

Abstract
Ganoderic acids produced by Ganoderma lucidum, a well-known traditional Chinese medicinal mushroom, exhibit antitumor and antimetastasis activities. Genetic modification of G. lucidum is difficult but critical for the enhancement of cellular accumulation of ganoderic acids. In this study, a homologous genetic transformation system for G. lucidum was developed for the first time using mutated sdhB, encoding the iron-sulfur protein subunit of succinate dehydrogenase, as a selection marker. The truncated G. lucidum gene encoding the catalytic domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase (HMGR) was overexpressed by using the Agrobacterium tumefaciens-mediated transformation system. The results showed that the mutated sdhB successfully conferred carboxin resistance upon transformation. Most of the integrated transfer DNA (T-DNA) appeared as a single copy in the genome. Moreover, deregulated constitutive overexpression of the HMGR gene led to a 2-fold increase in ganoderic acid content. It also increased the accumulation of intermediates (squalene and lanosterol) and the upregulation of downstream genes such as those of farnesyl pyrophosphate synthase, squalene synthase, and lanosterol synthase. This study demonstrates that transgenic basidiomycete G. lucidum is a promising system to achieve metabolic engineering of the ganoderic acid pathway.
INTRODUCTION
Medicinal mushrooms are a rich source of therapeutically useful, biologically active agents. Ganoderma lucidum, a well-known traditional Chinese medicinal mushroom, has been used as a medication for the prevention and treatment of various human diseases for centuries in Asia. Ganoderic acids, a kind of highly oxygenated C30 lanostane-type triterpenoids produced by G. lucidum, exhibit a number of significant pharmacological activities such as cytotoxicity to hepatoma cells and antitumor, antimetastasis, and anti-HIV activity (50, 53). The biosynthesis of ganoderic acids was considered to start from acetyl coenzyme A (acetyl-CoA) and termed as mevalonate (MVA) pathway, where MVA was considered to be the only precursor. This pathway involves the sequential conversion of farnesyl diphosphate to squalene, 2, 3-oxidosqualene, and lanosterol (50). Some structural genes in the early stages of ganoderic acid biosynthetic pathway that encode 3-hydroxy-3-methylglutaryl CoA reductase (HMGR), farnesyl-diphosphate synthase (FPS), squalene synthase (SQS), and lanosterol synthase (LS) have been isolated and characterized (33). Although previous reports indicated that lanosterol is subsequently converted to ganoderic acids in G. lucidum (35), the final steps from lanosterol to ganoderic acids including a series of oxidation, reduction, and acylation reactions remain unclear (50).
Ganoderic acids receive extensive attention due to their important bioactivities. Thus, several approaches are adopted to improve their accumulation in submerged fermentation of G. lucidum, such as the manipulation of fermentation conditions, the addition of inducers, and the development of new bioprocessing strategies (18, 26, 39, 40, 42, 52, 56). However, higher yield of ganoderic acids in G. lucidum mycelia is needed for commercial applications (33). Genetic engineering has been successfully used to enhance the yields of bioactive compounds (55) and some enzymes such as laccase (1, 13) and versatile peroxidase MnP2 (41) in basidiomycete Agaricomycetes. However, no study reported the enhancement of terpenes accumulation through genetic manipulation of basidiomycetes.
The availability of practical transformation systems is a limiting factor for the genetic improvement of basidiomycetous mushrooms for biotechnological applications (11, 12, 14). Basidiomycete mushrooms generally exhibit higher resistance to genetic transformation compared to ascomycetous fungi (37). Despite the great requirement for G. lucidum, only a few reports exist regarding its genetic transformation using heterologous resistance genes as selection marker (16, 34, 38). In addition, the application of these methods to express other genes except marker genes has never been reported. Based on our experience, poor expression of heterologous genes, instability, and the formation of false positives possibly contribute to this situation.
A suitable transformation system is required to increase ganoderic acid accumulation in G. lucidum via molecular manipulation. Generally, a suitable selection marker gene and a stable transformation method are necessary for the development of a stable transformation system. The homologous marker gene is more efficient in establishing transformation system of basidiomycete mushrooms (15), which is also expected to be maintained and expressed more stably in the mycelium (10). Agrobacterium tumefaciens-mediated transformation (ATMT) is an efficient fungal transformation method and that shows a greater degree of stability for transgenes (5, 7, 45). Therefore, a homologous selection marker gene, cbxr, conferred by sdhB mutation and ATMT were used here to obtain stable transformants of G. lucidum. A homologous drug resistance marker that confers dominant resistance to fungicide carboxin has been used in some mushrooms such as Pleurotus ostreatus (10), Lentinula edodes (12), and Coprinopsis cinerea (15). This marker gene encodes a mutant iron-sulfur protein subunit of succinate dehydrogenase (SdhB). To the best of our knowledge, there has been no report of a homologous transformation system for G. lucidum.
HMGR is a key enzyme in the MVA-isoprenoid pathway that leads to the synthesis of various intermediates (8, 22), used for the biosynthesis of various terpenes. Constitutive overexpression of HMGR gene highly increases accumulation of monoterpenes (27), sesquiterpenes (2, 28), diterpenes (4), and tetraterpenes (43). Mutations of the HMGR gene also decrease triterpenoid levels in Arabidopsis thaliana (24). These observations reinforce the pivotal function of HMGR in the biosynthesis of terpenoids. The G. lucidum HMGR gene was cloned and characterized, and its expression exhibited a positive correlation with triterpenoid content according to reverse transcription-PCR (RT-PCR) data (32, 49). However, no direct evidence was found for the regulatory function of HMGR gene in the biosynthesis of triterpenoids in G. lucidum. Therefore, the regulation of ganoderic acid biosynthesis by overexpression of HMGR gene remains to be elucidated.
In the present study, a homologous genetic transformation system for G. lucidum was developed and successfully used for the genetic engineering of G. lucidum for greater a accumulation of ganoderic acids. The MVA pathway in G. lucidum was deregulated by the overexpression of the catalytic subunit of HMGR gene under the control of the G. lucidum glyceraldehyde-3-phosphate dehydrogenase gene (gpd) promoter. The effect of genetic modification on ganoderic acid biosynthesis was investigated by quantitatively analyzing ganoderic acid accumulation, intermediate formation, and gene expression of the key enzymes.
MATERIALS AND METHODS
Strains and growth conditions.
The strain G. lucidum CGMCC 5.616 from China General Microbiological Fermentation Center was maintained on potato dextrose agar and used as the recipient host strain for transformation. For plasmid construction, Escherichia coli strain DH5α was used and grown on Luria-Bertani (LB) agar plates containing 100 μg of ampicillin or 50 μg of kanamycin/ml, when appropriate. Agrobacterium tumefaciens EHA105 was used as a transfer DNA (T-DNA) donor for fungal transformation. The details of culture medium and culture conditions for G. lucidum are as follows. Preculture medium consisted of the following components (g/liter): glucose, 35; peptone, 5; yeast extract, 2.5; KH2PO4·H2O, 1; MgSO4·7H2O, 0.5; and vitamin B1, 0.05. For the first preculture, 40 ml of medium at an initial pH of 5.5 was prepared in a 250-ml flask, and then 10 ml of mycelium suspension from a slant culture was inoculated, followed by incubation for 5 days at 30°C on a rotary shaker (120 rpm). For the second preculture, 45 ml of medium was prepared in a 250-ml flask and inoculated with 5 ml of preculture broth (with ca. 600 to 700 mg [dry weight] of cells per liter), followed by 3 days of incubation at 30°C on a rotary shaker (120 rpm), as reported earlier (40). CYM medium (1% maltose, 2% glucose, 0.2% yeast extract, 0.2% tryptone, 0.05% MgSO4, 0.46% KH2PO4, 0.6 M mannitol, 1% agar) was used for the regeneration of protoplasts from G. lucidum. LB agar was used for the transformation of A. tumefaciens EHA105. Minimal medium [MM; 10 mM K2HPO4, 10 mM KH2PO4, 2.5 mM NaCl, 2 mM MgSO4, 0.7 mM CaCl2, 9 μM FeSO4, 4 mM (NH4)2SO4, 10 mM glucose (pH 7.0)] was used for cultivation of A. tumefaciens EHA105. Induction medium [IM; MM containing 0.5% (wt/vol) glycerol, 200 μM acetosyringone, 40 mM 2-(N-morpholino)ethanesulfonic acid, and 0.6 M mannitol (pH 5.3)] was used with A. tumefaciens EHA105 to infect G. lucidum protoplasts.
Nucleic acid extraction and cDNA synthesis.
G. lucidum mycelia were harvested, frozen in liquid nitrogen, and ground to a fine powder with a mortar and pestle. The genomic DNA was extracted using the CTAB (cetyltrimethylammonium bromide) method, and the total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). The quality and quantity of DNA and RNA samples were determined by using ethidium bromide-stained agarose gel electrophoresis and a spectrophotometer. Residual genomic DNA in isolated total RNA was removed using RNase-free DNase I (MBI Fermentas, Canada) according to the manufacturer's protocol. Reverse transcription was achieved by using total RNA as the starting material and a Superscript RNase H− First-Strand synthesis kit (Invitrogen).
Isolation and mutation of the G. lucidum sdhB.
Initially, the G. lucidum sdhB partial sequence of the genomic DNA was isolated by PCR amplification using the degenerate primers sdhB-pF and sdhB-pR (Table 1) designed on the basis of the highly conserved amino acid regions of known SdhB. After amplification, a 780-bp DNA fragment was obtained. The amplification products were cloned into pMD18-T vector (TaKaRa, Dalian, China) and sequenced. An attempt was then made to obtain the complete sequence of the sdhB by SEFA-PCR (46). PCR amplification of the 5′ end of the genomic DNA and promoter region was performed using three gene specific primers (sdhB-5sp1, sdhB-5sp2, and sdhB-5sp3) (Table 1) according to the acquired 780-bp sequence. PCR amplification of the 3′ end of the genomic DNA and terminator region was performed using three specific primers (sdhB-3sp1, sdhB-3sp2, and sdhB-3sp3) (Table 1), which were also designed based on the acquired 780-bp sequence. Self-formed adaptor-PCR (SEFA-PCR) was conducted in accordance with the manufacturer's protocol (46). We cloned and sequenced an ∼2.3-kb PCR product, including the 5′-untranslated region, and a 1.1-kb product, including the 3′-untranslated region. To amplify the whole genomic DNA of the sdhB (including the promoter and the terminator), the oligonucleotide primers sdhB-DF and sdhB-DR (Table 1) were designed based on the sequences of DNA obtained by SEFA-PCR. The thermal cycling parameters were as follows: an initial denaturation at 95°C for 10 min, followed by 30 cycles of 95°C for 30 s, 66°C for 30 s, and 72°C for 3.5 min, with a final extension at 72°C for 10 min. The amplification products were cloned into pMD18-T vector and sequenced.
Table 1.
Oligonucleotide primers used in this study
| Primer | Sequence (5′–3′)a |
|---|---|
| sdhB-pF | CGGCTCGTGYGCVATGAAC |
| sdhB-pR | GGTTGAGRCCCTTBGGACA |
| sdhB-5sp1 | CCGTCGAGCTTCTTCCTGTCCTCCTGC |
| sdhB-5sp2 | TTGCTTGTAGAATTGGGTGAGGTCGGGC |
| sdhB-5sp3 | ATGGGTAATATGCGAACGNNNNNNNNNGAAGGG |
| sdhB-3sp1 | GTGCCCGACCTCACCCAATTCTACAAGC |
| sdhB-3sp2 | GTCATTGGCTATCCCGTTGCTCGTTTCG |
| sdhB-3sp3 | GGCGGACTCTCGGGTATGNNNNNNNNTCCTTC |
| sdhB-DF | TCTGCTCTTCCCGATTGCTGCATTTGT |
| sdhB-DR | CTATGTCTTGCCTTGTCTCGCGTCAACC |
| sdhB-MR | GAAGATCGTGAGGCAGCGGTATAGGC |
| sdhB-MF | GCCTATACCGCTGCCTCACGATCTTC |
| gpd-F | TCCAAAGCCGCTCTCATGGCATGGCAC |
| gpd-SOE-R | CCGCACGCATGTTGAGAGGGGGATGAAGAGTGAGTAAGAAG |
| hmgr-SOE-F | CCCTCTCAACATGCGTGCGGTCCGCAGCCAAG |
| hmgr-R | TCACTTCGCCTCGACGACGTACCC |
| cbx-F | TGCGTTCAGCATGTATGAG |
| cbx-R | CAATCTAGCCCTGCGTTA |
| gpd-F2 | AGTTTCTGTGGTGCTGTTGC |
| hmgr-R2 | CGAGTTAGCTTCGTCCGTCT |
| thmgr-F | TCCCGTGGAGCCTATTACC |
| thmgr-R | GGATGACCTCTTCGTCGTTC |
DNA letter code: Y = C or T; R = A or G; V = A, C, or G; B = T, C, or G; N = A, T, G, or C.
A point mutation (CAC to CTC) that causes an amino acid substitution (His216 to Leu) was introduced into the sdhB of G. lucidum. First, a 2,644-bp DNA fragment containing the sdhB promoter and coding sequence was amplified using the primers sdhB-DF and sdhB-MR (5′-GAAGATCGTGAGGCAGCGGTATAGGC-3′, where A indicates the site of base substitution). A 546-bp DNA fragment, including the sdhB coding and terminator sequence, was amplified using the primers sdhB-MF (5′-GCCTATACCGCTGCCTCACGATCTTC-3′, where T indicates the site of base substitution) and sdhB-DR. The template DNA used for amplification of the two fragments was genomic DNA of G. lucidum. Thermal cycling parameters for the former fragment were as follows: an initial denaturation at 95°C for 10 min, followed by 35 cycles of 95°C for 30 s, 66°C for 30 s, and 72°C for 3 min, with a final extension at 72°C for 10 min. The amplification conditions of the latter fragment were as follows: an initial denaturation at 95°C for 10 min, followed by 35 cycles of 95°C for 30 s, 66°C for 30 s, and 72°C for 1 min, with a final extension at 72°C for 10 min. The DNA fragments were separated on a 0.8% agarose gel and purified. Subsequently, the two fragments were combined by PCR to form one 3,171-bp fragment using the sdhB-DF and sdhB-DR primers and the two purified fragments as templates. The amplification was initiated with a 10-min denaturation at 95°C, followed by 35 cycles of 95°C for 30 s, 66°C for 30 s, and 72°C for 4 min. The PCR product was subcloned into the pMD18-T vector, designated pMD18TCBX, and sequenced to confirm the base substitution.
Vector construction.
T-DNA binary vectors were constructed on the backbone of pCAMBIA1301 (Cambia, Canberra, Australia). The mutated sdhB cassette was amplified from plasmid pMD18TCBX using the primers sdhB-DF and sdhB-DR (Table 1) and Pfu DNA polymerase (Fermentas). Binary plasmid vector pJW (Fig. 1) was made by digesting pBAMBIA1301 with both PmlI and XhoI to remove the hygromycin B phosphotransferase gene (hph) and the β-glucuronidase gene (gus) and inserting the mutated sdhB cassette by blunt end ligation. Another binary vector pJW-tHMGR (Fig. 1) was constructed as follows. The promoter sequence (1,210 bp) for the gpd of G. lucidum (48) was obtained by PCR amplification using the primers gpd-F and gpd-SOE-R (Table 1) with genome DNA as a template. The catalytic domain of the HMGR gene (1,908 bp) of G. lucidum (32) was amplified from genome DNA using the primers hmgr-SOE-F and hmgr-R (Table 1). DNA fragments were separated on a 0.8% agarose gel and purified. Subsequently, the two fragments were combined by PCR to form one 3,109-bp fragment (Pgpd-tHMGR) using the gpd-F and hmgr-R primers and the two purified fragments as templates. Intermediate plasmid pBAR-PGPD-tHMGR was generated by digesting pBARGPE1 (University of Kansas Medical Center, Kansas City, KS) with XhoI and inserting the Pgpd-tHMGR fragment after filling the sticky end. Finally, plasmid pJW-tHMGR was constructed by excising the PGPD-tHMGR-trpC terminator fragment from plasmid pBAR-PGPD-tHMGR using EcoRV and NotI and inserting this fragment by blunt end ligation at the AseI site in plasmid pJW.
Fig 1.

Structure of plasmids used for G. lucidum transformation. See Materials and Methods for descriptions of the construction of these plasmids.
Agrobacterium-mediated fungal transformation.
The transformation protocol was a modification of the method developed by de Groot et al. (5). Protoplasts of G. lucidum were prepared as described by Sun et al. (38). A. tumefaciens strain EHA105, containing the binary vector pJW or pJW-tHMGR, was grown at 28°C for 2 days in MM supplemented with kanamycin (50 μg/ml) and rifampin (50 μg/ml). The A. tumefaciens cells were diluted to an optical density at 600 nm (OD600) of 0.15 in IM in the presence or absence of 200 μM acetosyringone. After preincubation for 6 to 8 h at 28°C on a rotary shaker (220 rpm) to an OD600 of 0.6 to 0.8, 200 μl of the bacterial cell suspension was mixed with an equal volume of a protoplast suspension (107/ml) from G. lucidum and then spread onto nitrocellulose membranes. The membranes were placed on a cocultivation medium (same as IM except containing 1% low-melting-point agar) and incubated at 25°C for 3 days. After cocultivation, the membranes were transferred to select medium (CYM medium) containing 2 μg of carboxin (Sigma, St. Louis, MO)/ml as a selection agent for transformants and carbenicillin (400 μg/ml) to kill the A. tumefaciens cells. After 10 to 14 days of incubation at 30°C, hyphae from visible fungal colonies were transferred to fresh CYM medium containing carboxin (2 μg/ml), which was repeated three times to obtain stable transformants. Control experiments were performed without acetosyringone. G. lucidum mycelia of transformants were scraped from surface culture and stained on a glass slide by using DAPI (4′,6′-diamidino-2-phenylindole; Sigma) at a concentration of 10 μg/ml in 0.1 M phosphate-buffered saline (pH 6.8) for 2 min. The mycelia were then washed and stained by calcofluor white (Sigma) at a concentration of 1 μg/ml in Tris-HCl buffer (pH 8.5) for 2 min. The morphology of mycelia was observed and photographed using an Olympus SP-51OUZ digital camera (Olympus, Japan).
Insertion analysis for the genomic DNA of transformants.
For PCR analysis, the 0.49 bp of the mutant sdhB fragments were amplified by using the primers cbx-F and cbx-R (Table 1). The amplification products were cloned into pMD18-T vector and sequenced. The fusion fragment of gpd promoter and the tHMGR gene was detected by using the primers gpd-F2 and hmgr-R2 (Table 1).
For genomic Southern blot analysis, 10 μg of DNA samples from transformed and nontransformed strains was digested with BamHI and separated on 0.8% agarose gel. The mutant sdhB fragment (0.49 kb) amplified from plasmid pJW with the primers cbx-F and cbx-R was used as the probe. DNA probe labeling and hybridization were performed under conditions recommended for the digoxigenin (DIG) hybridization system by Roche (Mannheim, Germany).
Analyses of dry cell weight and ganoderic acids.
Mycelia were harvested by centrifuging a sample at 10,000 × g for 10 min, and the precipitated cells were washed for three times with distilled water and then dried at 50°C to constant weight. The dry cell weight was measured by the gravimetric method. Ganoderic acids were extracted and measured according to a method described elsewhere (40).
Extraction and analysis of squalene and lanosterol.
Cellular squalene and lanosterol were extracted according to the methods of Yue et al. (51) and Mantzouridou et al. (21), with some modifications. Dried mycelia (50 mg) were saponified by 2 ml of 10% (wt/vol) KOH–75% (vol/vol) ethanol solution at 50°C for 2 h. The mixture was extracted with 2 ml of hexane for three times. The hexane layer was collected and evaporated to dryness under N2. The residue was dissolved in 0.5 ml of acetonitrile for subsequent high-pressure liquid chromatography (HPLC) analysis. Agilent 1200 series HPLC apparatus equipped with an Agilent G1315B diode array detector and an Agilent Zorbax SB-C18 column (250 by 4.6 mm, 5 μm) were used. The separation of squalene and lanosterol was achieved by using a mobile phase of 100% acetonitrile at a constant flow rate of 1.5 ml/min and a mobile phase of 100% methanol at a constant flow rate of 1.0 ml/min, respectively. The squalene and lanosterol were monitored at wavelengths of 195 and 210 nm, respectively. The peaks of squalene and lanosterol were identified based on their retention time and UV spectra against those obtained from squalene and lanosterol standards (Sigma), respectively. The quantification was performed according to the external calibration graphs obtained from the peak areas versus different concentrations of squalene and lanosterol standards, respectively.
Measurement of hmgr, fps, sqs, and ls gene expressions by qRT-PCR.
The transcription levels of the hmgr, fps, sqs, and ls genes (which encode the enzymes HMGR, FPS, SQS, and LS, respectively) were analyzed by real-time quantitative RT-PCR (qRT-PCR). The sequences of the primers for the amplification of the 18S rRNA gene, fps, sqs, and ls were described previously (49, 52). For hmgr, the primers thmgr-F and thmgr-R (Table 1) were used. The 18S rRNA gene was used as the internal control gene because its expression was found to be stable under our experimental conditions. The expression level of the different genes was normalized with respect to the 18S rRNA expression level. For each gene, an expression level of 1 was assigned to the samples from the wild-type (WT) strain, and the expression levels in the control transgenic strain and the pJW-HMGR-transformed strain are presented as the fold changes relative to this reference level.
Statistical analysis.
Data are the averages of three independent sample measurements. The error bars indicate the standard deviations from the means of triplicates. The data were analyzed with the Student t test. The difference between contrasting treatments was considered significant when P was <0.05 in a two-tailed analysis.
Nucleotide sequence accession number.
The GenBank accession number of the G. lucidum gene sdhB examined in the present study, including its promoter and terminator, was JN377411.
RESULTS
Establishment of a homologous transformation system for G. lucidum with the selection marker cbxr conferred by sdhB mutation.
Mutation of sdhB confers dominant resistance to carboxin in basidiomycetes (10, 12, 15). Thus, sdhB (including the promoter and terminator) from the genome of G. lucidum was amplified. The genomic sequence was submitted in the DDBJ/EMBL/GenBank nucleotide sequence databases under accession no. JN377411. The sdhB gene was mutagenized, as described in Materials and Methods, to obtain a variant expected to mediate carboxin resistance upon transformation into G. lucidum. Fungal colonies appeared on the selection medium containing 2 μg of carboxin/ml after the transformation of G. lucidum protoplasts with plasmid pJW via ATMT. No resistant colonies were observed when the acetosyringone was omitted. The transformation frequency was 10 transformants to 15 transformants per 107 protoplasts. Ten transformants were repeatedly transferred to a nonselective medium to investigate the mitotic stability of the integrated T-DNA. Clones were transferred to selective CYM medium after five rounds of growth on a nonselective medium (without carboxin). All tested transformants continued to grow on the selective medium. Moreover, the transformants exhibited 100% mitotic stability for the cbxr selection marker.
PCR amplification and Southern blotting were performed on genomic DNA from the wild-type (WT) strain and randomly selected transformants to confirm the presence of cbxr in the carboxin-resistant colonies. The expected product size of 0.49 kb was amplified from the genomic DNA of the WT strain and all transformants using the cbx-F and cbx-R primers (data not shown). Sequencing analysis revealed that the transformants carried the cbxr (Fig. 2). The presence of transgenic cbxr in the genome of the transformants was further tested by molecular hybridization. DNA from the transformants, and the WT strain was digested with BamHI and probed with a cbxr fragment (Fig. 3). The analysis confirmed that cbxr was integrated into the genome of the analyzed G. lucidum transformants.
Fig 2.
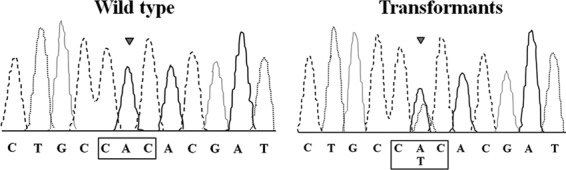
Detection of the introduced cbxr gene in transformants. PCR products amplified from genomic DNA of transformants or the wild-type strain were directly sequenced. Sequencing chromatograms are shown for mutation site of the sdhB in the wild type and in the carboxin-resistant transformants. Any two overlapping peaks indicate the nucleotide from the endogenous sdhB and the transformed cbxr. Arrowheads indicate the mutated nucleotides.
Fig 3.
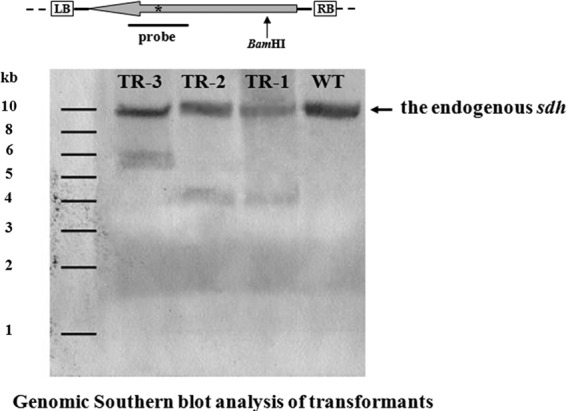
Detection of wild-type and transgenic mutant copies of sdhB in three randomly selected G. lucidum transformants. (Top) Schematic representation of the restriction enzyme-probe combination used to detect the transgenic mutated allele of sdhB. White boxes are the left and right border regions of T-DNA, respectively. The large gray arrow represents the carboxin resistance cassette cbxr. An asterisk (*) indicates the position of single nucleotide mutation responsible for carboxin resistance in sdhB. The position of the single BamHI site within the T-DNA is indicated by a black arrow. (Bottom) Random integration of T-DNA copies into the G. lucidum genome. WT, G. lucidum wild-type strain; TR-1 to TR-3, independent transformants obtained after ATMT using plasmid pJW. The genomic copy of the sdhB is indicated by arrow on the right.
Deregulated overexpression of an N-terminally truncated HMGR gene in G. lucidum.
The plasmid pJW-tHMGR, encoding a truncated G. lucidum HMGR that lacks the N-terminal membrane-binding domain of the enzyme, was used to transform the WT strain using the established transformation system. The truncated HMGR (tHMGR) gene was controlled by the strong constitutive gpd promoter of G. lucidum and the trpC terminator of Aspergillus nidulans in the plasmid pJW-tHMGR (Fig. 1). Resistance to carboxin was used to select putative transformants. The integration of the catalytic domain of the HMGR gene in the carboxin-resistant strains was confirmed by PCR. The PCR product showed a clear band for the fusion gpd promoter and the tHMGR gene fragment (938 bp) in the HMGR transformant (pJW-HMGR-transformed strain), but no corresponding band was observed in the WT strain and the control transgenic (TR) strain (Fig. 4). In addition, no morphological difference was observed between the HMGR transformant and the control. Total RNA was isolated from different strains. qRT-PCR analysis was conducted to compare HMGR gene expression levels in the transformant and WT. The results showed that HMGR gene was overexpressed in the HMGR transformant, which was ∼3-fold higher than the control on day 4 (see Fig. 6).
Fig 4.
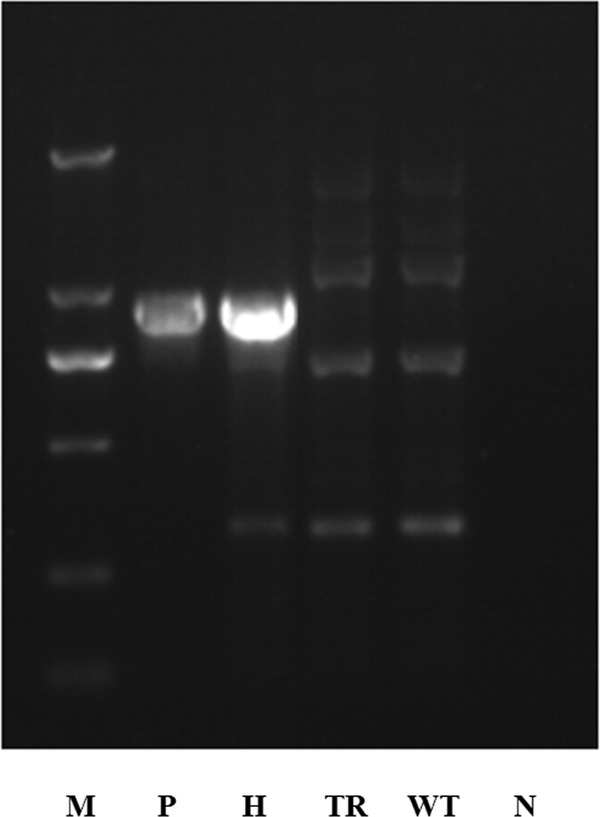
Identification and characterization of the tHMGR gene-overexpressing strains. Amplification patterns were obtained with primers for the fusion gpd promoter-tHMGR gene fragment in genomic DNA isolated from different strains. Lane N, negative control; lane WT, wide-type strain; lane TR, the strain transformed with pJW; lane H, the tHMGR overexpressed strain; lane P, pJW-tHMGR as a positive control; lane M, DL 2000 DNA marker.
Fig 6.

Transcriptional levels of biosynthetic genes in the WT strain (□), the strain transformed with pJW (▨), and the tHMGR overexpressed strain (■). Expression of the samples from the WT strain is defined as 1.0, and the expression levels in TR and HMGR strains are displayed as the fold increase over the reference sample. *, statistical significance (P < 0.05) compared to the WT strain. d, days.
Enhanced ganoderic acid biosynthesis in G. lucidum overexpressing the tHMGR gene.
The maximal biomass and contents of GA and intermediates (squalene and lanosterol) were assayed in the WT and TR strains and in three strains of tHMGR-overexpressing transformants. The maximal biomass and GA content of all strains were reached on day 12, and the maximal intermediates were obtained on day 4. The results, summarized in Table 2, confirm the consistency in HMGR overexpression. From the information obtained in Table 2, one overexpressing transformant (H3) was chosen with WT and TR strains to study the kinetic profiles of cell growth, ganoderic acid content, and intermediate (squalene and lanosterol) accumulation. Figure 5A shows that the overexpression of the tHMGR gene slightly affected cell growth compared to WT and TR strains. The maximum dry cell weights in the WT, TR, and HMGR strains were 8.73, 8.10, and 7.33 g/liter, respectively. The ganoderic acid content of the WT and TR strains did not change substantially during the entire fermentation, whereas the HMGR strain content significantly increased and reached a maximal value at the end of fermentation. The maximum content in the HMGR strain was 29.4 mg/g (dry weight), which was ∼2-fold of that obtained in the WT strain (14.1 mg/g [dry weight]) (Fig. 5B). Squalene and lanosterol are important intermediates in the ganoderic acid biosynthetic pathway. Thus, their accumulations in WT, TR, and HMGR strains were analyzed. The results showed that more squalene and lanosterol accumulated in the HMGR strain than in control strains throughout the cultivation (Fig. 5C and D). The overexpression of the tHMGR gene increased squalene content (86.6 μg/g [dry weight] versus 12.8 μg/g [dry weight] relative to the WT strain) at day 0. Although the content of squalene decreased to 26.4 μg/g (dry weight) in the HMGR strain at day 16, it still maintained a higher level than that of the WT (6.16 μg/g [dry weight]). The amount of lanosterol increased to a maximum of 2.97 mg/g (dry weight) at day 4 but dropped to 1.21 mg/g (dry weight) at the end of fermentation in the HMGR strain. The lanosterol content of the HMGR strain was 3.8- and 1.7-fold higher than that of the WT strain on days 4 and 16, respectively. The levels of HMGR, FPS, SQS, and LS mRNAs were also analyzed in different strains by qRT-PCR. Figure 6 shows that no significant differences existed in the gene expression between the WT and TR strains. The overexpression of the tHMGR gene caused the obvious accumulation of HMGR mRNA throughout the cultivation period. The transcription level of hmgr on day 12 in the HMGR strain was ∼10-fold higher than that of the WT strain. Some downstream genes in the isoprenoid biosynthetic pathway (fps, sqs, and ls) were also transcriptionally activated by the overexpression of tHMGR gene in the fermentation process. The highest transcription level of fps in the HMGR strain was 6.8-fold higher than that of the control on day 16. The highest change in the transcription profile for sqs was observed on day 12 when it was upregulated by 6.8-fold in the HMGR strain. The expression level of ls also increased by 9.1-fold on day 16 in the HMGR strain.
Table 2.
Maximum content of ganoderic acids, squalene, and lanosterol in different cell lines
| Cell line | Mean value ± SDa |
|||
|---|---|---|---|---|
| Biomass (g/liter) | GA content (mg/g DW) | Squalene content (μg/g DW) | Lanosterol content (mg/g DW) | |
| WT | 9.18 ± 0.29 | 14.13 ± 0.71 | 15.24 ± 0.83 | 1.96 ± 0.22 |
| TR | 8.80 ± 0.35 | 14.21 ± 1.58 | 9.82 ± 1.11* | 2.64 ± 0.43 |
| H1 | 7.31 ± 0.24* | 30.39 ± 1.98* | 46.77 ± 4.08* | 6.80 ± 0.14* |
| H2 | 7.31 ± 0.14* | 27.22 ± 1.03* | 46.00 ± 4.30* | 5.39 ± 0.26* |
| H3 | 7.72 ± 0.18* | 30.69 ± 1.27* | 47.88 ± 5.35* | 6.01 ± 0.41* |
The maximal contents for ganoderic acids (GA), lanosterol, and squalene were reached on days 12, 4, and 4, respectively. g DW, gram dry weight. *, significantly different from value for wild type (P < 0.05).
Fig 5.

Kinetic profiles of cell growth (A), content of ganoderic acids (B), and accumulation of squalene (C) and lanosterol (D) in the wild-type strain (○), the strain transformed with pJW (□), and the tHMGR overexpressed strain (△). The error bars indicate the standard deviations from three independent samples. *, statistical significance (P < 0.05) compared to the WT strain. d, days.
DISCUSSION
Structural genes such as hmgr, fps, sqs, and ls involved in ganoderic acid biosynthesis have been cloned from the basidiomycete G. lucidum (33). However, no report on the enhancement of ganoderic acid content by genetic engineering is available. In the present study, a homologous transformation system for G. lucidum was developed using a modified sdhB as a dominant selectable marker. This system was used to increase ganoderic acid accumulation through overexpression of G. lucidum tHMGR gene, which encodes the catalytic domain of HMGR.
Traditionally, obtaining stable transformants of basidiomycetes is more difficult compared to the process for obtaining transformants in filamentous ascomycetes, partially due to the inefficient expression of heterologous (prokaryotic) selection marker genes in some basidiomyctes (47). We initially attempted to transform the protoplasts of G. lucidum with plasmids pLG-hph (containing hygromycin resistance gene) (9) and pBARGPE1 (containing the bialaphos resistance gene) by electroporation and restriction enzyme-mediated integration (REMI) methods (16, 38). However, no stable transformants were obtained. The background growth was observed even at high concentrations of hygromycin (200 mg/liter). A. tumefaciens-mediated transformation of G. lucidum protoplasts with plasmid pCAMBIA1301 also failed to achieve hygromycin resistance transformants (data not shown), possibly because the constructs containing the heterologous gene may not be functional in G. lucidum. Previous studies reported that the hygromycin resistance gene and phleomycin resistance gene cannot work in the basidiomycetes Schizophyllum commune and Clitopilus passeckerianus unless the appropriate N terminus and appropriate introns were present within the coding sequences (14, 30). In the present study, a homologous selection marker gene cbxr was developed and used to transform basidiomycete by ATMT to establish a stable transformation system for G. lucidum. Transformation with plasmid pJW resulted in dominant resistance to carboxin. Southern blot analysis revealed that the cbxr cassette was stably integrated in the genome of the transformants. This success may be due to the fact that the homologous selection marker used for the G. lucidum transformation contains introns and is suitable for expression. Our results indicated that cbxr can be used as a dominant selective marker for the transformation of G. lucidum. The success of carboxin resistance selection indicates that it is a useful selection system based on host-derived gene rather than heterologous expression. The transformation efficiency obtained is comparable to that obtained using electroporation and REMI methods (16, 38). The mycelia were transformed in the present study via ATMT; however, no stable transformants were obtained, possibly due to the mycelial cell wall (29). Monokaryons were probably obtained from dikaryotic mycelia by protoplasting because the nuclei were separated and no clamp connections were observed in some mycelia of the transformants (data not shown). A similar case was reported in the mushroom Lyophyllum shimeji (20). Therefore, the transformation system described here is a practical method for the genetic improvement of this well-known traditional medicinal mushroom.
The expression of heterologous genes and cDNAs in basidiomycetes is hindered by several factors. In the S. commune model, the hygromycin B resistance gene is inactivated by methylation (23), AT-rich sequences hamper the expression of the bacterial Geneticin resistance gene and the hygromycin B resistance gene (30, 31), and introns are required for the efficient mRNA accumulation of the Agaricus bisporus hydrophobin gene ABH1 and the hygromycin B resistance gene (19, 30). Therefore, the tHMGR gene from the genome that contained introns was used to overexpress in G. lucidum, thereby avoiding the above-mentioned problems. The deregulated overexpression of tHMGR gene led to enhanced ganoderic acid accumulation, which was 2-fold higher than that of the control, suggesting that HMGR is a key enzyme for ganoderic acid biosynthesis in G. lucidum. Our study is the first report on the enhanced accumulation of triterpene via the overexpression of biosynthetic gene(s) in basidiomycetous mushroom. This report is similar to other examples in Saccharomyces cerevisiae (2, 28), Candida utilis, and Neurospora crassa (36, 43). Comparison of intermediate contents in different strains of G. lucidum revealed that more squalene (6.7-fold) and lanosterol (3.8-fold) accumulated in the tHMGR gene-overexpressing strain than in the WT. The results suggest that increased ganoderic acid accumulation may be due to the increased supply of precursors. Similar observations were also reported for S. cerevisiae, in which the overexpression of tHMGR gene also resulted in more accumulation of squalene and lanosterol (6, 21, 25). Constitutive expression of HMGR gene in tobacco led to a >100-fold increase in the level of cycloartenol, the analogue of lanosterol in fungi (3). The deregulated overexpression of tHMGR gene in G. lucidum also increased the transcription levels of the downstream genes in the MVA pathway, such as fps, sqs, and ls, indicating that downstream genes in the MVA pathway are transcriptionally activated by the overexpression of tHMGR gene. This result is similar to that previously described in Arabidopsis and Panax ginseng, in which the overexpression of HMG-CoA synthase and SQS genes resulted in an upregulation of the downstream genes in sterol and triterpene biosynthesis, respectively (17, 44). Therefore, changes in the production of MVA-derived precursors may stimulate the transcription of downstream genes involved in triterpene biosynthesis (17).
The time courses of ganoderic acid accumulation showed that the level of ganoderic acid started to increase sharply on day 8 in the HMGR strain. This result is consistent with the increased expressions of fps, sqs, and ls starting on day 8, suggesting that the enhanced ganoderic acid content in the transgenic strain may be related to increased transcription of biosynthetic genes. This observation was in accordance with previous reports that the increased production of ganoderic acids accompanied the upregulated expression of hmgr, fps, sqs, and ls (18, 26, 49).
In conclusion, we demonstrated here that the mutant G. lucidum sdhB is an excellent selection marker for the successful gene manipulation of this important medicinal mushroom, which is well known for its transformation resistance. Moreover, the overexpression of tHMGR gene leads to the upregulation of an existing MVA pathway and increased accumulation of antitumor ganoderic acid in G. lucidum. The homologous transformation system described here constitutes a useful tool for its molecular breeding. In addition, the engineered strain developed here represents a suitable platform for the further development of an industrial process for the hyperproduction of the antitumor secondary metabolite. Moreover, the related information is helpful for the genetic improvement and metabolic engineering of other mushrooms, which are an important source of biopharmaceuticals and functional foods (53, 54).
ACKNOWLEDGMENTS
This study received financial support from the National Natural Science Foundation of China (NSFC projects 20776084 and 30821005), the National Basic Research Program of China (973 Program 2012CB721006), and the National “863” Hi-Tech Program (2012AA021701).
Footnotes
Published ahead of print 31 August 2012
REFERENCES
- 1. Alves AM, et al. 2004. Highly efficient production of laccase by the basidiomycete Pycnoporus cinnabarinus. Appl. Environ. Microbiol. 70:6379–6384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Asadollahi MA, Maury J, Schalk M, Clark A, Nielsen J. 2010. Enhancement of farnesyl diphosphate pool as direct precursor of sesquiterpenes through metabolic engineering of mevalonate pathway in Saccharomyces cerevisiae. Biotechnol. Bioeng. 106:86–96 [DOI] [PubMed] [Google Scholar]
- 3. Chappell J, Wolf F, Proulx J, Cuellar R, Saunders C. 1995. Is the reaction catalyzed by 3-hydroxy-3-methylglutaryl coenzyme A reductase a rate-limiting step for isoprenoid biosynthesis in plants? Plant Physiol. 109:1337–1343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Dai ZB, Cui GH, Zhou SF, Zhang XA, Huang LQ. 2011. Cloning and characterization of a novel 3-hydroxy-3-methylglutaryl coenzyme A reductase gene from Salvia miltiorrhiza involved in diterpenoid tanshinone accumulation. J. Plant Physiol. 168:148–157 [DOI] [PubMed] [Google Scholar]
- 5. de Groot MJ, Bundock P, Hooykaas PJ, Beijersbergen AG. 1998. Agrobacterium tumefaciens-mediated transformation of filamentous fungi. Nat. Biotechnol. 16:839–842 [DOI] [PubMed] [Google Scholar]
- 6. Donald KA, Hampton RY, Fritz IB. 1997. Effects of overproduction of the catalytic domain of 3-hydroxy-3-methylglutaryl coenzyme A reductase on squalene synthesis in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 63:3341–3344 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Godio RP, Fouces R, Gudiña EJ, Martín JF. 2004. Agrobacterium tumefaciens-mediated transformation of the antitumor clavaric acid producer basidiomycete Hypholoma sublateritium. Curr. Genet. 46:287–294 [DOI] [PubMed] [Google Scholar]
- 8. Hampton R, Dimster-Denk D, Rine J. 1996. The biology of HMG-CoA reductase: the pros of contra-regulation. Trends Biochem. Sci. 21:142–145 [PubMed] [Google Scholar]
- 9. Hirano T, Sato T, Yaegashi K, Enei H. 2000. Efficient transformation of the edible basidiomycete Lentinus edodes with a vector using a glyceraldehyde-3-phosphate dehydrogenase promoter to hygromycin B resistance. Mol. Gen. Genet. 263:1047–1052 [DOI] [PubMed] [Google Scholar]
- 10. Honda Y, Matsuyama T, Irie T, Watanabe T, Kuwahara M. 2000. Carboxin resistance transformation of the homobasidiomycete fungus Pleurotus ostreatus. Curr. Genet. 37:209–212 [DOI] [PubMed] [Google Scholar]
- 11. Irie T, et al. 2001. Stable transformation of Pleurotus ostreatus to hygromycin B resistance using Lentinus edodes GPD expression signals. Appl. Microbiol. Biotechnol. 56:707–709 [DOI] [PubMed] [Google Scholar]
- 12. Irie T, et al. 2003. Construction of a homologous selectable marker gene for Lentinula edodes transformation. Biosci. Biotechnol. Biochem. 67:2006–2009 [DOI] [PubMed] [Google Scholar]
- 13. Kilaru S, et al. 2006. Expression of laccase gene lcc1 in Coprinopsis cinerea under control of various basidiomycetous promoters. Appl. Microbiol. Biotechnol. 71:200–210 [DOI] [PubMed] [Google Scholar]
- 14. Kilaru S, Collins CM, Hartley AJ, Bailey AM, Foster GD. 2009. Establishing molecular tools for genetic manipulation of the pleuromutilin-producing fungus Clitopilus passecherianus. Appl. Environ. Microbiol. 75:7196–7204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Kilaru S, et al. 2009. I nvestigating dominant selection markers for Coprinopsis cinerea: a carboxin resistance system and re-evaluation of hygromycin and phleomycin resistance vectors. Curr. Genet. 55:543–550 [DOI] [PubMed] [Google Scholar]
- 16. Kim S, Song J, Choi HT. 2004. Genetic transformation and mutant isolation in Ganoderma lucidum by restriction enzyme-mediated integration. FEMS Microbiol. Lett. 233:201–204 [DOI] [PubMed] [Google Scholar]
- 17. Lee MH, et al. 2004. Enhanced triterpene and phytosterol biosynthesis in Panax ginseng overexpressing squalene synthase gene. Plant Cell Physiol. 45:976–984 [DOI] [PubMed] [Google Scholar]
- 18. Liang CX, et al. 2010. Enhanced biosynthetic gene expressions and production of ganoderic acids in static liquid culture of Ganoderma lucidum under phenobarbital induction. Appl. Microbiol. Biotechnol. 86:1367–1374 [DOI] [PubMed] [Google Scholar]
- 19. Lugones LG, Scholtmeijer K, Klootwijk R, Wessels JGH. 1999. Introns are necessary for mRNA accumulation in Schizophyllum commune. Mol. Microb. 32:681–689 [DOI] [PubMed] [Google Scholar]
- 20. Maeta K, et al. 2008. Preparation and crossing of mating-capable monokaryons via protoplasting of the dikaryotic mycelia of a mycorrhizal mushroom, Lyophyllum shimeji. J. Wood Sci. 54:337–340 [Google Scholar]
- 21. Mantzouridou F, Tsimidou MZ. 2010. Observations on squalene accumulation in Saccharomyces cerevisiae due to the manipulation of HMG2 and ERG6. FEMS Yeast Res. 10:699–707 [DOI] [PubMed] [Google Scholar]
- 22. Maury J, Asadollahi MA, Møller K, Clark A, Nielsen J. 2005. Microbial isoprenoid production: an example of green chemistry through metabolic engineering. Adv. Biochem. Eng. Biotechnol. 100:19–51 [DOI] [PubMed] [Google Scholar]
- 23. Mooibroek H, Kuipers AGJ, Sietsma JH, Punt PJ, Wessels JGH. 1990. Introduction of hygromycin B resistance into Schizophyllum commune—preferential methylation of donor DNA. Mol. Gen. Genet. 222:41–48 [DOI] [PubMed] [Google Scholar]
- 24. Ohyama K, Suzuki M, Masuda K, Yoshida S, Muranaka T. 2007. Chemical phenotypes of the hmg1 and hmg2 mutants of Arabidopsis demonstrate the in planta role of HMG-CoA reductase in triterpene biosynthesis. Chem. Pharm. Bull. 55:1518–1521 [DOI] [PubMed] [Google Scholar]
- 25. Polakowski T, Stahl U, Lang C. 1998. Overexpression of a cytosolic hydroxymethylglutaryl-CoA reductase leads to squalene accumulation in yeast. Appl. Microbiol. Biotechnol. 49:66–71 [DOI] [PubMed] [Google Scholar]
- 26. Ren A, et al. 2010. Methyl jasmonate induces ganoderic acid biosynthesis in the basidiomycetous fungus Ganoderma lucidum. Bioresour. Technol. 101:6785–6790 [DOI] [PubMed] [Google Scholar]
- 27. Rico J, Pardo E, Orejas M. 2010. Enhanced production of a plant monoterpene by overexpression of the 3-hydroxy-3-methylglutary coenzyme A reductase catalytic domain in Saccharomyces cerevisiae. Appl. Environ. Microbiol. 76:6449–6454 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ro DK, et al. 2006. Production of the antimalarial drug precursor artemisic acid in engineered yeast. Nature 440:940–943 [DOI] [PubMed] [Google Scholar]
- 29. Ruiz-Díez B. 2002. Strategies for the transformation of filamentous fungi. J. Appl. Microbiol. 92:189–195 [DOI] [PubMed] [Google Scholar]
- 30. Scholtmeijer K, Wösten HAB, Springer J, Wessels JGH. 2001. Effect of introns and AT-rich sequences on expression of the bacterial hygromycin B resistance gene in the basidiomycete Schizophyllum commune. Appl. Environ. Microbiol. 67:481–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Schuren FHJ, Wessels JGH. 1998. Expression of heterologous genes in Schizophyllum commune is often hampered by the formation of truncated transcripts. Curr. Genet. 33:151–156 [DOI] [PubMed] [Google Scholar]
- 32. Shang CH, et al. 2008. Cloning and characterization of a gene encoding HMG-CoA reductase from Ganoderma lucidum and it functional identification in yeast. Biosci. Biotechnol. Biochem. 72:1333–1339 [DOI] [PubMed] [Google Scholar]
- 33. Shi L, Ren A, Mu DS, Zhao MW. 2010. Current progress in the study on biosynthesis and regulation of ganoderic acids. Appl. Microbiol. Biotechnol. 88:1243–1251 [DOI] [PubMed] [Google Scholar]
- 34. Shi L, et al. 2011. Development of a simple and efficient transformation system for the basidiomycetous medicinal fungus Ganoderma lucidum. World J. Microbiol. Biotechnol. 28:283–291 doi:10.1007/s11274-011-0818-z [DOI] [PubMed] [Google Scholar]
- 35. Shiao MS. 1992. Triterpenoid natural products in the fungus Ganoderma lucidum. J. Chin. Chem. Soc. 39:669–674 [Google Scholar]
- 36. Shimada H, et al. 1998. Increased carotenoid production by the food yeast Candida utilis through metabolic engineering of the isoprenoid pathway. Appl. Environ. Microbiol. 64:2676–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Stoop JMH, Mooibroek H. 1999. Advances in genetic analysis and biotechnology of the cultivated button mushroom, Agaricus bisporus. Appl. Microbiol. Biotechnol. 52:474–483 [Google Scholar]
- 38. Sun L, et al. 2001. Efficient transformation of the medicinal mushroom Ganoderma lucidum. Plant Mol. Biol. Rep. 19:383a–383j [Google Scholar]
- 39. Tang YJ, Zhang W, Zhong JJ. 2009. Performance analyses of a pH-shift and DOT-shift integrated fed-batch fermentation process for the production of ganoderic acid and Ganoderma polysaccharides by medicinal mushroom Ganoderma lucidum. Bioresour. Technol. 100:1852–1859 [DOI] [PubMed] [Google Scholar]
- 40. Tang YJ, Zhong JJ. 2002. Fed-batch fermentation of Ganoderma lucidum for hyperproduction of polysaccharide and ganoderic acid. Enzyme Microb. Technol. 31:20–28 [Google Scholar]
- 41. Tsukihara T, Honda Y, Sakai R, Watanabe T, Watanabe T. 2006. Exclusive overproduction of recombinant versatile peroxidase MnP2 by genetically modified white rot fungus Pleurotus ostreatus. J. Biotechnol. 126:431–439 [DOI] [PubMed] [Google Scholar]
- 42. Wagner R, et al. 2003. Current techniques for the cultivation of Ganoderma lucidum for the production of biomass, ganoderic acid and polysaccharides. Food Technol. Biotechnol. 41:371–382 [Google Scholar]
- 43. Wang GY, Keasling JD. 2002. Amplification of HMG-CoA reductase production enhances carotenoid accumulation in Neurospora crassa. Metab. Eng. 4:193–201 [DOI] [PubMed] [Google Scholar]
- 44. Wang H, et al. 2011. Overexpression of Brassica juncea wild-type and mutant HMG-CoA synthase 1 in Arabidopsis up-regulates genes in sterol biosynthesis and enhances sterol production and stress tolerance. Plant Biotechnol. J. doi:10.1111/j.1467-7652.2011.00631.x [DOI] [PubMed] [Google Scholar]
- 45. Wang J, Guo LQ, Zhang K, Wu Q, Lin JF. 2008. Highly efficient Agrobacterium-mediated transformation of Volvariella volvacea. Bioresour. Technol. 99:8524–8527 [DOI] [PubMed] [Google Scholar]
- 46. Wang S, He J, Cui Z, Li S. 2007. Self-formed adaptor PCR: a simple and efficient method for chromosome walking. Appl. Environ. Microbiol. 73:5048–5051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Wu J, O'Brien PA. 2009. Stable transformation of Rhizoctonia solani with a modified hygromycin resistance gene. Australas. Plant Pathol. 38:79–84 [Google Scholar]
- 48. Xu F, Zhao MW, Li YX. 2006. Cloning and sequence analysis of a glyceraldehyde-3-phosphate dehydrogenase gene from Ganoderma lucidum. J. Microbiol. 44:515–522 [PubMed] [Google Scholar]
- 49. Xu JW, Xu YN, Zhong JJ. 2010. Production of individual ganoderic acids and expression of biosynthetic genes in liquid static and shaking cultures of Ganoderma lucidum. Appl. Microbiol. Biotechnol. 85:941–948 [DOI] [PubMed] [Google Scholar]
- 50. Xu JW, Zhao W, Zhong JJ. 2010. Biotechnological production and application of ganoderic acids. Appl. Microbiol. Biotechnol. 87:457–466 [DOI] [PubMed] [Google Scholar]
- 51. Yue CJ, Jiang Y. 2009. Impact of methyl jasmonate on squalene biosynthesis in microalga Schizochytrium mangrovei. Process Biochem. 44:923–927 [Google Scholar]
- 52. Zhao W, Xu JW, Zhong JJ. 2011. Enhanced production of ganoderic acids in static liquid culture of Ganoderma lucidum under nitrogen-limiting conditions. Bioresour. Technol. 102:8185–8190 [DOI] [PubMed] [Google Scholar]
- 53. Zhong JJ, Tang YJ. 2004. Submerged cultivation of medicinal mushrooms for production of valuable bioactive metabolites. Adv. Biochem. Eng. Biotechnol. 87:25–59 [DOI] [PubMed] [Google Scholar]
- 54. Zhong JJ, Xiao JH. 2009. Secondary metabolites from higher fungi: discovery, bioactivity, and bioproduction. Adv. Biochem. Eng. Biotechnol. 113:79–150 [DOI] [PubMed] [Google Scholar]
- 55. Zhu H, Sun SJ, Zhang SS. 2011. Enhanced production of total flavones and exopolysaccharides via Vitreoscilla hemoglobin biosynthesis in Phellinus igniarius. Bioresour. Technol. 102:1747–1751 [DOI] [PubMed] [Google Scholar]
- 56. Zhu LW, Zhong JJ, Tang YJ. 2008. Significance of fungal elicitors on the production of ganoderic acid and Ganoderma polysaccharides by the submerged culture of medicinal mushroom Ganoderma lucidum. Process Biochem. 43:1359–1370 [Google Scholar]