

Abstract
Prader-Willi Syndrome (PWS) is a complex multisystem genetic disorder that shows great variability, with changing clinical features during a patient's life. The syndrome is due to the loss of expression of several genes encoded on the proximal long arm of chromosome 15 (15q11.2–q13). The complex phenotype is most probably caused by a hypothalamic dysfunction that is responsible for hormonal dysfunctions and for absence of the sense of satiety. For this reason a Prader-Willi (PW) child develops hyperphagia during the initial stage of infancy that can lead to obesity and its complications. During infancy many PW child display a range of behavioural problems that become more noticeable in adolescence and adulthood and interfere mostly with quality of life. Early diagnosis of PWS is important for effective long-term management, and a precocious multidisciplinary approach is fundamental to improve quality of life, prevent complications, and prolong life expectancy.
1. Introduction
PWS (OMIM number 176270) is a complex multisystem genetic disorder originally described in 1956 by three Swiss doctors, Prader et al. [1]. PWS was the first recognized disorder related to genomic imprinting in humans [2] and is caused by the lack of expression of paternally inherited imprinted genes on chromosome 15q11–q13. In several studied populations prevalence has been estimated to be 1/15,000–1/25,000.
The syndrome shows great variability, with changing clinical features during a patient's life. A newborn might suffer from severe hypotonia with feeding problems and global developmental delay. During infancy these characteristics impede the acquisition of gross motor and language milestones. A PW child develops hyperphagia during the initial stage of infancy that can lead to precocious obesity if left uncontrolled. This is most probably caused by a hypothalamic dysfunction, which impedes the sense of satiety. This hypothalamic dysfunction is also responsible for growth-hormone (GH) and thyroid-stimulating hormone (TSH) deficiencies, central adrenal insufficiency, and hypogonadism. During infancy, the PW child shows a characteristic problematic behavioral pattern, which has been reported to worsen with age. Patients sometimes present psychosis.
Early diagnosis of PWS is important for effective long-term management. In fact, the multidimensional problems of patients with PWS cannot be treated with a single intervention. A precocious multidisciplinary approach is fundamental to improve quality of life, prevent complications, and prolong life expectancy.
PWS can be considered an excellent example of how early diagnosis and meticulous management can markedly improve long-term outcome, despite the genetic background.
2. Diagnosis
PWS diagnosis is based on specific clinical features, and it is confirmed by genetic testing.
2.1. Clinical Features
Although diagnostic molecular testing for PWS is currently available, the clinical identification of patients remains a challenge as many features of PWS are nonspecific while others evolve over time or can be subtle [3].
In 1993, Holm et al. [4] proposed consensus clinical diagnostic criteria for PWS. In this consensus, features were divided into three groups: major criteria (1 point), minor criteria (1/2 point), and supportive criteria. Clinical diagnosis requires five points (at least four of them major) at age <3 years; eight points (at least five of them major) at age 3 years or older.
In 2001 Gunay-Aygun et al. [5] proposed a revision of these diagnostic criteria (Table 1) to help identify appropriate patients for DNA testing for PWS. The suggested age groupings are based on characteristic phases of the natural history of PWS. Some features, such as neonatal hypotonia and feeding problems in infancy, help diagnose the syndrome during the first years of life, whereas others, such as excessive eating, are useful during early childhood.
Table 1.
Published revised diagnostic criteria for PWS, Gunay-Aygun et al. [5].
| Age at assessment | Features sufficient to prompt DNA testing |
|---|---|
| Birth to 2 years | Hypotonia with poor suck. |
|
| |
| 2–6 years | (1) Hypotonia with history of poor suck. (2) Global developmental delay. |
|
| |
| 6–12 years | (1) History of hypotonia with poor suck (hypotonia often persists). (2) Global developmental delay. (3) Excessive eating (hyperphagia; obsession with food) with central obesity if uncontrolled. |
|
| |
| 13 years through adulthood | (1) Cognitive impairment, usually mild mental retardation. (2) Excessive eating (hyperphagia; obsession with food) with central obesity if uncontrolled. (3) Hypothalamic hypogonadism and/or typical behavior problems (including temper tantrums and obsessive-compulsive features). |
2.2. Genetic Findings
PWS, together with Angelman syndrome (AS), represents perhaps the best example of genomic imprinting in humans. Genomic imprinting is an epigenetic process by which the male and the female germ lines confer specific marks (imprints) onto certain gene regions. Probably <1% of our genes are imprinted with an expression pattern determined by the parent of origin [6–8].
The PWS region is found in a 5-6 Mb genomic region on the proximal long arm of chromosome 15 (15q11.2–q13; Figure 1). The complex phenotype is due to the loss of expression of several paternally genes on chromosome 15q11.2–q13 [9, 10].
Figure 1.
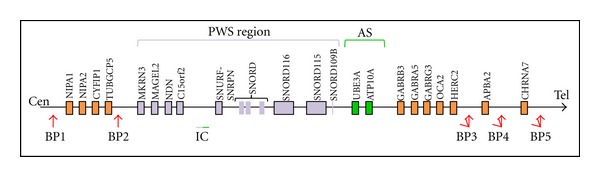
Schematic representation of the region 15q11.2-q13. The Prader-Willi syndrome (PWS) region is shown in violet, and Angelman syndrome (AS) region is shown in green. The red arrows indicate the three deletion breakpoints common to PWS and AS (BP1, BP2, and BP3). On rare occasions, there will be a distal breakpoint at BP4 or BP5. Type-1 deletions (T1D) extend from BP1 to BP3, and type-2 deletions (T2D) extend from BP2 to BP3.
Three main molecular mechanisms result in PWS.
Paternal microdeletion is responsible for 75–80% of cases.
Maternal uniparental disomy (UPD) is responsible for 20–25% of cases.
Imprinting defect (ID) is responsible for 1–3% of cases.
Other defects is such as balanced and unbalanced translocations, which, together with ID, are responsible for the majority of familial cases.
2.2.1. Genotype-Phenotype Correlation
Genotype-phenotype correlation is not possible, because no features are known to occur exclusively in individuals with one of the genetic classes. However, some studies identify significant statistical differences between the two largest genetic subtypes (deletion and UPD). For example, postterm delivery is more common in UPD patients. They are less likely to show hypopigmentation [11, 12], the typical characteristic facial appearance [11, 13], or possess jigsaw-puzzle skills [14]. In most studies, patients with UPD have a somewhat higher verbal IQ and milder behavioral problems [15–17]. However, psychosis [18] and autism spectrum disorder [19, 20] occur with significantly greater frequency among those with UPD. Individuals with the slightly larger, type-1 deletions (BP1–BP3) show poorer adaptive behavior, and lower intellectual ability and academic achievement than those with type-2 deletions (BP2-BP3) deletions [21, 22]. Two other studies found fewer clinically significant differences between individuals with these two deletion types [23, 24].
2.2.2. Genetic Counseling
Knowing the specific genetic etiology in individuals with PWS is essential for the appropriate genetic counseling of affected families. The risk of PWS recurrence in families with affected children is usually less than 1%, except for inherited mutation in the imprinting center (up to 50%), and translocation inherited mutation with break point in the 15q11.2–q13 region (up to 25%).
2.2.3. PW-Like Patients
The term “PW-like” is used to indicate a patient with clinical features that are very similar to PW, but without the confirmation of a classical genetic subtype that can cause the syndrome.
The literature reports many PW-like cases, but also many reports of the association between these patients and some particular genetic anomalies or syndromes.
The most frequently reported are the associations between PW-like and
2.3. Genetic Tests
Various types of genetic test can be used for PWS diagnosis and to characterize the different subtypes (Figure 2) [32].
Figure 2.
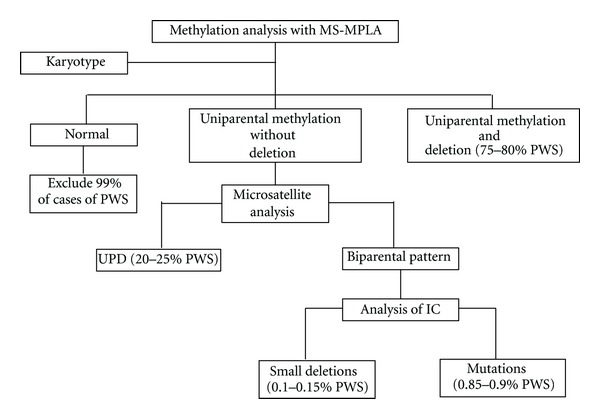
Algorithm for genetic testing for PWS. MS-MPLA: methylation-specific-multiplex ligation-dependent probe amplification analysis. UPD: uniparental disomy. IC: imprinting centre.
methylation-specific-multiplex ligation-dependent probe amplification analysis (MS-MLPA) combines both DNA methylation analysis and dosing analysis across the PWS region and can be considered the gold standard for PWS diagnosis in 99% of cases. This test shows the absence of the paternal allele using a methylation analysis to measure the amplitude of deletion (type 1 or type 2) and exclude suspect UPD or imprinting defects.
The karyotype is useful in association with MS-MLPA to point out balanced translocation whose breakpoint might be in the critical PWS region (15q11–q13).
Microsatellite analysis is necessary only if MS-MLPA does not show deletions in the critical PWS region, but just paternal allele absence at methylation analysis. It shows UDP (both alleles belonging to the mother) and also specifies if there is a heterodisomy (two mother alleles that are different from each other) or isodisomy (the same maternal allele in a double copy). If this analysis shows a biparental pattern, there is indication of mutation or microdeletions of the imprinting center.
3. Clinical Presentation
PWS is a multigenic pathology that shows great clinical variability that shows great clinical variability. Features change from patient to patient and even during the lifetime of the individual patient.
We have designated 4 age classes that can help the physician to outline ideal lifelong PW patient followup taking into consideration all the problems these patients might have to face.
3.1. From Birth to 3 Years
Pregnancy is generally normal, but some mothers may report decreased fetal activity, and newborns are often found in the breech position at time of delivery. Premature delivery may occur, and newborns that are adequate for gestational age frequently have low weight and length at birth.
The majority of newborns with PWS present marked neonatal axial hypotonia (babies are described as “floppy”); this is associated with lethargy, decreased movement, weak crying, and poor reflexes, including poor sucking, often resulting in failure to thrive [33, 34].
The baby can present dysmorphic characteristics, such as narrow bifrontal diameter, dolichocephaly, almond-shaped eyes, downturned angles of the mouth with abundant and thick saliva, and small hands and feet. These are less pronounced at birth but can become more evident with age.
Newborns generally present the clinical signs of hypogonadism. In males, the penis may be small; more characteristic is a hypoplastic scrotum that is small, poorly rugated, and poorly pigmented. Unilateral or bilateral cryptorchidism is present in 80–90% of males [35, 36]. In females, genital hypoplasia is often overlooked; however, the clitoris and labia, especially the labia minora, are generally small from birth.
Hypotonia and hypogonadism are the first manifestations of a primitive hypothalamic alteration, which many studies indicate to be at the base of PWS. This central deficiency leads to many manifestations, in particular a pituitary hormonal deficit (GH, TSH, central adrenal insufficiency [37]), satiety alteration, sleep disturbances, and a tendency for dysthermoregulation.
Although hypotonia slowly evolves over time, gross motor and language milestones are delayed. Early milestones are reached on average at double the normal age (e.g., sitting at 12 months, walking at 24 months, and saying words at 2 years)[38].
3.2. From 3 to 10 Years
During preschool age, PW patients develop a food obsession; children become overweight as a consequence of an insatiable appetite and compulsive eating, which can lead to morbid obesity in adolescence and adulthood if not kept under control.
PWS eating habits are complex and multifactorial. They are thought to be associated with abnormalities in the hypothalamic circuitry or the peripheral satiety signals [39, 40].
PWS individuals show differences in various gut hormones, including high levels of obestatin (an anorexigenic hormone) in infancy, with markedly elevated levels of ghrelin (an orexigenic hormone) in childhood and adulthood. The structural brain abnormalities present in these individuals might also contribute to appetite aberrations [41]. Functional MRI studies indicate that these individuals assign a high reward value to food with increased activation of the limbic and paralimbic areas of the brain that drive eating behavior, even after meal, showing that the brain influencing appetite in this syndrome.
In 2011, McAllister et al. [42] reviewed eating behavior in PWS. They concluded that a genetic abnormality might be the cause of fetal malnourishment or fetal starvation, leading to neonatal failure to thrive and also causing feeding problems. Ghrelin may be involved in the instigation of the binging and hyperphagic stage, and later development of atypical reward circuitry in response to food might be the result of altered pathways generated in the early binging stages in childhood, combined with insensitivity to satiation cues, such as leptin levels. Examining reward pathways by neural imaging in response to food in younger children, for example, may elucidate the development of the hyperresponsive circuitry [42].
Growth restriction is a frequently observed sequel of PWS; approximately 90% of affected individuals are short in stature, probably resulting from GH deficiency [37].
During the first 6 years of life, children with PWS often do not achieve normal levels of cognitive, motor, and language development. Indeed, according to one study, these individuals have a below-average IQ of about 70 [43–45]. A review of cognitive ability among 575 affected individuals confirms this, showing that just 5% of patients had a normal IQ (i.e., >85). Borderline mental retardation was observed in 28% of patients, while 34%, 27%, and 5%, respectively, were mildly, moderately, or severely mentally retarded [45].
During this period, many PW sufferers display a range of behavioral problems that include both excessive appetite and lack of food selectivity. There is also a high incidence of stubbornness, verbal perseverance, skin picking, and temper tantrums.
Despite hypogonadism generally causing incomplete, delayed, and sometimes disordered pubertal development, in approximately 15–20% of patients of both sexes, premature adrenarche or precocious puberty often occurs [35, 46].
3.3. From 10 to 18 Years
During adolescence, behavioral problems and hyperphagia become more noticeable.
In experimental settings, PWS individuals have been seen to consume around three-to-six times more than the normal caloric intake at a given meal [47, 48]. Overeating can lead to stomach rupture [49], and PWS individuals have also been known to steal and hoard food. These individuals have also been reported to eat inappropriate food, such as uncooked chicken, or even to eat nonfood items.
Central obesity results from the combination of uncontrolled food intake, a low metabolic rate, and a decreased activity level (resulting in a decreased total caloric requirement). Obesity-related complications appear, such as cardiorespiratory insufficiency, obstructive sleep apnea, thrombophlebitis, and chronic leg edema, and are the major causes of morbidity and mortality [50].
The severity of behavioral problems increases with age and body mass index and can then diminish in older adults. Psychosis is evident by young adulthood in at least 5–10% of individuals [51–53]. Behavioral and psychiatric problems interfere mostly with quality of life in adolescence and adulthood.
Hypogonadism causes incomplete, delayed, and sometimes disordered pubertal development. Primary amenorrhea or oligomenorrhea are present in females. Infertility is the rule in both sexes although a few instances of reproduction in females have been reported and presented [54, 55]. Although hypogonadism in PWS has long been believed to be entirely hypothalamic, resulting in low gonadotropins and subsequent low gonadal hormones, recent studies have suggested a combination of hypothalamic and primary gonadal deficiencies [56–58].
3.4. The Adult
The quality of life of adults with PWS is largely conditioned by the degree of obesity, the presence of its complications, and behavioral problems.
4. Management
Patient management has to be tailored, due to the clinical variability between patients, but also to variability in the same patient throughout this or her lifetime. Therapeutic decisions and clinical followup need to consider each problem that a patient might have to face. A multidisciplinary team, consisting of neonatologists, pediatricians, endocrinologists, orthopedic surgeons, psychologists, psychiatrists, physiotherapists and urologists, has to deal with all of the patient's medical and psychological problems.
4.1. Nutrition
The maintenance of adequate and appropriate nutrition is fundamental to the treatment of people with PWS at every age.
A correct approach should consider the two distinct nutritional stages of every PW patient:
stage 1: poor feeding and hypotonia, which can often cause a failure to thrive;
4.1.1. Feeding Support
During the first two months of life, most PWS infants are unable to suck an adequate quantity of milk from the breast or the feeding bottle, and they have to be fed by gavage. The use of a gastrostomy tube (generally using a button-style device) can be avoided in most cases, but if, after considering the risks and benefits of both approaches, a decision to use a gastrostomy tube is made, the device should be promptly removed when no longer needed.
Milk requirement is that of other infants of the same age and weight. Children should not be given simple sugars for a “sweet taste.”
Infants may require feeding support for several months. Caloric needs may sometimes be somewhat reduced in infants with PWS, who typically do not spontaneously demand feeding. The infant's diet must therefore be adjusted to maintain appropriate weight gain as determined by frequent weight checks. Increased caloric density can be helpful.
4.1.2. Dietary Control
Incorrect eating is one of the most serious disorders affecting the lives of children and adults with PWS. Hyperphagia is a serious chronic problem for children with PWS, together with their families, and it can severely limit independence in adult life due to the risk of life-threatening obesity.
Controlling food-related behavior is complex, aiming to limit the child's access to food, reduce exposure that can cause the child to think about food, and promote a daily routine that helps obtain good weight control. Relatives and friends also have to understand that “sneaking” food to the child with PWS is not a demonstration of affection as, on the contrary, it undermines the child's nutritional regimen and sense of wellbeing.
When hyperphagia occurs the caloric intake needs to be reduced. The diet should have a balanced distribution and be rich in fiber, and the caloric intake should be about 75–80% that of a healthy child of the same age.
During adulthood, the caloric intake should be below 1000–1200 kcal/day to maintain stable weight structure, or between 800 and 1000 kcal/day to lose weight. Dietary restriction should anyway be balanced, and complex carbohydrates should be preferred.
Pharmacological treatment, including available anorexigenic agents, has not been of benefit in treating hyperphagia although there are some published placebocontrolled studies [40, 60]. The potential benefits of newer agents, such as endocannabinoid antagonists, are still under examination in PWS. Recent concerns regarding psychiatric side effects need careful monitoring in these patients.
Restrictive bariatric surgery, such as gastric banding or bypass, has not been seen to reduce hyperphagia or achieve long-term weight reduction and are associated with unacceptable morbidity and mortality [61, 62]. Whereas some of the reports using biliopancreatic diversion reported successful weight loss, there have been frequent complications from the resulting intestinal malabsorption. Bariatric surgery should only be considered when the patient's excessive weight becomes life threatening. Strict diet control and postsurgical followup are mandatory.
4.2. Motor Program
The motor program has to consider the various necessities during a patient's life; in newborns the goal is to improve axial hypotonia, while in childhood and adulthood the patient's physical, socials and metabolic condition needs to be focused.
4.2.1. Physiotherapy
The newborn is characterized by a variable grade of hypotonia that influences many aspects of normal development (delayed gross motor milestones) and growth (feeding difficulties).
Even when hypotonia evolves slowly, we suggest the prompt introduction of personal training programs. These programs must be supervised by physiotherapists and maintained by parents. They help counteract the hypotonic PWS infant's difficulties in overcoming gravity during early life. This is a particularly sensitive period for motor development and skills acquisition and might have consequences regarding cognitive and social development.
4.2.2. Physical Activity
Physical activity and sports are a fundamental therapy for PWS patients.
During childhood, physical activity
improves physical functions,
promotes socialization,
helps improve caloric expenditure, together with diet,
is one of the best ways to limit access to food.
We suggest regular daily physical activity of around 30 minutes. Any kind of sport is possible, and parents should consider the abilities and tastes of their children when choosing.
Although hypotonia improves with age, it persists into adulthood, together with reduced muscle mass. Regular exercise is therefore an important part of everyday life. In a recent study, Vismara et al. provided an effective and simple home-based training program representing a continuum of the rehabilitation process outside the hospital, which is a crucial issue in chronic conditions. In fact, following six months of daily activity patients were seen to improve their physical function [63].
4.3. Endocrinological Aspects
As already stated, the hypothalamic dysfunction at the base of PWS contributes to the development of multiple endocrine disorders, such as adrenal insufficiency, GH deficiency, LH/FS disorder, and thyroid dysfunction.
4.3.1. Adrenal Insufficiency
Several reports show a mortality rate in PWS estimated at 3% yearly [64, 65]. Disturbances in the hypothalamus-hypophysis-adrenal (HHA) axis are thought to be responsible for these events or, at least, to represent concurrent factors consistent with an inadequate or late response during infection or relevant dehydration episodes. This hypothesis is supported by pathological findings. Adrenal atrophy has been documented autoptically in a number of such cases [66], and small volumes of the hypothalamic paraventricular nuclei with decreased cell number were demonstrated in PWS adults [67]. Although these aspects have been described in PWS, the genetic basis of putative central adrenal insufficiency (CAI) is far from being unraveled, as the molecular mechanisms leading to PWS phenotype are still largely unknown.
From a functional viewpoint, some studies reveal a differing prevalence for this problem. De Lind van Wijngaarden et al. using the metyrapone test found that the hypophyseal response to adrenal inhibition was insufficient in 15 out of 25 (60%) PWS patients [68]. Two recent reports on 41 and 57 PWS patients found no cases of CAI by employing a low-dose (LDTST) and a standard ACTH stimulation test (250 μg), respectively [69, 70]. The Study Group for Genetic Obesity of the Italian Society of Pediatric Endocrinology and Diabetology (SIEDP/ESPED) designed a study that confirms that clinically relevant CAI in pediatric PWS patients is rare (14.3% to LDTST), with one third of them (4.8%) also having suboptimal response to a second test. The authors suggest [71].
According to the literature, the administration of glucocorticoids during episodes of moderate/severe stress is recommended (hydrocortisone at 30–70 mg/m2/day divided into 3 doses Continuous replacement should be limited to cases with clinical signs of adrenal insufficiency, as for other nonsyndromic forms of CAI.
4.3.2. Growth Hormone (GH) Deficiency
There are many data indicating reduced GH secretion in PWS patients. Low peak GH response to stimulation tests, decreased spontaneous GH secretion, and low serum IGF-I levels have been documented in at least 15 studies involving about 300 affected children [37]. Clinical features of the condition also support the presence of GHD in PWS. Both PWS and GHD are characterized by short stature, obesity with extra fat deposits over the abdomen, abnormal body composition with reduced muscle mass and decreased bone density, and, in some patients, retarded bone age [4, 72]. Patients with PWS are therefore GH deficient although the degree of GH deficiency may vary from mild to severe.
Guidelines for GH therapy in PWS children have been drawn up by the Italian Drug Agency (AIFA) note number 39 and do not depend on the presence of GH deficiency.
In accordance with the literature [50], we suggest beginning GH therapy during the first year of life after performing the following:
polysomnography and ENT evaluation,
fasting glucose and OGTT (glucose 1, 75 g/Kg, to maximum 75 g),
blood sample for: IGF1, fT4, TSH,
spine X-ray,
cardiologic evaluation with echocardiography.
The recommended dosage is 0, 01-0, and 03 mg/Kg/day, adjusted on IGF1 levels, which should not exceed + 2SDS. It is preferable to start with a dose corresponding to 1/3 of the minimal dosage. GH therapy should not be started in the presence of obstructive sleep apnea syndrome (OSAS), adenotonsillar hypertrophy, severe obesity, glucidic intolerance, and/or instable scoliosis. Interruption of therapy should be evaluated if scoliosis, glucose intolerance, or OSAS deteriorate.
When the individual stops growing in height, evaluation of the GH secretory pattern is required through ITT, or rather using the GnRH-arginine test that has greater tolerance. For adolescents in therapy, evaluation of the GH secretory pattern must be preceded by a therapeutic washout of at least 1-2 months. In patients with GH deficit (lower than 3 ug/L) a therapeutic pattern not involving weight is recommended, starting with a standard dose of 0.2 mg/day, which can be modified based on IGF1 levels.
During GH therapy, it is fundamental to carry out periodic monitoring of
polysomnography and ENT evaluation at 6 weeks–3 months–6 months after the start, and whenever necessary,
fasting glucose and OGTT, IGF1, fT4, TSH: 1 month after the start, and whenever necessary,
orthopedic evaluation and spine X-ray whenever necessary,
cardiovascular function evaluation every 6 months, or whenever necessary.
In children with PWS, the aims of GH treatment are to improve growth during childhood, adult height, and body composition. There is much evidence that this therapy has multiple beneficial effects on growth and body composition. In particular, it decreases fat mass and increases muscle mass, and it may have a beneficial effect on weight gain, and possibly appetite, in individuals with PWS [37, 73]. Infants with PWS treated with GH therapy show improved head circumference, height, BMI, body composition (with improvement of lean muscle mass and delay of fat tissue accumulation), body proportions, acquisition of gross motor skills, language acquisition, and cognitive scores. Several studies have documented the safety and efficacy of GH treatment in adults with PWS on body composition and quality of life [74, 75].
4.3.3. Disorder of Gonadotropins
Hypothalamic dysfunction can cause LHRH-LH/FSH axis disorders. In general, the patient shows hypogonadism from birth, but in some PW children precocious adrenarche or early puberty can occur.
(a) Hypogonadism: hypogonadism is a consistent feature in both males and females with PWS, and hypogenitalism is present, even at birth. There is increasing evidence to implicate both central and peripheral origins for hypogonadism, at least in males [76].
At birth, both sexes can have clinical signs of hypogonadism: females show genital hypoplasia (clitoris and labia minora), while males have cryptorchidism, scrotal hypoplasia, and small testicular volume. Cryptorchidism is present in over 80% of boys from birth [35, 36]. At present there is no consensus in the literature as to the best treatment to use. Our experience suggests human chorionic gonadotropins (HCG) therapy from 6 to 12 months with a dosage of 250 U, while from 1 to 5 years a dosage of 500 U should be used twice a week for 6 weeks.
Before and during therapy regular clinical evaluation, testosterone dosage and testicular ultrasound are necessary. When therapy does not have positive results, surgical correction needs to be considered before 2 years of life, or as soon as possible when the child is older. During adolescence patients can show delayed or incomplete pubertal development, and it is therefore necessary to consider hormonal treatment for the induction, promotion, or maintenance of puberty. Before any therapy is carried out, the following need to be considered:
dosage of LH, FSH, estradiol (in females), and testosterone (in males) both basal and after GnRH
pelvic ultrasound in females,
testicular ultrasound,
dual-energy X-ray photon absorptiometry (DEXA),
evaluation of thrombophilic status in females.
For the induction and maintenance of puberty there is no consensus regarding best therapy. We suggest testosterone in males (25 mg once a month, which after 3 months can be increased to 50 mg once a month if well tolerated), and estrogen (incremental dosage up to complete development) and progesterone in females (in general after around 24 months from start of estrogen).
Patients with PWS have low bone mineral density (BMD) and are at risk of osteoporosis related to sex-steroid and GH deficiencies, and low muscular activity with elevated biochemical markers of bone turnover [77–80]. Reduced BMD in PWS is associated with high risk of fracture in the long bones, as well as the small bones, of the hands and feet, with some patients suffering multiple fractures [81]. These findings support the need for hormone therapy, particularly sex-steroid replacement, during adolescence, and maintenance during adulthood. Estrogen, and androgen status should be monitored yearly during adolescence and adulthood and BMD assessed as indicated by DEXA.
Infertility is the rule in both sexes although a few instances of reproduction in females have been reported [54, 55].
(b) Precocious sdrenarche and early puberty: Isolated premature pubarche has been reported in 14% and precocious puberty in 4% of males and females [35, 82, 83]. There is no consensus as to the management of either of these conditions. Some investigators have suggested the use of hydrocortisone in premature pubarche to decrease adrenal androgens when associated advancement of bone age is present; treatment with GnRH analogs must be reserved for selected patients [35].
In some patients with advanced bone age and hyperandrogenism we propose cyproterone acetate therapy. In these patients we generally obtain a slowdown of both bone age and androgen values. This drug is off-label, and parental consent is therefore necessary.
4.3.4. Thyroid Dysfunction
In PWS children both central and peripheral hypothyroidisms have been documented. These can be congenital or of late onset [84, 85].
Blood levels of fT4 and TSH should be kept under control: those levels must be monitored at birth, and thereafter yearly (or every 6 months during GH therapy).
In the presence of primary or secondary hypothyroidism L-thyroxine therapy is required.
We recommend a dose of 5-6 mcg/Kg/day (8 mcg/Kg/day in infants <1 year old), which can be modified following levels of fT4 and TSH in primary hypothyroidism, or just fT4 (that should be maintained in the upper zone of the normal range) in secondary hypothyroidism.
4.4. Psychological/Psychiatric Support
A characteristic behavioral profile with temper tantrums, stubbornness, controlling and manipulative behavior, compulsivity, and difficulty with change in routine becomes evident in early childhood in 70–90% of individuals with PWS. Behavioral and psychiatric problems most interfere with the quality of life in adolescence and adulthood.
Interventions concerning behavioral problems must be coordinated by specialists (psychologists, psychiatrists, and doctors), primary care providers, parents, and other family members.
Psychological support during infancy is fundamental for parents and children alike. Early logopedic therapy should be carried out to prevent language disorders. Due to their low cognitive level, PW children should receive learning support during school time.
In adolescence and adulthood some patients develop psychosis that requires pharmacological treatment [53, 86, 87]. In our experience, it is advisable to start treatment with psychotropic drugs at low doses, due to the possible hyper-responsiveness or paradoxical effects induced by commonly used. The drugs used are benzodiazepines, classical antipsychotics and atypical antipsychotics, mood stabilizers, and selective serotonin reuptake inhibitors. Molecules lacking orexigenic action, or with a lower capacity to induce increased appetite, such as risperidone and fluoxetine, are preferable. Topiramate might help combat the skin-picking phenomenon [60].
4.4.1. Casa Sora
The nonprofit (ONLUS) foundation “Casa Sora Per Voi” [88] is the first Italian project to offer PW children, adolescents, adults, and their families the possibility of formative stays. This project stems from the need of a controlled environment, where patients can learn to cope with their daily lives, socialize, and experience nature and animals.
Various specialists are involved, such as dieticians and psychologists, who plan numerous activities. Families, individuals, and healthcare professionals meet during structured daily programs for adults and children; the children are involved in programmed psychological activities, while their parents and affiliated professionals attend lectures and group discussions.
The house is in an isolated hilly location near lake Iseo and is completely surrounded by greenery and uncontaminated nature. Daily activities include outdoor physical exercise, group experiences in contact with nature in a serene and stimulating environment, and psychospecific recreational and artistic tasks.
4.5. Other Problematics
4.5.1. Orthopedic Problems
Scoliosis is a frequent feature observed in children with PWS (30%–70%) and may be explained partly by hypotonia and obesity [81, 89–91]. Regular clinical assessment is required at each visit, and periodic spinal X-rays are useful, whether or not the patient is receiving GH. Surgical treatment is indicated in severe early-onset scoliosis-kyphosis, and in adolescents near skeletal maturity. Due to the possibility of complications, surgical treatment requires a multidisciplinary team with expertise in the management of scoliosis and PWS.
4.5.2. Ophthalmological Problems
Early screening and correction for myopia, hypermetropia, or other eye problems are recommended. Strabismus is also frequent and may require surgery.
4.5.3. Orthodontic Problems
Abnormal enamel and frequent caries have been previously reported, but in a recent survey PWS patients presented with a more favorable oral health status than those in previous studies [92]. This status is worsened by poor salivary production, which requires education for both parents and children. Education for regular daily drinking and products designed to increase saliva flow might help prevent dental complications. Orthodontic treatment is often needed.
4.6. Medical Alerts
4.6.1. Anesthesiological Risk
PWS patients have a higher anesthesiological risk characterized by an exaggerated response to hypnotic drugs, ventilation difficulties due to dysmorphic facial conformation, hypoxia, breathing problems, and thermoregulation control. These phenomena are more frequent and severe in obese patients that can have obstructive apnea and right ventricular hypertrophy and failure due to pulmonary hypertension. These conditions require preventive cardiology and pulmonary evaluation. Moreover, following surgery patients should be sent to an intensive care unit, and some characteristics must be considered:
high pain thresholds,
thermoregulation disorders. Although there is no indication for a predisposition to malignant hyperthermia in PWS, the use of depolarizing neuromuscular-blocking drugs (e.g., succinylcholine) should be avoided unless absolutely necessary,
dense salivation could compromise airway patency, especially during extubation,
hypotonia reduces cough reflex efficiency in clearing airways.
4.6.2. Acute Gastric Distension
In the literature there are at least 8 cases of acute gastric distension, and at least 3 cases of death following gastric rupture in PWS patients. The basis of this serious and potentially mortal complication is the presence of hyperphagia, a high pain threshold, the inability to vomit, and delayed gastric emptying. It is very important to keep PWS patients under strict regular control in the presence of large quantities of available food (banquets, parties, supermarkets, etc.). Furthermore, in the presence of abdominal pain and/or vomiting, an abdominal X-ray has to be carried out to exclude gastric perforation.
5. Follow Up
The PW patients need a lot of specialistics controls:
axiological evaluation: length/height, weight, head circumference, BMI (Figure 3),
nutritionist/dietician evaluation,
blood sample: oral glucose tolerance test (OGTT), HbA1c, total cholesterol and HDL, triglyceride level, uricemia, thyroid function (TSH, fT4), IGF1,
evaluation of pubertal development: dosage of LH, FSH, estradiol (in females) and testosterone (in males) both basal and after GnRH,
bone age (left hand X-ray),
evaluation of bone density: blood sample for calcium (Ca), phosphor (P), and magnesium (Mg) levels, protidemia, PTH, vitamin D3, and to carry out a vertebro femoral DEXA,
polysomnography and ENT with fibroscopy,
orthopedic evaluation,
orthoptic/ophthalmology evaluation,
dental evaluation,
dermatological evaluation,
echocardiography and cardiological evaluation,
neuropsychiatric and physiatric evaluation.
Figure 3.
Standard BMI proposed by WHO from 0 to 2 years.
In consideration of the numerous problems that can take place during the PW patient's life, we have identified various age classes in which we define the best followup, that is resumed in Table 2.
Table 2.
Clinical followup in different age classes of PW patient.
| From birth to 3 years | From 3 to 10 years | From 10 to 18 years | Adult | |
|---|---|---|---|---|
| Axiological evaluation | 3-4 months | 6 months | 6–12 months | |
|
| ||||
| Nutritionist/dietician evaluation | 6–12 months | 6–12 months | 6–12 months | 6–12 months |
|
| ||||
| Blood sample (OGTT, HbA1c, total cholesterol and HDL, triglyceride level, uricemia, TSH, fT4, IGF1) | 6 month | 6–12 months | 6–12 months | 1 year |
|
| ||||
| Evaluation of pubertal development (dosage of LH, FSH, estradiol (in females) and testosterone (in males) both basal and after GnRH) | According to clinical evaluation | |||
|
| ||||
| Bone age (left hand X-ray) | GH therapy: 1 year | According to clinical evaluation | ||
|
| ||||
| Evaluation of bone density | ||||
| Blood sample for Ca, P and Mg, protidemia, PTH, vitamin D3. | 1-2 years | 1-2 years | 1-2 years | |
| DEXA | 1-2 years | 1-2 years | 1-2 years | |
|
| ||||
| Polysomnography | No GH therapy: 1 year GH therapy: 3–6 months |
1 year | 1 year | In case of problem |
|
| ||||
| ENT with fibroscopy | No GH therapy: 6–12 months GH therapy: 3–6 months |
In case of problem | In case of problem | |
|
| ||||
| Orthopedic evaluation | 1 year (consider the necessity of spine X-ray) | 6 months (consider the necessity of spine X-ray) | 6 months (consider the necessity of spine X-ray) | In case of problem |
|
| ||||
| Orthoptic/ophthalmology evaluation | 1 year | In case of problem | In case of problem | |
|
| ||||
| Dental evaluation | Annual after 2 years | 6 months | 6 months | In case of problem |
|
| ||||
| Dermatological evaluation | In case of skin picking | In case of skin picking | In case of skin picking | |
|
| ||||
| Echocardiography and cardiological evaluation | 2 years | |||
|
| ||||
| Neuropsychiatric and physiatric evaluation | 3 months | 1 year | 1 year | 1-2 years |
OGTT: oral glucose tolerance test, DEXA: dual-energy X-ray photon absorptiometry; ENT: ear-nose-throat.
6. Conclusion
Prader-Willi syndrome is a complex multisystem disorder. Patients can be affected by various problems; therefore precocious diagnosis is fundamental to guarantee optimal assistance. Each patient should undergo personally tailored treatment from birth. Therapeutic decisions and clinical followup need to consider all of these possible problems. A multidisciplinary team is required, made up of specialists such as neonatologists, geneticist, pediatricians, endocrinologists, orthopedic surgeons, psychologists, psychiatrists, physiotherapists, and urologists to deal with the numerous medical and psychological problems a PWS patient has to face. Only in this way we can improve quality of life, prevent complications, and prolong life expectancy in patients with PWS.
References
- 1.Prader A, Labhart A, Willi H. Ein syndrom von adipositas, kleinwuchs, kryptorchismus und oligophrenie nach myotonieartigem zustand im neugeborenenalter. Schweizerische Medizinische Wochenschrift. 1956;86:p. 1260. [Google Scholar]
- 2.Driscoll DJ, Waters MF, Williams CA, et al. A DNA methylation imprint, determined by the sex of the parent, distinguishes the Angelman and Prader-Willi syndromes. Genomics. 1992;13(4):917–924. doi: 10.1016/0888-7543(92)90001-9. [DOI] [PubMed] [Google Scholar]
- 3.Crinó A, Di Giorgio G, Livieri C, et al. A survey on Prader-Willi syndrome in the Italian population: prevalence of historical and clinical signs. Journal of Pediatric Endocrinology & Metabolism. 2009;22(10):883–893. doi: 10.1515/jpem.2009.22.10.883. [DOI] [PubMed] [Google Scholar]
- 4.Holm VA, Cassidy SB, Butler MG, et al. Prader-Willi syndrome: consensus diagnostic criteria. Pediatrics. 1993;91(2):398–402. [PMC free article] [PubMed] [Google Scholar]
- 5.Gunay-Aygun M, Schwartz S, Heeger S, O’Riordan MA, Cassidy SB. The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics. 2001;108(5, article E92) doi: 10.1542/peds.108.5.e92. [DOI] [PubMed] [Google Scholar]
- 6.Horsthemke B. Mechanisms of imprint dysregulation. American Journal of Medical Genetics C. 2010;154(3):321–328. doi: 10.1002/ajmg.c.30269. [DOI] [PubMed] [Google Scholar]
- 7.Horsthemke B, Buiting K. Chapter 8 genomic imprinting and imprinting defects in humans. Advances in Genetics. 2008;61:225–246. doi: 10.1016/S0065-2660(07)00008-9. [DOI] [PubMed] [Google Scholar]
- 8.Buiting K, Groß S, Lich C, Gillessen-Kaesbach G, El-Maarri O, Horsthemke B. Epimutations in Prader-Willi and Angelman syndromes: a molecular study of 136 patients with an imprinting defect. American Journal of Human Genetics. 2003;72(3):571–577. doi: 10.1086/367926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Glenn CC, Driscoll DJ, Yang TP, Nicholls RD. Genomic imprinting: potential function and mechanisms revealed by the Prader-Willi and Angelman syndromes. Molecular Human Reproduction. 1997;3(4):321–332. doi: 10.1093/molehr/3.4.321. [DOI] [PubMed] [Google Scholar]
- 10.Bittel DC, Butler MG. Prader-Willi syndrome: clinical genetics, cytogenetics and molecular biology. Expert Reviews in Molecular Medicine. 2005;7(14):1–20. doi: 10.1017/S1462399405009531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gillessen-Kaesbach G, Robinson W, Lohmann D, Kaya-Westerloh S, Passarge E, Horsthemke B. Genotype-phenotype correlation in a series of 167 deletion and non-deletion patients with Prader-Willi syndrome. Human Genetics. 1995;96(6):638–643. doi: 10.1007/BF00210291. [DOI] [PubMed] [Google Scholar]
- 12.Butler MG. Hypopigmentation: a common feature of Prader-Labhart-Willi syndrome. American Journal of Human Genetics. 1989;45(1):140–146. [PMC free article] [PubMed] [Google Scholar]
- 13.Cassidy SB, Forsythe M, Heeger S, et al. Comparison of phenotype between patients with Prader-Willi syndrome due to deletion 15q and uniparental disomy 15. American Journal of Medical Genetics. 1997;68(4):433–440. [PubMed] [Google Scholar]
- 14.Dykens EM. Are jigsaw puzzle skills “spared” in persons with Prader-Willi syndrome? Journal of Child Psychology and Psychiatry and Allied Disciplines. 2002;43(3):343–352. doi: 10.1111/1469-7610.00025. [DOI] [PubMed] [Google Scholar]
- 15.Dykens EM, Cassidy SB, King BH. Maladaptive behavior differences in Prader-Willi syndrome due to paternal deletion versus maternal uniparental disomy. American Journal on Mental Retardation. 1999;104(1):67–77. doi: 10.1352/0895-8017(1999)104<0067:MBDIPS>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 16.Roof E, Stone W, MacLean W, Feurer ID, Thompson T, Butler MG. Intellectual characteristics of Prader-Willi syndrome: comparison of genetic subtypes. Journal of Intellectual Disability Research. 2000;44(1):25–30. doi: 10.1046/j.1365-2788.2000.00250.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Torrado M, Araoz V, Baialardo E, et al. Clinical-etiologic correlation in children with Prader-Willi syndrome (PWS): an interdisciplinary study. American Journal of Medical Genetics A. 2007;143(5):460–468. doi: 10.1002/ajmg.a.31520. [DOI] [PubMed] [Google Scholar]
- 18.Holland AJ, Whittington JE, Butler J, Webb T, Boer H, Clarke D. Behavioral phenotypes associated with specific genetic disorders: evidence from a population-based study of people with Prader-Willi syndrome. Psychological Medicine. 2003;33(1):141–153. doi: 10.1017/s0033291702006736. [DOI] [PubMed] [Google Scholar]
- 19.Veltman MWM, Thompson RJ, Roberts SE, Thomas NS, Whittington J, Bolton PF. Prader-Willi syndrome: a study comparing deletion and uniparental disomy cases with reference to autism spectrum disorders. European Child and Adolescent Psychiatry. 2004;13(1):42–50. doi: 10.1007/s00787-004-0354-6. [DOI] [PubMed] [Google Scholar]
- 20.Whittington J, Holland A, Webb T, Butler J, Clarke D, Boer H. Cognitive abilities and genotype in a population-based sample of people with Prader-Willi syndrome. Journal of Intellectual Disability Research. 2004;48(2):172–187. doi: 10.1111/j.1365-2788.2004.00556.x. [DOI] [PubMed] [Google Scholar]
- 21.Butler MG, Bittel DC, Kibiryeva N, Talebizadeh Z, Thompson T. Behavioral differences among subjects with Prader-Willi syndrome and Type I or Type II deletion and maternal disomy. Pediatrics. 2004;113(3):565–573. doi: 10.1542/peds.113.3.565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hartley SL, MacLean WE, Butler MG, Zarcone J, Thompson T. Maladaptive behaviors and risk factors among the genetic subtypes of Prader-Willi syndrome. American Journal of Medical Genetics. 2005;136(2):140–145. doi: 10.1002/ajmg.a.30771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milner KM, Craig EE, Thompson RJ, et al. Prader-Willi syndrome: intellectual abilities and behavioural features by genetic subtype. Journal of Child Psychology and Psychiatry and Allied Disciplines. 2005;46(10):1089–1096. doi: 10.1111/j.1469-7610.2005.01520.x. [DOI] [PubMed] [Google Scholar]
- 24.Varela MC, Kok F, Setian N, Kim CA, Koiffmann CP. Impact of molecular mechanisms, including deletion size, on Prader-Willi syndrome phenotype: study of 75 patients. Clinical Genetics. 2005;67(1):47–52. doi: 10.1111/j.1399-0004.2005.00377.x. [DOI] [PubMed] [Google Scholar]
- 25.Nowicki ST, Tassone F, Ono MY, et al. The Prader-Willi phenotype of fragile X syndrome. Journal of Developmental and Behavioral Pediatrics. 2007;28(2):133–138. doi: 10.1097/01.DBP.0000267563.18952.c9. [DOI] [PubMed] [Google Scholar]
- 26.Fryns JP, Haspeslagh M, Dereymaeker AM, Volcke P, Van Den Berghe H. A peculiar subphenotype in the fra(X) syndrome: extreme obesity-short stature-stubby hands and feet-diffuse hyperpigmentation. Further evidence of disturbed hypothalamic function in the fra(X) syndrome? Clinical Genetics. 1987;32(6):388–392. doi: 10.1111/j.1399-0004.1987.tb03155.x. [DOI] [PubMed] [Google Scholar]
- 27.Vasudevan PC, Quarrell OWJ. Prader-Willi and Klinefelter syndrome: a coincidence or not? Clinical Dysmorphology. 2007;16(2):127–129. doi: 10.1097/MCD.0b013e32801472cf. [DOI] [PubMed] [Google Scholar]
- 28.Verhoeven WMA, De Vries BBA, Duffels SJH, Egger JIM, Noordam C, Tuinier S. Klinefelter’s syndrome and Prader-Willi syndrome: a rare combination. Psychopathology. 2007;40(5):356–360. doi: 10.1159/000106313. [DOI] [PubMed] [Google Scholar]
- 29.Gilhuis HJ, Van Ravenswaaij CMA, Hamel BJC, Gabreëls FJM. Interstitial 6q deletion with a Prader-Willi-like phenotype: a new case and review of the literature. European Journal of Paediatric Neurology. 2000;4(1):39–43. doi: 10.1053/ejpn.1999.0259. [DOI] [PubMed] [Google Scholar]
- 30.Xu F, Cheng DH, Di YF, et al. Identification of a cryptic 1p36. 3 microdeletion in a patient with Prader-Willi-like syndrome features. Chinese Journal of Medical Genetics. 2010;27(5):524–529. doi: 10.3760/cma.j.issn.1003-9406.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 31.Tsuyusaki Y, Yoshihashi H, Furuya N, et al. 1p36 deletion syndrome associated with Prader-Willi-like phenotype. Pediatrics International. 2010;52(4):547–550. doi: 10.1111/j.1442-200X.2010.03090.x. [DOI] [PubMed] [Google Scholar]
- 32.Ramsden SC, Clayton-Smith J, Birch R, Buiting K. Practice guidelines for the molecular analysis of Prader-Willi and Angelman syndromes. BMC Medical Genetics. 2010;11(1, article 70) doi: 10.1186/1471-2350-11-70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Õiglane-Shlik E, Žordania R, Varendi H, et al. The neonatal phenotype of Prader-Willi syndrome. American Journal of Medical Genetics A. 2006;140(11):1241–1244. doi: 10.1002/ajmg.a.31223. [DOI] [PubMed] [Google Scholar]
- 34.Trifirò G, Livieri C, Bosio L, et al. Neonatal hypotonia: don’t forget the Prader-Willi syndrome. Acta Paediatrica, International Journal of Paediatrics. 2003;92(9):1085–1089. doi: 10.1080/08035250310004810. [DOI] [PubMed] [Google Scholar]
- 35.Crinó A, Schiaffini R, Ciampalini P, et al. Hypogonadism and pubertal development in Prader-Willi syndrome. European Journal of Pediatrics. 2003;162(5):327–333. doi: 10.1007/s00431-002-1132-4. [DOI] [PubMed] [Google Scholar]
- 36.Eiholzer U, l’Allemand D, Rousson V, et al. Hypothalamic and gonadal components of hypogonadism in boys with Prader-Labhart-Willi syndrome. Journal of Clinical Endocrinology and Metabolism. 2006;91(3):892–898. doi: 10.1210/jc.2005-0902. [DOI] [PubMed] [Google Scholar]
- 37.Burman P, Ritzén EM, Lindgren AC. Endocrine dysfunction in Prader-Willi syndrome: a review with special reference to GH. Endocrine Reviews. 2001;22(6):787–799. doi: 10.1210/edrv.22.6.0447. [DOI] [PubMed] [Google Scholar]
- 38.Cassidy SB, Driscoll DJ. Prader-Willi syndrome. European Journal of Human Genetics. 2009;17(1):3–13. doi: 10.1038/ejhg.2008.165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Eiholzer U, Nordmann Y, L’Allemand D, Schlumpf M, Schmid S, Kromeyer-Hauschild K. Improving body composition and physical activity in Prader-Willi Syndrome. Journal of Pediatrics. 2003;142(1):73–78. doi: 10.1067/mpd.2003.mpd0334. [DOI] [PubMed] [Google Scholar]
- 40.Goldstone AP. Prader-Willi syndrome: advances in genetics, pathophysiology and treatment. Trends in Endocrinology and Metabolism. 2004;15(1):12–20. doi: 10.1016/j.tem.2003.11.003. [DOI] [PubMed] [Google Scholar]
- 41.Iughetti L, Bosio L, Corrias A, et al. Pituitary height and neuroradiological alterations in patients with Prader-Labhart-Willi syndrome. European Journal of Pediatrics. 2008;167(6):701–702. doi: 10.1007/s00431-007-0555-3. [DOI] [PubMed] [Google Scholar]
- 42.McAllister CJ, Whittington JE, Holland AJ. Development of the eating behaviour in Prader-Willi Syndrome: advances in our understanding. International Journal of Obesity. 2011;35(2):188–197. doi: 10.1038/ijo.2010.139. [DOI] [PubMed] [Google Scholar]
- 43.Dykens EM, Hodapp RM, Walsh K, Nash LJ. Profiles, correlates, and trajectories of intelligence in Prader-Willi syndrome. Journal of the American Academy of Child and Adolescent Psychiatry. 1992;31(6):1125–1130. doi: 10.1097/00004583-199211000-00022. [DOI] [PubMed] [Google Scholar]
- 44.Dykens EM, Leckman JF, Cassidy SB. Obsessions and compulsions in Prader-Willi syndrome. Journal of Child Psychology and Psychiatry and Allied Disciplines. 1996;37(8):995–1002. doi: 10.1111/j.1469-7610.1996.tb01496.x. [DOI] [PubMed] [Google Scholar]
- 45.Curfs LMG, Fryns JP. Prader-Willi syndrome: a review with special attention to the cognitive and behavioral profile. Birth Defects. 1992;28(1):99–104. [PubMed] [Google Scholar]
- 46.Crinò A, Di Giorgio G, Schiaffini R, et al. Central precocious puberty and growth hormone deficiency in a boy with Prader-Willi syndrome. European Journal of Pediatrics. 2008;167(12):1455–1458. doi: 10.1007/s00431-008-0679-0. [DOI] [PubMed] [Google Scholar]
- 47.Holland AJ, Treasure J, Coskeran P, et al. Measurement of excessive appetite and metabolic changes in Prader-Willi syndrome. International Journal of Obesity and Related Metabolic Disorders. 1993;17(9):527–532. [PubMed] [Google Scholar]
- 48.Fieldstone A. Food intake in prader-willi syndrome and controls with obesity after administration of a benzodiazepine receptor agonist. Obesity Research. 1998;6(1):29–33. doi: 10.1002/j.1550-8528.1998.tb00311.x. [DOI] [PubMed] [Google Scholar]
- 49.Stevenson DA, Heinemann J, Angulo M, et al. Gastric rupture and necrosis in Prader-Willi syndrome. Journal of Pediatric Gastroenterology and Nutrition. 2007;45(2):272–274. doi: 10.1097/MPG.0b013e31805b82b5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Goldstone AP, Holland AJ, Hauffa BP, Hokken-Koelega AC, Tauber M. Recommendations for the diagnosis and management of Prader-Willi syndrome. Journal of Clinical Endocrinology and Metabolism. 2008;93(11):4183–4197. doi: 10.1210/jc.2008-0649. [DOI] [PubMed] [Google Scholar]
- 51.Steinhausen HC, Eiholzer U, Hauffa BP, Malin Z. Behavioural and emotional disturbances in people with Prader-Willi syndrome. Journal of Intellectual Disability Research. 2004;48(1):47–52. doi: 10.1111/j.1365-2788.2004.00582.x. [DOI] [PubMed] [Google Scholar]
- 52.Dykens EM. Maladaptive and compulsive behavior in Prader-Willi syndrome: new insights from older adults. American Journal on Mental Retardation. 2004;109(2):142–153. doi: 10.1352/0895-8017(2004)109<142:MACBIP>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 53.Vogels A, De Hert M, Descheemaeker MJ, et al. Psychotic disorders in Prader-Willi syndrome. American Journal of Medical Genetics. 2004;127(3):238–243. doi: 10.1002/ajmg.a.30004. [DOI] [PubMed] [Google Scholar]
- 54.Akefeldt A, Tornhage CJ, Gillberg C. A woman with Prader-Willi syndrome gives birth to a healthy baby girl. Developmental Medicine and Child Neurology. 1999;41(11):789–790. doi: 10.1017/s0012162299221573. [DOI] [PubMed] [Google Scholar]
- 55.Schulze A, Mogensen H, Hamborg-Petersen B, Græm N, Østergaard JR, Brøndum-Nielsen K. Fertility in Prader-Willi syndrome: a case report with Angelman syndrome in the offspring. Acta Paediatrica, International Journal of Paediatrics. 2001;90(4):455–459. [PubMed] [Google Scholar]
- 56.Hirsch HJ, Eldar-Geva T, Benarroch F, Rubinstein O, Gross-Tsur V. Primary testicular dysfunction is a major contributor to abnormal pubertal development in males with Prader-Willi syndrome. Journal of Clinical Endocrinology and Metabolism. 2009;94(7):2262–2268. doi: 10.1210/jc.2008-2760. [DOI] [PubMed] [Google Scholar]
- 57.Eldar-Geva T, Hirsch HJ, Rabinowitz R, Benarroch F, Rubinstein O, Gross-Tsur V. Primary ovarian dysfunction contributes to the hypogonadism in women with prader-willi syndrome. Hormone Research. 2009;72(3):153–159. doi: 10.1159/000232489. [DOI] [PubMed] [Google Scholar]
- 58.Eldar-Geva T, Hirsch HJ, Benarroch F, Rubinstein O, Gross-Tsur V. Hypogonadism in females with Prader-Willi syndrome from infancy to adulthood: variable combinations of a primary gonadal defect and hypothalamic dysfunction. European Journal of Endocrinology. 2010;162(2):377–384. doi: 10.1530/EJE-09-0901. [DOI] [PubMed] [Google Scholar]
- 59.Butler MG. Management of obesity in Prader-Willi syndrome. Nature Clinical Practice Endocrinology & Metabolism. 2006;2(11):592–593. doi: 10.1038/ncpendmet0320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shapira NA, Lessig MC, Lewis MH, et al. Effects of topiramate in adults with Prader-Willi syndrome. American Journal on Mental Retardation. 2004;109(4):301–309. doi: 10.1352/0895-8017(2004)109<301:EOTIAW>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- 61.Papavramidis ST, Kotidis EV, Gamvros O. Prader-Willi syndrome-associated obesity treated by biliopancreatic diversion with duodenal switch. Case report and literature review. Journal of Pediatric Surgery. 2006;41(6):1153–1158. doi: 10.1016/j.jpedsurg.2006.01.065. [DOI] [PubMed] [Google Scholar]
- 62.Scheimann AO, Butler MG, Gourash L, Cuffari C, Klish W. Critical analysis of bariatric procedures in Prader-Willi syndrome. Journal of Pediatric Gastroenterology and Nutrition. 2008;46(1):80–83. doi: 10.1097/01.mpg.0000304458.30294.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Vismara L, Cimolin V, Grugni G, et al. Effectiveness of a 6-month home-based training program in Prader-Willi patients. Research in Developmental Disabilities. 2010;31(6):1373–1379. doi: 10.1016/j.ridd.2010.07.001. [DOI] [PubMed] [Google Scholar]
- 64.Nagai T, Obata K, Tonoki H, et al. Cause of sudden, unexpected death of Prader-Willi syndrome patients with or without growth hormone treatment. American Journal of Medical Genetics. 2005;136(1):45–48. doi: 10.1002/ajmg.a.30777. [DOI] [PubMed] [Google Scholar]
- 65.Tauber M, Diene G, Molinas C, Hébert M. Review of 64 cases of death in children with Prader-Willi syndrome (PWS) American Journal of Medical Genetics A. 2008;146(7):881–887. doi: 10.1002/ajmg.a.32131. [DOI] [PubMed] [Google Scholar]
- 66.Stevenson DA, Anaya TM, Clayton-Smith J, et al. Unexpected death and critical illness in Prader-Willi syndrome: report of ten individuals. American Journal of Medical Genetics. 2004;124(2):158–164. doi: 10.1002/ajmg.a.20370. [DOI] [PubMed] [Google Scholar]
- 67.Swaab DF, Purba JS, Hofman MA. Alterations in the hypothalamic paraventricular nucleus and its oxytocin neurons (putative satiety cells) in Prader-Willi syndrome: a study of five cases. Journal of Clinical Endocrinology and Metabolism. 1995;80(2):573–579. doi: 10.1210/jcem.80.2.7852523. [DOI] [PubMed] [Google Scholar]
- 68.de Lind Van Wijngaarden RFA, Otten BJ, Festen DAM, et al. High prevalence of central adrenal insufficiency in patients with Prader-Willi syndrome. Journal of Clinical Endocrinology and Metabolism. 2008;93(5):1649–1654. doi: 10.1210/jc.2007-2294. [DOI] [PubMed] [Google Scholar]
- 69.Nyunt O, Cotterill AM, Archbold SM, et al. Normal cortisol response on low-dose synacthen (1 αg) test in children with Prader Willi Syndrome. Journal of Clinical Endocrinology and Metabolism. 2010;95(12):E464–E467. doi: 10.1210/jc.2010-0647. [DOI] [PubMed] [Google Scholar]
- 70.Farholt S, Sode-Carlsen R, Christiansen JS, Østergaard JR, Høybye C. Normal cortisol response to high-dose synacthen and insulin tolerance test in children and adults with Prader-Willi syndrome. Journal of Clinical Endocrinology and Metabolism. 2011;96(1):E173–E180. doi: 10.1210/jc.2010-0782. [DOI] [PubMed] [Google Scholar]
- 71.Corrias A, Grugni G, Crinó A, et al. Assessment of central adrenal insufficiency in children and adolescents with Prader-Willi syndrome. Clinical Endocrinology. 2012;76(6):843–850. doi: 10.1111/j.1365-2265.2011.04313.x. [DOI] [PubMed] [Google Scholar]
- 72.Wollmann HA, Schultz U, Grauer ML, Ranke MB. Reference values for height and weight in Prader-Willi syndrome based on 315 patients. European Journal of Pediatrics. 1998;157(8):634–642. doi: 10.1007/s004310050901. [DOI] [PubMed] [Google Scholar]
- 73.Myers SE, Carrel AL, Whitman BY, Allen DB. Sustained benefit after 2 years of growth hormone on body composition, fat utilization, physical strength and agility, and growth in Prader-Willi syndrome. Journal of Pediatrics. 2000;137(1):42–49. doi: 10.1067/mpd.2000.105369. [DOI] [PubMed] [Google Scholar]
- 74.Mogul HR, Lee PDK, Whitman BY, et al. Growth hormone treatment of adults with Prader-Willi syndrome and growth hormone deficiency improves lean body mass, fractional body fat, and serum triiodothyronine without glucose impairment: results from the United States multicenter trial. Journal of Clinical Endocrinology and Metabolism. 2008;93(4):1238–1245. doi: 10.1210/jc.2007-2212. [DOI] [PubMed] [Google Scholar]
- 75.Sode-Carlsen R, Farholt S, Rabben KF, et al. One year of growth hormone treatment in adults with prader-willi syndrome improves body composition: results from a randomized, placebo-controlled study. Journal of Clinical Endocrinology and Metabolism. 2010;95(11):4943–4950. doi: 10.1210/jc.2010-0907. [DOI] [PubMed] [Google Scholar]
- 76.Siemensma EP, de Lind van Wijngaarden RF, Otten BJ, et al. Testicular failure in boys with Prader-Willi syndrome: longitudinal studies of reproductive hormones. Journal of Clinical Endocrinology and Metabolism. 2012;97(3):E452–E459. doi: 10.1210/jc.2011-1954. [DOI] [PubMed] [Google Scholar]
- 77.Brambilla P, Bosio L, Manzoni P, Pietrobelli A, Beccaria L, Chiumello G. Peculiar body composition in patients with Prader-Labhart-Willi syndrome. American Journal of Clinical Nutrition. 1997;65(5):1369–1374. doi: 10.1093/ajcn/65.5.1369. [DOI] [PubMed] [Google Scholar]
- 78.Höybye C, Frystyk J, Thorén M. The growth hormone-insulin-like growth factor axis in adult patients with Prader Willi syndrome. Growth Hormone and IGF Research. 2003;13(5):269–274. doi: 10.1016/s1096-6374(03)00017-0. [DOI] [PubMed] [Google Scholar]
- 79.van Mil EGAH, Westerterp KR, Gerver WJM, Van Marken Lichtenbelt WD, Kester ADM, Saris WHM. Body composition in Prader-Willi syndrome compared with nonsyndromal obesity: relationship to physical activity and growth hormone function. Journal of Pediatrics. 2001;139(5):708–714. doi: 10.1067/mpd.2001.118399. [DOI] [PubMed] [Google Scholar]
- 80.Butler MG, Haber L, Mernaugh R, Carlson MG, Price R, Feurer ID. Decreased bone mineral density in prader-willi syndrome: comparison with obese subjects. American Journal of Medical Genetics. 2001;103(3):216–222. [PMC free article] [PubMed] [Google Scholar]
- 81.Butler JV, Whittington JE, Holland AJ, Boer H, Clarke D, Webb T. Prevalence of, and risk factors for, physical ill-health in people with Prader-Willi syndrome: a population-based study. Developmental Medicine and Child Neurology. 2002;44(4):248–255. doi: 10.1017/s001216220100202x. [DOI] [PubMed] [Google Scholar]
- 82.Schmidt H, Schwarz HP. Premature adrenarche, increased growth velocity and accelerated bone age in male patients with Prader-Labhart-Willi syndrome. European Journal of Pediatrics. 2001;160(1):69–70. doi: 10.1007/s004310000633. [DOI] [PubMed] [Google Scholar]
- 83.Siemensma EPC, De Lind Van Wijngaarden RFA, Otten BJ, De Jong FH, Hokken-Koelega ACS. Pubarche and serum dehydroepiandrosterone sulphate levels in children with Prader-Willi syndrome. Clinical Endocrinology. 2011;75(1):83–89. doi: 10.1111/j.1365-2265.2011.03989.x. [DOI] [PubMed] [Google Scholar]
- 84.Vaiani E, Herzovich V, Chaler E, et al. Thyroid axis dysfunction in patients with Prader-Willi syndrome during the first 2 years of life. Clinical Endocrinology. 2010;73(4):546–550. doi: 10.1111/j.1365-2265.2010.03840.x. [DOI] [PubMed] [Google Scholar]
- 85.Savopoulos C, Hatzitolios A, Panagopoulou P, Kosmidou M, Tsirogianni E, Konstantinou V. Hypothyroidism in Prader-Willi syndrome: a case report and review of the literature. Journal of Endocrinological Investigation. 2007;30(9):804–805. doi: 10.1007/BF03350822. [DOI] [PubMed] [Google Scholar]
- 86.Einfeld SL, Smith A, Durvasula S, et al. Behavior and emotional disturbance in Prader-Willi syndrome. American Journal of Medical Genetics. 1999;82(2):123–127. doi: 10.1002/(sici)1096-8628(19990115)82:2<123::aid-ajmg4>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 87.Dykens E, Shah B. Psychiatric disorders in Prader-Willi syndrome: epidemiology and management. CNS Drugs. 2003;17(3):167–178. doi: 10.2165/00023210-200317030-00003. [DOI] [PubMed] [Google Scholar]
- 88.Casa Sora Per Voi ONLUS. casasorapervoi@gmail.com.
- 89.Odent T, Accadbled F, Koureas G, et al. Scoliosis in patients with prader-willi syndrome. Pediatrics. 2008;122(2):e499–e503. doi: 10.1542/peds.2007-3487. [DOI] [PubMed] [Google Scholar]
- 90.Kroonen LT, Herman M, Pizzutillo PD, MacEwen GD. Prader-Willi Syndrome: clinical concerns for the orthopaedic surgeon. Journal of Pediatric Orthopaedics. 2006;26(5):673–679. doi: 10.1097/01.bpo.0000226282.01202.4f. [DOI] [PubMed] [Google Scholar]
- 91.Accadbled F, Odent T, Moine A, et al. Complications of scoliosis surgery in Prader-Willi syndrome. Spine. 2008;33(4):394–401. doi: 10.1097/BRS.0b013e318163fa24. [DOI] [PubMed] [Google Scholar]
- 92.Bailleul-Forestier I, Verhaeghe V, Fryns JP, Vinckier F, Declerck D, Vogels A. The oro-dental phenotype in Prader-Willi syndrome: a survey of 15 patients. International Journal of Paediatric Dentistry. 2008;18(1):40–47. doi: 10.1111/j.1365-263X.2007.00857.x. [DOI] [PubMed] [Google Scholar]