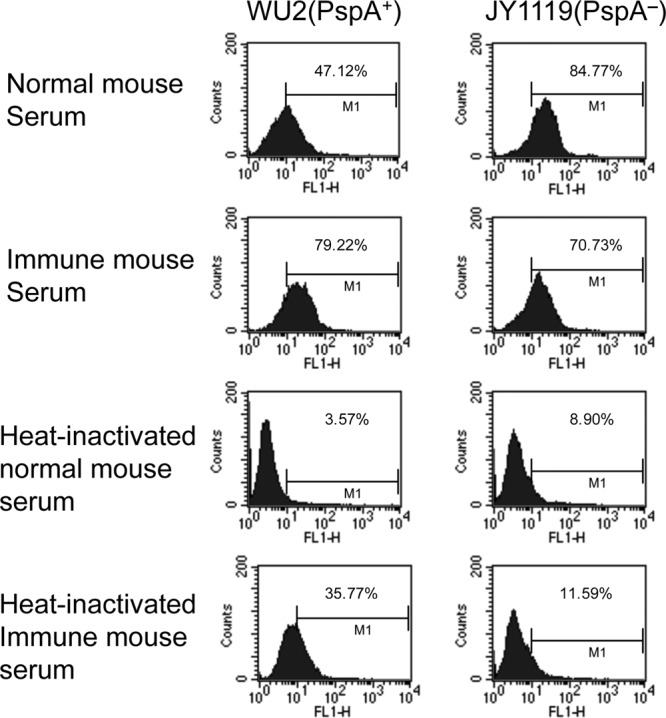
Antibody to PspA mediated phagocytosis of Streptococcus pneumoniae. PspA+ (WU2) and PspA− (JY1119) pneumococci were incubated with fresh normal mouse serum or fresh immune (anti-PspA) mouse serum prior to incubation with macrophages. As a no-complement control, the study was also conducted using the same normal mouse serum and immune mouse serum that had been heat inactivated to destroy complement. The data are presented as the fluorescence-activated cell sorter plots for which the percentage of macrophages in the positive gate for ingested pneumococci is given. All flow cytometry profiles were based on ≥15,000 gated events.
When heat-inactivated serum was used, we observed the lowest percentage (3.5%) of macrophages containing pneumococci when WU2 organisms were opsonized with only heat-inactivated normal mouse serum. Interestingly, the addition of heat-inactivated immune (anti-PspA) sera raised the percentage of macrophages containing pneumococci to 35%, but it was still not as high as the 47% containing pneumococci when complement alone was the opsonizing agent. In the presence of heat-inactivated normal mouse sera, JY1119 was phagocytosed only slightly better than WU2. As expected, the addition of antibody to PspA did not enhance the opsonization of JY1119, which lacks PspA expression. Taken together, the data shown in Fig. 4 indicate that both antibody to PspA and complement enhance phagocytosis, but that the maximum phagocytosis of a wild-type strain is seen when both antibody and complement are present.
Attenuated virulence of PspA− mutants in our mouse model of fatal sepsis.
The virulence of WU2, A66.1, and their new PspA− mutants were evaluated in a mouse sepsis model (Fig. 5A and B). Two hundred CFU of WU2, BR260.1, or JY1119 was injected i.v. into CBA/N mice. The mice were monitored to determine the time until each mouse became moribund, at which time they were each euthanized. All of the mice infected with WU2 became moribund and were euthanized within 46 h. However, most mice infected with BR260.1 and JY1119 survived for ≥200 h, and some mice infected with the mutant bacteria were still alive at 528 h, when the experiment was terminated (Fig. 5A). The survival times of groups infected with BR260.1 and JY1119 were statistically different from those of the group challenged with WU2 at P < 0.001 and P = 0.002, respectively. The survival times of groups infected with BR260.1 and JY1119 were not significantly different from each other (Fig. 5A).
Fig 5.

Virulence of PspA+ and PspA− pneumococci in mice. Virulence was monitored by comparing the survival time of mice infected with wild-type and mutant strains of S. pneumoniae. (A) WU2, BR260.1, JY1119, A66.1, or BR240.1 (200 CFU) was used to infect CBA/N mice. (B) A66.1 or BR240.1 (1 × 104 CFU) was used to infect BALB/cByJ mice. The survival times of mouse groups infected with mutant strains were compared to those of mouse groups infected with wild-type strains. (C) A66.1 or BR240.1 (2 × 105 CFU) was injected i.v. into groups of 4 to 5 BALB/cByJ mice. Mouse blood samples were collected from the retro-orbital plexus at the indicated time points. Samples were serially diluted and plated on blood agar to determine the numbers of viable bacteria (CFU) in the blood. (*, P < 0.05; **, P < 0.002).
Using this same sepsis model with CBA/N mice, the inactivation of PspA did not alter the virulence of A66.1; both A66.1- and BR240.1-infected groups showed similar survival times (P = 0.50) (Fig. 5A). However, when A66.1 and BR240.1 were used to challenge BALB/cByJ mice (1 × 104 CFU/mouse), the survival time for the BR240.1-infected group was significantly longer than that for the A66.1-infected group (P = 0.03) (Fig. 5B). The A66.1 strain apparently was so virulent in the CBA/N mouse that it was able to cause rapid death even without PspA. However, when much more resistant BALB/cByJ mice were used, it was possible to see the lower virulence of the PspA− mutant of A66.1.
Effect of PspA in blood clearance.
BALB/cByJ mice were injected i.v. with 2 × 105 CFU of A66.1 or BR240.1 and monitored for 24 h. During the first 6 h, BR240.1 was cleared more rapidly than A66.1. Moreover, unlike A66.1 the mutant BR240.1 bacteria had continuously decreasing numbers in the blood during the first 6-h period postinoculation. The numbers of A66.1, on the other hand, started increasing after 3 h postinoculation and were almost 100-fold greater than the numbers of BR240.1 CFU at 6 and 24 h postinoculation (Fig. 5C).
DISCUSSION
As an essential part of innate immunity, complement defends against invasive pathogens by several mechanisms. Many Gram-negative bacteria are lysed by a membrane attack complex inserted into the bacterial outer membrane. Gram-positive bacteria, such as pneumococci, are opsonized by the deposition of activated complement proteins (C3 and C4) and then engulfed by phagocytes through recognition of the complement on the opsonized bacterial surface by complement receptors (33, 35). Although previous studies have demonstrated that PspA expression reduces the C3 deposition on pneumococcal surfaces, no direct evidence has been reported that the reduction of complement deposition impairs the pneumococcal uptake by host phagocytes. With the in vitro phagocytosis assay used here, we observed that all three PspA− strains, JY1119, BR260.1, and BR240.1, were engulfed more easily than were their WU2 and A66.1 PspA+ wild-type parental strains. However, the efficiency of pneumococcal phagocytosis of both wild-type and mutant strains was dependent on the serum (complement) concentration and the ratio of bacteria to macrophage.
iC3b, a cleaved fragment of C3, is the major form of C3 found on the capsular serotype 3 strain of pneumococci (37). CR3 is the principal receptor for iC3b and is found on neutrophils, on monocytes/macrophages, and on activated lymphocytes, where it is involved in cell-cell adhesion, phagocytosis, chemotaxis, and cell activation (2). When iC3b serves as the ligand, CR3 mediates opsonization and phagocytosis of microorganisms (27, 46). To determine whether pneumococcal phagocytosis present in the absence of PspA is largely dependent on the direct interaction of iC3b and CR3, we determined the effect of monoclonal antibody M1/70 (blocking antibody of CD11b) (7, 24, 47) on pneumococcal phagocytosis by macrophages. Ingestion of pneumococci by macrophages was inhibited by the anti-CR3 MAb, and the magnitude of inhibition was directly dependent on the antibody concentration. The absolute, as well as the relative, effect of anti-CR3 was greater for the PspA− strain, which bound high levels of C3 compared to the PspA+ strain, which bound low levels of C3. Our results thus provide direct evidence to support the hypothesis that the pneumococcal phagocytosis in this in vitro system, and the difference in phagocytosis of PspA+ and PspA− pneumococci, is largely dependent on the interaction between iC3b and CR3. This finding does not preclude the possibility that other phagocyte surface receptors, such as Fcγ receptor, Toll-like receptors (23, 28), mannose receptors, and SIGN (specific intracellular adhesion molecule-grabbing nonintegrin)-R1 (18, 22), also contribute to the process of pneumococcal clearance from the host.
Although the amount of complement on the bacterial surface determines the efficiency of phagocytosis, other bacterial characteristics determined by genetic background also affect this process. WU2 and A66.1 have the same serotype 3 polysaccharide capsule and bound similar amounts of C3 on their surfaces. However, the phagocytosis rates of WU2 and its PspA− mutants were still increasing even at a bacterium/macrophage ratio of 240:1, whereas the phagocytosis of A66.1 and its PspA− mutant reached a maximum level at a ratio of 60:1. Consistent with this in vitro difference in WU2 and A66.1, the in vivo virulence study also confirmed that the A66.1 genetic background was more virulent than the WU2 background, with or without PspA. In the immunocompromised CBA/N mice, the attenuation of virulence by deletion of PspA was observed for WU2 but not for A66.1. Only with immunocompetent BALB/cByJ mice, where higher inocula were needed to cause death, could the reduced virulence of BR240.1, the PspA− variant of A66.1, be detected. WU2 is an unusually low virulence type 3 strain (11), and CBA/N is an unusually susceptible mouse strain (11, 50). As a result, A66.1 and BALB/cJ may be more representative of a human situation. However, WU2 showed virulence in CBA/N mice that was similar to the virulence shown by A66 in BALB/c mice. The fact that PspA played a strong role in the virulence of both in models of similarly fatal consequences was strongly supportive of a role for PspA in the virulence of type 3 pneumococci. PspA also plays an easily demonstrable, but smaller, role in virulence of type 2 (D39) pneumococci (8, 31).
Complement opsonization and phagocytosis are two essential steps in host innate immunity against bacterial infection. Our study suggested that pneumococci use PspA to help evade these host defense mechanisms. A consequence of PspA's ability to interfere with complement deposition and subsequent phagocytosis is the enabling of pneumococci to cause serious invasive disease. However, in the case of strains WU2 and A66.1, the presence of antibody to PspA could negate the advantage that PspA gives pneumococci in phagocytosis. It is already well established that antibody to PspA can protect mice against invasive disease (10). Our findings in this report raise the possibility that opsonization of pneumococci by immune sera to rPspA is useful as a surrogate for protection against invasive disease by immunization of humans with rPspA.
Many of the most important prior studies of the effect of PspA on complement activation and deposition used a PspA-null strain, JY1119, that we now know is lacking 3 kb upstream of pspA on the WU2 chromosome. Although the role of genes in the 3-kb sequence of the upstream region of pspA has not been defined, analysis of the genome of the TIGR4 strain (44) suggests that the sequence of that region has several open reading frames and encodes proteins. To determine if the deletion of this 3 kb of DNA affected any of the previous phenotypes associated with JY1119, two new PspA− strains of WU2 and A66.1 were constructed that were lacking PspA but contained the 3 kb of upstream DNA that had been missing from JY1119. Our results from this report indicated that an absence of PspA alone was responsible for increasing complement deposition, increasing phagocytosis in the presence of complement, and reducing virulence.
In addition to PspA affecting innate immunity mediated by complement, it is known to be a target of protective antibody elicited in the adaptive immune responses. It has been established that this antibody to PspA can enhance complement deposition on pneumococci (34, 39, 42, 43). In these studies, we have shown that antibody to PspA is able to mediate phagocytosis and that the PspA-dependent phagocytosis is maximal in the presence of complement. A phagocytic assay of the type described here could serve as a surrogate assay for protective immunity mediated by antibody to PspA.
ACKNOWLEDGMENTS
We thank Bernard P. Arulanandam for providing us the mouse macrophage cell line J774A.1. We are grateful to Simon L. Newman and Moon H. Nahm for their interest in these studies and their help and suggestions regarding the in vitro phagocytosis assays. Susan Kniebes and Nancy G. Abney are acknowledged for editing of the presubmission manuscript.
This work was supported by NIH grants AI21548 and T32 AI07041.
Footnotes
Published ahead of print 1 August 2012
REFERENCES
- 1. Abeyta M, Hardy GG, Yother J. 2003. Genetic alteration of capsule type but not PspA type affects accessibility of surface-bound complement and surface antigens of Streptococcus pneumoniae. Infect. Immun. 71:218–225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Agramonte-Hevia J, Gonzalez-Arenas A, Barrera D, Velasco-Velazquez M. 2002. Gram-negative bacteria and phagocytic cell interaction mediated by complement receptor 3. FEMS Immunol. Med. Microbiol. 34:255–266 [DOI] [PubMed] [Google Scholar]
- 3. Alper CA, Abramson N, Johnston RB, Jr, Jandl JH, Rosen FS. 1970. Increased susceptibility to infection associated with abnormalities of complement-mediated functions and of the third component of complement (C3). N. Engl. J. Med. 282:350–354 [DOI] [PubMed] [Google Scholar]
- 4. Arulanandam BP, Lynch JM, Briles DE, Hollingshead SK, Metzger DW. 2001. Intranasal vaccination with pneumococcal surface protein A and IL-12 augments antibody-mediated opsonization and protective immunity against Streptococcus pneumoniae infection. Infect. Immun. 69:6718–6724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Avery OT, Macleod CM, McCarty M. 1944. Studies on the chemical nature of the substance inducing transformation of pneumococcal types: induction of transformation by a desoxyribonucleic acid fraction isolated from pneumococcus type III. J. Exp. Med. 79:137–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Balachandran P, Brooks-Walter A, Virolainen-Julkunen A, Hollingshead SK, Briles DE. 2002. The role of pneumococcal surface protein C (PspC) in nasopharyngeal carriage and pneumonia and its ability to elicit protection against carriage of Streptococcus pneumoniae. Infect. Immun. 70:2526–2534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beller DI, Springer TA, Schreiber RD. 1982. Anti-Mac-1 selectively inhibits the mouse and human type three complement receptor. J. Exp. Med. 156:1000–1009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Berry AM, Paton JC. 2000. Additive attenuation of virulence of Streptococcus pneumoniae by mutation of the genes encoding pneumolysin and other putative pneumococcal virulence proteins. Infect. Immun. 68:133–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Briles DE, et al. 1989. Antipneumococcal effects of C-reactive protein and monoclonal antibodies to pneumococcal cell wall and capsular antigens. Infect. Immun. 57:1457–1464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Briles DE, et al. 2000. Immunization of humans with recombinant pneumococcal surface protein A (rPspA) elicits antibodies that passively protect mice from fatal infection with Streptococcus pneumoniae bearing heterologous PspA. J. Infect. Dis. 182:1694–1701 [DOI] [PubMed] [Google Scholar]
- 11. Briles DE, et al. 1986. Genetic control of susceptibility to pneumococcal infection. Curr. Top. Microbiol. Immunol. 124:103–120 [DOI] [PubMed] [Google Scholar]
- 12. Brooks-Walter A, Briles DE, Hollingshead SK. 1999. The pspC gene of Streptococcus pneumoniae encodes a polymorphic protein, PspC, which elicits cross-reactive antibodies to PspA and provides immunity to pneumococcal bacteremia. Infect. Immun. 67:6533–6542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Brown EJ, Hosea SW, Frank MM. 1983. The role of antibody and complement in the reticuloendothelial clearance of pneumococci from the bloodstream. Rev. Infect. Dis. 5(Suppl. 4):S797–S805 [DOI] [PubMed] [Google Scholar]
- 14. Brown JS, et al. 2002. The classical pathway is the dominant complement pathway required for innate immunity to Streptococcus pneumoniae infection in mice. Proc. Natl. Acad. Sci. U. S. A. 99:16969–16974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Crain MJ, et al. 1990. Pneumococcal surface protein A (PspA) is serologically highly variable and is expressed by all clinically important capsular serotypes of Streptococcus pneumoniae. Infect. Immun. 58:3293–3299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fallman M, Andersson R, Andersson T. 1993. Signaling properties of CR3 (CD11b/CD18) and CR1 (CD35) in relation to phagocytosis of complement-opsonized particles. J. Immunol. 151:330–338 [PubMed] [Google Scholar]
- 17. Fedson DS, Musher DM. 2004. Pneumococcal polysaccharide vaccine, p 529–588 In Plotkin SA, Orenstein WA. (ed), Vaccines. W. B. Saunders, Philadelphia, PA [Google Scholar]
- 18. Gordon S. 2002. Pattern recognition receptors: doubling up for the innate immune response. Cell 111:927–930 [DOI] [PubMed] [Google Scholar]
- 19. Hollingshead SK, et al. 2006. Pneumococcal surface protein A (PspA) family distribution among clinical isolates from adults over 50 years of age collected in seven countries. J. Med. Microbiol. 55:215–221 [DOI] [PubMed] [Google Scholar]
- 20. Hollingshead SK, Becker RS, Briles DE. 2000. Diversity of PspA: mosaic genes and evidence for past recombination in Streptococcus pneumoniae. Infect. Immun. 68:5889–5900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Johnston JW, et al. 2004. Lipoprotein PsaA in virulence of Streptococcus pneumoniae: surface accessibility and role in protection from superoxide. Infect. Immun. 72:5858–5867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kang HY, Srinivasan J, Curtiss R., 3rd 2002. Immune response to recombinant pneumococcal PspA antigen delivered by live attenuated Salmonella enterica serovar Typhimurium vaccine. Infect. Immun. 70:1739–1749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Koedel U, et al. 2003. Toll-like receptor 2 participates in mediation of immune response in experimental pneumococcal meningitis. J. Immunol. 170:438–444 [DOI] [PubMed] [Google Scholar]
- 24. Leon F, et al. 2006. Antibodies to complement receptor 3 treat established inflammation in murine models of colitis and a novel model of psoriasiform dermatitis. J. Immunol. 177:6974–6982 [DOI] [PubMed] [Google Scholar]
- 25. Li J, Glover DT, Szalai AJ, Hollingshead SK, Briles DE. 2007. PspA and PspC minimize immune adherence and transfer of pneumococci from erythrocytes to macrophages through their effects on complement activation. Infect. Immun. 75:5877–5885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li J, et al. 2010. Complement receptor 1 expression on mouse erythrocytes mediates clearance of Streptococcus pneumoniae by immune adherence. Infect. Immun. 78:3129–3135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lutz MA, Correll PH. 2003. Activation of CR3-mediated phagocytosis by MSP requires the RON receptor, tyrosine kinase activity, phosphatidylinositol 3-kinase, and protein kinase C zeta. J. Leukoc. Biol. 73:802–814 [DOI] [PubMed] [Google Scholar]
- 28. Malley R, et al. 2003. Recognition of pneumolysin by Toll-like receptor 4 confers resistance to pneumococcal infection. Proc. Natl. Acad. Sci. U. S. A. 100:1966–1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. McDaniel LS, McDaniel DO, Hollingshead SK, Briles DE. 1998. Comparison of the PspA sequence from Streptococcus pneumoniae EF5668 to the previously identified PspA sequence from strain Rx1 and ability of PspA from EF5668 to elicit protection against pneumococci of different capsular types. Infect. Immun. 66:4748–4754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. McDaniel LS, Scott G, Kearney JF, Briles DE. 1984. Monoclonal antibodies against protease-sensitive pneumococcal antigens can protect mice from fatal infection with Streptococcus pneumoniae. J. Exp. Med. 160:386–397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. McDaniel LS, et al. 1987. Use of insertional inactivation to facilitate studies of biological properties of pneumococcal surface protein A (PspA). J. Exp. Med. 165:381–394 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Miyaji EN, Dias WO, Tanizaki MM, Leite LC. 2003. Protective efficacy of PspA (pneumococcal surface protein A)-based DNA vaccines: contribution of both humoral and cellular immune responses. FEMS Immunol. Med. Microbiol. 37:53–57 [DOI] [PubMed] [Google Scholar]
- 33. Mold C. 1999. Role of complement in host defense against bacterial infection. Microbes Infect. 1:633–638 [DOI] [PubMed] [Google Scholar]
- 34. Moreno AT, et al. 2010. Immunization of mice with single PspA fragments induces antibodies capable of mediating complement deposition on different pneumococcal strains and cross-protection. Clin. Vaccine Immunol. 17:439–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morgan BP. 2000. The complement system: an overview, p 1–13 In Morgan BP. (ed), Complement methods and protocols. Humana Press, Totowa, NJ: [DOI] [PubMed] [Google Scholar]
- 36. Neeleman C, et al. 1999. Resistance to both complement activation and phagocytosis in type 3 pneumococci is mediated by binding of complement regulatory protein factor H. Infect. Immun. 67:4517–4524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nepomuceno RR, Ruiz S, Park M, Tenner AJ. 1999. C1qRP is a heavily O-glycosylated cell surface protein involved in the regulation of phagocytic activity. J. Immunol. 162:3583–3589 [PubMed] [Google Scholar]
- 38. Newman SL, Vogler LB, Feigin RD, Johnston RB., Jr 1978. Recurrent septicemia associated with congenital deficiency of C2 and partial deficiency of factor B and the alternative complement pathway. N. Engl. J. Med. 299:290–292 [DOI] [PubMed] [Google Scholar]
- 39. Ochs MM, et al. 2008. Vaccine-induced human antibodies to PspA augment complement C3 deposition on Streptococcus pneumoniae. Microb. Pathog. 44:204–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Quin LR, Moore QC, 3rd, McDaniel LS. 2007. Pneumolysin, PspA, and PspC contribute to pneumococcal evasion of early innate immune responses during bacteremia in mice. Infect. Immun. 75:2067–2070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Ren B, et al. 2004. The virulence function of Streptococcus pneumoniae surface protein A involves inhibition of complement activation and impairment of complement receptor-mediated protection. J. Immunol. 173:7506–7512 [DOI] [PubMed] [Google Scholar]
- 42. Ren B, Szalai AJ, Hollingshead SK, Briles DE. 2004. Effects of PspA and antibodies to PspA on activation and deposition of complement on the pneumococcal surface. Infect. Immun. 72:114–122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ren B, Szalai AJ, Thomas O, Hollingshead SK, Briles DE. 2003. Both family 1 and family 2 PspAs can inhibit complement deposition and confer virulence to a capsular 3 serotype Streptococcus pneumoniae. Infect. Immun. 71:75–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Tettelin H, et al. 2001. Complete genome sequence of a virulent isolate of Streptococcus pneumoniae. Science 293:498–506 [DOI] [PubMed] [Google Scholar]
- 45. Tu A-HT, Fulgham RL, McCory MA, Briles DE, Szalai AJ. 1999. Pneumococcal surface protein A (PspA) inhibits complement activation by Streptococcus pneumoniae. Infect. Immun. 67:4720–4724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Underhill DM, Ozinsky A. 2002. Phagocytosis of microbes: complexity in action. Annu. Rev. Immunol. 20:825–852 [DOI] [PubMed] [Google Scholar]
- 47. Vachon E, et al. 2007. CD44-mediated phagocytosis induces inside-out activation of complement receptor-3 in murine macrophages. Blood 110:4492–4502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Van Strijp JA, Van Kessel KP, van der Tol ME, Verhoef J. 1989. Complement-mediated phagocytosis of herpes simplex virus by granulocytes. Binding or ingestion. J. Clin. Investig. 84:107–112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Wardlaw T, White E, Hodge M. 2006. Pneumonia the forgotten killer of children. Unicef, World Health Organization, Geneva, Switzerland [Google Scholar]
- 50. Wu HY, Nahm MH, Guo Y, Russell MW, Briles DE. 1997. Intranasal immunization of mice with PspA (pneumococcal surface protein A) can prevent intranasal carriage, pulmonary infection, and sepsis with Streptococcus pneumoniae. J. Infect. Dis. 175:839–846 [DOI] [PubMed] [Google Scholar]
- 51. Yother J, Handsome GL, Briles DE. 1992. Truncated forms of PspA that are secreted from Streptococcus pneumoniae and their use in functional studies and cloning of the pspA gene. J. Bacteriol. 174:610–618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yother J, White JM. 1994. Novel surface attachment mechanism for the Streptococcus pneumoniae protein PspA. J. Bacteriol. 176:2976–2985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Yuste J, et al. 2006. Roles of the alternative complement pathway and C1q during innate immunity to Streptococcus pyogenes. J. Immunol. 176:6112–6120 [DOI] [PubMed] [Google Scholar]