

Abstract
Subtilase cytotoxin (SubAB), which is produced by certain strains of Shiga-toxigenic Escherichia coli (STEC), cleaves an endoplasmic reticulum (ER) chaperone, BiP/Grp78, leading to induction of ER stress and caspase-dependent apoptosis. SubAB alters the innate immune response. SubAB pretreatment of macrophages inhibited lipopolysaccharide (LPS)-induced production of both monocyte chemoattractant protein 1 (MCP-1) and tumor necrosis factor α (TNF-α). We investigated here the mechanism by which SubAB inhibits nitric oxide (NO) production by mouse macrophages. SubAB suppressed LPS-induced NO production through inhibition of inducible NO synthase (iNOS) mRNA and protein expression. Further, SubAB inhibited LPS-induced IκB-α phosphorylation and nuclear localization of the nuclear factor-κB (NF-κB) p65/p50 heterodimer. Reporter gene and chromatin immunoprecipitation (ChIP) assays revealed that SubAB reduced LPS-induced NF-κB p65/p50 heterodimer binding to an NF-κB binding site on the iNOS promoter. In contrast to the native toxin, a catalytically inactivated SubAB mutant slightly enhanced LPS-induced iNOS expression and binding of NF-κB subunits to the iNOS promoter. The SubAB effect on LPS-induced iNOS expression was significantly reduced in macrophages from NF-κB1 (p50)-deficient mice, which lacked a DNA-binding subunit of the p65/p50 heterodimer, suggesting that p50 was involved in SubAB-mediated inhibition of iNOS expression. Treatment of macrophages with an NOS inhibitor or expression of SubAB by E. coli increased E. coli survival in macrophages, suggesting that NO generated by macrophages resulted in efficient killing of the bacteria and SubAB contributed to E. coli survival in macrophages. Thus, we hypothesize that SubAB might represent a novel bacterial strategy to circumvent host defense during STEC infection.
INTRODUCTION
Shiga-toxigenic Escherichia coli (STEC) produces Shiga toxin 1 (Stx1) and Stx2, which are cytotoxic for colon cells, resulting in hemorrhagic colitis. Shiga toxins are significant virulence factors in STEC infection and may be responsible for life-threatening complications, such as hemolytic-uremic syndrome (HUS) (27, 43). However, it is not clear whether Shiga toxins are the only factors responsible for the morbidity and mortality associated with STEC-associated disease.
A new member of the AB5 toxin family, named subtilase cytotoxin (SubAB), was identified in E. coli O113:H21 strain 98NK2, which produces Stx2 and was responsible for an outbreak of HUS (42). SubAB binds to receptors on the cell membrane (59, 60) and thereby enters the cell, resulting in a site-specific cleavage of endoplasmic reticulum (ER) chaperone protein BiP/Grp78. Previous studies have shown that BiP/Grp78 cleavage by SubAB initiates an ER stress-induced unfolded protein response (UPR) (41, 54), resulting in transient inhibition of protein synthesis (34), G0/G1 cell cycle arrest (33, 34), downregulation of gap junction expression (24), and caspase-dependent apoptosis via mitochondrial membrane damage (32, 58). These actions of SubAB are responsible for cell death and may be involved in STEC-induced disease. Intriguingly, in addition to these activities, a series of recent studies showed that SubAB pretreatment of various cell lines inhibited lipopolysaccharide (LPS)- and tumor necrosis factor alpha (TNF-α)-induced NF-κB activation (17, 37). SubAB inhibition of TNF-α-induced NF-κB activation in rat renal tubular epithelial cells resulted from induction of CCAAT/enhancer-binding protein beta (C/EBPβ) and a mammalian target of rapamycin (mTOR)-dependent Akt phosphorylation pathways (37). However, an early event following SubAB-induced ER stress involved activation of NF-κB through an Akt-dependent pathway (61).
Nitric oxide (NO) is a short-lived free radical and an internal messenger that mediates a variety of functions, including vascular homeostasis, neurotransmission, and host defense (30). NO is synthesized from l-arginine by NO synthases (NOS) (2, 30). In mammals, three different isoforms of NOS exist (i.e., neuronal [nNOS], inducible [iNOS], and endothelial [eNOS]). nNOS and eNOS are primarily expressed in neurons and endothelial cells, respectively. In contrast, iNOS is a primary regulator of NO production in the innate immune system whose expression can be induced by LPS, gamma interferon (IFN-γ), interleukin-1β (IL-1β), IL-6, and TNF-α (2). iNOS gene expression is regulated through transcriptional control, particularly by NF-κB activation (29, 56, 57). Five mammalian NF-κB subunits, p65 (RelA), RelB, c-Rel, NF-κB1 (p50 and its precursor, p105), and NF-κB2 (p52 and its precursor, p100), form homo- or heterodimers to produce gene regulatory complexes with different properties (10, 46). In LPS-induced iNOS expression, the involvement of the NF-κB p65/p50 heterodimer is well documented (10). p65/p50 heterodimer is held in an inactive state in the cytoplasm by IκB, which is phosphorylated by the IκB kinase (IKK) complex (9), leading to IκB degradation and NF-κB activation (26). In mouse, the iNOS gene promoter contains two NF-κB binding sites, one upstream (GGGATTTTCC; nucleotides −971 to −962, designated the NF-κBu site) and one downstream (GGGACTCTCC; nucleotides −85 to −76, designated the NF-κBd site), both of which need to be occupied to obtain full induction of iNOS by LPS (56).
Phagocytic cells such as macrophages or neutrophils are important components of the innate immune response. Two major antimicrobial systems of phagocytic cells are the NADPH phagocyte oxidase (also known as phox) and iNOS pathways, which are responsible for the generation of superoxide (O2−) and NO, respectively (11). The NO produced by iNOS contributes to the bactericidal activities of macrophages. NO reacts with simultaneously generated reactive oxygen species (ROS), resulting in formation of reactive nitrogen species (RNS), such as peroxynitrite (ONOO−) and nitrogen dioxide (NO2). NO, RNS, and ROS have antimicrobial activities (39). NO potentiates hydrogen peroxide (H2O2)-induced killing of E. coli in part through the generation of ONOO− (3, 40). Nitrite exerts antimicrobial effects through generation of NO, which is toxic to STEC (35). Furthermore, NO suppresses Stx2 production by STEC through inhibition of stx2 mRNA expression (51). These findings suggest that NO and NO-derived RNS protect the host from STEC-induced disease.
In this study, we demonstrate in mouse macrophages that SubAB inhibited LPS-stimulated NO production through inhibition of NF-κB nuclear translocation and iNOS expression. Moreover, we show that NF-κB p50 is involved in inhibition of LPS-induced iNOS expression by SubAB. In addition, we report that SubAB enhanced E. coli survival in macrophages. This study provides the first evidence for direct inhibition by SubAB of LPS-induced, NF-κB activation-mediated NO production and for a novel bacterial strategy to survive in macrophages.
MATERIALS AND METHODS
Reagents.
LPS (from Escherichia coli O111:B4), penicillin-streptomycin, polymyxin B, and anti-α-tubulin monoclonal antibody were purchased from Sigma-Aldrich (St. Louis, MO). Dulbecco's modified Eagle's medium (DMEM) and fetal bovine serum (FBS) were obtained from Gibco BRL (Grand Island, NY). All primers were synthesized chemically by Invitrogen (Carlsbad, CA). Anti-NF-κB p65 polyclonal antibodies (C-20, sc-372), anti-NF-κB p50 polyclonal antibodies (H-119, sc-7178), and anti-NF-κB p50 monoclonal antibodies (E-10, sc-8414) were purchased from Santa Cruz Biotechnology; anti-NF-κB p50 polyclonal antibodies and mouse recombinant M-CSF were from eBioscience; anti-nucleoporin p62 and anti-BiP/Grp78 monoclonal antibodies were from BD Bioscience; anti-phospho-IκB-α monoclonal antibody (9246) and recombinant TNF-α were from Cell Signaling; and anti-GAPDH polyclonal antibodies were from GeneTex. L-NAME was from DOJINDO (Kumamoto, Japan). Other reagents were purchased from Wako Pure Chemical Industries (Osaka, Japan). iNOS-specific monoclonal antibody i2G4 was prepared as described previously (38).
Animals.
NF-κB1−/− mice (46) were purchased from the Jackson Laboratories (Bar Harbor, ME) and C57BL/6J wild-type mice from SLC (Hamamatsu, Japan). Animal experiments were approved by Chiba University Institutional Animal Care and Use Committee, Chiba, Japan.
Preparation of SubAB.
Recombinant His-tagged SubAB and a catalytically inactive SubAB mutant, SubAS272AB (mSubAB), were purified as reported previously (34). Heat-inactivated SubAB (Hi-SubAB) and heat-inactivated mSubAB (Hi-mSubAB) were prepared as described previously (59).
Cell culture and treatment.
RAW 264.7 cells, a murine macrophage cell line, were obtained from Riken Cell Bank (Tsukuba, Japan) and maintained in Dulbecco's modified Eagle's medium (DMEM) containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 0.1 mg/ml streptomycin (DMEM-10% FBS) at 37°C under 5% CO2. The preparation of bone marrow macrophages (BMMs) from NF-κB1−/− mice was based on the procedure of Guilbert and Stanley (16). Briefly, bone marrow cells were collected from the femurs and tibiae of mice and cultured in DMEM-10% FBS, supplemented with 20 ng/ml M-CSF, for 4 to 6 days before use in experiments. RAW 264.7 cells and BMMs were stimulated with LPS (0.01 to 10 μg/ml) or recombinant TNF-α (10 ng/ml) in the presence or absence of SubAB (0.005 to 1 μg/ml) or mSubAB (0.5 μg/ml) in DMEM containing 1% heat-inactivated FBS, 100 U/ml penicillin, and 0.1 mg/ml streptomycin (DMEM-1% FBS) as described in the figure legends.
Analysis of nitrite accumulation.
NO release from macrophage cells was determined by assaying nitrite levels, a relatively stable NO metabolite. The accumulation of nitrite in culture supernatants was quantified by Griess reaction, as described previously (47). Briefly, cells were stimulated as described in the figure legends and 50-μl aliquots of the culture supernatants were dispensed in triplicate into 96-well plates and mixed with 25 μl of Griess reagent A (1% sulfanilamide in 5% H3PO4). After 5 min of incubation, 25 μl of Griess reagent B [0.1% N-(1-naphthyl)-ethylenediamine] was added, followed by incubation for 10 min at room temperature. The absorbance of samples at 540 nm was compared with that of sodium nitrite standard on a microplate reader (Bio-Rad).
Cell viability assay.
RAW 264.7 cells were incubated with 0.5 μg/ml of SubAB, mSubAB, Hi-SubAB, and Hi-mSubAB for 24 or 48 h, and cell viability was evaluated by cell counting kit 8 (DOJINDO), according to the manufacturer's protocol.
Preparation of total cell lysate and cytoplasmic and nuclear protein extracts.
To prepare total cell lysate, cells were treated with LPS in the presence or absence of SubAB or mSubAB, washed with phosphate-buffered saline (PBS), and lysed with SDS sample buffer (62.5 mM Tris-HCl [pH 6.8], 1% SDS, 10% glycerol, 2.5% mercaptoethanol, and 0.001% bromophenol blue). Cytoplasmic and nuclear protein extracts were prepared as described previously (45). Briefly, cells were washed with ice-cold PBS and then incubated with hypotonic lysis buffer (10 mM HEPES [pH 8.0], 2 mM MgCl2, 1 mM CaCl2, and 10% glycerol) containing protease inhibitor (PI) cocktail (Complete ULTRA, mini, EDTA free) (Roche Diagnostics) on ice for 10 min. Nonidet P-40 was added to a final concentration of 0.6%, and the resulting preparation was vortexed for 10 s and then centrifuged at 10,000 × g for 30 s. Nuclear pellets were washed with hypotonic lysis buffer, and nuclear proteins were extracted by incubation of the nuclei with nuclear extract buffer (20 mM HEPES [pH 8.0], 0.42 M NaCl, 0.5 mM dithiothreitol [DTT], and 10% glycerol) containing PI cocktail for 15 min at 4°C, and cell debris was removed by centrifugation at 15,000 × g for 15 min at 4°C. The Bradford method (protein assay; Bio-Rad) was used to measure protein concentration in the extract, which was then stored in aliquots at −80°C.
Immunoprecipitation.
Coimmunoprecipitation of NF-κB p50 and p65 was carried out as described previously (60). Briefly, cytoplasmic proteins were incubated with anti-NF-κB p50 antibody (Santa Cruz Biotechnology) at 4°C overnight. Immunoprecipitates were collected by incubation with protein G-Sepharose (Invitrogen) for 1 h, followed by centrifugation for 1 min at 4°C. Immunocomplexes were washed with hypotonic lysis buffer three times, and proteins were dissolved in sodium dodecyl sulfate (SDS) sample buffer, subjected to SDS-polyacrylamide gel electrophoresis (PAGE) in 7.5% gels, transferred to polyvinylidene difluoride (PVDF) membranes, and then analyzed by Western blotting using anti-NF-κB p65 antibodies (Santa Cruz Biotechnology).
Immunostaining.
RAW 264.7 cells were seeded in 12-well plates containing coverslips and incubated at 37°C overnight. After treatment as described in the figure legends, the cells were fixed with 4% formaldehyde in PBS at room temperature for 15 min and then washed three times with PBS. Cells permeabilized with methanol for 10 min at −20°C were treated with PBS containing 5% goat serum and 0.05% Triton X-100 for 1 h. Cells were incubated with anti-NF-κB p65 polyclonal antibodies or anti-NF-κB p50 monoclonal antibody overnight at 4°C and washed three times with PBS, followed by incubation at room temperature for 1 h with Cy3-conjugated anti-rabbit IgG or Alexa 488-conjugated anti-mouse IgG. Cells on the coverslips were then washed three times with PBS, once with water, dried, and mounted on glass slides using ProLong Gold antifade reagent with DAPI (4′,6-diamidino-2-phenylindole) (Invitrogen). Stained cells were visualized using confocal microscopy (Olympus).
Immunoblotting analysis.
Total cell lysate and cytoplasmic and nuclear extracts lysed in SDS sample buffer were heated at 100°C for 5 min before proteins were analyzed by SDS-PAGE. After electrophoresis at room temperature, separated proteins were transferred to PVDF membranes at 100 V for 1 h. Membranes were blocked with 5% nonfat milk in TBS-T (20 mM Tris-HCl [pH 7.6], 137 mM NaCl, 0.1% Tween 20) for 30 min and then incubated with primary antibodies for 1 h at room temperature or overnight at 4°C. After being washed five times for 5 min with TBS-T, membranes were incubated with horseradish peroxidase-labeled secondary antibodies. Bands were visualized using Las 1000 (Fuji film).
Quantitative reverse-transcription PCR (qRT-PCR).
Total RNA was prepared from RAW 264.7 cells or BMMs using the PureLink RNA minikit (Invitrogen), according to the manufacturer's protocol. Total RNA was measured, and 1 to 5 μg were reverse transcribed using Ready-To-Go You-Prime first-strand beads (GE Healthcare) with oligo(dT)12-18 primer (Invitrogen). cDNA content was measured by real-time PCR with SYBR green reagent using an ABI PRISM 7300 sequence detection system (Applied Biosystems, Foster City, CA). Specific primers used for real-time PCR were as follows: mouse iNOS forward, 5′-GTTCTCAGCCCAACAATACAAGA-3′, and reverse, 5′-GTGGACGGGTCGATGTCAC-3′. Primers for GAPDH cDNA amplification were as described previously (13).
Plasmids, transient transfection, and reporter gene assay.
The mouse iNOS promoter-luciferase reporter plasmid (pGL2-iNOS) was generously provided by Charles J. Lowenstein (Johns Hopkins University) via Addgene Inc. (Addgene plasmid 19296). Deletion constructs were created by modified methods as reported (29). All constructs were then sequenced to characterize them definitively. Endotoxin-free plasmid DNA was purified using Qiagen plasmid plus midikit (Qiagen), according to the manufacturer's protocol. RAW 264.7 cells (1 × 104 or 2.5 × 104 cells) transfected with a luciferase reporter plasmid were cultured in 24-well plates in DMEM-1% FBS. Reporter constructs (pGL2-iNOS; 0.5 μg or HIV-NF-κB luciferase reporter construct [9]; 1.5 μg) were mixed with 20 ng of phRL-TK (Promega) in 50 μl of Opti-MEM I reduced serum medium (GIBCO/Invitrogen). The solution was mixed with 1 μl of FuGENE 6 (Roche Diagnostics) and incubated at room temperature for 30 min; the two vectors in 50-μl solutions were cotransfected into RAW 264.7 cells after the medium was replaced with 300 μl of fresh DMEM-10% FBS. Subsequently, the cells were incubated at 37°C for 24 or 72 h. After transfection with plasmid, the medium was replaced with 300 μl of fresh DMEM-1% FBS and cells were stimulated with LPS (10 μg/ml) in the presence or absence of SubAB or mSubAB. After incubation for 4, 8, or 24 h, cells were washed with cold PBS and lysed by adding 100 μl of passive lysis buffer (Promega). Aliquots of 20 μl of cell lysate were used to assay for luciferase activity with the dual-luciferase reporter assay kit (Promega), according to the manufacturer's guidelines.
ChIP-qPCR assays.
Chromatin immunoprecipitation (ChIP)-qPCR assays were performed to examine the binding of NF-κB p65 and p50 subunits to iNOS promoter, as reported previously (22). Briefly, cells were fixed with 1% formaldehyde for 10 min at 37°C, collected by scraping, and lysed in SDS buffer (50 mM Tris-HCl [pH 8.1], 10 mM EDTA, 1% SDS) containing protease inhibitor cocktail (Roche Diagnostics). Lysates were sonicated to fragment the chromatin by using Bioruptor (Cosmo Bio Co., Tokyo, Japan) and then diluted with ChIP dilution buffer (20 mM Tris-HCl [pH 8.1], 1 mM EDTA, 150 mM NaCl, and 0.3% Triton X-100). Immunoprecipitation analysis was carried out using control rabbit IgG and anti-NF-κB p65 or anti-NF-κB p50 antibodies. Cross-links were reversed at 65°C for 6 h, and proteins were digested with proteinase K (0.4 mg/ml) for 1 h at 55°C. Immunoprecipitated DNA was recovered by the QIAQiuck PCR purification kit (Qiagen). DNA fragments were amplified by PCR with specific primers as follows: 5′-CACACAGACTAGGAGTGTCCATCAT-3′ and 5′-CATAACTGTTCCCAAAGGGAGAGT-3′. DNA content was measured by real-time PCR with SYBR green reagent using an ABI PRISM 7300 sequence detection system (Applied Biosystems, Foster City, CA). Results are expressed as the percent input for each ChIP fraction.
Macrophage infection.
Transformants of E. coli strain BL21(DE3) (Invitrogen) carrying the SubAB and mSubAB genes on pET23b (BL21/pET-SubAB and BL21/pET-mSubAB, respectively) were generated as reported previously (34). For macrophage infection, RAW 264.7 cells were cultured in antibiotic-free DMEM containing 10% heat-inactivated FBS and infected as described previously (47). Briefly, cells were seeded at 1 × 106 cells per well in 24-well tissue culture dishes for 24 h and then infected with BL21/pET23b, BL21/pET-SubAB, or BL21/pET-mSubAB at a multiplicity of infection (MOI) of 10. The plate was centrifuged for 5 min at 1,000 × g to synchronize the infection and incubated for 20 min, and then the cells were washed and fresh medium containing 100 μg/ml of gentamicin was added to kill extracellular bacteria. After 2 h, the medium was discarded, the cells were washed, and the medium was changed to include 20 μg/ml of gentamicin with or without L-NAME (10 mM), SubAB, or mSubAB. The infected monolayers were lysed on the tissue culture dishes by the addition of 0.1% sodium deoxycholate in phosphate-buffered saline. The number of surviving bacteria was determined by bacterial plate counts (CFU).
Statistical analysis.
Student's t test was used to determine significant difference when only two treatment groups were being compared. Analysis of variance (ANOVA) with the Student-Newman-Keuls test was used to analyze significant differences among multiple groups.
RESULTS
SubAB inhibits LPS-induced NO production by RAW 264.7 cells.
To investigate the effect of SubAB on LPS-induced NO production in RAW 264.7 cells, cells were incubated with LPS for 24 h in the presence or absence of SubAB, a catalytically inactive SubAB mutant (mSubAB), heat-inactivated SubAB, or heat-inactivated mSubAB, and then supernatants were collected for the measurement of nitrite, a relatively stable metabolite of NO (Fig. 1A). When RAW 264.7 cells were incubated with LPS, a significant amount of NO production was observed. LPS-induced NO production was markedly decreased in SubAB-treated RAW 264.7 cells. mSubAB treatment did not inhibit LPS-induced NO production; rather, it slightly increased NO production. Heat-inactivated SubAB or heat-inactivated mSubAB did not affect LPS-induced NO production by RAW 264.7 cells. In addition, LPS-induced NO production was completely reduced in the presence of an iNOS-specific inhibitor 1400W dihydrochloride, indicating that iNOS is essential for LPS-induced NO production by RAW 264.7 cells. These results suggest that SubAB-induced ER stress inhibits LPS-induced NO production by RAW 264.7 cells, and mSubAB binding to RAW 264.7 cells slightly enhances LPS-induced NO production. In addition, SubAB treatment suppressed LPS-induced NO production in a dose-dependent manner in RAW 264.7 cells, with 0.5 μg/ml of SubAB inhibiting LPS-induced NO production by RAW 264.7 cells (Fig. 1B). Posttreatment with SubAB after LPS treatment for 0 to 180 min significantly suppressed LPS-induced NO production by RAW 264.7 cells, suggesting that SubAB effectively inhibits NO production (Fig. 1C). The effect of SubAB appeared to be diminished with time. To examine the effect of mSubAB binding on NO production in RAW 264.7 cells, cells were treated with PBS control, LPS (10 ng/ml), SubAB, or mSubAB in the presence or absence of polymyxin B, which inactivates LPS (36, 62). In the absence of polymyxin B, both LPS and mSubAB, but not SubAB, induced NO production. In the presence of polymyxin B, LPS-induced NO production was completely inhibited, but mSubAB-induced NO production was only partially reduced (Fig. 1D). These results suggest that mSubAB binding to cells may promote NO production. mSubAB is known to bind surface receptors, even though it is catalytically inactive. A recent study reported that SubAB inhibits TNF-α-induced NF-κB activation in rat renal tubular epithelial cells (37). To examine the effect of SubAB on TNF-α-induced NO production by RAW 264.7 cells, cells were treated with recombinant mouse or human TNF-α in the presence or absence of SubAB. However, both recombinant mouse and human TNF-α did not induce NO production by RAW 264.7 (data not shown). Next, we tested the effect of SubAB on RAW 264.7 cell viability. At 0.5 μg/ml, SubAB did not affect cell viability at 24 h, but cell death was detected after 48 h (Fig. 1E). Heat-inactivated SubAB or mSubAB or heat-inactivated mSubAB did not affect cell viability. Therefore, we used 0.5 μg/ml SubAB in our experiments.
Fig 1.

SubAB inhibits LPS-induced nitrite production by RAW 264.7 cells. (A) RAW 264.7 cells (1 × 105 cells/well) in a 96-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with LPS (10 μg/ml) in the presence or absence of SubAB (0.5 μg/ml), its catalytically inactive mutant, mSubAB (MT), heat-inactivated SubAB (Hi-WT), heat-inactivated mSubAB (Hi-MT) (0.5 μg/ml), or 1400W dihydrochloride for 24 h. The accumulation of nitrite in culture supernatants was quantified by Griess assay, as described in Materials and Methods. (B) RAW 264.7 cells (1 × 105 cells/well) in a 96-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with LPS (10 μg/ml) in the presence or absence of the indicated concentrations of SubAB (WT) for 24 h. The accumulation of nitrite in culture supernatants was quantified by Griess assay as described above. (C) RAW 264.7 cells (1 × 105 cells/well) in a 96-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with LPS (10 μg/ml) and then posttreated with SubAB (0.5 μg/ml) after LPS stimulation for 0, 15, 30, 60, 180, and 300 min. After a total incubation time of 24 h, nitrite was quantified by Griess assay as described above. (D) RAW 264.7 cells (1 × 105 cells/well) in a 96-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with LPS (10 ng/ml), SubAB (WT), or mSubAB (MT) at 0.5 μg/ml for 24 h in DMEM-1% FBS in the presence or absence of polymyxin B (10 μg/ml). The accumulation of nitrite in culture supernatants was quantified as described above. (E) RAW 264.7 cells (1 × 105 cells/well) in a 96-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with SubAB (WT), mSubAB (MT), heat-inactivated SubAB (Hi-WT), or heat-inactivated mSubAB (Hi-MT) (0.5 μg/ml) for 24 or 48 h. Cell viability was evaluated by cell counting kit 8 (DOJINDO) according to the instruction manual. Data for all these experiments are means ± standard deviations (SD) of values from three independent assays in triplicate experiments. Statistical significance: *, P < 0.01; **, P < 0.05; N.S., not significant.
SubAB inhibits LPS-induced iNOS expression in RAW 264.7 cells.
To investigate the mechanism of the inhibitory effects of SubAB on LPS-induced NO production in RAW 264.7 cells, LPS-induced iNOS expression in the presence and absence of SubAB was analyzed by immunoblotting. LPS induced iNOS protein expression in a dose-dependent manner; it was dramatically blocked by SubAB (Fig. 2A). LPS also induced iNOS expression in RAW 264.7 cells in a time-dependent manner, which was attenuated in the presence of SubAB but not mSubAB (Fig. 2B). In addition, densitometric analysis also showed that SubAB suppressed LPS-induced iNOS expression. To test whether the inhibitory effect involved ER stress, we investigated the effects of ER stress-inducing agents that prevent disulfide bond formation (DTT), ER-associated degradation (MG132), or N-glycosylation (tunicamycin) or alter calcium homeostasis (thapsigargin) on LPS-induced iNOS expression. After cells were incubated with LPS in the presence or absence of SubAB, mSubAB, or ER stress-inducing agents for 6 h, iNOS expression was analyzed. Although SubAB and ER stress-inducing agents inhibited LPS-induced iNOS expression, mSubAB and vehicle control dimethyl sulfoxide (DMSO) were inactive, suggesting that LPS-induced iNOS expression was inhibited by ER stress (Fig. 2C). We next examined the inhibitory effect of SubAB on LPS-induced iNOS mRNA expression in RAW 264.7 cells. RAW 264.7 cells were treated with LPS for 4 or 24 h in the presence of SubAB or mSubAB, and then total mRNA was used for quantitative PCR analysis. For 4 h of incubation, LPS treatment significantly induced iNOS mRNA expression in RAW 264.7 cells, while SubAB, but not mSubAB, inhibited LPS-induced iNOS mRNA expression (Fig. 2D, left). For 24 h of incubation, SubAB treatment inhibited LPS-induced iNOS mRNA expression, which was slightly increased by mSubAB (Fig. 2D, right). These results indicate that SubAB strongly suppressed LPS-induced iNOS mRNA expression.
Fig 2.

SubAB inhibits LPS-induced iNOS expression by RAW 264.7 cells. (A) RAW 264.7 cells (5 × 104 cells/well) in a 24-well dish were grown in DMEM-1% FBS for 24 h. Cells were treated with LPS (0, 0.01, 0.1, 1, or 10 μg/ml) in the presence or absence of SubAB (0.5 μg/ml) for 24 h. Cell lysates were analyzed by immunoblotting with anti-iNOS or anti-α-tubulin antibodies as a control. Data are representative of at least three separate experiments. (B) RAW 264.7 cells (5 × 104 cells/well) were treated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for the indicated times. Cell lysates were collected from cells and analyzed by immunoblotting as described above. Data are representative of three separate experiments. (C) RAW 264.7 cells (2 × 105 cells/well) were incubated with LPS (10 μg/ml) for 6 h in the presence or absence of DTT (1 mM), tunicamycin (Tm; 1 μg/ml), MG132 (MG; 1 μM), SubAB (WT; 0.5 μg/ml), mSubAB (MT; 0.5 μg/ml), or DMSO as a control. Cell lysates were analyzed by immunoblotting with anti-iNOS, anti-α-tubulin, or anti-BiP/Grp78 antibodies as described above. Data are representative of three separate experiments. (D) RAW 264.7 cells (5 × 104 cells/well) were treated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 4 or 24 h. Total RNA extracted from the cells was subjected to real-time PCR using specific primers for iNOS as described in Materials and Methods. iNOS expression was normalized to GAPDH (iNOS/GAPDH). Data are means ± SD of values from three separate experiments. Statistical significance: *, P < 0.01; **, P < 0.05; N.S., not significant.
SubAB inhibits LPS-induced transcriptional activation of the iNOS promoter in RAW 264.7 cells.
In macrophages, LPS-induced iNOS expression is triggered by activation of the Toll-like receptor 4 (TLR4) signaling pathway, followed by binding of transcription factors (i.e., NF-κB, STAT-1, AP-1, C/EBP, CREB, IRF-1, Oct-1) to the iNOS promoter (2, 29, 57). To investigate the effect of SubAB on LPS-induced iNOS promoter activity, RAW 264.7 cells were transiently transfected with mouse iNOS promoter-luciferase reporter plasmid (pGL2-iNOS), and then transcriptional activity was determined by relative luciferase activity 24 h after LPS stimulation in the presence or absence of SubAB or mSubAB. As shown in Fig. 3, LPS treatment significantly increased relative luciferase activity, which was suppressed in the presence of SubAB but not mSubAB. These results indicate that SubAB inhibits LPS-induced transcriptional activation of the iNOS promoter. To determine which sites in the iNOS promoter are responsible for inhibition of iNOS expression by SubAB, we transfected RAW 264.7 cells with iNOS promoter deletion plasmids (see the diagram in Fig. 3). SubAB suppressed LPS-induced luciferase activity of deletion mutants pGL2-iNOS/−724 and pGL2-iNOS/−93. However, inhibitory effects of SubAB on LPS-induced luciferase activity were completely lost in pGL2-iNOS/−40-transfected cells. Stimulatory effects of LPS were also lost in this mutant. These results suggest that the inhibitory effects of SubAB on iNOS promoter activity are contained in the region between −93 bp and −40 bp, which has NF-κB-, NF–IL-6-, and Oct-binding sites (29, 57). To understand the importance of the region for iNOS transcription, RAW 264.7 cells were transfected with pGL2-iNOS full/Δ−46–−203, with a deletion of the region from position −203 to position −46. These cells showed slight LPS-stimulated luciferase activity, suggesting that this region plays an important role in iNOS transcriptional activity in the presence of LPS.
Fig 3.

SubAB inhibits LPS-induced iNOS promoter activity in RAW 264.7 cells. RAW 264.7 cells (2.5 × 104 cells/well) were cotransfected with 0.5 μg of pGL2-iNOS and 20 ng of phRL-TK as an internal control. After transfection, cells were incubated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 24 h. Relative changes in luciferase expression were measured as described in Materials and Methods. Luciferase activity was normalized for Renilla luciferase activity. Data are means ± SD of values from three independent assays in triplicate experiments. Statistical significance: *, P < 0.05 versus LPS treatment.
Of note, mSubAB increased the effects of LPS on iNOS transcriptional activity of the intact promoter. This stimulation was not observed in any of the promoter mutants, where, in some instances, slight inhibition was observed.
SubAB inhibits LPS-induced NF-κB nuclear translocation in RAW 264.7 cells.
The results of iNOS promoter assays suggested that SubAB affects NF-κB, NF–IL-6, or Oct binding to the iNOS promoter. Since previous studies showed that SubAB inhibited LPS- or TNF-α-induced NF-κB activation through inhibition of IκB-α degradation (17, 37), we examined the effect of SubAB on LPS-induced IκB-α phosphorylation and NF-κB nuclear translocation in RAW 264.7 cells. As shown in Fig. 4A, LPS treatment induced NF-κB p65 and p50 nuclear localization, which was inhibited by pretreatment with SubAB but not mSubAB. In addition, densitometric analysis also showed that SubAB significantly suppressed LPS-induced NF-κB p65 and p50 nuclear localization. NF-κB p65 and p50 subunits most commonly form an NF-κB p65/p50 heterodimer with transactivation activity (10, 50). To confirm the inhibitory effect of SubAB on nuclear translocation of the NF-κB p65/p50 heterodimer, we next performed coimmunoprecipitation with NF-κB p50 antibody in a cytoplasmic fraction from RAW 264.7 cells. In LPS-treated cells, the amount of NF-κB p65/p50 heterodimer was reduced but not in the presence of SubAB. mSubAB did not affect the LPS-induced NF-κB p65/p50 heterodimer. As shown in Fig. 4B, confocal microscopy demonstrated that the NF-κB p65/p50 heterodimer was localized in cytoplasm in untreated RAW 264.7 cells. In LPS-treated cells, the NF-κB p65/p50 heterodimer was translocated into nuclei, while treatment with SubAB, but not mSubAB, blocked the translocation to the nuclei. These data suggest that SubAB inhibits LPS-induced nuclear translocation of the NF-κB p65/p50 heterodimer.
Fig 4.

SubAB inhibits LPS-induced NF-κB nuclear translocation in RAW 264.7 cells. (A) After RAW 264.7 cells (5 × 106 cells/dish) were treated with SubAB (0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 3 h in the presence of polymyxin B (10 μg/ml), cells were treated with LPS (10 μg/ml) for 30 min. Cells were collected and lysed in lysis buffer, and cytoplasmic extracts (CE) and nuclear protein extracts (NE) were prepared as described in Materials and Methods. Proteins in CE or NE were analyzed by immunoblotting using anti-NF-κB p65, anti-NF-κB p50, anti-nucleoporin p62 (nuclear protein control), and anti-GAPDH (cytoplasmic protein control) antibodies. Nuclear translocation of p65 and p50 was normalized to nucleoporin p62. CE were immunoprecipitated with anti-NF-κB p50 monoclonal antibody and subjected to analysis by immunoblotting using anti-NF-κB p65 polyclonal antibodies. The relative amounts of p65 and p50 are based on densitometric quantification. Data are means ± SD of values from three independent experiments. Statistical significance: *, P < 0.01; **, P < 0.05. (B) After RAW 264.7 cells were treated with SubAB (0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 3 h in the presence of polymyxin B (10 μg/ml), cells were treated with LPS (10 μg/ml) for 30 min. Cells were fixed for immunofluorescence staining with anti-NF-κB p65 (red) or anti-NF-κB p50 (green) antibodies as described in Materials and Methods. A merged picture shows colocalization in RAW 264.7 cells. The nuclei were stained with DAPI. Solid arrows show colocalization of NF-κB p65 and p50. Bars represent 20 μm. Experiments were repeated two times with similar results. (C) RAW 264.7 cells were transiently transfected with HIV-NF-κB luciferase reporter construct, as well as the reference plasmid phRL-TK. Cells were treated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 4, 8, or 24 h. Relative changes in luciferase expression were measured as described in Materials and Methods. Luciferase activity was normalized to Renilla luciferase activity. Results are expressed as the fold induction of luciferase activities in PBS-treated cells. Data are means ± SD of values from three independent experiments, with n = 3 per experiment. Statistical significance: *, P < 0.01; **, P < 0.05; N.S., not significant. (D) RAW 264.7 cells (5 × 104 cells/well) were incubated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for the indicated times. Cell lysates were analyzed by immunoblotting using anti-phospho-IκB α, anti-α-tubulin, or anti-BiP/Grp78 antibodies (top). Data are representative of three independent experiments. IκB α phosphorylation was normalized to α-tubulin using densitometry. Relative densities of phospho-IκB α (phospho-IκB/tubulin) were compared with densities obtained at 0 min for each of three treated cells (bottom). Data are means ± SD of values from triplicate experiments. **, P < 0.05.
To determine the inhibitory effect of SubAB on the LPS-induced DNA-binding activity of NF-κB, RAW 264.7 cells were transiently transfected with the HIV-3×-κB luciferase reporter construct, which contains a consensus sequence for NF-κB binding, and then LPS-induced transcriptional activity was determined by relative luciferase activity at 4, 8, and 24 h in the presence or absence of SubAB or mSubAB. LPS treatment gradually increased NF-κB-mediated luciferase activity in a time-dependent manner, which was significantly suppressed in the presence of SubAB at 8 or 24 h (Fig. 4C).
To examine whether SubAB affects IκB-α phosphorylation as a trigger of IκB-α degradation, we next investigated IκB-α phosphorylation by immunoblotting. LPS treatment of RAW 264.7 cells increased phosphorylation of IκB-α within 1 h, with a subsequent decrease by 3 h. In the presence of SubAB, BiP cleavage was observed after 30 min of incubation, followed by suppression of LPS-induced IκB-α phosphorylation. A densitometric analysis also showed that SubAB significantly suppressed LPS-induced IκB-α phosphorylation at 0.5 and 1 h. In contrast, mSubAB treatment did not affect LPS-induced IκB-α phosphorylation (Fig. 4D). These results suggest that SubAB suppresses LPS-induced NF-κB nuclear localization through inhibition of phosphorylation-induced IκB-α degradation.
SubAB inhibits LPS-induced NF-κB binding to the iNOS promoter in RAW 264.7 cells.
The NF-κB p65/p50 heterodimer has been shown to bind to the NF-κB binding site at a position from −85 to −76 adjacent to the TATA box of the iNOS promoter, which was designated the NF-κBd site (56). To examine whether SubAB treatment suppresses LPS-induced NF-κB p65/p50 heterodimer binding to the NF-κBd site of the iNOS promoter, we performed ChIP-qPCR assay in RAW 264.7 cells using anti-p65 or anti-p50 antibodies. As shown in Fig. 5, LPS treatment dramatically increased p65 and p50 binding to the NF-κBd site. Consistent with the NF-κB reporter assay, SubAB treatment downregulated LPS-induced NF-κB p65/p50 heterodimer binding to the NF-κBd site, whereas mSubAB induced additional binding. Our findings indicate that SubAB treatment suppresses LPS-induced iNOS expression in RAW 264.7 cells via inhibition of NF-κB p65/p50 heterodimer binding to the NF-κBd site in the iNOS promoter; in contrast, mSubAB increases binding.
Fig 5.

SubAB suppresses LPS-induced NF-κB p65/p50 heterodimer binding to the iNOS promoter. RAW 264.7 cells (2 × 106 cells/dish) were treated with or without LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 4 h. Cells were fixed with formaldehyde, lysed in SDS buffer, and then sonicated to fragment the chromatin. Immunoprecipitation was performed using anti-p65 (p65 IP) or anti-p50 (p50 IP) antibodies or normal rabbit IgG. Normal IgG IP refers to immunoprecipitation of chromatin with a nonimmune IgG. After purification of immunoprecipitated DNA, targeted promoter regions of iNOS were amplified by qPCR. Results are expressed as the percent input for each ChIP fraction, and fold changes were calculated to normal IgG IP. Data are representative of three independent experiments.
NF-κB p65/p50 heterodimer is involved in SubAB-mediated inhibition of LPS-induced iNOS expression.
To further determine the role of NF-κB p65/p50 heterodimer in LPS-induced iNOS expression, we prepared bone marrow macrophages (BMMs) from wild-type or NF-κB1−/− mice and examined the effect of SubAB on LPS-induced NO production and iNOS expression. First, we evaluated SubAB-mediated inhibition of LPS-induced NO production in BMMs by Griess assay. Compared with RAW 264.7 cells, SubAB treatment more effectively inhibited LPS-induced NO production by BMMs (Fig. 6A). To further investigate an involvement of p50 in SubAB-mediated inhibition of LPS-induced iNOS expression, we next examined the inhibitory effect of SubAB treatment on iNOS expression in wild-type and NF-κB1−/− BMMs by immunoblotting. We first confirmed p50 deficiency in NF-κB1−/− BMMs and the same expression levels of endogenous p65 in wild-type and NF-κB1−/− BMMs (Fig. 6B). As shown in Fig. 6C, LPS-induced iNOS expression in NF-κB1−/− BMMs was slightly attenuated in comparison with wild-type BMMs after 8 h of incubation but not after 24 h of incubation. In addition, densitometric analysis showed that, in wild-type BMMs, SubAB dramatically inhibited LPS-induced iNOS expression by 77% after 8 h of incubation and 95% after 24 h of incubation. On the other hand, SubAB inhibition of LPS-induced iNOS expression in NF-κB1−/− BMMs at 8 and 24 h was significantly decreased at 59% and 71%, respectively. Wild-type BMMs were significantly more responsive for SubAB-mediated inhibition of LPS-induced iNOS expression than NF-κB1−/− BMMs at 8 h and 24 h (P < 0.01 and P < 0.01, respectively). These results suggest that effects of the NF-κB p50 subunit are partially responsible for SubAB-mediated inhibition of LPS-induced iNOS expression.
Fig 6.

NF-κB p50 is involved in SubAB-mediated inhibition of LPS-induced iNOS expression. (A) RAW 264.7 cells (5 × 104 cells/well) in a 48-well dish were grown in DMEM-1% FBS for 24 h. Mouse BMMs (5 × 104 cells/well) in a 48-well dish were grown in DMEM-1% FBS containing 20 ng/ml M-CSF for 24 h. Cells were treated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 24 h. The accumulation of nitrite in culture supernatants was quantified by Griess assay as described in the legend to Fig. 1. Data are means ± SD of values from triplicate experiments. *, P < 0.01. (B) Wild-type (NF-κB1+/+) or NF-κB1 knockout (NF-κB1−/−) BMMs (5 × 104 cells/well) in a 48-well dish were grown in DMEM-1% FBS containing 20 ng/ml M-CSF for 24 h. Cell lysates were analyzed by immunoblotting with anti-p65, anti-p50, or anti-α-tubulin antibodies. (C) Wild-type (NF-κB1+/+) or NF-κB1 knockout (NF-κB1−/−) BMMs (5 × 104 cells/well) in a 48-well dish were grown in DMEM-1% FBS containing 20 ng/ml M-CSF for 24 h. Cells were treated with LPS (10 μg/ml) in the presence or absence of SubAB (WT; 0.5 μg/ml) or mSubAB (MT; 0.5 μg/ml) for 8 and 24 h. Cell lysates were analyzed by immunoblotting with anti-iNOS or anti-α-tubulin antibodies. iNOS expression was normalized to α-tubulin using densitometry (iNOS/tubulin) and is depicted in bar graphs. Data are means ± SD of values from triplicate experiments. *, P < 0.01.
SubAB contributes to E. coli survival in RAW 264.7 cells.
The ability of NO to kill microbes makes it an important part of primordial host defense. Because we found that SubAB inhibited LPS-induced NO production by macrophages, we hypothesized that SubAB contributes to the viability of E. coli in macrophages. Therefore, to investigate the effect of SubAB on E. coli survival in macrophages, RAW 264.7 cells were infected with a nonpathogenic E. coli strain, BL21(DE3), carrying pET23b in the presence or absence of an NOS inhibitor (L-NAME) and purified SubAB or mSubAB, and then bacteria in RAW 264.7 cells were quantified at 2 h or 16 h. During the initial period of infection for 2 h, similar number of E. coli were taken up by RAW 264.7 cells in the presence or absence of PBS control, L-NAME, SubAB, or mSubAB (1.4 × 106, 1.6 × 106, 1.6 × 106, or 1.6 × 106 CFU/well, respectively). As shown in Fig. 7A, treatment of L-NAME and SubAB, but not mSubAB, increased intracellular survival of E. coli in RAW 264.7 cells at 16 h. E. coli-induced NO production by RAW 264.7 cells was reduced by L-NAME and SubAB but not mSubAB. Next, we examined the effect of expression of SubAB or mSubAB on E. coli BL21(DE3) survival within RAW 264.7 cells. During the initial 2 h of infection with BL21/pET-SubAB or BL21/pET-mSubAB, the same number of the E. coli CFU (0.7 × 106 CFU/well) were taken up by RAW 264.7 cells. As shown in Fig. 7B, expression of SubAB by E. coli, but not that of mSubAB, increased intracellular survival in RAW 264.7 cells (top) and inhibited E. coli-induced iNOS expression and NO production (bottom) at 16 h. These results suggest that NO generated by macrophages results in killing of the bacteria and SubAB contributes to E. coli survival in macrophages.
Fig 7.
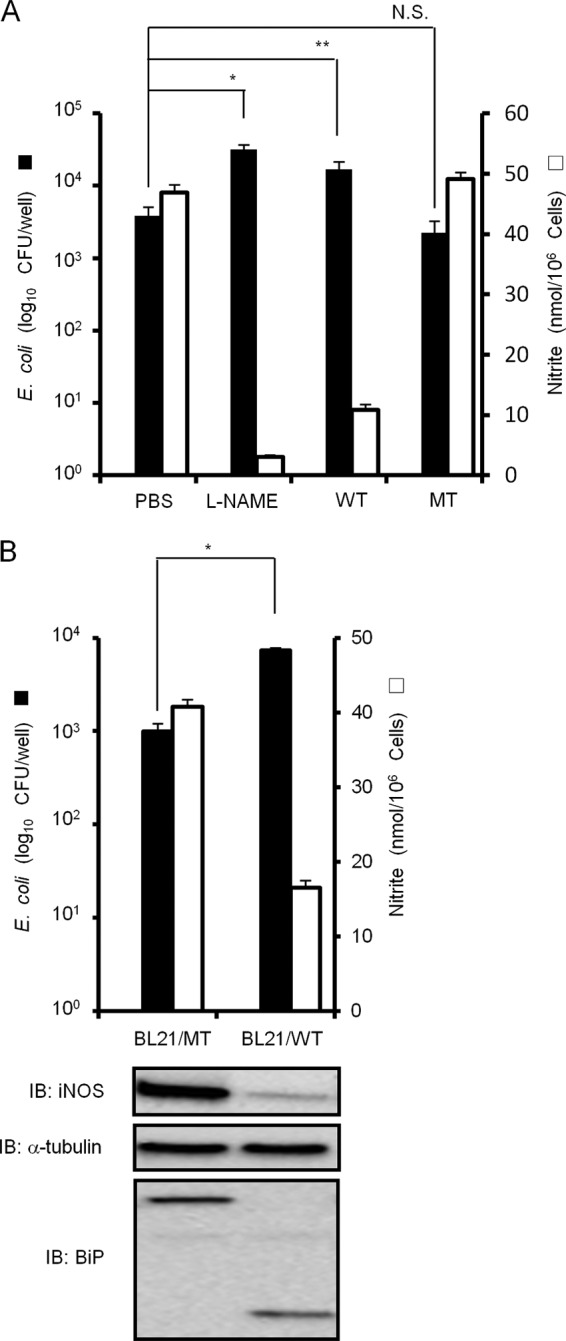
SubAB contributes to E. coli survival in RAW 264.7 cells. (A) RAW 264.7 cells (1 × 106 cells/well) in a 24-well dish were grown in antibiotic-free DMEM-10% FBS for 24 h. Cells were infected with E. coli BL21/pET23b in the presence or absence of L-NAME (10 mM), SubAB (WT; 0.5 μg/ml), or mSubAB (MT; 0.5 μg/ml) as described in Materials and Methods. Intracellular bacteria were quantified 16 h later. Data are means ± SD of values from triplicate experiments. *, P < 0.01; N.S., not significant. (B) RAW 264.7 cells (1 × 106 cells/well) in a 24-well dish were infected with E. coli BL21/pET-SubAB (BL21/WT) or BL21/pET-mSubAB (BL21/MT) as described above. After 16 h, intracellular bacteria were quantified by CFU (top). Cell lysates were analyzed by immunoblotting using anti-iNOS, anti-α-tubulin, or anti-BiP/Grp78 antibodies (bottom). Data are means ± SD of values from triplicate experiments. *, P < 0.01.
DISCUSSION
The reactive free radical, NO, is a major inflammatory mediator which is involved in host defense against bacterial infection and is produced following activation of phagocytes, such as neutrophils and macrophages (2, 28). In this study, LPS-induced NO production by RAW 264.7 cells was strongly inhibited by SubAB but not by mSubAB (Fig. 1). Because iNOS expression is responsible for LPS-induced NO production in macrophages (25, 29, 56, 57), we first investigated the effect of SubAB on LPS-induced iNOS protein and mRNA expression. In agreement with a previous report (25), iNOS mRNA and protein expressions were significantly induced within 4 h after treatment with LPS. SubAB, but not mSubAB, inhibited LPS-induced iNOS expression by an ER stress-mediated mechanism (Fig. 2B to D). These results indicate that SubAB inhibited LPS-induced NO production by RAW 264.7 cells through ER stress-mediated inhibition of iNOS expression. Lowenstein et al. (29) and Xie et al. (57) identified the murine iNOS promoter sequence and showed that E. coli-derived LPS induced iNOS transcriptional activation. The published sequences of iNOS promoter in murine cells exhibit homologies to binding sites for numerous transcription factors (i.e., NF-κB, STAT-1, AP-1, C/EBP, CREB, IRF-1, Oct-1), which were known to be involved in the LPS- and cytokine-mediated induction of transcription (28, 29, 57). Our results from reporter assays using mouse iNOS promoter-luciferase reporter plasmid or iNOS promoter-deletion plasmids showed that the −93 bp region from the transcription initiation site of iNOS promoter, which contains an NF-κB-binding site, is responsible for SubAB-mediated inhibition of iNOS (Fig. 3). These findings suggest that SubAB controls LPS-induced NF-κB nuclear translocation and binding to iNOS promoter.
NF-κB is known to be a primary regulator of iNOS expression by an LPS-dependent pathway (29, 56, 57). Several reports showed that ER stress activates NF-κB (6, 23) but can also inhibit inflammatory responses in association with suppression of the NF-κB pathway (8, 19–21, 49). A recent study showed that SubAB pretreatment of macrophages inhibited LPS-induced NF-κB activation and MCP-1 and TNF-α production through inhibition of IκB-α degradation (17). Consistent with these studies, our results demonstrated that LPS-induced NF-κB nuclear translocation and activation were blocked by SubAB (Fig. 4A to C). Although these data raise the possibility that SubAB inhibits IκB degradation by effects on proteasomal function or IKK-mediated IκB phosphorylation, we now present evidence that SubAB treatment of RAW 264.7 cells suppressed LPS-induced IκB-α phosphorylation, a trigger for its degradation (Fig. 4D).
In addition, we observed that mSubAB treated in RAW 264.7 cells slightly enhanced LPS-induced NO production (Fig. 1A), iNOS mRNA expression (Fig. 2D), and iNOS transcription (Fig. 3). These results suggest that mSubAB binding to the target cells induced activation of an NF-κB signaling pathway. Our previous study demonstrated that SubAB binds to functional receptors, i.e., L1CAM, α2β1 integrin, and Met (60). Interestingly, these membrane proteins are known to modulate NF-κB activity (4, 14, 48). Thus, SubAB binding to these receptors could stimulate NF-κB signaling, followed by increased LPS-induced iNOS expression. However, the stimulatory effect of toxin interaction with its receptors is diminished by SubAB-induced ER stress after BiP cleavage, suggesting that NF-κB activation by toxin binding does not affect iNOS expression in the presence of wild-type SubAB.
NF-κB subunits can form various dimers, but the classical, best-characterized complex is composed of p50 and p65 (1, 50, 53). Mouse iNOS promoter contains two NF-κB-binding sites, NF-κBu site (−971 to −962) and NF-κBd site (−85 to −76), which play an important role in iNOS expression. NF-κB subunits (p65, p50, p52, and c-Rel) bind to the NF-κBd site in response to LPS stimulation. In particular, p50 may have a more pivotal role in NF-κB transactivation activity at the NF-κBd site since p50 binding was demonstrated by electrophoretic mobility shift assay (EMSA) (7, 56). NF-κB p50 displayed higher-affinity DNA binding than p65 (12), suggesting that p50 is an essential DNA-binding subunit of the p65/p50 heterodimer. In NF-κB1−/− BMMs, SubAB-mediated iNOS inhibition was partially abolished (Fig. 6C), indicating that the p65/p50 heterodimer is, at least, involved in SubAB-mediated iNOS inhibition. However, it is still possible that SubAB may have an inhibitory effect on other NF-κB dimers composed of p65, c-Rel, and p52 or other transcription factors which induce iNOS expression by LPS.
A number of pathogenic bacteria have developed a variety of mechanisms to evade host defense mechanisms and enhance their virulence (44). Previous studies showed that Stx or other bacterial factors of STEC, such as EspA, B, and D, downregulate NF-κB activation (15, 18). In addition, a recent study showed that an undefined effector of O157 inhibited iNOS expression (52). Therefore, to exclude the effect of the iNOS inhibitors, we used nonpathogenic E. coli strain BL21(DE3) for macrophage infection. Inhibition of NO production by treatment with NOS inhibitor L-NAME resulted in increased E. coli survival in RAW 264.7 cells (Fig. 7A), indicating that NO acts as a intracellular antibacterial molecule. Further, treatment with purified SubAB or expression of SubAB by E. coli also increased the intracellular survival of E. coli in RAW 264.7 cells (Fig. 7A and B). NO is a key component of the host immune response and is encountered by pathogenic bacteria during their lives outside and within the host (11). The effects of NO and ROS can be synergistic and efficiently kill microorganisms by causing double-stranded DNA breakage, bacterial Fe2+ release, and depletion of the antioxidant glutathione (31, 40, 55). The reaction of O2− and NO leads to the generation of peroxynitrite (ONOO−), which can decompose to species with an extremely potent oxidizing potential, capable of killing E. coli (40). Thus, our findings suggested that NO represents an important antimicrobial agent of special significance to E. coli, and reduction of NO production by SubAB results in increased STEC viability and infectivity. Moreover, a recent study reported that NO inhibits Stx synthesis in STEC (47, 51). Thus, increasing NO production in infected patients might represent an alternative strategy to limit the development of HUS. This finding suggests that reduction of NO production during the host immune response may result in Stx synthesis by STEC and maintenance of STEC virulence. Taken together, SubAB by inhibiting NO production may enhance STEC pathogenicity.
LPS is a major component of the outer membrane of Gram-negative bacteria, such as STEC. LPS interacts with TLR4 on macrophages during STEC infection, leading to increased phagocytosis (5), production of cytokines, and/or ROS/RNS generation (25). Interestingly, posttreatment with SubAB after LPS stimulation for 3 h also significantly inhibited NO production (Fig. 1C). These findings further support our proposal that SubAB-producing STEC can inhibit NO production effectively during infection.
In conclusion, we provide molecular mechanisms for the inhibitory effect of SubAB on LPS-induced NO production in mouse macrophages as shown in Fig. 8. This inhibition occurs through a process involving SubAB-mediated reduction of p65/p50 heterodimer binding to the NF-κB binding site on iNOS promoter. As NO is an essential factor for antimicrobial host defense in E. coli infections, we propose that SubAB might be involved in the upregulation of STEC virulence. Future studies involving macrophage infection with isogenic SubAB-deficient STEC or an animal infection model will help to determine the apparent role of SubAB in STEC survival.
Fig 8.
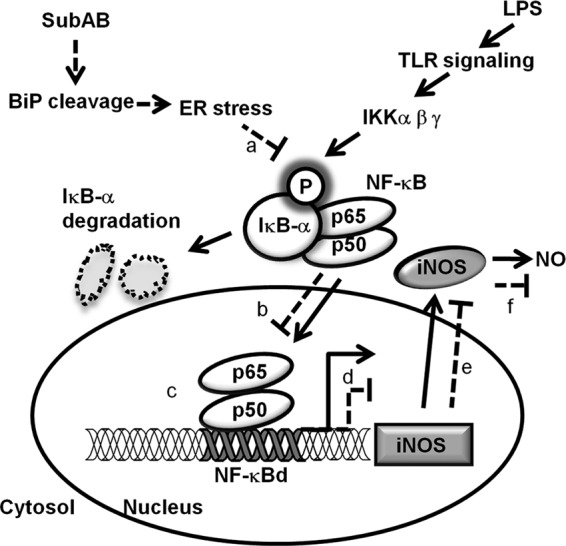
Proposed model of the inhibitory effect of SubAB on NO production in macrophages. SubAB-mediated inhibition of LPS-induced iNOS can be summarized as follows. The solid lines represent mechanisms of LPS-induced NO production. LPS induces IκB-α phosphorylation through a TLR-signaling pathway, followed by IκB-α degradation and NF-κB p65/p50 heterodimer nuclear translocation. Binding of NF-κB p65/p50 heterodimer to the NF-κBd site in the iNOS promoter is responsible for iNOS expression and NO production. The dashed lines show inhibitory effects of SubAB on LPS-induced NO production. First, SubAB cleaves BiP/Grp78, leading to ER stress, which suppresses IκB-α phosphorylation (a). Suppression of IκB-α phosphorylation prevents IκB-α degradation and NF-κB p65/p50 heterodimer translocation to nuclei (b), followed by reduction of NF-κB p65/p50 heterodimer binding to the NF-κBd site in iNOS promoter (c), resulting in inhibition of iNOS mRNA expression (d). Inhibition of LPS-induced iNOS mRNA expression by SubAB suppresses iNOS protein expression (e). Finally, SubAB-mediated inhibition of LPS-induced iNOS expression leads to inhibition of NO production by macrophages (f), which permits survival of intracellular E. coli.
ACKNOWLEDGMENTS
This work was supported by grants in aid for scientific research from the Ministry of Education, Science, and Culture of Japan and Improvement of Research Environment for Young Researchers from Japan Science and Technology Agency. Joel Moss was supported by the Intramural Research Program, National Institutes of Health, National Heart, Lung, and Blood Institute.
We thank T. Hirayama (Institute of Tropical Medicine, Nagasaki University) and N. Morinaga (Chiba University) for helpful discussions and suggestions. We acknowledge the expert technical assistance of D. Nakagomi, C. Noritake, and A. Kiuchi (Chiba University).
Footnotes
Published ahead of print 4 September 2012
REFERENCES
- 1. Baeuerle PA, Baltimore D. 1996. NF-kappa B: ten years after. Cell 87:13–20 [DOI] [PubMed] [Google Scholar]
- 2. Bogdan C. 2001. Nitric oxide and the immune response. Nat. Immunol. 2:907–916 [DOI] [PubMed] [Google Scholar]
- 3. Brunelli L, Crow JP, Beckman JS. 1995. The comparative toxicity of nitric oxide and peroxynitrite to Escherichia coli. Arch. Biochem. Biophys. 316:327–334 [DOI] [PubMed] [Google Scholar]
- 4. Chung CH, Lin KT, Chang CH, Peng HC, Huang TF. 2009. The integrin alpha2beta1 agonist, aggretin, promotes proliferation and migration of VSMC through NF-kB translocation and PDGF production. Br. J. Pharmacol. 156:846–856 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cooper PH, Mayer P, Baggiolini M. 1984. Stimulation of phagocytosis in bone marrow-derived mouse macrophages by bacterial lipopolysaccharide: correlation with biochemical and functional parameters. J. Immunol. 133:913–922 [PubMed] [Google Scholar]
- 6. Deng J, et al. 2004. Translational repression mediates activation of nuclear factor kappa B by phosphorylated translation initiation factor 2. Mol. Cell. Biol. 24:10161–10168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Deng WG, Tang ST, Tseng HP, Wu KK. 2006. Melatonin suppresses macrophage cyclooxygenase-2 and inducible nitric oxide synthase expression by inhibiting p52 acetylation and binding. Blood 108:518–524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Du S, et al. 2009. Suppression of NF-kappaB by cyclosporin A and tacrolimus (FK506) via induction of the C/EBP family: implication for unfolded protein response. J. Immunol. 182:7201–7211 [DOI] [PubMed] [Google Scholar]
- 9. Ducut Sigala JL, et al. 2004. Activation of transcription factor NF-kappaB requires ELKS, an IkappaB kinase regulatory subunit. Science 304:1963–1967 [DOI] [PubMed] [Google Scholar]
- 10. Fagerlund R, Kinnunen L, Kohler M, Julkunen I, Melen K. 2005. NF-κB is transported into the nucleus by importin α3 and importin α4. J. Biol. Chem. 280:15942–15951 [DOI] [PubMed] [Google Scholar]
- 11. Fang FC. 2004. Antimicrobial reactive oxygen and nitrogen species: concepts and controversies. Nat. Rev. Microbiol. 2:820–832 [DOI] [PubMed] [Google Scholar]
- 12. Fujita T, Nolan GP, Ghosh S, Baltimore D. 1992. Independent modes of transcriptional activation by the p50 and p65 subunits of NF-kappa B. Genes Dev. 6:775–787 [DOI] [PubMed] [Google Scholar]
- 13. Furukawa T, et al. 2011. Fatal hemorrhage induced by subtilase cytotoxin from Shiga-toxigenic Escherichia coli. Microb. Pathog. 50:159–167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Gavert N, Ben-Shmuel A, Lemmon V, Brabletz T, Ben-Ze'ev A. 2010. Nuclear factor-kappaB signaling and ezrin are essential for L1-mediated metastasis of colon cancer cells. J. Cell Sci. 123:2135–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gobert AP, et al. 2007. Shiga toxin produced by enterohemorrhagic Escherichia coli inhibits PI3K/NF-kappaB signaling pathway in globotriaosylceramide-3-negative human intestinal epithelial cells. J. Immunol. 178:8168–8174 [DOI] [PubMed] [Google Scholar]
- 16. Guilbert LJ, Stanley ER. 1986. The interaction of 125I-colony-stimulating factor-1 with bone marrow-derived macrophages. J. Biol. Chem. 261:4024–4032 [PubMed] [Google Scholar]
- 17. Harama D, et al. 2009. A subcytotoxic dose of subtilase cytotoxin prevents lipopolysaccharide-induced inflammatory responses, depending on its capacity to induce the unfolded protein response. J. Immunol. 183:1368–1374 [DOI] [PubMed] [Google Scholar]
- 18. Hauf N, Chakraborty T. 2003. Suppression of NF-kappa B activation and proinflammatory cytokine expression by Shiga toxin-producing Escherichia coli. J. Immunol. 170:2074–2082 [DOI] [PubMed] [Google Scholar]
- 19. Hayakawa K, et al. 2009. Acquisition of anergy to proinflammatory cytokines in nonimmune cells through endoplasmic reticulum stress response: a mechanism for subsidence of inflammation. J. Immunol. 182:1182–1191 [DOI] [PubMed] [Google Scholar]
- 20. Hayakawa K, et al. 2008. Blunted activation of NF-kappaB and NF-kappaB-dependent gene expression by geranylgeranylacetone: involvement of unfolded protein response. Biochem. Biophys. Res. Commun. 365:47–53 [DOI] [PubMed] [Google Scholar]
- 21. Hayakawa K, et al. 2010. ER stress depresses NF-kappaB activation in mesangial cells through preferential induction of C/EBP beta. J. Am. Soc. Nephrol. 21:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Hiramatsu Y, et al. 2010. c-Maf activates the promoter and enhancer of the IL-21 gene, and TGF-beta inhibits c-Maf-induced IL-21 production in CD4+ T cells. J. Leukoc. Biol. 87:703–712 [DOI] [PubMed] [Google Scholar]
- 23. Hu P, Han Z, Couvillon AD, Kaufman RJ, Exton JH. 2006. Autocrine tumor necrosis factor alpha links endoplasmic reticulum stress to the membrane death receptor pathway through IRE1alpha-mediated NF-kappaB activation and down-regulation of TRAF2 expression. Mol. Cell. Biol. 26:3071–3084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Huang T, et al. 2009. Downregulation of gap junction expression and function by endoplasmic reticulum stress. J. Cell Biochem. 107:973–983 [DOI] [PubMed] [Google Scholar]
- 25. Jacobs AT, Ignarro LJ. 2001. Lipopolysaccharide-induced expression of interferon-beta mediates the timing of inducible nitric-oxide synthase induction in RAW 264.7 macrophages. J. Biol. Chem. 276:47950–47957 [DOI] [PubMed] [Google Scholar]
- 26. Karin M, Ben-Neriah Y. 2000. Phosphorylation meets ubiquitination: the control of NF-[kappa]B activity. Annu. Rev. Immunol. 18:621–663 [DOI] [PubMed] [Google Scholar]
- 27. Karmali MA. 1989. Infection by verocytotoxin-producing Escherichia coli. Clin. Microbiol. Rev. 2:15–38 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kleinert H, Pautz A, Linker K, Schwarz PM. 2004. Regulation of the expression of inducible nitric oxide synthase. Eur. J. Pharmacol. 500:255–266 [DOI] [PubMed] [Google Scholar]
- 29. Lowenstein CJ, et al. 1993. Macrophage nitric oxide synthase gene: two upstream regions mediate induction by interferon gamma and lipopolysaccharide. Proc. Natl. Acad. Sci. U. S. A. 90:9730–9734 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lowenstein CJ, Snyder SH. 1992. Nitric oxide, a novel biologic messenger. Cell 70:705–707 [DOI] [PubMed] [Google Scholar]
- 31. Marcinkiewicz J. 1997. Nitric oxide and antimicrobial activity of reactive oxygen intermediates. Immunopharmacology 37:35–41 [DOI] [PubMed] [Google Scholar]
- 32. Matsuura G, et al. 2009. Novel subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli induces apoptosis in Vero cells via mitochondrial membrane damage. Infect. Immun. 77:2919–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Morinaga N, Yahiro K, Matsuura G, Moss J, Noda M. 2008. Subtilase cytotoxin, produced by Shiga-toxigenic Escherichia coli, transiently inhibits protein synthesis of Vero cells via degradation of BiP and induces cell cycle arrest at G1 by downregulation of cyclin D1. Cell Microbiol. 10:921–929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Morinaga N, et al. 2007. Two distinct cytotoxic activities of subtilase cytotoxin produced by Shiga-toxigenic Escherichia coli. Infect. Immun. 75:488–496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Morita H, et al. 2004. Antimicrobial action against verotoxigenic Escherichia coli O157:H7 of nitric oxide derived from sodium nitrite. Biosci. Biotechnol. Biochem. 68:1027–1034 [DOI] [PubMed] [Google Scholar]
- 36. Morrison DC, Jacobs DM. 1976. Binding of polymyxin B to the lipid A portion of bacterial lipopolysaccharides. Immunochemistry 13:813–818 [DOI] [PubMed] [Google Scholar]
- 37. Nakajima S, et al. 2011. Selective abrogation of BiP/GRP78 blunts activation of NF-kappaB through the ATF6 branch of the UPR: involvement of C/EBPbeta and mTOR-dependent dephosphorylation of Akt. Mol. Cell. Biol. 31:1710–1718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Nakamura Y, et al. 2006. Potentiation by high potassium of lipopolysaccharide-induced nitric oxide production from cultured astrocytes. Neurochem. Int. 48:43–49 [DOI] [PubMed] [Google Scholar]
- 39. Okamoto T, et al. 2010. A new paradigm for antimicrobial host defense mediated by a nitrated cyclic nucleotide. J. Clin. Biochem. Nutr. 46:14–19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pacelli R, et al. 1995. Nitric oxide potentiates hydrogen peroxide-induced killing of Escherichia coli. J. Exp. Med. 182:1469–1479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Paton AW, et al. 2006. AB5 subtilase cytotoxin inactivates the endoplasmic reticulum chaperone BiP. Nature 443:548–552 [DOI] [PubMed] [Google Scholar]
- 42. Paton AW, Srimanote P, Talbot UM, Wang H, Paton JC. 2004. A new family of potent AB(5) cytotoxins produced by Shiga toxigenic Escherichia coli. J. Exp. Med. 200:35–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Paton JC, Paton AW. 1998. Pathogenesis and diagnosis of Shiga toxin-producing Escherichia coli infections. Clin. Microbiol. Rev. 11:450–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Poirier K, et al. 2008. Escherichia coli O157:H7 survives within human macrophages: global gene expression profile and involvement of the Shiga toxins. Infect. Immun. 76:4814–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Schreiber E, Matthias P, Muller MM, Schaffner W. 1989. Rapid detection of octamer binding proteins with ‘mini-extracts,’ prepared from a small number of cells. Nucleic Acids Res. 17:6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sha WC, Liou HC, Tuomanen EI, Baltimore D. 1995. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell 80:321–330 [DOI] [PubMed] [Google Scholar]
- 47. Shimizu T, Tsutsuki H, Matsumoto A, Nakaya H, Noda M. 2012. The nitric oxide reductase of enterohaemorrhagic Escherichia coli plays an important role for the survival within macrophages. Mol. Microbiol. 85:492–512 [DOI] [PubMed] [Google Scholar]
- 48. Tacchini L, De Ponti C, Matteucci E, Follis R, Desiderio MA. 2004. Hepatocyte growth factor-activated NF-kappaB regulates HIF-1 activity and ODC expression, implicated in survival, differently in different carcinoma cell lines. Carcinogenesis 25:2089–2100 [DOI] [PubMed] [Google Scholar]
- 49. Takano Y, et al. 2007. Suppression of cytokine response by GATA inhibitor K-7174 via unfolded protein response. Biochem. Biophys. Res. Commun. 360:470–475 [DOI] [PubMed] [Google Scholar]
- 50. Thanos D, Maniatis T. 1995. Identification of the rel family members required for virus induction of the human beta interferon gene. Mol. Cell. Biol. 15:152–164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Vareille M, de Sablet T, Hindre T, Martin C, Gobert AP. 2007. Nitric oxide inhibits Shiga-toxin synthesis by enterohemorrhagic Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 104:10199–10204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Vareille M, et al. 2008. Heme oxygenase-1 is a critical regulator of nitric oxide production in enterohemorrhagic Escherichia coli-infected human enterocytes. J. Immunol. 180:5720–5726 [DOI] [PubMed] [Google Scholar]
- 53. Verma IM, Stevenson JK, Schwarz EM, Van Antwerp D, Miyamoto S. 1995. Rel/NF-kappa B/I kappa B family: intimate tales of association and dissociation. Genes Dev. 9:2723–2735 [DOI] [PubMed] [Google Scholar]
- 54. Wolfson JJ, et al. 2008. Subtilase cytotoxin activates PERK, IRE1 and ATF6 endoplasmic reticulum stress-signalling pathways. Cell Microbiol. 10:1775–1786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Woodmansee AN, Imlay JA. 2003. A mechanism by which nitric oxide accelerates the rate of oxidative DNA damage in Escherichia coli. Mol. Microbiol. 49:11–22 [DOI] [PubMed] [Google Scholar]
- 56. Xie QW, Kashiwabara Y, Nathan C. 1994. Role of transcription factor NF-kappa B/Rel in induction of nitric oxide synthase. J. Biol. Chem. 269:4705–4708 [PubMed] [Google Scholar]
- 57. Xie QW, Whisnant R, Nathan C. 1993. Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med. 177:1779–1784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Yahiro K, Morinaga N, Moss J, Noda M. 2010. Subtilase cytotoxin induces apoptosis in HeLa cells by mitochondrial permeabilization via activation of Bax/Bak, independent of C/EBF-homologue protein (CHOP), Ire1alpha or JNK signaling. Microb. Pathog. 49:153–163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Yahiro K, et al. 2006. Identification and characterization of receptors for vacuolating activity of subtilase cytotoxin. Mol. Microbiol. 62:480–490 [DOI] [PubMed] [Google Scholar]
- 60. Yahiro K, et al. 2011. Identification of subtilase cytotoxin (SubAB) receptors whose signaling, in association with SubAB-induced BiP cleavage, is responsible for apoptosis in HeLa cells. Infect. Immun. 79:617–627 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Yamazaki H, et al. 2009. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J. Immunol. 183:1480–1487 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yuhas Y, Kaminsky E, Mor M, Ashkenazi S. 1996. Induction of nitric oxide production in mouse macrophages by Shiga toxin. J. Med. Microbiol. 45:97–102 [DOI] [PubMed] [Google Scholar]