

Abstract
On the basis of positive preclinical data, we evaluated the safety and immunogenicity of an alphavirus replicon HIV-1 subtype C gag vaccine (AVX101), expressing a nonmyristoylated form of Gag, in two double-blind, randomized, placebo-controlled clinical trials in healthy HIV-1-uninfected adults. Escalating doses of AVX101 or placebo were administered subcutaneously to participants in the United States and Southern Africa. Because of vaccine stability issues, the first trial was halted prior to completion of all dose levels and a second trial was implemented. The second trial was also stopped prematurely due to documentation issues with the contract manufacturer. Safety and immunogenicity were evaluated through assessments of reactogenicity, reports of adverse events, and assessment of replication-competent and Venezuelan equine encephalitis (VEE) viremia. Immunogenicity was measured using the following assays: enzyme-linked immunosorbent assay (ELISA), chromium 51 (51Cr)-release cytotoxic T lymphocyte (CTL), gamma interferon (IFN-γ) ELISpot, intracellular cytokine staining (ICS), and lymphoproliferation assay (LPA). Anti-vector antibodies were also measured. AVX101 was well tolerated and exhibited only modest local reactogenicity. There were 5 serious adverse events reported during the trials; none were considered related to the study vaccine. In contrast to the preclinical data, immune responses in humans were limited. Only low levels of binding antibodies and T-cell responses were seen at the highest doses. This trial also highlighted the difficulties in developing a novel vector for HIV.
INTRODUCTION
Despite advances in HIV treatment and prevention, the virus continues to spread, especially in sub-Saharan Africa, where the need for a safe, accessible vaccine for the prevention of HIV infection is critical.
Venezuelan equine encephalitis (VEE) virus belongs to the family of alphaviruses—single-stranded, positive-sense RNA viruses with two open reading frames, one encoding the capsid and envelope structural proteins and the other encoding the nonstructural proteins that transcribe and replicate the viral RNA. Although alphaviruses can infect humans, the primary host of the VEE virus is the horse. Foreign genes (e.g., HIV gag) can be inserted in place of the VEE structural protein gene region in a cDNA plasmid, and an RNA transcript from such a plasmid, when introduced into cells, will be amplified and express the foreign genes. This self-amplifying RNA (replicon) will direct the synthesis of very large amounts of the foreign gene product within the cell, typically reaching levels of 15 to 20% of total cell protein (15). The replicon RNA can be packaged in vitro into virus-like replicon particles (VRP) by supplying the structural protein genes of VEE virus in trans, from helper RNA molecules that are amplified by the VEE virus nonstructural proteins (Fig. 1). VRP are morphologically identical to live alphavirus particles and retain similar cell tropisms but are safe, single-cycle, propagation-defective vectors that are genetically restricted to a single round of infection and expression due to deletion of the structural protein genes from the replicon RNA. The use of VRP based on VEE virus as a vaccine vector is especially attractive because they express heterologous proteins to high levels, target expression to dendritic cells, and are capable of inducing both humoral and cellular immune responses to the vectored gene products (3, 4, 8, 10, 11, 16, 18, 19).
Fig 1.
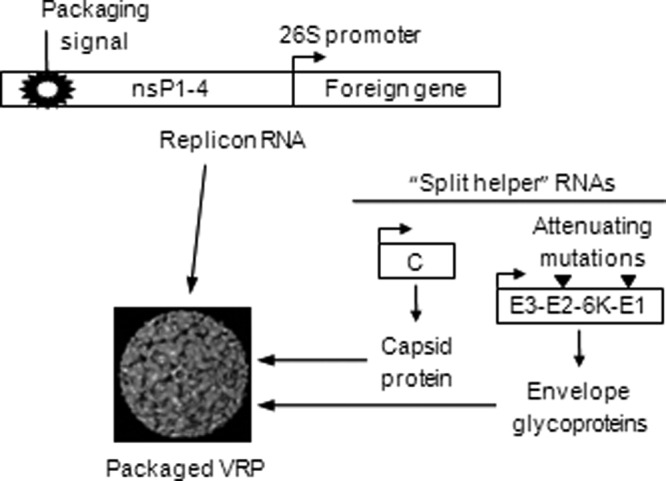
Virus-like replicon particle vaccine and packaging system.
AVX101 is an alphavirus replicon vaccine in which the alphavirus structural proteins have been replaced with HIV-1 subtype C gag. Preclinical immunogenicity studies of AVX101 in mice and rabbits revealed consistent antibody responses to Gag. In mice receiving two doses, 3 weeks apart, of 1 × 105 infectious units (IU) by subcutaneous (s.c.) footpad injections, the geometric mean reciprocal antibody titers 1 week after the second injection were 5,747 and 6,451 for two different good manufacturing practice (GMP) lots of AVX101 (compared to <40 for the controls). In mice, cellular immune responses were robust and consistently induced Gag-specific cytotoxic T lymphocyte (CTL) responses, as measured by chromium 51 (51Cr)-release CTL assay and gamma interferon (IFN-γ) enzyme-linked immunosorbent spot assay (ELISpot) assay (2). In a standard 51Cr release assay of splenic lymphocytes from mice immunized with two doses of 105 IU AVX101, a Gag-specific CTL response was observed for cells stimulated with either a recombinant vaccinia virus expressing an HIV subtype C gag gene or with an H-2Kd-restricted Gag peptide, representing the immunodominant CTL epitope. A Gag-specific CD8+ T-cell response was also observed using an IFN-γ ELISpot assay with a mean of 219 spot-forming cells (SFC)/106 cells (compared to 0 for the control) (7).
Based on these positive preclinical results, a clinical trial program was initiated. The results reported here include data from two separate clinical trial protocols (HVTN 040 and HVTN 059). The primary objective of these double-blind, randomized clinical trials was to evaluate the safety and tolerability of escalating doses of AVX101. The secondary objective was evaluation of immunogenicity as measured by IFN-γ ELISpot, 51Cr-release CTL, lymphoproliferation, and intracellular cytokine staining (ICS) assays, binding antibody by enzyme-linked immunosorbent assay (ELISA), and anti-VEE virus antibodies using a VRP neutralization assay.
HVTN 040 was designed to evaluate the safety of four escalating doses (1 × 104, 1 × 105, 1 × 106, and 1 × 107 IU) of AVX101 in healthy, HIV-1-uninfected adults. During the study, 9-month stability testing showed that the two higher vaccine doses (1 × 106 and 1 × 107 IU) stored at −20°C were below the release specification due to loss of titer when stored at −20°C but not when stored at −80°C. The storage conditions were changed from −20°C to −80°C for the two lower doses (104 and 105 IU) of the existing clinical trial material to minimize potential further decreases in potency at these dose levels, and the original protocol, HVTN 040, was modified to include only the two lower doses, 1 × 104 and 1 × 105 IU.
Improvements in the methods for manufacturing AVX101 enabled production of larger quantities and higher concentrations of the vaccine, allowing evaluation of dosage levels up to 1 × 108 IU per injection. Therefore, HVTN 059 was designed to obtain additional safety and immunogenicity data by evaluating higher doses of AVX101 in healthy HIV-1-uninfected adults. This second protocol repeated testing of the 1 × 105 IU dose and included additional dose levels of 1 × 106, 1 × 107 and 1 × 108 IU. HVTN 040 and HVTN 059 had identical objectives and outcomes and were conducted at the same clinical sites. In November 2005, potential issues with the contract manufacturer's documentation and environmental monitoring program were identified, and further injections were terminated, reducing the number of persons enrolled at the highest dose level. This report includes results from both the 040 and 059 clinical trials.
MATERIALS AND METHODS
Participants.
A total of 144 participants were to be enrolled, half in Africa (South Africa and Botswana) and half at sites in the United States, and randomized to receive either AVX101 or placebo in a 5:1 ratio. Participants were eligible for enrollment if they were between the ages of 18 and 60 for HVTN 040 or 18 and 50 years for HVTN 059, were HIV-1 uninfected, were in good general health, demonstrated an understanding of the purpose of the study, and provided informed consent. Female participants were required to agree to use birth control, not to breastfeed, and not to become pregnant during the course of the study.
Interventions.
The AVX101 vaccine contains an RNA replicon that was constructed by replacing the structural genes of an attenuated strain of VEE virus with the gag gene from a South African isolate (DU422) of subtype C HIV-1. The gag gene cDNA was modified by site-specific mutagenesis to remove the amino-terminal myristoylation site of Gag in order to prevent the formation of Gag-derived virus-like particles. The recombinant replicon was packaged into VRP by transfecting Vero cells with replicon RNA and two helper RNAs that express the structural proteins of the attenuated strain of VEE virus.
AVX101 was supplied by AlphaVax, Inc., Research Triangle Park, NC, as a sterile, clear solution in single-dose vials formulated at 5 different concentrations (1 × 104, 1 × 105, 1 × 106, 1 × 107, and 1 × 108 IU per 0.5 ml) and containing phosphate-buffered saline, pH 7.2, human serum albumin (HSA), sodium gluconate, and sucrose as a cryopreservative. The matching placebo (phosphate-buffered saline, pH 7.2, HSA, sodium gluconate, and sucrose) was also supplied by AlphaVax, Inc., as a sterile, clear solution in single-dose vials. Both AVX101 and placebo were manufactured at a GMP contract manufacturing facility. The same placebo was used in both trials, and results for all placebo recipients in the two trials were combined for data analysis.
All injections were 0.5 ml, administered subcutaneously in the upper arm after preparation of the site with alcohol. Participants and clinic staff were blinded as to the study product received. Only the clinic pharmacist had access to the randomization scheme, to allow for product preparation.
Participants received escalating doses of AVX101. For each dose level, participants were enrolled at sites in the United States, and safety data from the day 14 visit were evaluated for all participants at each dose level before proceeding with enrollment of participants at the same dose level at sites in Africa and at the next dose level at sites in the United States. The trial schemas are combined in Table 1.
Table 1.
Study schemaa
| Groupb | Dose (IU) | No. of participantsc |
Injection schedule in mo (days) |
|||
|---|---|---|---|---|---|---|
| US | SA | 0 | 1 (28) | 3 (84) | ||
| 1 | 1 × 104 | 10 | 10 | Vaccine | Vaccine | Vaccine |
| 2 | 2 | Placebo | Placebo | Placebo | ||
| 2 | 1 × 105 | 20 | 20 | Vaccine | Vaccine | Vaccine |
| 4 | 4 | Placebo | Placebo | Placebo | ||
| 3 | 1 × 106 | 10 | 10 | Vaccine | Vaccine | Vaccine |
| 2 | 2 | Placebo | Placebo | Placebo | ||
| 4 | 1 × 107 | 10 | 10 | Vaccine | Vaccine | Vaccine |
| 2 | 2 | Placebo | Placebo | Placebo | ||
| 5 | 1 × 108 | 10 | 10 | Vaccine | Vaccine | Vaccine |
| 2 | 2 | Placebo | Placebo | Placebo | ||
IU, infectious units; US, United States; SA, Southern Africa (South Africa and Botswana).
Group 1 and half of group 2 were enrolled in HVTN 040; the remainder of group 2, as well as groups 3 to 5, was enrolled in HVTN 059.
There were 72 participants (60 given the vaccine and 12 given the placebo) for each geographical group, for a total of 144 participants (120 given the vaccine and 24 given the placebo).
Safety.
Participants received injections of study vaccine or placebo on days 0, 28, and 84. Participants returned to the clinic for follow-up visits 2 weeks after each injection and again at months 6, 9, and 12 after the first injection. Reactogenicity assessments were performed for all participants before and after each injection, beginning 25 to 45 min postinjection and continuing daily for 7 days. Participants were asked to record their body temperature at the same time each day for 7 days, and clinical assessments were made during telephone or clinic visits on days 1, 2, 3, and 7 postvaccination. Assessments performed included systemic reactogenicity (body temperature, malaise and/or fatigue, myalgia, headache, chills, arthralgia, nausea, vomiting) and local reactogenicity (injection site pain, tenderness, erythema or induration, and axillary lymph node tenderness or enlargement). In addition, participants were queried about adverse experiences at each visit or telephone contact.
Any participant who reported a fever greater than 38°C, or other moderate symptoms consistent with a viral illness (e.g., headache or malaise) during the 7 days following vaccination, or neurological symptoms (e.g., nuchal rigidity, ataxia, convulsions, coma, paralysis) within the window of the 2-week postvaccination visit, provided a serum sample to confirm the absence of replication-competent VEE viremia.
Immunogenicity.
The assays for 51Cr-release, ELISpot, and ELISA were performed at the South African Immunology Laboratory–National Institute for Communicable Diseases (Johannesburg, South Africa) on specimens from Southern African participants. For the U.S. participant specimens these assays were performed at Duke University Medical Center (Durham, NC). All assays were conducted in a blinded manner.
Binding antibodies by ELISA.
Binding antibodies to commercially available Gag protein (p55 Gag; Quality Biologicals) were assessed by ELISA using single serum dilutions (1/50 or 1/100) on samples taken at baseline, 2 weeks after the second and third vaccinations and at the final visit. Samples that were positive in the initial ELISA were tested by endpoint titration ELISA using six 2- to 7-fold serial dilutions of serum beginning at a 1/50 or 1/100 dilution. The magnitude of responses is reported as the difference in optical density (OD) in antigen-containing and non-antigen-containing wells at the 1:50 dilution.
51Cr-release CTL assay.
A standard 51Cr-release CTL assay was performed on fresh peripheral blood mononuclear cells (PBMC) at baseline and 2 weeks after the second and third vaccinations, using a 50:1 effector-to-target cell (E:T) ratio. Prevaccination samples were utilized to generate Epstein-Barr virus (EBV)-transformed B-lymphocyte cell lines (BLCL) from each trial participant. BLCL were infected with a recombinant vaccinia virus expressing HIV Gag, loaded with 51Cr, and used as targets for PBMC effectors isolated from the participant's autologous fresh whole blood. Responses are reported as percent HIV-specific 51Cr release.
IFN-γ ELISpot assay.
Bulk T-cell responses were assessed by IFN-γ ELISpot using cryopreserved PBMC at 200,000 cells per well, stimulated overnight with Gag peptide pools. Protocol-specific peptide pools included HIV-1 15-mer peptides overlapping by 11 amino acids (80 to 95% purity). Peptide pools covered the DU422 isolate Gag protein sequence with a total of 121 peptides. Peptides were provided at >80% purity from the NIH AIDS Reagents and Reference Repository Program. For these protocols, we used 3 peptide pools as follows: DU422-gag pool 1, Gag 1 peptides 1 to 40; DU422-gag pool 2, Gag 2 peptides 41 to 80; and DU422-gag pool 3, Gag 3 peptides 81 to 121.
ELISpot assays were performed at baseline and 2 weeks after the second and third vaccinations. Responses are reported as the number of spot-forming cells (SFC) per 106 PBMC recognizing any of the three Gag peptide pools evaluated.
Intracellular cytokine staining (ICS) assay.
For participants in HVTN 059, flow cytometry was used to examine HIV-specific CD4+ and CD8+ T-cell responses using ICS, following stimulation with Gag peptides that span the protein sequence encoded by the vaccine construct. ICS assays were performed at the Fred Hutchinson Cancer Research Center Laboratories. Assays were performed at baseline and 2 weeks after the second and third vaccinations. Responses are reported as percentages of CD4+ or CD8+ T cells recognizing any peptide pool.
LPA.
For participants in HVTN 040, a lymphocyte proliferation assay (LPA) in response to purified Gag protein and/or Gag peptides was performed on cryopreserved PBMC collected at baseline, 2 weeks after the second and third vaccinations, and at the final visit. The LPA was conducted at the Fred Hutchinson Cancer Research Center Laboratories.
Antibodies to VEE virus.
To evaluate the development of anti-vector immune responses, neutralizing antibodies to VEE virus were measured in serum obtained at baseline, 2 weeks after the second and third vaccinations, and at the final visit, using a green fluorescent protein (GFP) VRP neutralization assay performed at AlphaVax, Inc. Samples were screened at a 1:10 dilution, and those with a positive response (>50% neutralization in the number of cells expressing GFP) were retested using serial 2-fold dilutions. The magnitude of responses in responders is reported as the sample dilution at which GFP levels were reduced by 50% compared to GFP levels in virus control wells after subtraction of background GFP in cell control wells.
Sample size.
The sample size at each vaccine dose level was selected such that the stopping rule for not escalating the dose (2 or more vaccine-related grade IV adverse experiences) would be met with high probability if the true toxicity rate was above 15 to 20% and such that dose escalation would occur with high probability if the true toxicity rate was less than 5%. The studies planned for 20 participants to receive each of the 1 × 104, 1 × 106, 1 × 107, and 1 × 108 IU vaccine dose levels and for 40 participants to receive the 1 × 105 IU vaccine dose level, evenly divided among the U.S. and Southern African participants for each dose level. Based on the planned 20 vaccine recipients evaluable for the toxicity endpoint, if the true rate of toxicity is less than 5%, then there is at least a 0.74 probability that dose escalation would occur. If the true rate of toxicity is 10%, 20%, or 30%, respectively, then there are probabilities of 0.61, 0.93, and 0.99, respectively, to stop the dose escalation. Sample size calculations were not performed for the immunogenicity endpoints because they were secondary.
Randomization.
For both HVTN 040 and HVTN 059, the randomization sequence was obtained by computer-generated random numbers and provided to each site using standard statistical and data management center (SDMC) procedures in place at the Statistical Center for HIV/AIDS Research and Prevention (SCHARP). For each region separately (United States and Southern Africa), the lists allocated participants to each dose group and to active or placebo within each dose group, using permuted blocks of 6 treatments (5 active, 1 placebo). The randomization code was kept at each institution by the pharmacist of record with primary responsibility for drug dispensing. Syringes containing vaccine or placebo were provided to the investigators and clinic staff in a blinded fashion by the pharmacist at the clinical study site.
Statistical methods.
Data were analyzed according to a slightly modified intention-to-treat approach, wherein all subjects receiving the first vaccination were included in the analysis, and analyzed based on initial randomization assignment regardless of the number of injections received. Immunogenicity of the vaccine was measured by the rate of positive response (for each group and time point) for each assay type described above. Ninety-five percent confidence intervals for the positive response rates were computed with the Wilson method (1). Fisher's exact tests were used to compare positive response rates between study groups. For ELISA, ICS, and LPA data, immunogenicity of the vaccine was also measured by the median magnitude of response among those with a positive response. These magnitudes were compared between study groups with the Mann-Whitney test. To improve statistical power for detecting vaccine-induced immune responses and simplifying the presentation, the placebo groups for the HVTN 040 and 059 trials were combined for assessing vaccine immunogenicity. This introduces minimal risk of bias because the placebos were identical for the two trials and the same lab performed the assays. Ten vaccine groups were analyzed separately: the 1 × 104 and 1 × 105 IU doses for HVTN 040 in the United States and in Southern Africa, the 1 × 105, 1 × 106, 1 × 107, and 1 × 108 IU doses for HVTN 059 in the United States, and the 1 × 105 and 1 × 106 IU doses for HVTN 059 in Southern Africa. Immunogenicity was evaluated on days 0, 42, 98, 168, and 364 of visits, with day 98 the primary time point. All statistical tests were two-sided and were deemed statistically significant if P was ≤0.05.
ELISA.
Response to Gag was considered positive if the difference in duplicate antigen-containing and non-antigen-containing wells had an OD of >0.2. The positivity criterion for the ELISA was established through validation studies comparing sensitivity and specificity of the assay using samples from HIV-infected donors, recipients of HIV vaccines, and normal healthy donors. The criterion of 0.2 OD units was selected to ensure a low false-positive rate while maintaining a reasonable level of sensitivity.
51Cr-release CTL assay.
The CD8+ CTL response was considered positive if all of the following three conditions held: (i) specific lysis for the HIV-1 antigen-expressing targets relative to the control was ≥10%; (i) this specific lysis decreased by at least 50% after removal of CD8+ cells; and (iii) this specific lysis was ≥5% after removal of CD4+ cells. Response rates were computed for HIV-1 antigen-expressing targets.
IFN-γ ELISpot assay.
To determine a positive response to a specific peptide pool, the bootstrap test described by Moodie et al. (12) was used to test the null hypothesis—that the mean result for the experimental wells was equal to twice the mean for the negative-control wells, versus the alternative that the experimental mean was greater—based on log10-transformed data. This method adjusts for the multiple peptide pools by calculating step-down, maxT-adjusted P values. Peptide pools with adjusted one-sided P values of ≤0.05 were considered positive. In addition to a significant P value, the mean background-subtracted response for the peptide pool must have been >10 SFC/200,000 PBMC for the peptide pool to be considered positive. This criterion ensures demonstration of a minimum level of biological activity. If the response to any of the three peptide pools was positive, then the overall response was considered positive.
Intracellular cytokine staining (ICS) assay.
To assess positivity, two-by-two contingency tables were constructed comparing the HIV-1 peptide-stimulated and negative-control data for each peptide pool and the two T-cell subsets (CD4 or CD8) expressing IFN-γ or interleukin 2 (IL-2). Cytokine subsets with negative staining for both IFN-γ and IL-2 were not of interest and therefore were not included in the analysis. In these contingency tables, the four entries are the number of cells positive and the number of cells negative for IL-2 or IFN-γ for both the stimulated and the negative-control data. A one-sided Fisher exact test was applied to each table, to test whether the number of cytokine-producing cells for the stimulated data was equal to that for the negative-control data. Since multiple individual tests (for each peptide pool and for each T-cell subset) were conducted simultaneously, a multiplicity adjustment was made using the discrete Bonferroni method (17). The adjusted P values were used to determine positivity, with values of ≤0.00001 indicating a positive response. If at least one peptide pool for a specific gene was positive, then the overall response to the gene was considered positive. If any peptide pool was positive for a T-cell subset, then the overall response for that T-cell subset was considered positive.
Lymphocyte proliferation assay (LPA).
For each peptide pool, a positive lymphocyte proliferation response was defined as a stimulation index greater than 3. If the response to any peptide pool was positive, then the overall response was considered positive.
RESULTS
Enrollment and demographics.
For HVTN 040, the first participant was enrolled in July 2003, the last participant was enrolled in May 2004, and follow-up was complete in July 2005. For HVTN 059, the first participant was enrolled in October 2004, the last participant was enrolled in October 2005, and follow-up was completed in September 2006. As noted above, in November 2005 a decision was made to halt all further injections of study product due to product manufacturing issues.
The numbers of participants who received each vaccine injection are shown in Table 2. Of the 132 enrolled participants, 109 (83%) received all 3 planned injections. Of those who did not complete the study, only 3 (2%) discontinued due to adverse events (discussed in detail below). Enrollment of the third dose level in Southern Africa was under way when the decision to halt further injections was made, and thus only 2 of 10 participants in this group received a second dose of vaccine and none received a third dose.
Table 2.
Exposure by dose level and regiona
| Group | Dose (IU) | Injection type | No. of participants who received injection no. [mo (day)] |
|||||
|---|---|---|---|---|---|---|---|---|
| 1 [0] |
2 [1 (28)] |
3 [3 (84)] |
||||||
| US | SA | US | SA | US | SA | |||
| 1 | 1 × 104 | Vaccine | 10 | 10 | 10 | 9 | 9 | 9 |
| Placebo | 2 | 2 | 2 | 2 | 2 | 2 | ||
| 2 | 1 × 105 | Vaccine | 20 | 20 | 18 | 20 | 18 | 19 |
| Placebo | 4 | 4 | 4 | 4 | 4 | 4 | ||
| 3 | 1 × 106 | Vaccine | 10 | 10 | 9 | 10 | 9 | 8 |
| Placebo | 2 | 2 | 2 | 2 | 2 | 1 | ||
| 4 | 1 × 107 | Vaccine | 10 | 10 | 10 | 2 | 10 | 0 |
| Placebo | 2 | 2 | 2 | 0 | 2 | 0 | ||
| 5 | 1 × 108 | Vaccine | 10 | 0 | 10 | 0 | 9 | 0 |
| Placebo | 2 | 0 | 1 | 0 | 1 | 0 | ||
| Total | Vaccine | 60 | 50 | 57 | 41 | 55 | 36 | |
| Placebo | 12 | 10 | 11 | 8 | 11 | 7 | ||
IU, infectious units; US, United States; SA, Southern Africa (South Africa and Botswana).
A summary of the demographics is provided in Table 3. The vaccine and control groups were well balanced with regard to demographics. The median age was 29.5 years (range, 20 to 55 years).
Table 3.
Demographics and vaccination frequencies
| Parametera | Result for vaccine and dose (IU) |
|||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| HVTN 040 |
HVTN 059 |
|||||||||
| Placebo (n = 8) | 1 × 104 (n = 20) | 1 × 105 (n = 20) | Total (n = 48) | Placebo (n = 14) | 1 × 105 (n = 20) | 1 × 106 (n = 20) | 1 × 107 (n = 20) | 1 × 108 (n = 10) | Total (n = 84) | |
| Sex | ||||||||||
| Male | 4 (50%) | 12 (60%) | 9 (45%) | 25 (52%) | 9 (64%) | 10 (50%) | 13 (65%) | 10 (50%) | 5 (50%) | 47 (56%) |
| Female | 4 (50%) | 8 (40%) | 11 (55%) | 23 (48%) | 5 (36%) | 10 (50%) | 7 (35%) | 10 (50%) | 5 (50%) | 37 (44%) |
| Race | ||||||||||
| White non-Hispanic | 4 (50%) | 7 (35%) | 5 (25%) | 16 (33%) | 3 (21%) | 10 (50%) | 5 (25%) | 9 (45%) | 4 (40%) | 31 (37%) |
| Black/African American non-Hispanic | 3 (38%) | 12 (60%) | 14 (70%) | 29 (60%) | 8 (57%) | 10 (50%) | 14 (70%) | 11 (55%) | 3 (30%) | 46 (55%) |
| Hispanic | 1 (13%) | 1 (5%) | 0 (0%) | 2 (4%) | 2 (14%) | 0 (0%) | 0 (0%) | 0 (0%) | 3 (30%) | 5 (6%) |
| Native Hawaiian/Pacific Islander or multiracialb | 0 (0%) | 0 (0%) | 1 (5%) | 1 (2%) | 1 (7%) | 0 (0%) | 1 (5%) | 0 (0%) | 0 (0%) | 2 (2%) |
| Age (yr) | ||||||||||
| 18–20 | 1 (13%) | 0 (0%) | 1 (5%) | 2 (4%) | 1 (7%) | 2 (10%) | 0 (0%) | 2 (10%) | 0 (0%) | 5 (6%) |
| 21–30 | 2 (25%) | 9 (45%) | 13 (65%) | 24 (50%) | 9 (64%) | 10 (50%) | 10 (50%) | 8 (40%) | 4 (40%) | 41 (49%) |
| 31–40 | 1 (13%) | 3 (15%) | 2 (10%) | 6 (13%) | 3 (21%) | 3 (15%) | 4 (20%) | 5 (25%) | 2 (20%) | 17 (20%) |
| 41–50 | 2 (25%) | 7 (35%) | 4 (20%) | 13 (27%) | 1 (7%) | 5 (25%) | 6 (30%) | 5 (25%) | 4 (40%) | 21 (25%) |
| Over 50 | 2 (25%) | 1 (5%) | 0 (0%) | 3 (6%) | ||||||
| Median | 39.5 | 32.0 | 27.0 | 29.5 | 25.5 | 26.5 | 30.5 | 29.5 | 32.0 | 28.5 |
| Range | 20–55 | 21–54 | 20–48 | 20–55 | 20–48 | 20–48 | 21–50 | 18–45 | 24–46 | 18–50 |
| Vaccination frequencies | ||||||||||
| Day 0 | 8 (100%) | 20 (100%) | 20 (100%) | 48 (100%) | 14 (100%) | 20 (100%) | 20 (100%) | 20 (100%) | 10 (100%) | 84 (100%) |
| Day 28 | 8 (100%) | 19 (95%) | 18 (90%) | 45 (94%) | 11 (79%) | 20 (100%) | 19 (95%) | 12 (60%) | 10 (100%) | 72 (86%) |
| Day 84 | 8 (100%) | 18 (90%) | 17 (85%) | 43 (90%) | 10 (71%) | 20 (100%) | 17 (85%) | 10 (50%) | 9 (90%) | 66 (79%) |
Results are no. (%) unless otherwise noted.
Data represent Native Hawaiian/Pacific Islander subjects for HVTN 040 and multiracial subjects for HVTN 059.
Safety.
A summary of reactogenicity symptoms is provided in Fig. 2. Overall, 64 (48.5%) subjects reported mild local reactions, with no obvious relationship to dose level or number of injections received. Only 3 participants (2.3%) reported moderate pain or tenderness at the injection site. Systemic reactogenicity symptoms were reported by 78 (59.1%) subjects, most of which were mild in intensity, with the patterns of response similar between the vaccine and control groups.
Fig 2.

Maximum local (A) and systemic (B) reactogenicity following each dose (1, 2, or 3) at each dose level.
There were 5 serious adverse events reported during the trials, none of which were considered related to the study vaccine. These reports included hospitalization following a car accident 1 month after receiving the 3rd injection of 1 × 104 IU of AVX101, hospitalization for epigastric discomfort (attributed to pregnancy) which began 4 months after the 3rd injection of 1 × 106 IU of AVX101, hospitalization for suicidal ideation which began 4 months after the 3rd injection of 1 × 107 IU of AVX101, hospitalization for elevated liver function tests and hyperglycemia 6 months after the 3rd injection of the control, and severe hypertension noted 2 weeks after the first injection of 1 × 106 IU of AVX101. The participant who became pregnant delivered a full-term healthy baby. After delivery, the participant was diagnosed as HIV infected, but her baby remained uninfected.
Additional events reported to the sponsor on an expedited basis, but not considered serious, were infectious hepatitis A or E, severe fatigue, and severe headache, each in one participant.
In addition to the participant with a severe headache mentioned above, there were 28 other participants who met the protocol requirements to have blood drawn and tested for VEE viremia. All cultures were negative.
Three participants discontinued their vaccinations due to adverse events. One was a 55-year-old white male with a history of Bell's palsy who had a recurrence of Bell's palsy 2 weeks after the first injection of AVX101 (1 × 105 IU). He was treated with Valtrex (valacyclovir) and prednisone. This event was considered possibly related to the vaccine. The second was a 30-year-old black male with neutropenia 2 weeks after receiving the first injection of AVX101 (1 × 105 IU). He had met the study entrance criteria at the time of enrollment; however, his white blood cell count and neutrophil count were low. His absolute neutrophil count fluctuated, increasing before the second scheduled injection at month 1 but dropping again after that injection. It was decided to discontinue vaccinations prior to the third scheduled injection, and the event was considered probably not related to the vaccine. The third was a 32-year-old white female who requested not to receive the third injection of AVX101 (1 × 108 IU) due to fatigue. Approximately 1 week following the second injection she developed a streptococcal infection and subsequently received an intramuscular injection of Rocephin (ceftriaxone) which resulted in a severe rash within 24 h. After 10 days the rash resolved, but the fatigue continued. This event was considered probably not related to the vaccine.
There were no deaths reported during the trials.
Immunogenicity.
The numbers of randomized participants included in the immunogenicity analyses are described in Table S1 in the supplemental material. For the ELISA, 51Cr-release CTL, LPA, and ELISpot assays, evaluable immunogenicity data at day 98 (the primary immunogenicity time point) were available for 70 to 100% of participants in the various dose groups. For ICS the rate was lower (40 to 100% across the dose levels). The most common reasons for missing data were a missed visit or an invalid blood draw (93.7%).
A dose-dependent antibody response to AVX101 was noted (Fig. 3 and Table 4). There were no positive Gag ELISA responses in subjects who received placebo or vaccine at the 1 × 104 IU dose, and only one vaccine recipient had a positive response at the 1 × 105 IU dose. For the three highest dose levels there was a progressive increase in the proportion of subjects with a positive response, and the response rates in the 1 × 107 IU (80%) and 1 × 108 IU (100%) dose groups were significantly greater than those in the placebo group (P = 0.0003 and P < 0.0001) and the 1 × 106 IU dose group (P = 0.017 and P < 0.0001). At the highest dose, the average ELISA magnitude (background-subtracted OD) was 0.55. The cellular immune response as measured by 51Cr-release, IFN-γ ELISpot, and ICS assays was quite modest and often did not demonstrate a clear dose response. Data from the 51Cr-release assay showed three subjects with a positive CD8+ CTL response at day 98, all from Southern Africa, at vaccine doses of 1 × 105 or 1 × 106 IU, and a positive CD4+ CTL response in one subject in the United States at a vaccine dose of 1 × 106 IU (Fig. 4). Only one subject had a positive IFN-γ ELISpot response, a U.S. vaccine recipient receiving 1 × 107 IU (Fig. 5). ICS was performed only for HVTN 059 participants in the United States. Only one subject had a positive CD8+ response, at a dose of 1 × 106 IU. In contrast, 2 of 8, 2 of 9, 5 of 10, and 4 of 10 vaccine recipients at doses of 1 × 105, 1 × 106, 1 × 107, and 1 × 108 IU, respectively, had a positive CD4+ response at day 98 (Fig. 6). Although a response could be considered positive if cells expressed only IFN-γ, only IL-2, or both IFN-γ and IL-2, all positive responses detected both IFN-γ and IL-2. The ICS data support higher response rates for the two highest dose groups compared to the control group (P = 0.0026, P = 0.13).
Fig 3.

ELISA responses 2 weeks after 3rd vaccination (day 98). A solid dot represents a positive response, and an open dot is for a negative response. Numbers at the top show the number of responders/number of assay results and the percentage with a positive result. For each box plot, 25% of the data points lie below the box and 25% lie above the box, and the horizontal line inside the box is the median. The vertical lines (whiskers) above and below the box extend to the most extreme data point, which is no more than 1.5 times the height of the box.
Table 4.
ELISA response rates by regiona
| Dose | No. with positive ELISA/total no. tested |
|||
|---|---|---|---|---|
| United States |
Southern Africa |
|||
| 2 wk post 2nd vac (day 42) | 2 wk post 3rd vac (day 98) | 2 wk post 2nd vac (day 42) | 2 wk post 3rd vac (day 98) | |
| 105 | 0/10 | 0/10 | 0/10 | 1/10 |
| 106 | 2/10 | 3/10 | 0/8 | 0/6 |
| 107 | 3/10 | 8/10 | NA | NA |
| 108 | 8/10 | 10/10 | NA | NA |
vac, vaccination; NA, not applicable.
Fig 4.

51Cr release. The left panel shows CD4 responses 2 weeks after 3rd vaccination (day 98), presented as the percentage of specific lysis after CD8 depletion. The right panel shows the CD8 responses at the same time point, presented as the percentage of specific lysis after CD4 depletion. A solid dot represents a positive response, and an open dot represents a negative response. Numbers at the top show the number of responders/number of assay results. For each box plot, 25% of the data points lie below the box and 25% lie above the box, and the and the horizontal line inside the box is the median. The vertical lines (whiskers) above and below the box extend to the most extreme data point, which is no more than 1.5 times the height of the box.
Fig 5.

ELISpot 2 weeks after 3rd vaccination (day 98). Data were obtained only for U.S. participants in study HVTN 059. A solid dot represents a positive response, and an open dot represents a negative response. Numbers at the top show the number of responders/number of assay results and the percentage with a positive result. For each box plot, 25% of the data points lie below the box and 25% lie above the box, and the horizontal line inside the box is the median. The vertical lines (whiskers) above and below the box extend to the most extreme data point, which is no more than 1.5 times the height of the box.
Fig 6.

ICS 2 weeks after 3rd vaccination (day 98). Data were obtained only for U.S. participants in study HVTN 059. A solid dot represents a positive response, and an open dot represents a negative response. Numbers at the top show the number of responders/number of assay results and the percentage with a positive result. For each box plot, 25% of the data points lie below the box and 25% lie above the box, and the horizontal line inside the box is the median. The vertical lines (whiskers) above and below the box extend to the most extreme data point, which is no more than 1.5 times the height of the box.
LPA was performed only for participants in HVTN 040, in which only 1 × 104 and 1 × 105 IU doses were administered. Two subjects had positive responses to the Gag-1 pool at day 98, one each in the 1 × 104 and 1 × 105 IU vaccine dose groups. Three subjects had day 98 responses to the Gag-2 pool, 2 in the 1 × 104 IU group and 1 in the 1 × 105 IU group. Twelve subjects had day 98 responses to the p24 pool (2 in the placebo group and 5 in each of the 1 × 104 and 1 × 105 IU groups), and 14 subjects had a day 98 response to the p55 pool (2 in the placebo group, 7 in the 1 × 104 IU group and 5 in the 1 × 105 IU group). The LPA response rates were similar in the vaccine groups and the control group (P > 0.5 for each peptide pool).
Anti-VEE virus antibody.
At baseline, only one subject (a U.S. participant in the 1 × 105 IU dose group in HVTN 040) had a positive anti-VEE virus antibody titer (1:10). At day 98, a positive anti-VEE titer was detected in 0/18, 0/19, 3/38, 6/18, 10/10, and 10/10 subjects in the placebo, 1 × 104, 1 × 105, 1 × 106, 1 × 107, and 1 × 108 IU dose groups, respectively. The geometric mean titers (GMT) for positive responses were 10, 160, 1,194, and 4,159 in the 1 × 105, 1 × 106, 1 × 107, and 1 × 108 IU dose groups, respectively.
DISCUSSION
These phase I trials of a recombinant VRP vaccine expressing a subtype C gag gene, modified to express nonmyristoylated Gag, demonstrated that the AVX101 vaccine was well tolerated in healthy adults at doses of up to 1 × 108 IU and that despite promising preclinical immunogenicity data, there were only modest immune responses among these trial participants. These trials also highlighted the difficulties in developing a novel vector for HIV. The trials were conducted sequentially over 3 years (2004 to 2006). Each trial was terminated early: one trial due to loss of vaccine titer during storage and the other due to potential issues with the contract manufacturer.
The vaccine was well tolerated and exhibited only modest local reactogenicity, similar to the modest reactogenicity seen in a phase I clinical trial of a VRP vaccine expressing cytomegalovirus (CMV) antigens (5). Few severe adverse events were reported during the studies. Subjects were enrolled in the two studies, in the United States and Southern Africa, successfully, and safety assessments in the two populations were similar.
The ELISA data revealed consistent dose-dependent antibody responses in participants who received the higher doses of vaccine (1 × 106 to 1 × 108 IU), with 3/10, 8/10 and 10/10 U.S. participants responding; however, the magnitude was much lower than was seen in preclinical studies. Using an assay in which the endpoint titer was the last dilution eliciting an OD of >0.20, the ELISA GMT ranged from 4,200 to 6,450 after two injections of 1 × 105 IU of AVX101 in four studies in mice and was 508, 1,280, and 3,620 after three injections of AVX101 in rabbits at doses of 1 × 106, 1 × 107, and 2 × 108 IU, respectively (2). In contrast, using an assay in which the OD at a 1:50 serum dilution was determined, the ELISA magnitudes at a dose of 1 × 108 IU in the clinical trial were only slightly above the positivity cutoff-adjusted OD of >0.2. In addition, because the two higher doses (1 × 107 and 1 × 108 IU) were not studied in Southern African participants, any conclusions about the anti-Gag antibody response are limited to U.S. participants.
Similarly, despite promising results in preclinical studies, AVX101 was poorly immunogenic in stimulating Gag-specific cellular immune responses in people. Preclinical immunogenicity studies of AVX101 in mice demonstrated robust cellular responses. In mice given two injections of 1 × 105 IU of AVX101 by subcutaneous footpad injection, positive ELISpot responses were detected in 100% of mice, with responses ranging from 75 to 420 SFC per 105 splenocytes, and 51Cr-release CTL assays showed >30% specific lysis at a 50:1 E:T ratio using splenocytes stimulated with Gag peptides and >30% specific lysis at a 6:1 E:T ratio using splenocytes stimulated with a vaccine-Gag virus (2). Additionally, a prototype VRP vaccine expressing simian immunodeficiency virus (SIV) gag and env genes was associated with high levels of immunogenicity in nonhuman primates (NHP) (8). In that trial, rhesus macaques received VRP-expressing proteins encoded in the SIVsm H-4i molecular clone. Four inoculations were administered over a period of 21 weeks: two subcutaneous injections both at 1 × 105 IU and two intravenous (i.v.) injections at 1 × 107 and 5 × 108 IU. Humoral and cellular immune responses were measured, and animals were challenged with SIVsm E660 given i.v. Antibody responses to Env were not detected following the two s.c. injections, but binding antibodies were detected after the first i.v. inoculation (the GMT measured 3 weeks following the last vaccination was 17,947). Two of the four vaccinated monkeys showed strong cellular immune responses to both SIV proteins, one had a low-level response to either Gag or Env, and the fourth vaccinated monkey had no detectable cellular response. Four weeks after immunization, animals were challenged with SIVsm E660 administered i.v. All animals were infected; however, two of the four control animals showed signs of illness by 4 weeks postchallenge and were eventually sacrificed. All four of the vaccinated animals were protected from disease progression with a reduction in viral load of 200-fold compared to controls, and there was an inverse relationship between viral load and humoral and cellular immune response.
In contrast to these positive data from a different VRP vaccine in nonhuman primates, and the encouraging preclinical data with AVX101, the data from this clinical trial demonstrated a dose-dependent anti-Gag antibody response but only a marginal T-cell response to Gag in humans. Only low levels of binding antibodies to p55 were achieved at the highest doses (1 × 107 and 1 × 108 IU), notwithstanding the expectation that infection of dendritic cells by the VRP vaccine would result in a strong immunogenic response. T-cell responses as detected by a 7-day 51Cr release CTL assay were seen in only a few participants and only at the 1 × 105 and 1 × 106 IU doses in participants from Southern Africa, and as measured by ICS assay, CD4+ T-cell responses appeared more frequent than CD8+ T-cell responses. By ELISpot assay only one participant, who received the 1 × 107 IU dose, had detectable T-cell responses.
The reasons for the differences between the preclinical and human phase I data are unclear. Preexisting anti-vector immunity is not the reason, since anti-VEE virus antibodies were present at enrollment in only one subject. SIV Gag has been shown in several studies to be more immunogenic than HIV-1 Gag. In addition, the gag gene cDNA in AVX101 was modified by site-specific mutagenesis to remove the amino-terminal myristoylation site in order to prevent the formation of Gag-derived virus-like particles (VLP) during production in the Vero packaging cells (6, 9, 14, 20). It was unclear at the time of the project's initiation whether the downstream purification process would be sufficient to remove the large majority of contaminating VLP from the VRP vaccine harvest. Thus, the modification was employed to simplify the purification and characterization of the AVX101 vaccine particle components. Based in part on the results of the current clinical trial, an HIV Gag VRP vaccine has been designed using a gag gene with a normal myristoylation site. The immunogenicities of VRP vaccines expressing nonmyristoylated (myr−) Gag (i.e., AVX101) and myristoylated (myr+) Gag were compared in nonhuman primates (Chinese rhesus macaques) injected at weeks 0, 4, and 24. Gag-specific T-cell responses as measured by IFN-γ ELISpot were positive in 5 of 5 Gag (myr+) VRP-vaccinated animals by 4 weeks after the first dose and remained positive at all later time points. In contrast, only 4 of 5 AVX101-vaccinated animals had an inconsistent positive response, at only one or two time points after the second dose. Mean responses were 4- to 5-fold higher in Gag (myr+) VRP-vaccinated animals than in AVX101-vaccinated animals after the 2nd (514 versus 126 SFC/106 PBMC) and 3rd (514 versus 126 SFC/106 PBMC) injections (Olmsted et al., unpublished data). Irrespective of the vaccine received, all animals mounted anti-Gag antibody responses after two doses of vaccine and in contrast to the vaccine-specific differences in T-cell responses, the Gag-specific ELISA titers induced by either vaccine were of similar magnitude. The results from this comparative immunogenicity study are encouraging and may warrant clinical evaluation of the Gag (myr+) VRP vaccine to determine in human subjects whether a VRP vaccine expressing Gag (myr+) would elicit more frequent and higher-magnitude Gag-specific T-cell responses than did AVX101.
This clinical study with AVX101 also demonstrated a dose-dependent antibody response to the VEE virus vector. This response was consistent with results in preclinical studies, and it is possible that the anti-vector responses played a role in the limited anti-Gag immune response. However, anti-VRP antibody responses do not appear to inhibit the ability of repeated injections of VRP vaccines to boost immune responses to other antigens in humans. In a clinical trial with a CMV VRP vaccine, anti-vector antibodies developed in most subjects after a first dose and all subjects after a second dose, but CMV gB-specific antibody titers were boosted after the second and third doses of vaccine (5). Similarly, in a cancer immunotherapy clinical trial comparing two dose levels of a carcinoembryonic antigen (CEA) VRP vaccine, after a single dose of vaccine the anti-vector antibody titer increased dramatically and remained elevated throughout the period of repeated immunizations. Despite this, there was a significant increase in immune response for at least one postvaccination time point versus prevaccination for all three assays (anti-CEA antibody, IFN-γ ELISpot, and ICS) in the high-dose cohort (13).
The HVTN 040 and 059 trials also demonstrated the difficulty inherent in process development for biological products such as virus-vectored vaccines. Given the fact that the vast majority of products that enter clinical trials do not successfully complete phase III testing and become licensed, it is not feasible to complete all process development activities, such as manufacturing scale-up and long-term stability testing, before initiating clinical trials. With the AVX101 vaccine, a single dose-response clinical trial (HVTN 040) was originally planned. Because of issues with stability, the HVTN 040 trial was halted after enrolling only the first two dose level groups. After the first study was started, improvements in scale-up of the manufacturing process provided an opportunity to test a higher dose level in the second study (HVTN 059). However, because of issues with documentation at the contract research organization (CRO) where the vaccine was manufactured, the second trial was also halted prior to completion of dosing participants in Southern Africa. Stability issues, including loss of potency and critical storage conditions, may come to light only once trials are under way, as was the case for HVTN 040. Further, many candidate HIV-1 prophylactic vaccines have their start in small academic or biotech environments. For many, a contract manufacturer is the only option for production of clinical-grade material. Although these manufacturers may have passed audits and have documented procedures in place, the developer of the vaccine does not have complete control over the facility and processes. As such, problems may arise at any point, including once a trial is under way, that can result in halting a study.
Results in animals often are not completely predictive of results in humans, but there is no alternative that is more predictive. There are no in vitro models of immunogenicity that have demonstrated good predictive ability. These hurdles, as well as the challenges inherent in the manufacture of these complicated vaccines, face all vaccine developers. Despite these problems, and the premature stopping of both trials, these studies were able to address the primary and secondary objectives, to evaluate the safety and tolerability and immunogenicity of increasing doses of AVX101 given by s.c. injection. AVX101 was well tolerated, but the cellular and humoral immune responses were disappointingly modest compared with the promising preclinical results.
Supplementary Material
ACKNOWLEDGMENTS
We acknowledge the following contributors: Robert Olmsted, Elizabeth Reap, Maureen Maughan, and Sergey Dryga at AlphaVax, Inc., Research Triangle Park, NC, Adi Ferrara, Walter Clinton, Karisse Torres, Drienna Holman, Huguette Redinger, and Renee Holt at Fred Hutchinson Cancer Research Center, Seattle, WA, and Scharla Estep at the Division of AIDS, NIAID, NIH.
Financial support was provided by the Division of AIDS, National Institute of Allergy and Infectious Diseases, National Institutes of Health, including grants 5 U01 AI550771-02, N01-AI-30029, 5-U01-AI-47976, 5-U01-AI-47980, 5-U01-AI-46747, 5-U01-AI-47985, 5-U01-AI-48013, 5-U01-AI-48023, 5-U01-AI-46747, U01 AI046703, and 3U01 AIO46747.
The AVX101 and placebo products were supplied by AlphaVax, Inc., Research Triangle Park, NC, and the studies were conducted under an investigational new drug application for which AlphaVax was the sponsor. J. Chulay was an employee of AlphaVax at the time the study was conducted and has stock ownership in the company. This paper was written by some authors in their capacity as NIH employees, but the views expressed in this paper do not necessarily represent those of the NIH.
This study has been registered at ClinicalTrials.gov under trial numbers NCT00063778 and NCT00097838.
Footnotes
Published ahead of print 22 August 2012
Supplemental material for this article may be found at http://cvi.asm.org/.
REFERENCES
- 1. Agresti A, Coull BA. 1998. Approximate is better than “exact” for interval estimation of binomial proportions. Am. Stat. 52:199–256 [Google Scholar]
- 2. AlphaVax, Inc 2004. AVX101 clinical investigators brochure. AlphaVax, Inc., Research Triangle Park, NC [Google Scholar]
- 3. Balasuriya UB, et al. 2002. Alphavirus replicon particles expressing the two major envelope proteins of equine arteritis virus induce high level protection against challenge with virulent virus in vaccinated horses. Vaccine 20:1609–1617 [DOI] [PubMed] [Google Scholar]
- 4. Balasuriya UB, et al. 2000. Expression of the two major envelope proteins of equine arteritis virus as a heterodimer is necessary for induction of neutralizing antibodies in mice immunized with recombinant Venezuelan equine encephalitis virus replicon particles. J. Virol. 74:10623–10630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bernstein DI, et al. 2009. Randomized, double-blind, phase 1 trial of an alphavirus replicon vaccine for cytomegalovirus in CMV seronegative adult volunteers. Vaccine 28:484–493 [DOI] [PubMed] [Google Scholar]
- 6. Bryant M, Ratner L. 1990. Myristoylation-dependent replication and assembly of human immunodeficiency virus 1. Proc. Natl. Acad. Sci. U. S. A. 87:523–527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Chulay J, et al. 2006. Safety and immunogenicity of an alphavirus replicon HIV Gag vaccine (AVX101) in healthy HIV-uninfected adults. Antivir. Ther. 11(Suppl 2:P11–09 [Google Scholar]
- 8. Davis NL, et al. 2000. Vaccination of macaques against pathogenic simian immunodeficiency virus with Venezuelan equine encephalitis virus replicon particles. J. Virol. 74:371–378 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Gottlinger HG, Sodroski JG, Haseltine WA. 1989. Role of capsid precursor processing and myristoylation in morphogenesis and infectivity of human immunodeficiency virus type 1. Proc. Natl. Acad. Sci. U. S. A. 86:5781–5785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Lee JS, et al. 2002. Immune protection against staphylococcal enterotoxin-induced toxic shock by vaccination with a Venezuelan equine encephalitis virus replicon. J. Infect. Dis. 185:1192–1196 [DOI] [PubMed] [Google Scholar]
- 11. Lee JS, et al. 2001. Candidate vaccine against botulinum neurotoxin serotype A derived from a Venezuelan equine encephalitis virus vector system. Infect. Immun. 69:5709–5715 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Moodie Z, Huang Y, Gu L, Hural J, Self SG. 2006. Statistical positivity criteria for the analysis of ELISpot assay data in HIV-1 vaccine trials. J. Immunol. Methods 315:121–132 [DOI] [PubMed] [Google Scholar]
- 13. Morse MA, et al. 2010. An alphavirus vector overcomes the presence of neutralizing antibodies and elevated numbers of Tregs to induce immune responses in humans with advanced cancer. J. Clin. Invest. 120:3234–3241 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pal R, et al. 1990. Myristoylation of gag proteins of HIV-1 plays an important role in virus assembly. AIDS Res. Hum. Retroviruses 6:721–730 [DOI] [PubMed] [Google Scholar]
- 15. Pushko P, et al. 1997. Replicon-helper systems from attenuated Venezuelan equine encephalitis virus: expression of heterologous genes in vitro and immunization against heterologous pathogens in vivo. Virology 239:389–401 [DOI] [PubMed] [Google Scholar]
- 16. Reap E, et al. 2001. Preclinical immunogenicity studies on pilot lots of a prototype VEE replicon vector HIV-1 subtype C vaccine for South Africa, abstr 175. Abstr. AIDS Vaccine, Philadelphia, PA [Google Scholar]
- 17. Tarone RE. 1990. A modified Bonferroni method for discrete data. Biometrics 46:515–522 [PubMed] [Google Scholar]
- 18. Wilson JA, Hart MK. 2001. Protection from Ebola virus mediated by cytotoxic T lymphocytes specific for the viral nucleoprotein. J. Virol. 75:2660–2664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wilson JA, et al. 2000. Epitopes involved in antibody-mediated protection from Ebola virus. Science 287:1664–1666 [DOI] [PubMed] [Google Scholar]
- 20. Zhou W, Parent LJ, Wills JW, Resh MD. 1994. Identification of a membrane-binding domain within the amino-terminal region of human immunodeficiency virus type 1 Gag protein which interacts with acidic phospholipids. J. Virol. 68:2556–2569 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.