

Abstract
The l-pantoyl lactone (l-PL) dehydrogenase (LPLDH) gene (lpldh) has been cloned from Rhodococcus erythropolis AKU2103, and addition of 1,2-propanediol (1,2-PD) was shown to be required for lpldh expression in this strain. In this study, based on an exploration of the nucleotide sequence around lpldh, a TetR-like regulator gene, which we designated lplR, was found upstream of lpldh, and three putative open reading frames existed between the two genes. Disruption of lplR led to 22.8 times higher lpldh expression, even without 1,2-PD induction, than that in wild-type R. erythropolis AKU2103 without 1,2-PD addition. Introduction of a multicopy vector carrying lplR (multi-lplR) into the wild-type and ΔlplR strains led to no detectable LPLDH activity even in the presence of 1,2-PD. The results of an electrophoretic mobility shift assay revealed that purified LplR bound to a 6-bp inverted-repeat sequence located in the promoter/operator region of the operon containing lpldh. These results indicated that LplR is a negative regulator in lpldh expression. Based on the clarification of the expression mechanism of lpldh, recombinant cells showing high LPLDH activity were constructed and used as a catalyst for the conversion of l-PL to ketopantoyl lactone. Finally, a promising production process of d-PL from dl-PL was constructed.
INTRODUCTION
Flavin mononucleotide (FMN)-dependent l-pantoyl lactone (l-PL) dehydrogenase (LPLDH) (EC 1.1.99.27) has been isolated from Rhodococcus erythropolis AKU2103 and shown to be dramatically induced by the addition of 1,2-propanediol (1,2-PD) during culture (10). Its gene (lpldh) has been cloned and overexpressed by introduction of an overexpression vector containing lpldh and its 1,940-bp upstream region into R. erythropolis AKU2103 (22). These results suggested that a strong promoter is involved in the expression of lpldh. LPLDH activity has also been detected in R. erythropolis PR4 (unpublished data), whose whole genomic sequence (NCBI reference sequence [RefSeq] accession no. NC_012490) has been determined, and a putative TetR family transcriptional regulator gene has been found in the upstream region of a putative oxidoreductase gene (the homologous gene of lpldh). The TetR regulator was first identified in Escherichia coli and was shown to control the expression of genes encoding a tetracycline efflux pump (3, 12). Without tetracycline in the cell, the TetR protein binds to the operator region and inhibits the biosynthesis of the resistance protein TetA. When tetracycline is present, it binds to TetR, leading to a change of the conformation, and consequently, TetR leaves the operator region and the resistance protein TetA can be expressed (18). So far, the TetR family proteins have been widely observed in Gram-positive and Gram-negative bacteria and have been shown to be encoded by both chromosomal and plasmid DNA. They generally act as repressors of transcription and regulate multifarious cellular activities, including morphogenesis (7), drug efflux pumping (18), antibiotic biosynthesis (16), phenylacetic acid metabolism (14), and biofilm formation (1).
To overcome the 50% limitation of molar yield in d-PL production from racemic PL, we have constructed a novel production process using LPLDH (Fig. 1) (22). The stereospecific dehydrogenation of l-PL to ketopantoyl lactone (KPL), catalyzed by LPLDH, and asymmetric reduction of ketopantoic acid (KPA) to d-pantoic acid (d-PA) (23) are involved in this process. For industrial application of the process, microorganisms possessing more sufficient LPLDH activity would be required.
Fig 1.

Enzymatic process for d-PL synthesis from dl-PL via KPL and KPA.
LPLDH of R. erythropolis is now the only known enzyme that is useful for l-PL oxidation. The physiological function of LPLDH in R. erythropolis AKU2103 and the regulation mechanism of lpldh expression were not determined until now. In this study, to clarify the mechanism by which 1,2-PD induces lpldh expression, we explored the nucleotide sequence around lpldh. Here, we show the regulation mechanism of lpldh expression by the LplR protein, a TetR-like regulator. In addition, LPLDH was overproduced and applied to the stereoinversion of l-PL to d-PL.
MATERIALS AND METHODS
Chemicals and reagents.
d-PL and l-PL were obtained from Tokyo Chemical Industry Co. (Japan) and Daiichi Fine Chemicals (Japan), respectively. KPL was purchased from Sigma-Aldrich. Restriction enzymes, Ex Taq DNA polymerase, and PrimeStar Max DNA polymerase were purchased from TaKaRa-Bio (Japan). All other reagents used in this work were of analytical grade and commercially available.
Microorganisms and cultivation.
R. erythropolis AKU2103 (Graduate School of Agriculture, Kyoto University, Kyoto, Japan), which could be obtained as NBRC12539 (NITE Biological Resource Center, Japan), was used as the DNA source. The cultivation was carried out as previously described (22). E. coli BL21(DE3) was used as the overexpression host for LplR. It was grown in LB medium (pH 7.0) containing 1% NaCl, 0.5% yeast extract (Oriental Yeast, Japan) and 1% tryptone (Becton, Dickinson) at 37°C. When necessary, 100 μg/ml ampicillin, 50 μg/ml kanamycin, and/or 0.1 mM isopropyl-β-d-thiogalactopyranoside (IPTG) was added.
General DNA techniques.
The genomic DNA of R. erythropolis AKU2103 was isolated with a DNeasy Blood and Tissue Kit (Qiagen). Plasmid DNA was isolated with a QIAprep spin miniprep Kit (Qiagen). The nucleotide sequence was determined by the dideoxy chain termination method (20) with a CEQ dye terminator cycle-sequencing kit (Beckman Coulter Inc.) and an automated sequencer DNA analysis system, CEQ2000XL (Beckman Coulter Inc.).
Exploration of open reading frames (ORFs) around lpldh.
For exploration of the nucleotide sequence flanking lpldh, the primers LPLDH-U1 to -U8 and -D1 to -D8 (see Table S1 in the supplemental material) were designed based on the nucleotide sequence flanking the homologous gene of lpldh in R. erythropolis PR4 (gene ID RER_18000 in RefSeq accession no. NC_012490NC_012490). The nucleotide sequence upstream of lpldh in R. erythropolis AKU2103 was amplified with the primer pairs LPLDH-U1/U2, -U3/U4, -U5/U6, and -U7/U8, and the downstream region was cloned with the primer pairs LPLDH-D1/D2, -D3/D4, -D5/D6, and -D7/D8. All of the PCR products were ligated into pT7Blue T vector and sequenced as described above.
Disruption of lplR.
A 3-kb fragment, LplR-AD, containing ΔlplR and its flanking region was amplified by overlap extension PCR (8, 9, 22) (Fig. 2A). Using PrimeStar Max DNA polymerase and genomic DNA of wild-type R. erythropolis AKU2103 as the template, the fragments LplR-AB and LplR-CD were amplified with the primer pairs LplR-A/B and LplR-C/D, respectively (see Table S1 in the supplemental material). After electrophoresis, the fragments were recovered with an illustra GFX PCR DNA and Gel Band Purification Kit (GE Healthcare, United Kingdom). Fragment LplR-AD was amplified using the primers LplR-A/D with a mixture of the fragments LplR-AB and LplR-CD as the template. A part of lplR was deleted in the amplified fragment LplR-AD. After cloning of the fragment LplR-AD into pT7Blue T vector and digestion with BamHI and XbaI, the fragment was ligated into the broad-host-range plasmid pK18mobsacB (19, 21) digested with the same restriction enzymes. The resulting plasmid, pK18ΔLplR, was used to construct the ΔlplR strain using a method described previously (22).
Fig 2.

ORF map of the 10-kb region around lpldh in R. erythropolis AKU2103. (A) Construction of DNA fragments used for lplR disruption. The primers used for RT-PCR are shown by arrows (RT1 to RT3) in the map. (B) Schematic presentation of DNA fragments used for EMSA analyses. The overlapping region of fragments Bind-ag and Bind-cj is shown with thick lines. In panels A and B, the gray pattern indicates ORFs and the hatched pattern indicates the regions remaining in the ΔlplR mutant. (C) Sequence of the intergenic region between lplR and orfD. The −35 and −10 regions and the Shine-Dalgarno (SD) sequence are underlined. The TSS is shown as +1. The IR sequence is shown in boldface. The unmatched base in the IR sequence is shown in italics. The start codons of lplR and orfD are underlined with arrows. The borders of the DNA fragment used in EMSA are indicated by bent arrows (Bind-a to -j) above the sequence. The IR sequences in the primers used to amplify DNA fragments for EMSA are boxed, and the nucleotide replacements in the primers Bind-c2, -c4, -c6, -g2, -g4, and -g6 are shown at the top.
RT-PCR.
Total RNA was isolated from overnight-cultured R. erythropolis AKU2103 strains with an RNeasy Mini Kit (Qiagen). The cDNA was amplified with a PrimeScript High Fidelity Reverse transcription (RT)-PCR Kit (TaKaRa-Bio) using random 6-mer primers and total RNA of the wild-type or ΔlplR strain of R. erythropolis AKU2103 as a template. Amplification of the fragment containing lpldh and the upstream ORFs D to F was performed using the primer pair RT1/RT2 or RT2/RT3 (Fig. 2A; see Table S1 in the supplemental material) with genomic DNA or cDNA as the template.
Determination of the relative expression level of lpldh.
Using the cDNA obtained as described above as the template, real-time PCR was performed to quantify the relative expression level of lpldh in the wild-type or ΔlplR strain of R. erythropolis AKU2103 with a LightCycler Quick System 350S (Roche Diagnostics, Germany) and a LightCycler FastStart DNA Master SYBR green I Kit (Roche Diagnostics). A fragment in the middle of lpldh was amplified with the primers LPLDH-RTF/LPLDH-RTR (see Table S1 in the supplemental material), and a part of the β-actin gene was amplified as an internal standard with the primers actin-F/actin-R (see Table S1 in the supplemental material).
Determination of the TSS.
5′-Rapid amplification of cDNA ends (RACE) was performed to determine the transcription start site (TSS) of the operon containing lpldh. The 5′ terminus of mRNA derived from the operon containing lpldh was reverse transcribed with the primer 5′-RACE-P (see Table S1 in the supplemental material) using the total RNA of wild-type R. erythropolis AKU2103 as a template. Then, the degradation of the hybrid RNA and the self-ligation of the amplified single-stranded cDNA were carried out according to the instruction manual included in the 5′-Full RACE Core Set (TaKaRa-Bio). Two-time PCR amplification was performed using the primer pairs 5′-RACE-F1/R1 and 5′-RACE-F2/R2, respectively, according to the instruction manual. Finally, the fragment obtained was sequenced as described above to determine the TSS.
Construction of expression vectors.
lplR amplified with the primers LplR-F/R (see Table S1 in the supplemental material) was cloned into plasmid pK4 (6) to construct pKLplR. The lpldh expression plasmid, pKLPLDH, was constructed as described previously (22). pKLplR or pKLPLDH was introduced into the wild-type or ΔlplR strain of R. erythropolis AKU2103 by electroporation as described previously (24), except that medium containing 225 μg/ml kanamycin was used for selection of the successful transformants.
To obtain the purified LplR protein, lplR was amplified with the primers LplR-His-F/R (see Table S1 in the supplemental material) and inserted downstream of the IPTG-inducible T7 promoter of E. coli expression plasmid pET-21a(+) (Novagen, Germany) digested with NdeI and HindIII. Then, the resulting pETLplR-His6 was introduced into E. coli BL21(DE3).
Electrophoretic mobility shift assay (EMSA).
Recombinant E. coli cells harboring pETLplR-His6 were harvested from 10 ml culture by centrifugation and suspended in 1.2 ml of the binding and washing solution included in the MagExtractor Fusion Protein Purification Kit (Toyobo, Japan). The suspension was disrupted with an ultrasonic oscillator (Insonater 201 M; Kubota, Japan). The binding and elution of the His tag fusion LplR protein were performed according to the instructions in the kit.
The DNA fragments used in EMSA were amplified using the primer pairs Bind-a and Bind-f, -g, -h, or -i and Bind-j and Bind-b, -c, -d, or -e (see Table S1 in the supplemental material) with the genomic DNA of R. erythropolis AKU2103 as a template. Binding of the purified His tag fusion LplR protein to the DNA fragment was performed in 10 μl of a mixture containing 10 mM HEPES, 10 mM Tris-HCl, 50 mM KCl, 1 mM EDTA, 1 mM dithiothreitol, 10% (vol/vol) glycerol, 0.5 mg/ml bovine serum albumin, 0.4 pmol DNA fragment, and 0.1 to 4 pmol purified His tag fusion LplR, pH 7.4. After incubation at 37°C for 30 min, the mixture was run on a 6.2% (wt/vol) polyacrylamide gel with Tris-glycine buffer at 4°C. The gel was stained with SYBR Gold nucleic acid gel stain (Invitrogen). The primers Bind-g2, -g4, and -g6 containing 2 (TGGCAT→TGGCGC), 4 (TGGCAT→GAGTAC), and 6 (TGGCAT→GAATCG) nucleotide replacements (italics) in the right half of the inverted-repeat (IR) sequence and primers Bind-c2, -c4, and -c6 containing 2 (TGTCAT→TGTTGT), 4 (TGTCAT→TGGTGG), and 6 (TGTCAT→CAGTGG) nucleotide replacements in the left half of the IR sequence, respectively, were used in the amplification of DNA fragments used in EMSA (see Table S1 in the supplemental material).
Preparation of CFE and enzyme assay.
The cell extract (CFE) was prepared and the LPLDH activity assay was performed as previously described (22).
Stereoinversion of l-PL.
Stereoinversion of l-PL was performed as described previously (22), except that dl-PL was also used as a substrate.
Nucleotide sequence accession number.
The nucleotide sequence of the 4-kb fragment upstream of lpldh and the 5-kb fragment downstream of lpldh was deposited under accession no. AB689131.
RESULTS
Sequence analysis around lpldh.
The nucleotide sequence of an approximately 4-kb fragment upstream of lpldh and an approximately 5-kb fragment downstream of lpldh was determined. A computer-aided homology search analysis using the BLASTN program revealed that 7 putative ORFs were present in the upstream fragment and 3 putative ORFs were present in the downstream fragment (Fig. 2A). Recently, the whole or partial genomic sequences of several Rhodococcus strains were determined and deposited in the sequence database. Including lpldh, the homologous genes of the 11 ORFs exist, in the same order and orientation, only in the genomic sequence of R. erythropolis PR4 (gene ID RER_17930 to RER_18030). The proteins showing the highest identities to the deduced amino acid sequences of the 11 ORFs are shown in Table 1. Two putative ORFs, orfC and lplR, have an orientation opposite that of the downstream ORFs (D to F, lpldh, and G to I). The results of the alignment revealed that the deduced amino acid sequence of lplR showed 99% identity with the TetR family transcriptional regulator of R. erythropolis PR4 and the AcrR family transcriptional regulator of R. erythropolis SK121 and 71% identity with the TetR family transcriptional regulator of Rhodococcus opacus B4 and the AcrR family transcriptional regulator of Rhodococcus jostii RHA1. Thus, the putative LplR protein is thought to be a transcriptional regulator.
Table 1.
Putative features and similarities of the ORFs in R. erythropolis AKU2103 to the proteins deposited in GenBank
| Putative ORF | Gene length (nt) | Size of product (amino acids) | Proteins deposited in database (GenBank accession no.) showing closest similarity to the deduced amino acid sequencesa | % Identity at amino acid sequence level |
|---|---|---|---|---|
| orfA | 213 | 70 | Molybdenum-pterin binding protein of R. erythropolis PR4 (YP_002765240); conserved hypothetical protein of R. erythropolis SK121 (ZP_04384566) | 100 |
| orfB | 204 | 67 | Hypothetical protein RER_17940 of R. erythropolis PR4 (YP_002765241) | 100 |
| orfC | 834 | 277 | Methyltransferase domain protein of R. erythropolis SK121 (ZP_04384392) | 99 |
| lplR | 600 | 199 | AcrR family transcriptional regulator of R. erythropolis SK121 (ZP_04384490); TetR family transcriptional regulator of R. erythropolis PR4 (YP_002765243) | 99 |
| orfD | 168 | 55 | Hypothetical protein RER_17970 of R. erythropolis PR4 (YP_002765244) | 100 |
| orfE | 288 | 95 | Hypothetical protein RER_17980 of R. erythropolis PR4 (YP_002765245) | 100 |
| orfF | 1,218 | 405 | Hypothetical protein RER_17990 of R. erythropolis PR4 (YP_002765246); radical SAM domain protein of R. erythropolis SK121(ZP_04384594) | 100 |
| lpldh | 1,179 | 392 | FMN-dependent dehydrogenase of R. erythropolis SK121 (ZP_04384552) | 100 |
| orfG | 1,932 | 643 | 2,4-Dienoyl-CoA reductase of R. erythropolis SK121 (ZP_04384358) | 99 |
| orfH | 828 | 275 | Carveol dehydrogenase of R. erythropolis SK121 (ZP_04384472); oxidoreductase of R. erythropolis PR4 (YP_002765249) | 100 |
| orfI | 1,950 | 649 | FAD/FMN-binding oxidoreductase family of R. erythropolis SK121 (EEN88181) | 98 |
CoA, coenzyme A; FAD, flavin adenine dinucleotide.
Effects of lplR disruption.
The lplR disruption mutant was constructed by E. coli-mediated conjugation. After two-time homologous recombination, lplR was replaced by ΔlplR in the lplR disruption mutant. The lplR disruption was confirmed by PCR analysis. As shown in Fig. 3A, a 1,200-bp fragment containing the complete lplR, orfD, and orfE genes was amplified in the wild-type strain, while a 700-bp fragment was amplified in the ΔlplR strain. Sequence analysis also confirmed that a middle portion (477 bp) of lplR was deleted in the ΔlplR strain. The disruption of lplR did not affect the growth of the ΔlplR strain (data not shown). The LPLDH activities of the wild-type and ΔlplR strains of R. erythropolis AKU2103 were measured (Fig. 3B). Almost no activity (<0.1 mU/mg) was detected in the noninduction wild-type strain. However, the ΔlplR strain showed almost the same high LPLDH activity regardless of 1,2-PD induction (16.3 and 15.8 mU/mg with and without addition of 1,2-PD, respectively) (Fig. 3B). The activity was higher than that of the 1,2-PD-treated wild-type strain (9.6 mU/mg). The expression level of lpldh also showed the same tendency. In the wild-type strain, the expression of lpldh under the induction conditions was 6.6 times higher than that under the noninduction conditions. On the other hand, under the noninduction conditions, disruption of lplR led to 22.8 times higher expression of lpldh than in the noninduction wild-type strain (Fig. 3C). All of these results indicated that LplR is a negative regulator of lpldh expression. To confirm that the putative LplR transcription factor regulates the expression of lpldh, the effects of LplR overproduction in the wild-type and ΔlplR strains of R. erythropolis AKU2103 were also investigated. As in the wild-type strain cultured without 1,2-PD, almost no LPLDH activity was detected in either the recombinant ΔlplR strain or the recombinant wild-type strain bearing pKLplR (Fig. 3B).
Fig 3.

Effects of lplR disruption and overexpression of lplR. (A) PCR analysis of lplR of R. erythropolis AKU2103 strains with primers amplifying lplR and the two downstream ORFs D and E. M, marker. (B) LPLDH activity of CFE of each strain of R. erythropolis AKU2103. (C) Relative expression levels of lpldh in each strain of R. erythropolis AKU2103. +/−, with or without 1,2-PD induction; WT, wild-type strain of R. erythropolis AKU2103; ΔlplR, lplR disruption mutant of R. erythropolis AKU2103; WT (pKLplR), recombinant wild-type R. erythropolis AKU2103 harboring pKLplR; ΔlplR (pKLplR), recombinant ΔlplR strain of R. erythropolis AKU2103 harboring pKLplR; ΔlplR (pKLPLDH), recombinant ΔlplR strain of R. erythropolis AKU2103 harboring pKLPLDH.
Mapping the TSS.
The results of the RT-PCR proved that lpldh and the three upstream ORFs D to F are transcribed into the same mRNA. As shown in Fig. 4, using primers RT1 and RT2, a 3-kb fragment was amplified with either genomic DNA or cDNA as the template. However, no amplification was observed when total RNA was used as the template. When the primers RT2 and RT3 were used, no amplification was observed with cDNA or total RNA as the template, while a 4-kb fragment was amplified when genomic DNA was used as the template. These results suggested that lpldh and the three upstream ORFs are in the same operon.
Fig 4.
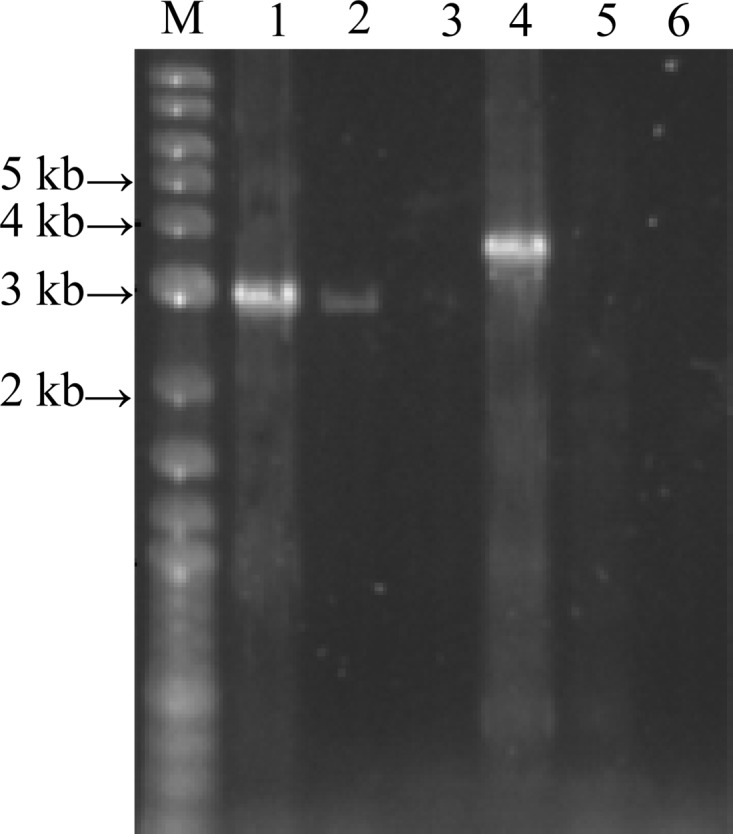
RT-PCR of lpldh and the three upstream ORFs D to F. Primers RT1 and RT2 were used in lanes 1 to 3; primers RT2 and RT3 were used in lanes 4 to 6. Genomic DNA was used as a template in lanes 1 and 4; cDNA obtained from R. erythropolis AKU2103 cultured in the presence of 1,2-PD (induction conditions) was used in lanes 2 and 5; total RNA not to be reverse transcribed was used as a control in lanes 3 and 6. M, marker.
The 5′ end of the mRNA transcribed from the operon containing lpldh was determined by 5′-RACE. Only one TSS was detected, and its transcription began at a G that was 22 bp upstream of the G of the start codon GTG of orfD (Fig. 2C).
Binding of LplR to the intergenic region between lplR and orfD.
We speculated that the intergenic region between lplR and orfD should be important for lpldh expression by acting as a potential binding site of LplR. When two fragments, Bind-ai and Bind-bj (Fig. 2B and C), containing the whole intergenic region were used in EMSA, complete shifted bands were observed (Fig. 5A and B). To determine the essential portion for LplR binding, six other DNA fragments (Fig. 2B and C) containing different portions of the intergenic region were employed in EMSA. When the fragment Bind-ah, Bind-ag, or Bind-cj was used, shifted bands were observed, while no shifted band appeared when the fragment Bind-af, Bind-dj, or Bind-ej was used (Fig. 5A and B). Therefore, the overlapping region of fragments Bind-ag and Bind-cj, that is, region Bind-cg, was considered to be indispensable for LplR binding. A 6-bp nonperfectly matched (1-bp) IR sequence was found in this overlapping region (Fig. 2C). Site-directed mutation was introduced into the IR sequence of the fragments Bind-ag and Bind-cj. The fragment Bind-ag with 2, 4, or 6 mutations in the right half-sequence showed no shifted band, while fragment Bind-cj containing 2, 4, or 6 mutations in the left half-sequence showed no effect on band shift (Fig. 5C).
Fig 5.

Binding of purified LplR to DNA fragments containing different portions of the intergenic region between lplR and orfD. (A) Binding of LplR to the fragments Bind-ai (lanes 1 and 2), Bind-ah (lanes 3 and 4), Bind-ag (lanes 5 and 6), and Bind-af (lanes 7 and 8). (B) Binding of LplR to the fragments Bind-bj (lanes 1 and 2), Bind-cj (lanes 3 and 4), Bind-dj (lanes 5 and 6), and Bind-ej (lanes 7 and 8). (C) Binding of LplR to the fragment Bind-cj (lanes 1 and 2); the fragment Bind-cj containing 2 mutations (lanes 3 and 4), 4 mutations (lanes 5 and 6), or 6 mutations (lanes 7 and 8) in the left half-sequence of the IR sequence; the fragment Bind-ag (lanes 9 and 10); and the fragment Bind-ag containing 2 mutations (lanes 11 and 12), 4 mutations (lanes 13 and 14), or 6 mutations (lanes 15 and 16) in the right half-sequence of the IR sequence. No LplR was added in the odd-numbered lanes, and 4 pmol purified LplR was added in the even-numbered lanes. M, marker.
Stereoinversion of l-PL.
To obtain a strain showing high LPLDH activity, the lpldh expression plasmid pKLPLDH was introduced into the ΔlplR strain. Like the original ΔlplR strain, the recombinant ΔlplR strain harboring pKLPLDH showed almost the same LPLDH activity with or without 1,2-PD induction (Fig. 3B).
Conversion of l-PL with the recombinant ΔlplR strain harboring pKLPLDH was carried out. As shown in Table 2, 0.384 M (50.0 mg/ml) l-PL could be completely converted to KPL with recombinant ΔlplR strain cells. When the l-PL concentration was 1.15 M (150 mg/ml), the conversion yield dramatically decreased to 85%. Because KPL is spontaneously hydrolyzed to KPA under neutral pH conditions, reduction of the accumulated KPA was performed by E. coli(pETSmKPR/pACGD) (23), which coexpressed KPA reductase and cofactor (NADPH)-regenerating enzyme (glucose dehydrogenase) genes. Irrespective of the conversion yield of l-PL to KPL, all of the KPA in the reaction mixture was reduced to d-PA. The resulting d-PA could be easily lactonized to d-PL with acid treatment. dl-PL was also used as a substrate in the conversion. Up to 0.768 M (100 mg/ml) dl-PL could be converted to d-PL with a combination of the recombinant R. erythropolis ΔlplR strain and E. coli(pETSmKPR/pACGD) in 96 h.
Table 2.
Stereoinversion of l-PL to d-PL with frozen recombinant ΔlplR strain of R. erythropolis AKU2103 harboring pKLPLDH and E. coli(pETSmKPR/pACGD)
| Substrate | Concn. (M) | Amt of Rhodococcus added (g wet wt)f | Time E. coli added (h) | Yield (%)g |
||
|---|---|---|---|---|---|---|
| d-PL | KPL | l-PL | ||||
| l-PL | 0.384 | 0.18 | Not added | 0 | >99.9 | 0 |
| 0.768 (0.384 + 0.384a) | 0.18 + 0.18 (72 h) | 168 | 94.5 | 0 | 5.5 | |
| 0.960 (0.384 + 0.192 + 0.192 + 0.192b) | 0.18 + 0.18 (72 h) | 168 | 95.2 | 0 | 4.8 | |
| 1.15 (0.384 + 0.192 + 0.192 + 0.192 + 0.192c) | 0.18 + 0.18 (72 h) | 168 | 85.5 | 0 | 14.5 | |
| dl-PL | 0.384 | 0.18 + 0.18 (30 h) | 36 | >99.9 | 0 | 0 |
| 0.768 (0.384 + 0.384d) | 0.18 + 0.18 (30 h) | 84 | >99.9 | 0 | 0 | |
| 1.15 (0.576 + 0.576e) | 0.18 + 0.18 (54 h) | 168 | 79.6 | 0 | 20.4 | |
l-PL (0.384 M) was added at 0 and 72 h.
l-PL (0.384 M) was added at 0 h, and 0.192 M l-PL was added at 36, 72, and 96 h.
l- PL (0.384 M) was added at 0 h, and 0.192 M l-PL was added at 36, 72, 96, and 120 h.
dl-PL (0.384 M) was added at 0 and 24 h.
dl-PL (0.576 M) was added at 0 and 48 h.
Frozen recombinant ΔlplR strain of R. erythropolis AKU2103 harboring pKLPLDH (0.18 g) was added at 0 h, and another 0.18 g was added at the time shown in parentheses.
The conversion of KPL under neutral pH conditions was performed for 12 h after addition of E. coli(pETSmKPR/pACGD).
DISCUSSION
The expression of lpldh in recombinant R. erythropolis AKU2103 harboring pKLPLDH has been shown to require addition of 1,2-PD (22). We therefore considered that there may be a strong regulator of the expression of lpldh in R. erythropolis AKU2103, and here, we performed a genetic analysis of the region around lpldh. Ten putative complete ORFs were found in the region around lpldh. A putative regulator gene, lplR, was found upstream of lpldh, and three ORFs (D to F) existed between these two genes (Fig. 2A). The putative LplR showed high similarity to members of the TetR protein family. Generally, members of the TetR family are preferentially negative regulators of the gene cluster adjacent to them (17). We therefore considered that the LplR protein might be related to expression of lpldh. While lpldh and lplR are not adjacent ORFs, lpldh and the three ORFs D to F (located between lpldh and lplR) were transcribed into the same mRNA. Thus, LplR seems to regulate lpldh expression by repressing transcription of the operon, including lpldh. To confirm this presumption, a disruption mutant of lplR was constructed, and it was found that, irrespective of the addition of 1,2-PD, the ΔlplR strain of R. erythropolis AKU2103 showed almost the same level of LPLDH activity, which was remarkably higher than that induced in the wild-type R. erythropolis AKU2103. The expression level of lpldh in the wild-type and ΔlplR strains of R. erythropolis AKU2103 also proved that disruption of lplR resulted in high-level expression of lpldh. This may have been due to the disruption of lplR preventing the repression of lpldh expression in the cells. Introduction of a multicopy vector carrying lplR (multi-lplR) into the wild-type and ΔlplR strains of R. erythropolis AKU2103 resulted in a drastic decrease of LPLDH activities. Since a large amount of LplR was produced in the recombinant strains bearing pKLplR, the transcription of the operon containing lpldh might be strongly repressed by LplR, even with the 1,2-PD addition. All of these results suggested that LplR is a negative regulator for lpldh expression.
The TSS of the operon containing lpldh was determined by 5′-RACE. Only one TSS was detected before the start codon of orfD. The putative −35 and −10 region sequences (GTCATTN16TACAGT) were selected by comparison with a promoter for the nit gene of Rhodococcus rhodochrous J1 (TTCATGN15TACTGT) (11) and a promoter for the casA gene of Streptomyces sp. (TTCACCN15TACCGT) (15). The putative promoter region showed sequence similarity to the E. coli promoter region (TTGACAN16–18TATAAT) (5). Nevertheless, no significant homology was detected with the promoter region for the dsz gene cluster of R. erythropolis IGTS8 (13) or for the cmr gene of Rhodococcus fascians (2). Generally, TetR-like regulators are reported to repress gene transcription by binding to the IR sequence in the promoter/operator region. An IR sequence (−37 to −32 and −21 to −16) (Fig. 2C) was found in the intergenic region between lplR and orfD. DNA fragments containing different parts of the IR sequence were used in EMSA. When fragments containing the intact IR sequence were used, shifted bands were observed. However, no shifted band was observed when the fragments contained the partial IR sequence. Therefore, the IR sequence is indispensable in binding of LplR to the promoter/operator region. Furthermore, any mutation in the right half-sequence of the IR sequence (near orfD) led to unbinding of LplR from the fragment Bind-ag, while no change was observed even with 6 mutations in the left half-sequence of the IR sequence (near lplR) in the fragment Bind-cj (Fig. 5C). These results indicated that the nucleotide length, not the nucleotide sequence of the left half-sequence, is important for binding of LplR. Förster et al. also reported that binding of AtuR to one of the inverted-repeat half-sequences did not depend on the second half-sequence (4).
The recombinant ΔlplR strain harboring pKLPLDH showed high LPLDH activity, even without 1,2-PD induction. Stereospecific oxidation of l-PL to KPL with the recombinant ΔlplR strain was achieved. Up to 1.15 M (150 mg/ml) l-PL could be converted to d-PL with an 85.5% molar yield. Our previous best result was an 80% molar yield with 1.15 M (150 mg/ml) l-PL as the substrate and using 1,2-PD-induced cells as the catalyst (22). So far, dl-PL is the most general intermediate of d-PL production discovered. dl-PL has also been used as a substrate in l-PL conversion. dl-PL (0.768 M) could be converted to d-PL completely with a recombinant ΔlplR strain of R. erythropolis AKU2103 harboring pKLPLDH and E. coli(pETSmKPR/pACGD). Thus, production of d-PL by stereoinversion of l-PL in dl-PL with these two strains is feasible.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by a Grant-in-Aid for Scientific Research, no. 23380051 (to M.K.), from the Japan Society for the Promotion of Science (JSPS) and by the Targeted Proteins Research Program (TPRP) of the Ministry of Education, Culture, Sports, Science and Technology (MEXT) of Japan.
Footnotes
Published ahead of print 31 August 2012
Supplemental material for this article may be found at http://aem.asm.org/.
REFERENCES
- 1. Conlon KM, Humphreys H, O'Gara JP. 2002. icaR encodes a transcriptional repressor involved in environmental regulation of ica operon expression and biofilm formation in Staphylococcus epidermidis. J. Bacteriol. 184: 4400– 4408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Desomer J, Vereecke D, Crespi M, Van Montagu M. 1992. The plasmid-encoded chloramphenicol-resistance protein of Rhodococcus fascians is homologous to the transmembrane tetracycline efflux proteins. Mol. Microbiol. 6: 2377– 2385 [DOI] [PubMed] [Google Scholar]
- 3. Eckert B, Beck CF. 1989. Topology of the transposon Tn10-encoded tetracycline resistance protein within the inner membrane of Escherichia coli. J. Biol. Chem. 264: 11663– 11670 [PubMed] [Google Scholar]
- 4. Förster-Fromme K, Jendrossek D. 2010. atuR is a repressor of acyclic terpene utilization (Atu) gene cluster expression and specifically binds to two 13 bp inverted repeat sequences of the atuA-atuR intergenic region. FEMS Microbiol. Lett. 308: 166– 174 [DOI] [PubMed] [Google Scholar]
- 5. Harley CB, Reynolds RP. 1987. Analysis of E. coli promoter sequences. Nucleic Acids Res. 15: 2343– 2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hashimoto Y, et al. 1992. Development of a host-vector system in a Rhodococcus strain and its use for expression of the cloned nitrile hydratase gene cluster. J. Gen. Microbiol. 138: 1003– 1010 [DOI] [PubMed] [Google Scholar]
- 7. Hillerich B, Westpheling J. 2008. A new TetR family transcriptional regulator required for morphogenesis in Streptomyces coelicolor. J. Bacteriol. 190: 61– 67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Horton RM, Cai ZL, Ho SN, Pease LR. 1990. Gene splicing by overlap extension: tailor-made genes using the polymerase chain reaction. Biotechniques 8: 528– 535 [PubMed] [Google Scholar]
- 9. Horton RM, Hunt HD, Ho SN, Pullen JK, Pease LR. 1989. Engineering hybrid genes without the use of restriction enzymes: gene splicing by overlap extension. Gene 77: 61– 68 [DOI] [PubMed] [Google Scholar]
- 10. Kataoka M, Shimizu S, Yamada H. 1992. Purification and characterization of a novel FMN-dependent enzyme. Membrane-bound L-(+)-pantoyl lactone dehydrogenase from Nocardia asteroides. Eur. J. Biochem. 204: 799– 806 [DOI] [PubMed] [Google Scholar]
- 11. Komeda H, Hori Y, Kobayashi M, Shimizu S. 1996. Transcriptional regulation of the Rhodococcus rhodochrous J1 nitA gene encoding a nitrilase. Proc. Natl. Acad. Sci. U. S. A. 93: 10572– 10577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Lederer T, Takahashi M, Hillen W. 1995. Thermodynamic analysis of tetracycline-mediated induction of Tet repressor by a quantitative methylation protection assay. Anal. Biochem. 232: 190– 196 [DOI] [PubMed] [Google Scholar]
- 13. Li MZ, Squires CH, Monticello DJ, Childs JD. 1996. Genetic analysis of the dsz promoter and associated regulatory regions of Rhodococcus erythropolis IGTS8. J. Bacteriol. 178: 6409– 6418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Mohamed Mel-S, Ismail W, Heider J, Fuchs G. 2002. Aerobic metabolism of phenylacetic acids in Azoarcus evansii. Arch. Microbiol. 178: 180– 192 [DOI] [PubMed] [Google Scholar]
- 15. Nakai R, Horinouchi S, Beppu T. 1988. Cloning and nucleotide sequence of a cellulase gene, casA, from an alkalophilic Streptomyces strain. Gene 65: 229– 238 [DOI] [PubMed] [Google Scholar]
- 16. Novakova R, Bistakova J, Homerova D, Rezuchova B, Kormanec J. 2002. Cloning and characterization of a polyketide synthase gene cluster involved in biosynthesis of a proposed angucycline-like polyketide auricin in Streptomyces aureofaciens CCM 3239. Gene 297: 197– 208 [DOI] [PubMed] [Google Scholar]
- 17. Ramos JL, et al. 2005. The TetR family of transcriptional repressors. Microbiol. Mol. Biol. Rev. 69: 326– 356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Saenger W, Orth P, Kisker C, Hillen W, Hinrichs W. 2000. The tetracycline repressor—a paradigm for a biological switch. Angew. Chem. Int. ed Engl. 39: 2042– 2052 [DOI] [PubMed] [Google Scholar]
- 19. Sánchez MB, Martínez JL. 2010. SmQnr contributes to intrinsic resistance to quinolones in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother. 54: 580– 581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sanger F, Nicklen S, Coulson AR. 1977. DNA sequencing with chain-terminating inhibitors. Proc. Natl. Acad. Sci. U. S. A. 74: 5463– 5467 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Schäfer A, Tauch A, Jäger Kalinowski WJ, Thierbach G, Pühler A. 1994. Small mobilizable multi-purpose cloning vectors derived from the Escherichia coli plasmids pK18 and pK19: selection of defined deletions in the chromosome of Corynebacterium glutamicum. Gene 145: 69– 73 [DOI] [PubMed] [Google Scholar]
- 22. Si D, et al. 2012. L-Pantoyl lactone dehydrogenase from Rhodococcus erythropolis: genetic analyses and application to the stereospecific oxidation of L-pantoyl lactone. Appl. Microbiol. Biotechnol. 95: 431– 440 [DOI] [PubMed] [Google Scholar]
- 23. Si D, Urano N, Shimizu S, Kataoka M. 2012. Cloning and overexpression of ketopantoic acid reductase gene from Stenotrophomonas maltophilia and its application to stereospecific production of d-pantoic acid. Appl. Microbiol. Biotechnol. 93: 1619– 1625 [DOI] [PubMed] [Google Scholar]
- 24. Urano N, et al. 2011. Genetic analysis around aminoalcohol dehydrogenase gene of Rhodococcus erythropolis MAK154: a putative GntR transcription factor in transcriptional regulation. Appl. Microbiol. Biotechnol. 89: 739– 774 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.